Tatjana Harting1, Mandy Stubbendorff 2, Saskia Willenbrock1, Siegfried Wagner1, Patrik Schadzek3, Anaclet Ngezahayo3, Hugo Murua Escobar1, Ingo Nolte1
1 Small Animal Clinic, University of Veterinary Medicine Hannover, Foundation, D-30559 Hannover
2 Division of Medicine Clinic III, Hematology, Oncology and Palliative Medicine, University of Rostock, D-18057 Rostock
3 Evotec AG, D-22419 Hamburg
4 Institute of Biophysics, Leibniz University, D-30419 Hannover, Germany
Correspondence: Professor Ingo Nolte, Small Animal Clinic, University of Veterinary Medicine Hannover, Foundation, Bünteweg 9, D-30559 Hannover, Germany; E-mail: [email protected]
Received: 12 July 2016
Accepted: 5 September 2016
Published Online: 5 October 2016
Abstract
The Warburg effect describes the ability of cancer cells to produce energy via aerobic glycolysis instead of oxidative phosphorylation of pyruvate. This deviation in mitochondrial metabolism inhibits apoptosis, allowing increased proliferation under conditions of reduced oxygen levels. Dichloroacetate (DCA) was successfully used in several human cancer cell lines to reactivate oxidative phosphorylation in mitochondria. The aim of this study was the characterization and response of canine cancer cell lines after DCA exposure. The effect of 10 mM DCA was characterized in vitro on a set of six canine prostate adenocarcinoma and transitional cell carcinoma (TCC) derived cell lines. Cell counts, lactate levels, apoptosis, expression of apoptotic proteins, survival factors and different miRNAs were analyzed. Additionally, metabolic activity, mitochondrial activity and proliferation were investigated. DCA significantly decreased cell number of all but one utilized cell lines and leads to a significant reduction of lactate release. Decreased survivin levels were found in all cell lines, two of which presented a significant reduction in metabolic activity. Increased miR-375 levels were measured in all TCC cell lines. Reactivation of pyruvate dehydrogenase and an elevated mitochondrial activity appear to induce the transition from aerobic glycolysis back to oxidative phosphorylation. Further, these results display that DCA treatment has a suppressant effect on proliferation of canine cancer cells.
Keywords: dichloroacetate, canine prostate adenocarcinoma, canine transitional cell carcinoma, Warburg effect
DOI: 10.3892/ijo.2016.3720
INTRODUCTION
In the last few years cancer treatments such as chemotherapy, radiation therapy and surgery, as they are used in human cancer treatment, became more important in veterinary medicine. Conventional chemotherapeutic agents target dividing cells, cancerous as well as non-neoplastic cells, causing several side effects as myelosuppression, diarrhea, vomitus and anorexia [1]. Further, due to advanced disease stage and resistance of prostate and bladder cancer, treatment is difficult and often associated with poor prognosis [2,3]. Therefore, new alternatives which are more effective have to be investigated.
Dichloroacetate (DCA), a small and cost-efficient molecule, affects different metabolic pathways by inhibiting pyruvate dehydrogenase kinase (PDK) [4,5]. This implicates that pyruvate dehydrogenase (PDH) is potentially indirectly activated by DCA which yields a metabolic shift to favor oxidation of pyruvate to acetyl-co-enzyme-A in mitochondria [5]. Despite this fact in the past decades DCA has been used in the treatment of a multitude of disorders like congenital lactate acidosis [6,7], hypercholesterolemia [8], hyperglycemia [9], congestive heart failure [10] and only recently in cancer research [11–16]. DCA was tested in different in vitro approaches in the field of human oncology including colorectal cancer [17,18], endometrial cancer [14], oral squamous cell carcinoma [19] and breast cancer [20]. In a clinical trial analyzing patients affected by glioblastoma and other solid tumors, DCA decreased tumor growth and angiogenesis [15]. With the exception of several studies investigating pharmacokinetic effects of DCA in dogs [21–23], there are currently no publications on the effect of DCA in canine cancer. However, DCA was successfully used in dogs with lactic acidosis [24] and is reported to be well tolerated in dogs [22]. Severe side effects such as death and paralysis were observed only in a high-dose long-term study [21].
Under aerobic conditions non-neoplastic cells refer to glucose oxidation via mitochondria which oxidize pyruvate to acetyl-co-enzyme-A [25]. PDH enables pyruvate to enter mitochondria. Energy production of cancer cells is primarily shifted from glucose oxidation to aerobic glycolysis which leads to increased cytosolic lactate production despite the fact that enough oxygen is available [26]. This behavior is referred to as Warburg effect. The biochemist Otto Warburg first reported these characteristics in 1926 and hypothesized that mitochondrial failure could be the reason [26]. Carcinogenesis preferably sets on in hypoxic tissues where glucose consumption is low. Accordingly, hypoxia inducible factor 1α is activated and leads to an upregulation of glucose transporters and PDK. Activation of PDK results in inhibition of PDH and thus in glycolysis [27,28]. Due to this metabolic change and the decrease of mitochondrial depolarization, cancer cells have a survival advantage and are not affected by intrinsic apoptosis pathways [29,30].
For preclinical assessment of anticancer drugs in vitro experiments with cell lines are important approaches in human as well as in veterinary research. In vitro investigations offer the possibility to receive more information on efficaciousness and sensitivity of several tumor entities [31–33]. In this study several established [34] and new cell lines were used.
To our knowledge this is the first study in which the effect of DCA on canine prostate adenocarcinoma and transitional cell carcinoma (TCC) cells have been investigated. The influence of DCA on cell counts, lactate levels, mitochondrial activity, apoptosis and metabolic activity was determined. In addition the effect of DCA on PDH and on apoptosis involved proteins was evaluated. The effect of DCA on several microRNAs (miR) has not been determined before. Further there is no literature on the influence of DCA on bladder cancer in human or in veterinary medicine.
Materials and methods
Cell lines and cell culture
Three canine prostate adenocarcinoma (DT08/46, CT1258, DT15/08) and three canine TCC (DT08/40, DT15/06, DT15/09) were used for experiments. Two TCC cell lines (DT08/40 and DT15/09) derived from prostate tissue and one TCC (DT15/06) from female bladder tissue. The cell lines were classified as prostate adenocarcinoma or TCC after pathohistological examination of the initial tissues. All cell lines were established in the Small Animal Clinic, University of Veterinary Medicine Hannover, Germany. The cell lines were cultured in 75 cm2 flask (TPP, Faust Lab Science, Klettgau, Germany) with 10 ml medium 199 (Gibco™, Thermo Fisher Scientific, Darmstadt, Germany), 10% fetal calf serum (Hyclone®, Thermo Fisher Scientific), 2% penicillin-streptomycin (Biochrom, Berlin, Germany), a medium change was performed every 48 h. The cells were allowed to grow to a density of 90% before splitting 1:3. The cells were incubated at 37°C and 5% CO2 in humidified air. For experiments the cells were treated with 10 mM DCA (Sigma-Aldrich GmbH, Taufkirchen, Germany) over 48 h. For cell splitting or after treatment period the cells were trypsinized with TrypLE™ Express (Gibco™, Thermo Fisher Scientific) and cell number was counted with an automated Cellometer™ Auto T4 (Nexcelom Bioscience, Lawrence, MA, USA) and compared with negative control. Cells were washed with PBS (Biochrom) and stored at −80°C for further examinations (quantitative RT-PCR and protein analysis). DCA was dissolved in deionized water, filter-sterilized and pH was adjusted to 7.4 with NaOH. The dosis of 10 mM was selected according to previous studies with human HeLa cells [14]. Even if this concentration might not be safely reached in vivo, this concentration was chosen to allow the comparability to other human in vitro studies.
Lactate levels
For lactate level measurements supernatant from cell culture was centrifuged at 1,000 rpm for 10 min to remove floating cells and debris and 1.3 ml were transferred to a sodium fluoride vessel (Sarstedt, Nümbrecht, Germany). To eliminate alterations due to phenol red and lactate natively from fetal calf serum, medium was used as negative control and deducted from measurements. Colorimetric determination of lactate was performed with Cobas® C311 (Hitachi, Tokyo, Japan). To relate the assessed lactate levels to cell number and volume, the total lactate content was normalized to intracellular protein concentration which was determined with Pierce™ BCA assay (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Metabolic activity
Cells were seeded in a 96-well plate (Falcon, Corning, Amsterdam, The Netherlands) with 200 μl medium 199, 10% fetal calf serum, 2% penicillin-streptomycin and incubated at 37°C and 5% humidified CO2. Medium was changed every 24 h and for measurement 20 μl MTT (CellTiter96® Aqueous One Solution assay, Promega, Mannheim, Germany) was added into each well. Absorbance was determined after 2 h with a Synergy2 plate reader (BioTek, Bad Friedrichshall, Germany). Measurements were performed every 24 h over a period of four days. Data was analyzed with Gen5™ 1.11 Software (BioTek) and normalized to negative control of medium.
Flow cytometry
For determination of apoptosis 105 cells were cultured in a 6-well-plate (TPP, Faust Lab Science) with 4 ml medium and treated with 10 mM DCA as described above for 48 h. After this period, cells were trypsinized and centrifuged together with medium containing non-adherent and dead cells at 1,000 rpm for 6 min. Supernatant was discarded and cells were resuspended in 500 μl assay buffer. Staining was performed with 5 μl Annexin-FITC and 1 μl Sytox (Annexin V-FITC Detection Kit Plus, PromoCell, Heidelberg, Germany). After 5-min incubation at room temperature, 104 cells were analyzed with BD FACScalibur™ (BD Biosciences, Heidelberg, Germany) and CellQuest™ Pro 6.0 software (BD Biosciences). Annexin and Sytox were detected in FL-1. Data analysis was performed with FlowJo Version 10.0.8r1 (FlowJo, Ashland, OR, USA). Gates were set by mean of positive controls (cells permeabilized with Saponin) and negative controls of each cell line (non-treated viable cells).
RNA isolation and quantitative RT-PCR
Total RNA was isolated from 106 cells using the NucleoSpin Small RNA kit (Macherey Nagel, Düren, Germany) as described in the manufacturer’s protocol. cDNA was prepared with 35 ng total RNA by reverse transcription using TaqMan® MicroRNA Reverse Transkription kit (Applied Biosystems™, Thermo Fisher Scientific) according to the manufacturer’s instructions. Relative quantification of microRNA expression of treated cells in comparison to negative control was performed with Eppendorf realplex4 Cycler (Eppendorf, Wesseling-Berzdorf, Germany) using 1.33 μl cDNA in a total volume of 20 μl containing TaqMan® Universal Master Mix NoAmpErase® UNG (Applied Biosystems, Thermo Fisher Scientific) and TaqMan® MicroRNA assays for Mir-141 (ID 245445_mat), Mir-145 (ID 002278), Mir-375 (ID 000564) purchased from Thermo Fisher Scientific. Procedure was maintained as described in the manufacturer’s protocol. Data was normalized to the housekeeping gene RNU6B (ID 001093) and analysis was performed with Rest2009 (Qiagen, Hilden, Germany).
Luminex magnetic bead analysis
Protein expression analysis was performed with xMAP® Luminex Bead Technology using a Luminex 200™ instrument (Luminex Corp., Hertogenbosch, The Netherlands). Data are shown as total amount (survivin pg/ml) or net MFI (other targets) and were shown with xPONENT 3.1 software (Luminex Corp.). Sample results with lower values as MFI < background MFI + 2× standard deviation were excluded from analysis. Quantitation of survivin was performed with ProcartaPlex Human Survivin Simplex kit (eBioscience, Frankfurt am Main, Germany) using cell culture supernatant as described in the manufacturer’s protocol. Survivin quantity was normalized to protein concentration which was established using Pierce BCA assay (Thermo Fisher Scientific). PDH and apoptotic proteins (BAD and JNK) were detected with multiplex assays from Merck Millipore (Multi-species PDH Complex Magnetic Bead Panel and 7-Plex Early Apoptosis Magnetic Bead kit, Darmstadt, Germany). Samples were processed as described in the manufacturer’s instructions. Additionally, samples for PDH measurements were filtered with centrifugal ultrafree filter units with a pore size of 0.65 μm (Merck Millipore) at 7,000 rpm for 4 min.
Mitochondrial activity
Cells grown on 8-well μ-dishes (Ibidi, Martinsried, Germany) treated with and without 10 mM DCA were fixed with 4% paraformaldehyde, washed three times with HBSS containing calcium and magnesium and stained with 4 μM MitoSox (Invitrogen, Thermo Fisher Scientific) for 15 min. After staining, cells were washed three times with HBSS (Gibco, Thermo Fisher Scientific) and cell nucleus was counterstained with DAPI (dilution of 1:1,000, Sigma-Aldrich GmbH) for 5 min. Fluorescence imaging was performed with an inverted confocal laser scanning microscope (Eclipse TE2000-E, Nikon, Düsseldorf, Germany) using a 60× water immersion objective (Nikon). Images were taken with EZ-C1 1.80 software (Nikon). The excitation occurred with a diode laser at 408 nm (DAPI) and with a helium/neon laser at 543 nm (MitoSox). Total cell fluorescence of MitoSox deducting background was analyzed with ImageJ and normalized to cell counts.
Immunofluorescence staining of Ki67 and TUNEL
Cells were seeded, fixed and washed as described above. After washing with HBSS, cells were permeabilized with 0.2% Triton X-100 for 20 min, washed and incubated overnight with a canine specific rabbit-polyclonal Ki67 antibody with a dilution of 1:150 (Life Technologies, Thermo Fisher Scientific). For staining, a monoclonal anti-rabbit Alexa Fluor® 555 antibody (Cell Signaling Technology, Leiden, The Netherlands) was incubated for 1 h with a dilution of 1:250 and cells were counterstained with DAPI (1:1,000) for 5 min. Fluorescence imaging protocol was the same as described above. Total cell fluorescence was established as described above. For TUNEL staining Apoptag Fluorescein Direct kit (Merck Millipore) was used according to the manufacturer’s instructions. The excitation occurred with an argonlaser at 488 nm and imaging was performed as described above. The percentage of TUNEL positive cells was evaluated.
Statistical analysis
Statistical analysis of data was performed with SAS software 7.1 (SAS Institute Inc., Cary, NC, USA). For comparison of two means, two-tailed t-test was used. The confidence value was set to 5% (P<0.05) and was considered statistically significant.
Results
Cell counts after DCA treatment
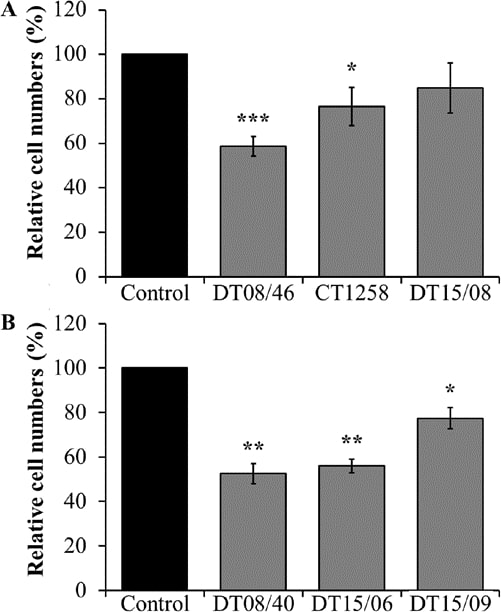
In comparison to non-treated negative controls, the prostate adenocarcinoma cell lines DT08/46 (P<0.0001) and CT1258 (P=0.0122) showed significantly lower cell numbers after treatment with 10 mM DCA over 48 h (Fig. 1A). The third prostate cell line DT15/08 DCA did not show a significant reduction (P=0.0748) but a tendency to a lower cell amount, respectively a lower proliferation rate in native cell culture was observed (Fig. 1A). As shown in Fig. 1B the same decreasing effect of DCA was noted in all examined TCC cell lines DT08/40 (P=0.0031), DT15/06 (P=0.0016) and in DT15/09 (P=0.0143).
Lactate levels in supernatant of cell culture after DCA exposure
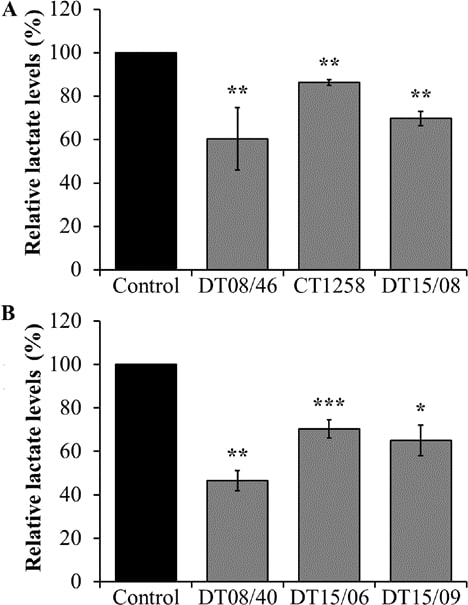
To assess the lactate release lowering effect of DCA treatment after 48 h, lactate amount in cell culture supernatant was measured, respectively. In all cell lines of both canine cancer entities, 10 mM DCA had a significant lactate lowering effect (Fig. 2).
Metabolic activity after treatment with DCA over a period of 96 h
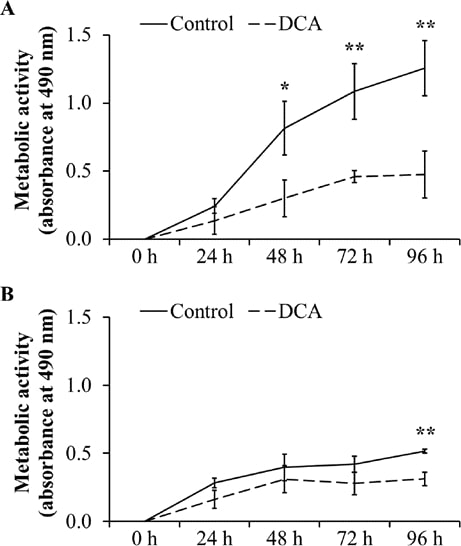
To confirm the negative effect on cell proliferation, the metabolic activity as indicator for proliferation and cell viability was analyzed by MTT assays. As shown in Fig. 3A, the metabolic activity in DT15/08 was significantly reduced in comparison to the respective negative control after 48 h of DCA treatment (P=0.0202). Following 96 h of continuous DCA treatment the metabolic activity was significant (P=0.007). Similar effect was observed in DT08/40 (Fig. 3B) after 96 h (P=0.0025) and in DT15/06 (P=0.0419) after 96 h (data not shown) of DCA treatment. The cell lines DT08/46, CT1258 and DT15/09 showed no statistically significant effect of DCA on metabolic activity (data not shown).
Proliferation after DCA treatment over 48 h
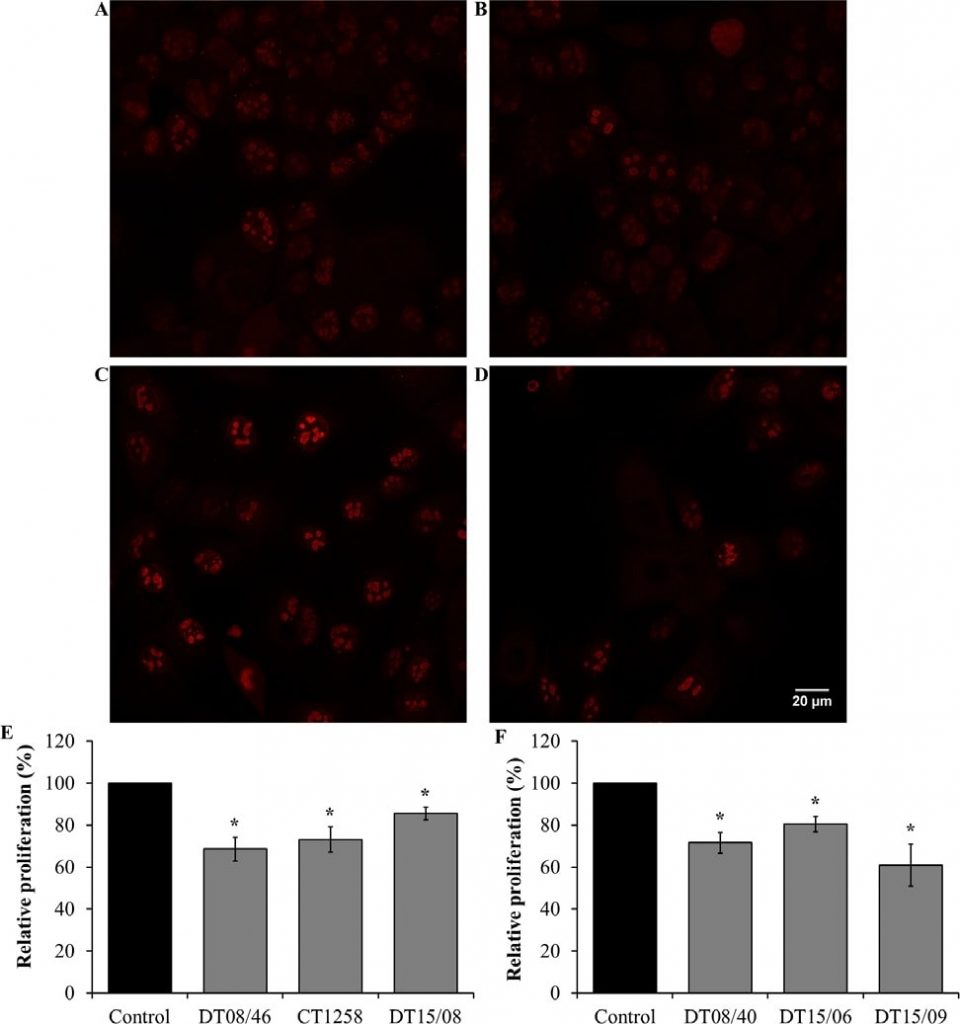
Cell proliferation in 10 mM DCA exposed cell lines were analyzed by Ki67 staining and visualized by confocal fluorescence microscopy. DCA decreased the amount of Ki67 indicating reduced cell proliferation after 48 h in all evaluated cell lines with significance (P<0.05) (Fig. 4).
Effect of DCA on apoptosis
The effect of 10 mM DCA on apoptosis and viability was assessed with Annexin and Sytox using flow cytometry. Regarding apoptosis and dead cells no statistical significant effects were noted in any of the cell lines (data not shown). Confirmation of FACS apoptosis rates was done by TUNEL staining in order to eliminate negative effects of trypsinization on cultured cells. Imaging was performed using confocal fluorescence microscopy. The results confirm the apoptosis ratio in all DCA treated cell lines except DT15/06. Significant decreased apoptosis levels were observed in DT15/08 (P=0.022) and DT15/09 (P=0.0265). DT15/06 showed significantly increased (P=0.041) apoptosis values (data not shown). The other cell lines displayed no significant apoptosis values.
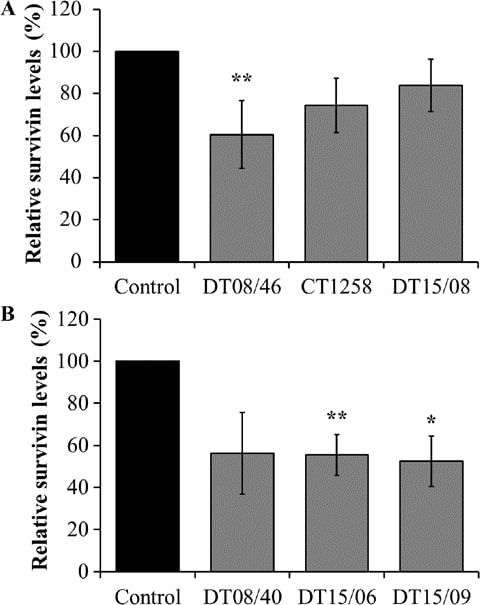
Survivin expression analyzed with xMAP® Magnetic Bead Technology
DCA significantly decreased survivin production in the prostate adenocarcinoma cell line DT08/46 (P=0.0053) and in the TCC cell lines DT15/06 (P=0.0027) and DT15/09 (P=0.0206). The cell lines CT1258 (P=0.0761), DT15/08 (P=0.1551) and DT08/40 (P=0.0598) showed no significant effects in survivin production (Fig. 5).
Bcl-2-antagonist-of-cell-death (BAD) and c-jun N-terminal kinases (JNK) expression analyzed with xMAP Magnetic Bead Technology
After 48-h DCA incubation, the phosphorylated and thus inactive form of BAD increased only significantly in DT08/46 (P=0.0285), CT1258 (P=0.0039) and DT15/06 (P=0.0235). Further no effect of DCA could be observed on active phosphorylated JNK with exception of DT08/46 (P=0.0044) which displayed decreased values (data not shown).
Pyruvate-dehydrogenase (PDH) expression analyzed with xMAP Magnetic Bead Technology
For confirmation of decreased lactate levels as consequence of lower glycolysis, phosphorylated PDH (PDH-P) quantity (Ser232, Ser293, Ser300) was measured with Luminex Magnetic Bead Technology. Decreased levels of PDH-P and thus increased active enzymes are associated with an increased pyruvate oxidation and acetyl coenzyme A metabolism which leads to increased TCA cycle activity [35]. In PDH-P (Ser232) a statistically significant decrease in comparison to non-treated controls was observed in all cell lines except DT08/46. This cell line showed a higher but non-significant PDH-P (Ser232) level. The PDH-P at residue Ser293 was significantly affected by DCA only in DT15/08 (P=0.0082) and DT15/06 (P=0.0122) cell lines. In all other cell lines no effect could be observed after DCA treatment. PDH-P at Ser300 was compromised by DCA significantly in DT08/46 (P=0.0060), CT1258 (P=0.0215), DT15/08 (P=0.0002) and DT15/06 (P=0.0097) (Table I).
Table I
PDH-P expression after DCA exposure with relative mean values ± SD (%).
| Cell line | PDH-P Ser232 | P-value | PDH-P Ser293 | P-value | PDH-P Ser300 | P-value |
|---|---|---|---|---|---|---|
| Control | 100 | 100 | 100 | |||
| Prostate adenocarcinoma | ||||||
| DT08/46 | 121.2±31.5 | 0.2715 | 127.7±27.8 | 0.0900 | 47.7±28.0 | 0.0060b |
| CT1258 | 11.0±5.1 | 0.0011b | 73.7±30.5 | 0.2740 | 24.3±19.5 | 0.0215a |
| DT15/08 | 18.0±17.6 | 0.0026b | 39.7±19.2 | 0.0082b | 18.4±6.8 | 0.0002c |
| Transitional cell carcinoma | ||||||
| DT08/40 | 14.3±10.2 | 0.0047b | 84.5±22.8 | 0.3594 | 48.6±32.1 | 0.1090 |
| DT15/06 | 28.2±17.7 | 0.0039b | 72.3±5.4 | 0.0122a | 39.1±10.4 | 0.0097b |
| DT15/09 | 20.5±24.8 | 0.0308a | 99.8±71.3 | 0.9967 | 34.1±41.0 | 0.1085 |
PDH-P, phosphorylated pyruvate dehydrogenase; Ser, serine; DCA, dichloroacetate; ±, plus minus.
a P<0.05;
b P<0.01;
c P<0.001;
SD, standard deviation; %, percent.
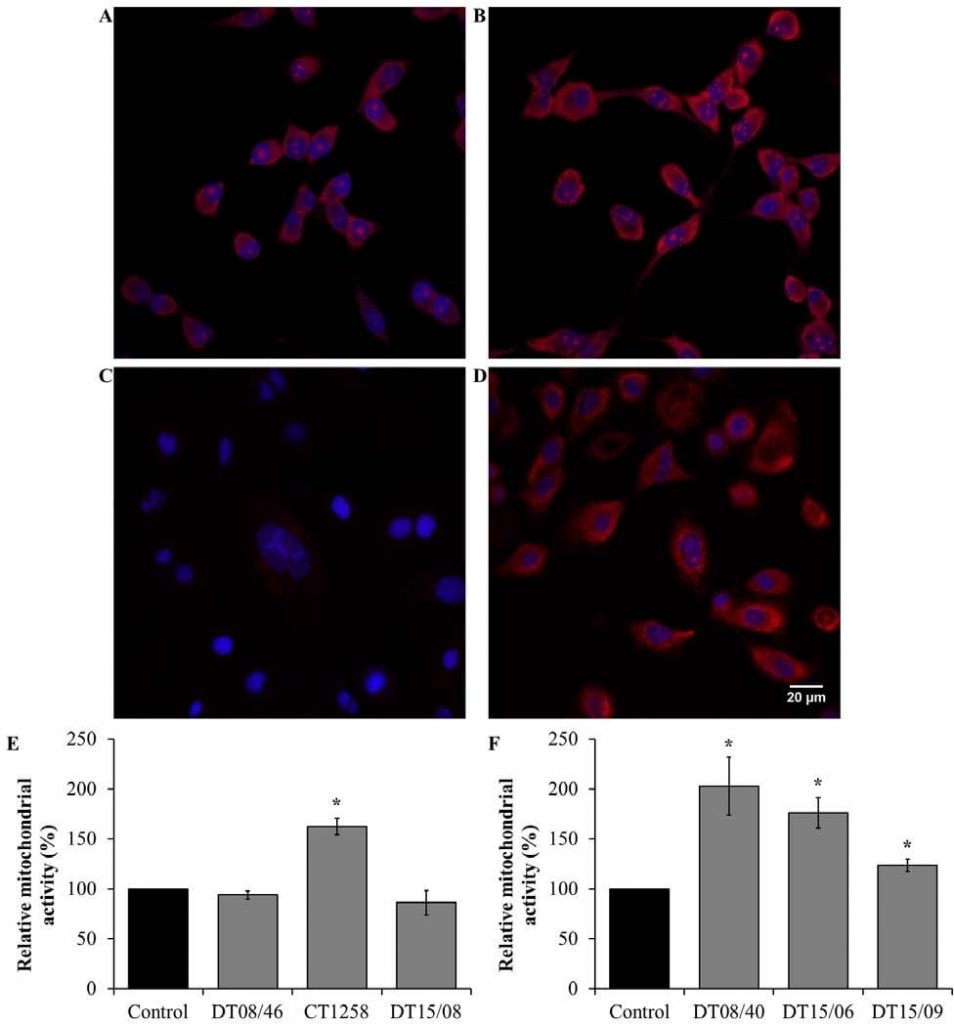
Mitochondrial activity after DCA treatment
Verification of the metabolic alteration to glucose oxidation mitochondrial activity was done by detection of mitochondrial derived reactive oxygen species (ROS). In the prostate adenocarcinoma cell lines (Fig. 6E) a significantly increased mitochondrial activity was observed in CT1258 (P=0.0153). The cell line DT08/46 (P=0.2829) and DT15/08 (P=0.3082) displayed no decreasing effect after DCA treatment. In contrast DCA was able to increase mitochondrial activity significantly (P<0.05) in all TCC cell lines (Fig. 6F).
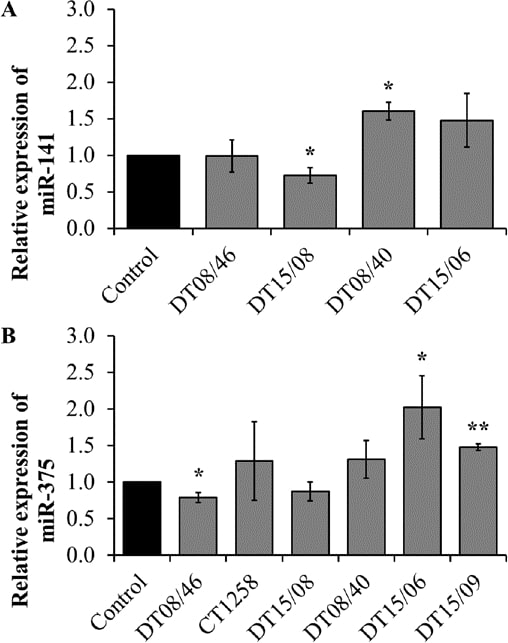
Quantitative miRNA RT-PCR
To assess if the observed lower proliferation rates correlate with changes in micro-RNAs, three miR involved in proliferation or apoptosis were evaluated. miR-141 expression revealed significant changes with decreased levels in DT15/08 (P=0.0451) and increased levels in DT08/40 (P=0.0135) compared to non-treated controls, whereas DT08/46 (P=0.9285) and DT15/06 (P=0.1520) showed no difference in comparison to the controls. In CT1258 and DT15/09 miR-141 was downregulated and excluded from analysis. DT08/46 (P=0.0322) showed a lower and DT15/06 (P=0.0267) as well as DT15/09 (P=0.0031) a higher expression of miR-375. The prostate adenocarcinoma cell line CT1258 displayed no significant changes in all examined microRNAs, respectively. The expression of miR-145 was not influenced by DCA treatment in any of the cell lines (excluded from analysis due to Ct-values >30). In summary prostate adenocarcinoma cell lines tend to show a downregulation of miR-141 (DT15/08) and miR-375 (DT08/46, DT15/08), the TCC cell lines showed upregulation of both (Fig. 7).
Discussion
DCA reduced the cell number in prostate adenocarcinoma and in TCC cell lines. This can occur on the basis of a reduced proliferation or increased apoptosis. In this study a proliferation lowering effect was observed indicated by decreased Ki67 in all cell lines and lower metabolic activities in DT15/08 and DT08/40. This finding is in accordance with previous results presented by Bonnet et al in several human cell lines [35] and Sun et al in breast cancer [12]. Further, decreased cell proliferation after DCA exposure was also observed in human prostate carcinoma [36], colorectal [37], colon [11] and lung cancer [38]. However, induction of mitochondria dependent apoptosis by DCA, as previously reported by several research groups [14,35,39] could not be observed in this study. However, this phenomenon was also observed by Feuerecker et al in murine and human neuroblastoma cells (40). Stockwin et al reported that high DCA concentrations are required for apoptosis induction [38]. The inconsistent results in one cell line (DT15/06) between flow cytometry and TUNEL staining do not allow an evaluation if DCA affects apoptosis. The results of apoptotic protein expression JNK and BAD are consistent with the effects observed in all other cell lines and support the hypothesis that DCA showed no influence on apoptosis in canine prostate and TCC cancer cells.
Survivin, an inhibitor of apoptosis and tumor promoter [41,42], was significantly decreased in all cell lines which displayed increased mitochondrial activity (DT15/06, DT15/09). Decreased survivin levels are reported to induce apoptosis via intrinsic pathways by activation of caspase-3 [41] and were observed in endometrial cancer cell lines after DCA exposure [14]. Unexpectedly, increased apoptosis could not be observed in these cell lines with exception of DT15/06 (P=0.041) which showed a slight but inconsistent increase in cell death. This leads to the conclusion that decreased survivin levels possibly entail decreased proliferation. In addition, a deregulation of further genes which are involved in apoptosis induction is conceivable and would declare, why decreased survivin levels did not result in apoptosis.
DCA is a PDK inhibitor which indirectly activates PDH. Due to decreased values of PDH-P, pyruvate can be oxidized by mitochondria [4,5]. Decreased PDH phosphorylation was observed in several human cancer cell lines [16,37,43]. In accordance with published results in human cell lines, this study confirmed decreasing PDH phosphorylation in all DCA-treated cells which are affirmed by reduced lactate release in all cell lines. This indicates that DCA promotes glucose oxidation in canine cancer cells as well as in human cancer cells. Furthermore, increased levels of ROS in mitochondria of all cell lines except two prostate adenocarcinoma cell lines were proved in this study. Increased ROS species in mitochondria which occur due to cellular respiration, confirm the decreased PDH-P values and the lactate reduction. It might be that increased mitochondrial activity does not result in apoptosis, but in an increased cellular respiration impeding the survival of cancerous cells with a changed metabolism. This could clarify the increased viability of cells after DCA exposure. Increased viability after DCA exposure was also observed by McPherson et al who reported on increased pyruvate oxidation and viability of embryos in an aged mouse model [44].
miR-375 upregulation has been reported to have anti-proliferating effects in many cells such as gastric cancer [45], pancreatic cancer [46], fetal cardiomyocyte-like cells [47] and colon cancer [48]. In prostate carcinogenesis microRNA-375 has variable effects depending on tumor phenotype. Costa-Pinheiro et al proved anti-proliferative effects in PC-3 cells due to upregulation as well as increased apoptosis in 22Rv1 cells following miR-375 knockdown [49]. Our results illustrate, the up- or downregulation of miR-375 after DCA treatment is not consistent in prostate adenocarcinoma cell lines, which is in accordance with the findings described above. In comparison to prostate cancer cells, all TCC cell lines showed increased miR-375 levels. There is no literature describing miR-375 effects in bladder cancer. It could be possible that these results are in accordance with anti-proliferative effects which are described in many other cells. The same findings were observed in microRNA-141. An upregulation of miR-141 inhibited cancer proliferation and cell cycle progression in neuroblastoma cells [50]. Furthermore miR-141 is downregulated in bladder cancer with muscle invasion [51]. In our study miR-141 and miR-375 were found upregulated in all TCC cell lines after DCA treatment indicating that these changes cause lower proliferation rates. In prostate cancer miR-141 is reported to be upregulated [52]. In this study the miR-141 levels decreased slightly, but we suppose that this effect is too mild to affect cancer cells. However, an analysis allowing to determine if a direct link between DCA and changes in different microRNAs is causative for the observed biologic responses would require a comprehensive transcriptomic approach.
In conclusion, this study illustrates that canine cancer cell lines are responding to DCA treatment and the effect can be reduced to decreased proliferation rates, increased pyruvate oxidation and mitochondrial activity. The results also show differences between the examined cancer entities. Thus, TCC cell lines seem to respond consistently and are more susceptible to DCA treatment than prostate adenocarcinoma cell lines. In comparison to most human cell lines DCA did not affect apoptosis which may constitute that DCA might be useful for tumor growth restriction in canine cancer, but not size reduction.
Moreover, DCA can be advantageous in sensitizing canine cancer cells to other anticancer drugs and therefore it might be appropriate for combination therapies [53]. To ensure higher DCA concentrations in cancerous tissues and to avoid severe generalized side effects an intralesional therapy, comparable to intravesical chemotherapy in human [54] and dogs with TCC [55,56], could be a further possibility. The concentration of 10 mM DCA was chosen to allow the comparability to other human in vitro studies [14,18,37]. For clinical studies the DCA concentration has to be re-evaluated with regard to compatibility and negative side effects. Therefore, further studies with DCA concentrations in lower concentrations should be examined.
Abbreviations:
| DCA | dichloroacetate |
| PDH | pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| TCC | transitional cell carcinoma |
| RNA | ribonucleic acid |
| PCR | polymerase chain reaction |
| JNK | c-jun N-terminal kinases |
| BAD | Bcl-2-antagonist-of-cell-death |
| ROS | reactive oxygen species |
| PBS | phosphate-buffered saline |
REFERENCES
1 Vail DM: Supporting the veterinary cancer patient on chemotherapy: Neutropenia and gastrointestinal toxicity. Top Companion Anim Med. 24:122–129. 2009.2 Cornell KK, Bostwick DG, Cooley DM, Hall G, Harvey HJ, Hendrick MJ, Pauli BU, Render JA, Stoica G, Sweet DC, et al: Clinical and pathologic aspects of spontaneous canine prostate carcinoma: A retrospective analysis of 76 cases. Prostate. 45:173–183. 2000.
3 Mutsaers AJ, Widmer WR and Knapp DW: Canine transitional cell carcinoma. J Vet Intern Med. 17:136–144. 2003.
4 Sutendra G and Michelakis ED: Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 3:382013.
5 Stacpoole PW: The pharmacology of dichloroacetate. Metabolism. 38:1124–1144. 1989.
6 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW and Shuster JJ: Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 121:e1223–e1228. 2008.
7 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, et al: Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 117:1519–1531. 2006.
8 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA and Stacpoole PW: Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis. 33:285–293. 1979.
9 Stacpoole PW, Moore GW and Kornhauser DM: Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med. 298:526–530. 1978.
10 Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, et al: Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 3:420–430. 2010.
11 Sánchez-Aragó M, Chamorro M and Cuezva JM: Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis. 31:567–576. 2010.
12 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG and Blackburn AC: Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 120:253–260. 2010.
13 Saed GM, Fletcher NM, Jiang ZL, Abu-Soud HM and Diamond MP: Dichloroacetate induces apoptosis of epithelial ovarian cancer cells through a mechanism involving modulation of oxidative stress. Reprod Sci. 18:1253–1261. 2011.
14 Wong JY, Huggins GS, Debidda M, Munshi NC and De Vivo I: Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 109:394–402. 2008.
15 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2:31ra342010.
16 Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al: Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 69:734–744. 2016.
17 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD and Coomber BL: Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis. 20:63–74. 2015.
18 Madhok BM, Yeluri S, Perry SL, Hughes TA and Jayne DG: Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 102:1746–1752. 2010.
19Ruggieri V, Agriesti F, Scrima R, Laurenzana I, Perrone D, Tataranni T, Mazzoccoli C, Lo Muzio L, Capitanio N and Piccoli C: Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget. 6:1217–1230. 2015.
20 Xintaropoulou C, Ward C, Wise A, Marston H, Turnbull A and Langdon SP: A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget. 6:25677–25695. 2015.
21 Cicmanec JL, Condie LW, Olson GR and Wang SR: 90-Day toxicity study of dichloroacetate in dogs. Fundam Appl Toxicol. 17:376–389. 1991.
22 Maisenbacher HW III, Shroads AL III, Zhong G, Daigle AD, Abdelmalak MM, Samper IS, Mincey BD, James MO and Stacpoole PW: Pharmacokinetics of oral dichloroacetate in dogs. J Biochem Mol Toxicol. 27:522–525. 2013.
23 Lukas G, Vyas KH, Brindle SD, Le Sher AR and Wagner WE Jr: Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration. J Pharm Sci. 69:419–421. 1980.
24 Park R, Arieff AI, Leach W and Lazarowitz VC: Treatment of lactic acidosis with dichloroacetate in dogs. J Clin Invest. 70:853–862. 1982.
25 Racker E: History of the Pasteur effect and its pathobiology. Mol Cell Biochem. 5:17–23. 1974.
26 Warburg O, Wind F and Negelein E: Über den Stoffwechsel von Tumoren im Körper. Klin Wochenschr. 5:829–832. 1926.
27 Kim JW, Tchernyshyov I, Semenza GL and Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3:177–185. 2006.
28 Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC and Thompson CB: The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 21:1037–1049. 2007.
29 Zhao Y, Butler EB and Tan M: Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4:e5322013.
30 Plas DR and Thompson CB: Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 13:75–78. 2002.
31 Simon D, Knebel JW, Baumgartner W, Aufderheide M, Meyer-Lindenberg A and Nolte I: In vitro efficacy of chemotherapeutics as determined by 50% inhibitory concentrations in cell cultures of mammary gland tumors obtained from dogs. Am J Vet Res. 62:1825–1830. 2001.
32 Knapp DW, Chan TC, Kuczek T, Reagan WJ and Park B: Evaluation of in vitro cytotoxicity of nonsteroidal anti-inflammatory drugs against canine tumor cells. Am J Vet Res. 56:801–805. 1995.
33 Sartin EA, Barnes S, Toivio-Kinnucan M, Wright JC and Wolfe LG: Heterogenic properties of clonal cell lines derived from canine mammary carcinomas and sensitivity to tamoxifen and doxorubicin. Anticancer Res. 13:229–236. 1993.
34 Winkler S, Murua Escobar H, Eberle N, Reimann-Berg N, Nolte I and Bullerdiek J: Establishment of a cell line derived from a canine prostate carcinoma with a highly rearranged karyotype. J Hered. 96:782–785. 2005.
35 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 11:37–51. 2007.
36 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C and Rosser CJ: Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 68:1223–1231. 2008.
37 Ho N and Coomber BL: Pyruvate dehydrogenase kinase expression and metabolic changes following dichloroacetate exposure in anoxic human colorectal cancer cells. Exp Cell Res. 331:73–81. 2015.
38 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG and Newton DL: Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 127:2510–2519. 2010.
39 Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C and Chen HZ: Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int J Oncol. 38:409–417. 2011.
40 Feuerecker B, Seidl C, Pirsig S, Bruchelt G and Senekowitsch-Schmidtke R: DCA promotes progression of neuroblastoma tumors in nude mice. Am J Cancer Res. 5:812–820. 2015.
41 Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC and Altieri DC: Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 396:580–584. 1998.
42 Ambrosini G, Adida C and Altieri DC: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 3:917–921. 1997.
43 Abemayor E, Kovachich GB and Haugaard N: Effects of dichloroacetate on brain pyruvate dehydrogenase. J Neurochem. 42:38–42. 1984.
44 McPherson NO, Zander-Fox D and Lane M: Stimulation of mitochondrial embryo metabolism by dichloroacetic acid in an aged mouse model improves embryo development and viability. Fertil Steril. 101:1458–1466. 2014.
45 Zhou N, Qu Y, Xu C and Tang Y: Upregulation of microRNA-375 increases the cisplatin-sensitivity of human gastric cancer cells by regulating ERBB2. Exp Ther Med. 11:625–630. 2016.
46 Zhou J, Song S, He S, Zhu X, Zhang Y, Yi B, Zhang B, Qin G and Li D: MicroRNA-375 targets PDK1 in pancreatic carcinoma and suppresses cell growth through the Akt signaling pathway. Int J Mol Med. 33:950–956. 2014.
47 Wang L, Song G, Liu M, Chen B, Chen Y, Shen Y, Zhu J and Zhou X: MicroRNA-375 overexpression influences P19 cell proliferation, apoptosis and differentiation through the Notch signaling pathway. Int J Mol Med. 37:47–55. 2016.
48 Zaharie F, Muresan MS, Petrushev B, Berce C, Gafencu GA, Selicean S, Jurj A, Cojocneanu-Petric R, Lisencu CI, Pop LA, et al: Exosome-carried microRNA-375 inhibits cell progression and dissemination via Bcl-2 blocking in colon cancer. J Gastrointestin Liver Dis. 24:435–443. 2015.
49 Costa-Pinheiro P, Ramalho-Carvalho J, Vieira FQ, Torres-Ferreira J, Oliveira J, Gonçalves CS, Costa BM, Henrique R and Jerónimo C: MicroRNA-375 plays a dual role in prostate carcinogenesis. Clin Epigenetics. 7:422015.
50 Wang Z, Lei H and Sun Q: MicroRNA-141 and its associated gene FUS modulate proliferation, migration and cisplatin chemosensitivity in neuroblastoma cell lines. Oncol Rep. 35:2943–2951. 2016.
51 Mahdavinezhad A, Mousavi-Bahar SH, Poorolajal J, Yadegarazari R, Jafari M, Shabab N and Saidijam M: Evaluation of miR-141, miR-200c, miR-30b Expression and Clinicopathological features of bladder cancer. Int J Mol Cell Med. 4:32–39. 2015.
52 Brase JC, Johannes M, Schlomm T, Fälth M, Haese A, Steuber T, Beissbarth T, Kuner R and Sültmann H: Circulating miRNAs are correlated with tumor progression in prostate cancer. Int J Cancer. 128:608–616. 2011.
53 Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang PC, Zhao YL, Xu Y, Jia L, Zhang X, et al: Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction. Mol Pharm. 9:634–644. 2012.
54 Porten SP, Leapman MS and Greene KL: Intravesical chemotherapy in non-muscle-invasive bladder cancer. Indian J Urol. 31:297–303. 2015.
55 Song D, Wientjes MG, Gan Y and Au JL: Bladder tissue pharmacokinetics and antitumor effect of intravesical 5-fluorouridine. Clin Cancer Res. 3:901–909. 1997.
56 Abbo AH, Jones DR, Masters AR, Stewart JC, Fourez L and Knapp DW: Phase I clinical trial and pharmacokinetics of intravesical mitomycin C in dogs with localized transitional cell carcinoma of the urinary bladder. J Vet Intern Med. 24:1124–1130. 2010.