Татьяна Хартинг1, Мэнди Штуббендорф2, Саския Вилленброк1, Зигфрид Вагнер1, Патрик Шадзек3, Анаклет Нгезахайо3, Уго Муруа Эскобар1, Инго Нольте1
1 Клиника мелких животных, Университет ветеринарной медицины Ганновера, Фонд, D-30559 Ганновер
2 Отделение медицинской клиники III, гематология, онкология и паллиативная медицина, Университет Ростока, D-18057 Росток
Evotec AG, D-22419 Гамбург
Институт биофизики, Университет Лейбница, D-30419 Ганновер, Германия
Переписка: Профессор Инго Нольте, Клиника мелких животных, Университет ветеринарной медицины Ганновера, Фонд, Bünteweg 9, D-30559 Ганновер, Германия; E-mail: [email protected]
Получено: 12 июля 2016 г.
Принято: 5 сентября 2016 г.
Опубликовано онлайн: 5 октября 2016 г
Аннотация
Эффект Варбурга описывает способность раковых клеток производить энергию посредством аэробного гликолиза вместо окислительного фосфорилирования пирувата. Это отклонение в митохондриальном метаболизме подавляет апоптоз, позволяя увеличивать пролиферацию в условиях пониженного уровня кислорода. Дихлорацетат (ДХА) был успешно использован в нескольких линиях раковых клеток человека для реактивации окислительного фосфорилирования в митохондриях. Целью данного исследования была характеристика и реакция линий раковых клеток собак после воздействия DCA. Влияние 10 мМ DCA было охарактеризовано in vitro на набор из шести клеточных линий аденокарциномы простаты и переходноклеточной карциномы (ППК), полученных от собак. Анализировались количество клеток, уровень лактата, апоптоз, экспрессия апоптотических белков, факторов выживания и различных миРНК. Кроме того, исследовались метаболическая активность, митохондриальная активность и пролиферация. DCA значительно уменьшил количество клеток во всех клеточных линиях, кроме одной, и привел к значительному снижению высвобождения лактата. Снижение уровня survivin было обнаружено во всех клеточных линиях, в двух из которых наблюдалось значительное снижение метаболической активности. Повышенные уровни miR-375 были определены во всех клеточных линиях TCC. Реактивация пируватдегидрогеназы и повышенная митохондриальная активность, по-видимому, вызывают переход от аэробного гликолиза обратно к окислительному фосфорилированию. Кроме того, эти результаты показывают, что лечение DCA оказывает подавляющее действие на пролиферацию раковых клеток собак.
Ключевые слова: дихлорацетат, аденокарцинома предстательной железы собак, переходноклеточная карцинома собак, эффект Варбурга
DOI: 10.3892/ijo.2016.3720
ВВЕДЕНИЕ
В последние несколько лет такие методы лечения рака, как химиотерапия, лучевая терапия и хирургия, используемые при лечении рака у людей, стали более важными в ветеринарии. Обычные химиотерапевтические средства направлены на делящиеся клетки, как раковые, так и неопластические, вызывая ряд побочных эффектов, таких как миелосупрессия, диарея, рвота и анорексия [1]. Кроме того, из-за поздней стадии заболевания и резистентности рака простаты и мочевого пузыря лечение затруднено и часто связано с плохим прогнозом [2,3]. Поэтому необходимо искать новые альтернативы, которые были бы более эффективными.
Дихлорацетат (ДХА), небольшая и экономичная молекула, влияет на различные метаболические пути, ингибируя киназу пируватдегидрогеназы (PDK) [4,5]. Это предполагает, что пируватдегидрогеназа (PDH) потенциально косвенно активируется DCA, что приводит к метаболическому сдвигу в пользу окисления пирувата до ацетил-коэнзима-А в митохондриях [5]. Несмотря на это, в последние десятилетия DCA используется для лечения множества заболеваний, таких как врожденный лактат-ацидоз [6,7], гиперхолестеринемия [8], гипергликемия [9], застойная сердечная недостаточность [10] и только недавно в исследованиях рака [11-16]. DCA был протестирован в различных in vitro подходах в области онкологии человека, включая колоректальный рак [17,18], рак эндометрия [14], плоскоклеточную карциному полости рта [19] и рак молочной железы [20]. В клиническом исследовании, проведенном на пациентах, страдающих глиобластомой и другими солидными опухолями, DCA уменьшил рост опухоли и ангиогенез [15]. За исключением нескольких исследований, посвященных изучению фармакокинетических эффектов ДКА у собак [21-23], в настоящее время нет публикаций о действии ДКА при раке у собак. Однако DCA успешно применялся у собак с молочнокислым ацидозом [24] и, как сообщается, хорошо переносится собаками [22]. Тяжелые побочные эффекты, такие как смерть и паралич, наблюдались только в долгосрочном исследовании с применением высоких доз [21].
В аэробных условиях неопластические клетки переходят к окислению глюкозы через митохондрии, которые окисляют пируват до ацетил-коэнзима-А [25]. PDH позволяет пирувату поступать в митохондрии. Производство энергии в раковых клетках в первую очередь смещается от окисления глюкозы к аэробному гликолизу, что приводит к увеличению производства цитозольного лактата, несмотря на то, что кислорода достаточно [26]. Такое поведение называют эффектом Варбурга. Биохимик Отто Варбург впервые сообщил об этих особенностях в 1926 году и предположил, что причиной может быть нарушение работы митохондрий [26]. Канцерогенез предпочтительно начинается в гипоксических тканях, где потребление глюкозы низкое. Соответственно, активируется гипоксия-индуцибельный фактор 1α, что приводит к повышению уровня транспортеров глюкозы и PDK. Активация PDK приводит к ингибированию PDH и, следовательно, гликолиза [27,28]. Благодаря этому метаболическому изменению и снижению митохондриальной деполяризации раковые клетки имеют преимущество в выживании и не подвержены внутренним путям апоптоза [29,30].
Для доклинической оценки противораковых препаратов эксперименты in vitro с клеточными линиями являются важными подходами как в человеческих, так и в ветеринарных исследованиях. Исследования in vitro дают возможность получить больше информации об эффективности и чувствительности нескольких опухолевых образований [31-33]. В данном исследовании были использованы несколько установленных [34] и новых клеточных линий.
Насколько нам известно, это первое исследование, в котором изучалось влияние DCA на клетки аденокарциномы простаты и переходноклеточной карциномы (ППК) собак. Было определено влияние DCA на количество клеток, уровень лактата, митохондриальную активность, апоптоз и метаболическую активность. Кроме того, оценивалось влияние DCA на PDH и на белки, участвующие в апоптозе. Влияние DCA на некоторые микроРНК (miR) ранее не определялось. Также отсутствует литература о влиянии ДКА на рак мочевого пузыря у человека или в ветеринарии.
Материалы и методы
Клеточные линии и культура клеток
Для экспериментов были использованы три линии аденокарциномы простаты собак (DT08/46, CT1258, DT15/08) и три линии ТКК собак (DT08/40, DT15/06, DT15/09). Две клеточные линии ТКК (DT08/40 и DT15/09) были получены из ткани простаты, а одна ТКК (DT15/06) — из ткани женского мочевого пузыря. Клеточные линии были классифицированы как аденокарцинома простаты или ТСС после патогистологического исследования исходных тканей. Все клеточные линии были созданы в клинике мелких животных, Университет ветеринарной медицины Ганновера, Германия. Клеточные линии культивировали в колбах площадью 75 см2 (TPP, Faust Lab Science, Klettgau, Германия) с 10 мл среды 199 (Gibco™, Thermo Fisher Scientific, Darmstadt, Германия), 10% фетальной телячьей сыворотки (Hyclone®, Thermo Fisher Scientific), 2% пенициллина-стрептомицина (Biochrom, Berlin, Германия), смена среды проводилась каждые 48 ч. Клеткам давали вырасти до плотности 90% перед разделением 1:3. Клетки инкубировали при 37°C и 5%CO2 в увлажненном воздухе. Для экспериментов клетки обрабатывали 10 мМ DCA (Sigma-Aldrich GmbH, Taufkirchen, Германия) в течение 48 ч. Для деления клеток или после периода лечения клетки трипсинизировали с помощью TrypLE™ Express (Gibco™, Thermo Fisher Scientific) и подсчитывали количество клеток с помощью автоматического Cellometer™ Auto T4 (Nexcelom Bioscience, Lawrence, MA, США) и сравнивали с отрицательным контролем. Клетки промывали PBS (Biochrom) и хранили при -80°C для дальнейших исследований (количественная RT-PCR и анализ белков). DCA растворяли в деионизированной воде, стерилизовали на фильтре и корректировали pH до 7,4 с помощью NaOH. Доза 10 мМ была выбрана в соответствии с предыдущими исследованиями с клетками HeLa человека [14]. Даже если эта концентрация не может быть безопасно достигнута in vivo, она была выбрана для сопоставимости с другими исследованиями человека in vitro.
Уровни лактата
Для измерения уровня лактата супернатант клеточной культуры центрифугировали при 1000 об/мин в течение 10 мин для удаления плавающих клеток и мусора и 1,3 мл переносили в сосуд с фторидом натрия (Sarstedt, Nümbrecht, Германия). Для исключения изменений, вызванных феноловым красным и лактатом из фетальной телячьей сыворотки, среду использовали в качестве отрицательного контроля и вычитали из измерений. Колориметрическое определение лактата проводилось с помощью Cobas® C311 (Hitachi, Токио, Япония). Чтобы соотнести оцененные уровни лактата с количеством и объемом клеток, общее содержание лактата нормировали на концентрацию внутриклеточного белка, которую определяли с помощью анализа Pierce™ BCA (Thermo Fisher Scientific) в соответствии с инструкциями производителя.
Метаболическая активность
Клетки высевали в 96-луночный планшет (Falcon, Corning, Амстердам, Нидерланды) с 200 мкл среды 199, 10% фетальной телячьей сыворотки, 2% пенициллина-стрептомицина и инкубировали при 37°C и 5% увлажненномCO2. Среду меняли каждые 24 ч, и для измерения в каждую лунку добавляли 20 мкл МТТ (CellTiter96® Aqueous One Solution assay, Promega, Mannheim, Germany). Абсорбцию определяли через 2 ч с помощью планшетного ридера Synergy2 (BioTek, Бад Фридрихсхалль, Германия). Измерения проводились каждые 24 ч в течение четырех дней. Данные анализировали с помощью программного обеспечения Gen5™ 1.11 (BioTek) и нормировали на отрицательный контроль среды.
Проточная цитометрия
Для определения апоптоза105 клеток культивировали в 6-луночном планшете (TPP, Faust Lab Science) с 4 мл среды и обрабатывали 10 мМ DCA, как описано выше, в течение 48 ч. После этого периода клетки трипсинизировали и центрифугировали вместе со средой, содержащей неприлипшие и мертвые клетки, при 1 000 об/мин в течение 6 мин. Супернатант отбрасывали, а клетки ресуспендировали в 500 мкл буфера для анализа. Окрашивание проводили с помощью 5 мкл Annexin-FITC и 1 мкл Sytox (Annexin V-FITC Detection Kit Plus, PromoCell, Гейдельберг, Германия). После 5-минутной инкубации при комнатной температуре104 клетки анализировали с помощью BD FACScalibur™ (BD Biosciences, Гейдельберг, Германия) и программного обеспечения CellQuest™ Pro 6.0 (BD Biosciences). Аннексин и Sytox были обнаружены в FL-1. Анализ данных проводился с помощью FlowJo Version 10.0.8r1 (FlowJo, Ashland, OR, USA). Границы устанавливали по среднему значению положительных контролей (клетки, пермеабилизированные сапонином) и отрицательных контролей каждой клеточной линии (жизнеспособные клетки, не подвергавшиеся обработке).
Выделение РНК и количественный RT-PCR
Тотальную РНК выделяли из106 клеток с помощью набора NucleoSpin Small RNA kit (Macherey Nagel, Düren, Германия), как описано в протоколе производителя. кДНК готовили из 35 нг тотальной РНК путем обратной транскрипции с использованием набора TaqMan® MicroRNA Reverse Transkription kit (Applied Biosystems™, Thermo Fisher Scientific) в соответствии с инструкциями производителя. Относительное количественное определение экспрессии микроРНК обработанных клеток по сравнению с отрицательным контролем проводили на циклере Eppendorf realplex4 Cycler (Eppendorf, Wesseling-Berzdorf, Германия) с использованием 1.33 мкл кДНК в общем объеме 20 мкл, содержащем TaqMan® Universal Master Mix NoAmpErase® UNG (Applied Biosystems, Thermo Fisher Scientific) и TaqMan® MicroRNA assays для Mir-141 (ID 245445_mat), Mir-145 (ID 002278), Mir-375 (ID 000564), приобретенных у Thermo Fisher Scientific. Процедура проводилась в соответствии с описанием в протоколе производителя. Данные были нормализованы к гену домашнего хозяйства RNU6B (ID 001093), анализ проводился с помощью программы Rest2009 (Qiagen, Хильден, Германия).
Анализ магнитных бусинок Luminex
Анализ экспрессии белков проводили с помощью технологии xMAP® Luminex Bead Technology на приборе Luminex 200™ (Luminex Corp., Хертогенбош, Нидерланды). Данные представлены в виде общего количества (сурвивин пг/мл) или чистого MFI (другие мишени) и были показаны с помощью программного обеспечения xPONENT 3.1 (Luminex Corp.). Результаты проб с более низкими значениями, как MFI < фонового MFI + 2× стандартное отклонение, были исключены из анализа. Количественное определение сурвивина проводили с помощью набора ProcartaPlex Human Survivin Simplex kit (eBioscience, Франкфурт-на-Майне, Германия) с использованием супернатанта клеточной культуры, как описано в протоколе производителя. Количество сурвивина нормировали на концентрацию белка, которую определяли с помощью анализа Pierce BCA (Thermo Fisher Scientific). PDH и апоптотические белки (BAD и JNK) определяли с помощью мультиплексного анализа от Merck Millipore (Multi-species PDH Complex Magnetic Bead Panel и 7-Plex Early Apoptosis Magnetic Bead kit, Дармштадт, Германия). Образцы обрабатывали в соответствии с инструкциями производителя. Кроме того, образцы для измерения PDH фильтровали с помощью центрифужных ультрачистых фильтров с размером пор 0,65 мкм (Merck Millipore) при 7 000 об/мин в течение 4 мин.
Митохондриальная активность
Клетки, выращенные на 8-луночных μ-лунках (Ibidi, Martinsried, Германия), обработанные 10 мМ DCA и без него, фиксировали 4% параформальдегидом, трижды промывали HBSS, содержащим кальций и магний, и окрашивали 4 мкМ MitoSox (Invitrogen, Thermo Fisher Scientific) в течение 15 мин. После окрашивания клетки трижды промывали HBSS (Gibco, Thermo Fisher Scientific) и ядра клеток контрастировали DAPI (разведение 1:1,000, Sigma-Aldrich GmbH) в течение 5 мин. Флуоресцентную визуализацию проводили с помощью инвертированного конфокального лазерного сканирующего микроскопа (Eclipse TE2000-E, Nikon, Дюссельдорф, Германия) с использованием объектива 60× с водной иммерсией (Nikon). Изображения получали с помощью программного обеспечения EZ-C1 1.80 (Nikon). Возбуждение происходило с помощью диодного лазера при 408 нм (DAPI) и гелий/неонового лазера при 543 нм (MitoSox). Общая флуоресценция клеток MitoSox, вычитая фон, анализировалась с помощью ImageJ и нормировалась на количество клеток.
Иммунофлуоресцентное окрашивание Ki67 и TUNEL
Клетки высевали, фиксировали и промывали, как описано выше. После промывки HBSS клетки пермеабилизировали 0,2% Triton X-100 в течение 20 мин, промывали и инкубировали в течение ночи со специфическим кроличьим поликлональным антителом Ki67 в разведении 1:150 (Life Technologies, Thermo Fisher Scientific). Для окрашивания моноклональное антирабитовое антитело Alexa Fluor® 555 (Cell Signaling Technology, Лейден, Нидерланды) инкубировали в течение 1 ч с разведением 1:250 и клетки контрастировали DAPI (1:1,000) в течение 5 мин. Протокол визуализации флуоресценции был таким же, как описано выше. Общая флуоресценция клеток определялась, как описано выше. Для окрашивания TUNEL использовали набор Apoptag Fluorescein Direct kit (Merck Millipore) в соответствии с инструкциями производителя. Возбуждение происходило с помощью аргонлазера при 488 нм, а визуализация проводилась, как описано выше. Оценивался процент TUNEL-позитивных клеток.
Статистический анализ
Статистический анализ данных проводился с помощью программного обеспечения SAS 7.1 (SAS Institute Inc., Cary, NC, USA). Для сравнения двух средних использовали двухфакторный t-тест. Доверительная вероятность была установлена на уровне 5% (P<0,05) и считалась статистически значимой.
Результаты
Количество клеток после обработки DCA
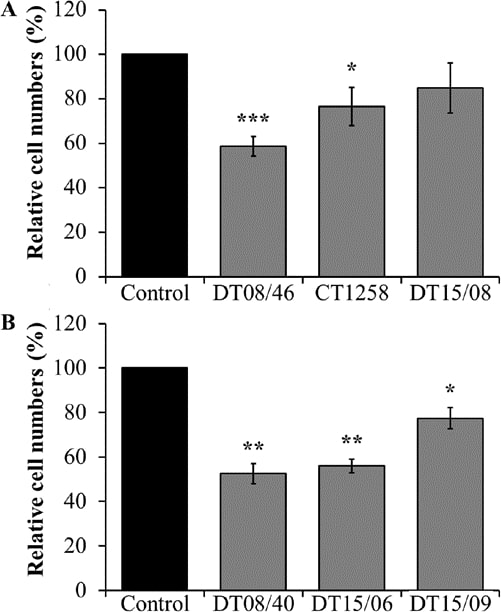
По сравнению с необработанным отрицательным контролем, клеточные линии аденокарциномы простаты DT08/46 (P<0,0001) и CT1258 (P=0,0122) показали значительно меньшее количество клеток после обработки 10 мМ DCA в течение 48 ч (рис. 1A). Третья клеточная линия простаты DT15/08 DCA не показала значительного снижения (P=0,0748), но наблюдалась тенденция к снижению количества клеток, соответственно, снижению скорости пролиферации в нативной культуре клеток (рис. 1A). Как показано на рис. 1В, такой же снижающий эффект ДКА был отмечен во всех исследованных клеточных линиях ТКК DT08/40 (P=0,0031), DT15/06 (P=0,0016) и в DT15/09 (P=0,0143).
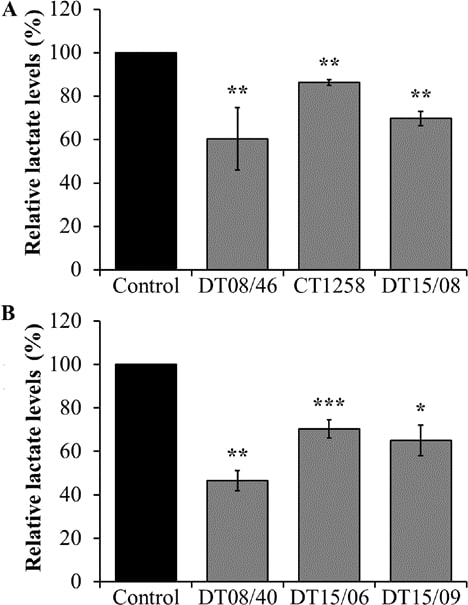
Уровень лактата в супернатанте клеточной культуры после воздействия DCA
Для оценки эффекта снижения высвобождения лактата при обработке DCA через 48 ч измеряли количество лактата в супернатанте клеточной культуры, соответственно. Во всех клеточных линиях обоих видов собачьего рака 10 мМ DCA оказывал значительный эффект снижения лактата (рис. 2).
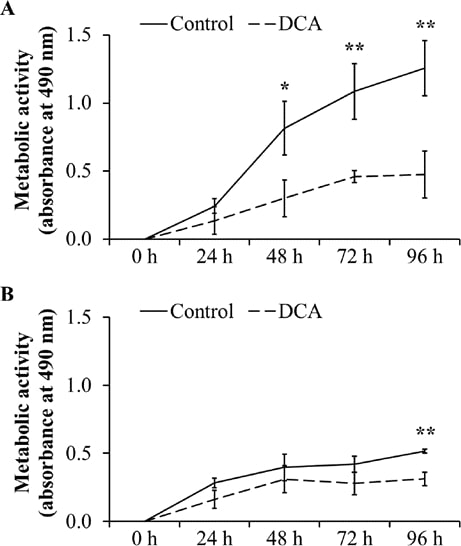
Метаболическая активность после обработки DCA в течение 96 ч
Для подтверждения негативного влияния на пролиферацию клеток метаболическая активность как индикатор пролиферации и жизнеспособности клеток была проанализирована с помощью МТТ-анализа. Как показано на рис. 3А, метаболическая активность в DT15/08 была значительно снижена по сравнению с соответствующим отрицательным контролем после 48 ч обработки ДКА (P=0,0202). После 96 ч непрерывной обработки ДКА метаболическая активность значительно снизилась (P=0,007). Аналогичный эффект наблюдался у DT08/40 (рис. 3B) через 96 ч (P=0,0025) и у DT15/06 (P=0,0419) через 96 ч (данные не показаны) обработки ДКА. Клеточные линии DT08/46, CT1258 и DT15/09 не показали статистически значимого влияния DCA на метаболическую активность (данные не показаны).
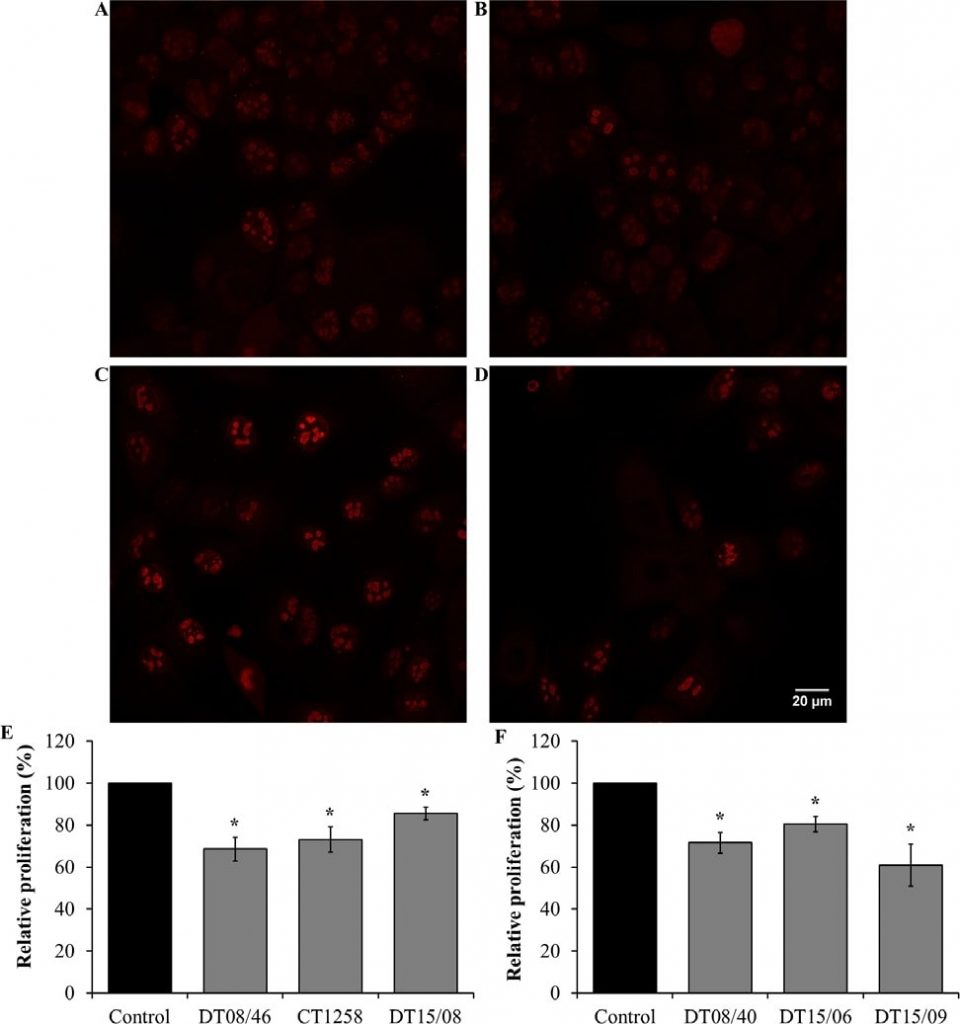
Пролиферация после обработки DCA в течение 48 ч
Пролиферация клеток в линиях клеток, подвергнутых воздействию 10 мМ DCA, анализировалась с помощью окрашивания Ki67 и визуализировалась с помощью конфокальной флуоресцентной микроскопии. DCA уменьшил количество Ki67, что указывает на снижение пролиферации клеток через 48 ч во всех оцененных клеточных линиях со значимостью (P<0,05) (рис. 4).
Влияние DCA на апоптоз
Влияние 10 мМ DCA на апоптоз и жизнеспособность оценивали с помощью Annexin и Sytox с использованием проточной цитометрии. В отношении апоптоза и мертвых клеток не было отмечено статистически значимого эффекта ни в одной из клеточных линий (данные не показаны). Подтверждение уровня апоптоза по FACS проводилось путем окрашивания TUNEL, чтобы исключить негативное влияние трипсинизации на культивируемые клетки. Визуализация проводилась с помощью конфокальной флуоресцентной микроскопии. Результаты подтверждают коэффициент апоптоза во всех клеточных линиях, обработанных DCA, за исключением DT15/06. Значительное снижение уровня апоптоза наблюдалось в DT15/08 (P=0,022) и DT15/09 (P=0,0265). DT15/06 показал значительно повышенные (P=0,041) значения апоптоза (данные не показаны). Другие клеточные линии не показали значительных значений апоптоза.
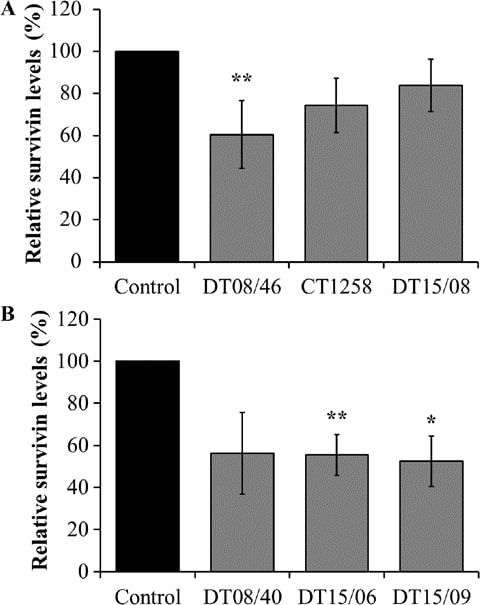
Анализ экспрессии сурвивина с помощью технологии xMAP® Magnetic Bead Technology
DCA значительно снизил выработку сурвивина в клеточной линии аденокарциномы простаты DT08/46 (P=0,0053) и в клеточных линиях ТСС DT15/06 (P=0,0027) и DT15/09 (P=0,0206). Клеточные линии CT1258 (P=0,0761), DT15/08 (P=0,1551) и DT08/40 (P=0,0598) не показали значительного влияния на выработку сурвивина (рис. 5).
Экспрессия Bcl-2-антагониста клеточной смерти (BAD) и c-jun N-terminal kinases (JNK) проанализирована с помощью технологии xMAP Magnetic Bead Technology
После 48-часовой инкубации с DCA фосфорилированная и, следовательно, неактивная форма BAD значительно увеличилась только в DT08/46 (P=0,0285), CT1258 (P=0,0039) и DT15/06 (P=0,0235). Кроме того, не наблюдалось влияния DCA на активный фосфорилированный JNK, за исключением DT08/46 (P=0,0044), который показал пониженные значения (данные не показаны).
Анализ экспрессии пируватдегидрогеназы (PDH) с помощью технологии магнитных бусинок xMAP
Для подтверждения снижения уровня лактата вследствие снижения гликолиза, количество фосфорилированной PDH (PDH-P) (Ser232, Ser293, Ser300) было измерено с помощью технологии Luminex Magnetic Bead Technology. Снижение уровня PDH-P и, следовательно, увеличение активных ферментов связано с повышенным окислением пирувата и метаболизмом ацетил коэнзима А, что приводит к увеличению активности цикла ТСА [35]. В PDH-P (Ser232) статистически значимое снижение по сравнению с необработанным контролем наблюдалось во всех клеточных линиях, кроме DT08/46. Эта клеточная линия показала более высокий, но незначительный уровень PDH-P (Ser232). На PDH-P в остатке Ser293 значительно повлиял DCA только в клеточных линиях DT15/08 (P=0,0082) и DT15/06 (P=0,0122). Во всех остальных клеточных линиях после обработки DCA не наблюдалось никакого эффекта. PDH-P на Ser300 была значительно нарушена под воздействием ДКА в DT08/46 (P=0,0060), CT1258 (P=0,0215), DT15/08 (P=0,0002) и DT15/06 (P=0,0097) (Таблица I).
Таблица I
Экспрессия PDH-P после воздействия DCA с относительными средними значениями ± SD (%).
| Клеточная линия | PDH-P Ser232 | P-value | PDH-P Ser293 | P-value | PDH-P Ser300 | P-value |
|---|---|---|---|---|---|---|
| Контроль | 100 | 100 | 100 | |||
| Аденокарцинома предстательной железы | ||||||
| dT08/46 | 121.2±31.5 | 0.2715 | 127.7±27.8 | 0.0900 | 47.7±28.0 | 0.0060b |
| cT1258 | 11.0±5.1 | 0.0011b | 73.7±30.5 | 0.2740 | 24.3±19.5 | 0.0215a |
| dT15/08 | 18.0±17.6 | 0.0026b | 39.7±19.2 | 0.0082b | 18.4±6.8 | 0.0002c |
| Переходноклеточная карцинома | ||||||
| dT08/40 | 14.3±10.2 | 0.0047b | 84.5±22.8 | 0.3594 | 48.6±32.1 | 0.1090 |
| dT15/06 | 28.2±17.7 | 0.0039b | 72.3±5.4 | 0.0122a | 39.1±10.4 | 0.0097b |
| dT15/09 | 20.5±24.8 | 0.0308a | 99.8±71.3 | 0.9967 | 34.1±41.0 | 0.1085 |
PDH-P, фосфорилированная пируватдегидрогеназа; Ser, серин; DCA, дихлорацетат; ±, плюс минус.
a P<0,05;
b P<0,01;
c P<0,001;
SD, стандартное отклонение; %, процент.
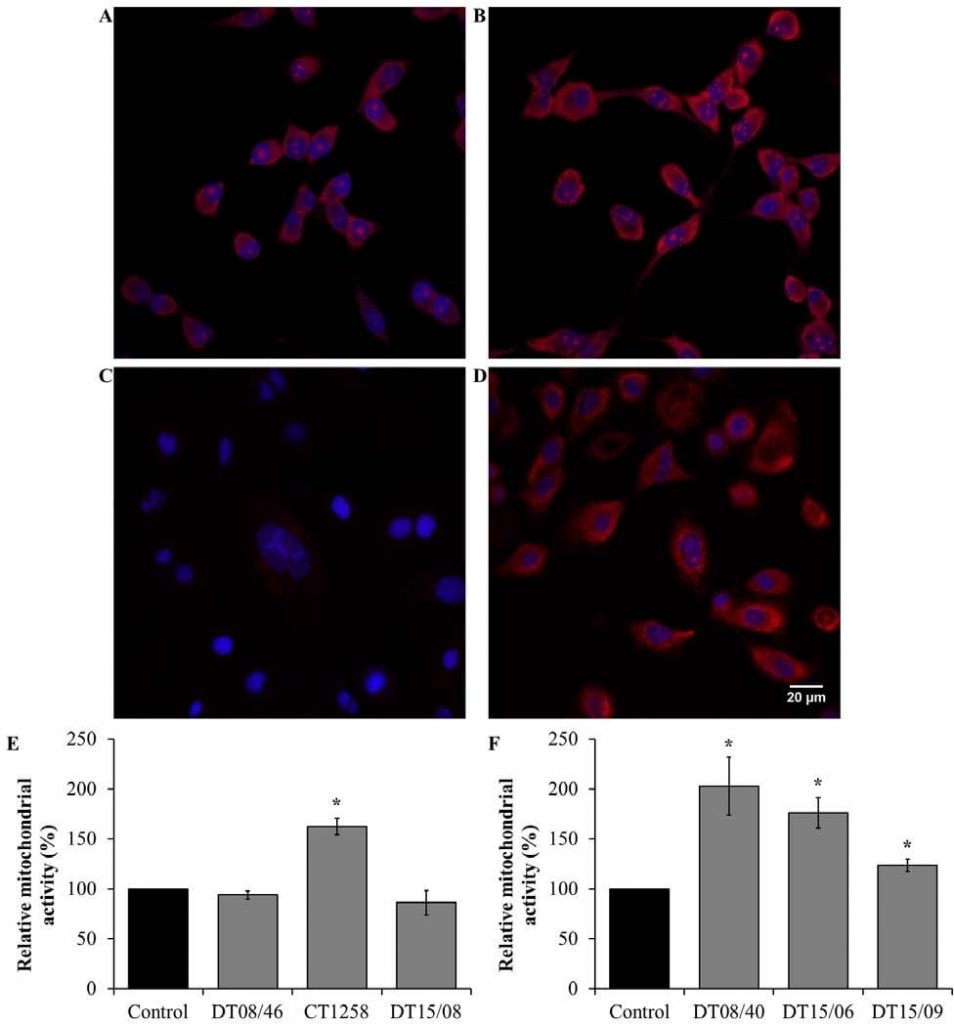
Митохондриальная активность после обработки DCA
Проверка метаболических изменений митохондриальной активности окисления глюкозы проводилась путем обнаружения митохондриальных производных реактивных форм кислорода (ROS). В клеточных линиях аденокарциномы простаты (рис. 6E) значительно повышенная митохондриальная активность наблюдалась в CT1258 (P=0,0153). Клеточные линии DT08/46 (P=0,2829) и DT15/08 (P=0,3082) не показали эффекта снижения после обработки ДКА. Напротив, DCA смог значительно повысить митохондриальную активность (P<0,05) во всех клеточных линиях TCC (рис. 6F).
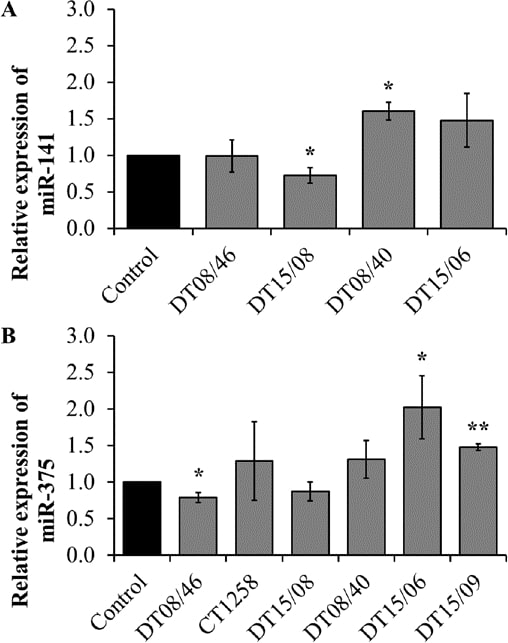
Количественная РТ-ПЦР миРНК
Чтобы оценить, коррелирует ли наблюдаемая более низкая скорость пролиферации с изменениями в микро-РНК, были оценены три miR, вовлеченные в пролиферацию или апоптоз. Экспрессия miR-141 выявила значительные изменения со снижением уровня в DT15/08 (P=0,0451) и повышением уровня в DT08/40 (P=0,0135) по сравнению с контролем без лечения, тогда как DT08/46 (P=0,9285) и DT15/06 (P=0,1520) не показали различий по сравнению с контролем. В CT1258 и DT15/09 miR-141 был даунрегулярен и исключен из анализа. DT08/46 (P=0,0322) показал более низкую, а DT15/06 (P=0,0267) и DT15/09 (P=0,0031) — более высокую экспрессию miR-375. Клеточная линия аденокарциномы простаты CT1258 не показала значительных изменений во всех исследованных микроРНК, соответственно. На экспрессию miR-145 не повлияла обработка DCA ни в одной из клеточных линий (исключены из анализа из-за значений Ct >30). В целом, клеточные линии аденокарциномы простаты демонстрируют тенденцию к снижению уровня miR-141 (DT15/08) и miR-375 (DT08/46, DT15/08), а клеточные линии ТСС — к повышению уровня обеих (рис. 7).
Обсуждение
DCA уменьшил количество клеток в аденокарциноме простаты и в клеточных линиях TCC. Это может происходить за счет снижения пролиферации или усиления апоптоза. В данном исследовании наблюдался эффект снижения пролиферации, о чем свидетельствует уменьшение Ki67 во всех клеточных линиях и снижение метаболической активности в DT15/08 и DT08/40. Этот вывод согласуется с предыдущими результатами, представленными Bonnet и др. на нескольких клеточных линиях человека [35] и Sun и др. на раке молочной железы [12]. Кроме того, снижение пролиферации клеток после воздействия DCA также наблюдалось при карциноме простаты человека [36], колоректальном [37], толстой кишки [11] и раке легкого [38]. Однако индукция митохондрий-зависимого апоптоза под действием DCA, о чем ранее сообщали несколько исследовательских групп [14,35,39], в данном исследовании не наблюдалась. Однако это явление также наблюдалось Feuerecker и др. в клетках нейробластомы мыши и человека (40). Стоквин и др. сообщили, что для индукции апоптоза необходимы высокие концентрации DCA [38]. Несоответствие результатов проточной цитометрии и окрашивания TUNEL в одной линии клеток (DT15/06) не позволяет оценить, влияет ли DCA на апоптоз. Результаты экспрессии апоптотических белков JNK и BAD согласуются с эффектами, наблюдаемыми во всех других клеточных линиях, и подтверждают гипотезу о том, что DCA не оказывает влияния на апоптоз в клетках рака простаты и ТСС собак.
Сурвивин, ингибитор апоптоза и промотор опухоли [41,42], был значительно снижен во всех клеточных линиях, которые демонстрировали повышенную митохондриальную активность (DT15/06, DT15/09). Сообщается, что снижение уровня сурвивина индуцирует апоптоз по внутреннему пути через активацию каспазы-3 [41] и наблюдалось в клеточных линиях рака эндометрия после воздействия ДКА [14]. Неожиданно, увеличение апоптоза не наблюдалось в этих клеточных линиях, за исключением DT15/06 (P=0,041), которая показала небольшое, но непоследовательное увеличение гибели клеток. Это позволяет сделать вывод, что снижение уровня сурвивина, возможно, влечет за собой снижение пролиферации. Кроме того, можно предположить дерегуляцию других генов, участвующих в индукции апоптоза, что объясняет, почему снижение уровня сурвивина не привело к апоптозу.
DCA является ингибитором PDK, который косвенно активирует PDH. Из-за снижения уровня PDH-P пируват может быть окислен митохондриями [4,5]. Снижение фосфорилирования PDH наблюдалось в нескольких линиях клеток рака человека [16,37,43]. В соответствии с опубликованными результатами в клеточных линиях человека, данное исследование подтвердило снижение фосфорилирования PDH во всех клетках, обработанных ДКА, что подтверждается снижением высвобождения лактата во всех клеточных линиях. Это указывает на то, что DCA способствует окислению глюкозы в раковых клетках собак, а также в раковых клетках человека. Кроме того, в данном исследовании было доказано повышение уровня ROS в митохондриях всех клеточных линий, кроме двух линий клеток аденокарциномы простаты. Увеличение количества ROS в митохондриях, которое происходит в результате клеточного дыхания, подтверждает снижение значений PDH-P и уменьшение лактата. Возможно, повышенная митохондриальная активность приводит не к апоптозу, а к усилению клеточного дыхания, препятствующего выживанию раковых клеток с измененным метаболизмом. Это может объяснить повышенную жизнеспособность клеток после воздействия DCA. Повышение жизнеспособности после воздействия DCA также наблюдали McPherson и др., которые сообщили о повышенном окислении пирувата и жизнеспособности эмбрионов в модели стареющей мыши [44].
сообщалось, что повышение уровня miR-375 оказывает антипролиферативное действие на многие клетки, такие как рак желудка [45], рак поджелудочной железы [46], фетальные кардиомиоцитоподобные клетки [47] и рак толстой кишки [48]. В канцерогенезе предстательной железы микроРНК-375 оказывает различное действие в зависимости от фенотипа опухоли. Costa-Pinheiro et al. доказали антипролиферативный эффект в клетках PC-3 за счет ап-регуляции, а также усиление апоптоза в клетках 22Rv1 после нокдауна miR-375 [49]. Наши результаты показывают, что повышение или понижение регуляции miR-375 после обработки DCA не является последовательным в клеточных линиях аденокарциномы простаты, что согласуется с результатами, описанными выше. По сравнению с клетками рака простаты, все клеточные линии ТСС показали повышенный уровень miR-375. Литература, описывающая эффекты miR-375 при раке мочевого пузыря, отсутствует. Возможно, эти результаты соответствуют антипролиферативным эффектам, которые описаны для многих других клеток. Такие же результаты были получены и в отношении микроРНК-141. Повышение уровня miR-141 ингибировало пролиферацию рака и прогрессию клеточного цикла в клетках нейробластомы [50]. Кроме того, miR-141 снижена в раке мочевого пузыря с мышечной инвазией [51]. В нашем исследовании miR-141 и miR-375 были обнаружены повышенными во всех клеточных линиях TCC после лечения DCA, что указывает на то, что эти изменения вызывают снижение уровня пролиферации. Сообщается, что при раке предстательной железы miR-141 повышается [52]. В данном исследовании уровень miR-141 снизился незначительно, но мы полагаем, что этот эффект слишком слабый, чтобы повлиять на раковые клетки. Однако анализ, позволяющий определить, является ли прямая связь между ДКА и изменениями в различных микроРНК причиной наблюдаемых биологических реакций, потребует комплексного транскриптомного подхода.
В заключение, данное исследование показывает, что линии раковых клеток собак реагируют на лечение DCA, и эффект может быть сведен к снижению скорости пролиферации, увеличению окисления пирувата и митохондриальной активности. Результаты также показывают различия между исследованными раковыми образованиями. Так, клеточные линии ТСС, по-видимому, последовательно реагируют и более восприимчивы к лечению ДКА, чем клеточные линии аденокарциномы простаты. По сравнению с большинством клеточных линий человека DCA не влияет на апоптоз, что может свидетельствовать о том, что DCA может быть полезен для ограничения роста опухоли при собачьем раке, но не для уменьшения ее размеров.
Более того, DCA может быть полезен для сенсибилизации раковых клеток собак к другим противораковым препаратам, поэтому он может быть пригоден для комбинированной терапии [53]. Чтобы обеспечить более высокую концентрацию DCA в раковых тканях и избежать тяжелых генерализованных побочных эффектов, дальнейшей возможностью может стать интрализиональная терапия, сравнимая с внутрипузырной химиотерапией у людей [54] и собак с ТСС [55,56]. Концентрация 10 мМ DCA была выбрана для обеспечения сопоставимости с другими исследованиями in vitro на человеке [14,18,37]. Для клинических исследований концентрация ДКК должна быть заново оценена с точки зрения совместимости и негативных побочных эффектов. Поэтому следует изучить дальнейшие исследования с более низкими концентрациями DCA.
Сокращения:
| DCA | дихлорацетат |
| PDH | пируватдегидрогеназа |
| PDK | киназа пируватдегидрогеназы |
| ТКК | переходноклеточная карцинома |
| РНК | рибонуклеиновая кислота |
| ПЦР | полимеразная цепная реакция |
| JNK | c-jun N-терминальная киназа |
| BAD | Bcl-2-антагонист клеточной смерти |
| ROS | реактивные виды кислорода |
| PBS | фосфатно-буферный солевой раствор |
ССЫЛКИ
1 Vail DM: Поддержка ветеринарного онкологического пациента при химиотерапии: Нейтропения и желудочно-кишечная токсичность. Top Companion Anim Med. 24:122-129. 2009.2 Cornell KK, Bostwick DG, Cooley DM, Hall G, Harvey HJ, Hendrick MJ, Pauli BU, Render JA, Stoica G, Sweet DC, et al: Clinical and pathologic aspects of spontaneous canine prostate carcinoma: A retrospective analysis of 76 cases. Prostate. 45:173-183. 2000.
3 Mutsaers AJ, Widmer WR and Knapp DW: Canine transitional cell carcinoma. J Vet Intern Med. 17:136-144. 2003.
4 Sutendra G and Michelakis ED: Киназа пируватдегидрогеназы как новая терапевтическая мишень в онкологии. Front Oncol. 3:382013.
5 Stacpoole PW: The pharmacology of dichloroacetate. Metabolism. 38:1124-1144. 1989.
6 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW and Shuster JJ: Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 121:e1223-e1228. 2008.
7 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, et al: Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Педиатрия. 117:1519-1531. 2006.
8 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA and Stacpoole PW: Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Атеросклероз. 33:285-293. 1979.
9 Stacpoole PW, Moore GW and Kornhauser DM: Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med. 298:526-530. 1978.
10 Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, et al: Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 3:420-430. 2010.
11 Sánchez-Aragó M, Chamorro M and Cuezva JM: Отбор раковых клеток с подавленными митохондриями запускает прогрессирование рака толстой кишки. Carcinogenesis. 31:567-576. 2010.
12 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG and Blackburn AC: Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 120:253-260. 2010.
13 Saed GM, Fletcher NM, Jiang ZL, Abu-Soud HM and Diamond MP: Dichloroacetate induces apoptosis of epithelial ovarian cancer cells through a mechanism involving modulation of oxidative stress. Reprod Sci. 18:1253-1261. 2011.
14 Wong JY, Huggins GS, Debidda M, Munshi NC and De Vivo I: Дихлорацетат индуцирует апоптоз в клетках рака эндометрия. Gynecol Oncol. 109:394-402. 2008.
15 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2:31ra342010.
16 Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al: Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 69:734-744. 2016.
17 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD and Coomber BL: Dichloroacetate affect proliferation but not survival of human colorectal cancer cells. Апоптоз. 20:63-74. 2015.
18 Madhok BM, Yeluri S, Perry SL, Hughes TA and Jayne DG: Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 102:1746-1752. 2010.
19RuggieriV, Agriesti F, Scrima R, Laurenzana I, Perrone D, Tataranni T, Mazzoccoli C, Lo Muzio L, Capitanio N and Piccoli C: Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget. 6:1217-1230. 2015.
20 Xintaropoulou C, Ward C, Wise A, Marston H, Turnbull A и Langdon SP: Сравнительный анализ ингибиторов пути гликолиза в моделях клеточных линий рака молочной железы и яичников. Oncotarget. 6:25677-25695. 2015.
21 Cicmanec JL, Condie LW, Olson GR and Wang SR: 90-Day toxicity study of dichloroacetate in dogs. Fundam Appl Toxicol. 17:376-389. 1991.
22 Maisenbacher HW III, Shroads AL III, Zhong G, Daigle AD, Abdelmalak MM, Samper IS, Mincey BD, James MO and Stacpoole PW: Pharmacokinetics of oral dichloroacetate in dogs. J Biochem Mol Toxicol. 27:522-525. 2013.
23 Lukas G, Vyas KH, Brindle SD, Le Sher AR and Wagner WE Jr: Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration. J Pharm Sci. 69:419-421. 1980.
24 Park R, Arieff AI, Leach W and Lazarowitz VC: Treatment of lactic acidosis with dichloroacetate in dogs. J Clin Invest. 70:853-862. 1982.
25 Racker E: История эффекта Пастера и его патобиология. Mol Cell Biochem. 5:17-23. 1974.
26 Warburg O, Wind F and Negelein E: Über den Stoffwechsel von Tumoren im Körper. Klin Wochenschr. 5:829-832. 1926.
27 Kim JW, Tchernyshyov I, Semenza GL and Dang CV: HIF-1-опосредованная экспрессия киназы пируватдегидрогеназы: Метаболический переключатель, необходимый для клеточной адаптации к гипоксии. Cell Metab. 3:177-185. 2006.
28 Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC и Thompson CB: Транскрипционный фактор HIF-1alpha играет важную роль в регуляции аэробного и анаэробного гликолиза, зависящей от факторов роста. Genes Dev. 21:1037-1049. 2007.
29 Zhao Y, Butler EB and Tan M: Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4:e5322013.
30 Plas DR and Thompson CB: Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 13:75-78. 2002.
31 Simon D, Knebel JW, Baumgartner W, Aufderheide M, Meyer-Lindenberg A and Nolte I: In vitro эффективность химиотерапевтических препаратов, определенная по 50% ингибирующей концентрации в клеточных культурах опухолей молочной железы, полученных от собак. Am J Vet Res. 62:1825-1830. 2001.
32 Knapp DW, Chan TC, Kuczek T, Reagan WJ и Park B: Оценка in vitro цитотоксичности нестероидных противовоспалительных препаратов против опухолевых клеток собак. Am J Vet Res. 56:801-805. 1995.
33 Sartin EA, Barnes S, Toivio-Kinnucan M, Wright JC and Wolfe LG: Heterogenic properties of clonal cell lines derived from canine mammary carcinomas and sensitivity to tamoxifen and doxorubicin. Anticancer Res. 13:229-236. 1993.
34 Winkler S, Murua Escobar H, Eberle N, Reimann-Berg N, Nolte I and Bullerdiek J: Establishment of a cell line derived from a canine prostate carcinoma with a highly rearranged karyotype. J Hered. 96:782-785. 2005.
35 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 11:37-51. 2007.
36 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C and Rosser CJ: Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 68:1223-1231. 2008.
37 Ho N and Coomber BL: Pyruvate dehydrogenase kinase expression and metabolic changes following dichloroacetate exposure in anoxic human colorectal cancer cells. Exp Cell Res. 331:73-81. 2015.
38 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG and Newton DL: Дихлорацетат натрия избирательно действует на клетки с дефектами митохондриального ЭТЦ. Int J Cancer. 127:2510-2519. 2010.
39 Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C и Chen HZ: Дихлорацетат смещает метаболизм с гликолиза на окисление глюкозы и демонстрирует синергетическое ингибирование роста с цисплатином в клетках HeLa. Int J Oncol. 38:409-417. 2011.
40 Feuerecker B, Seidl C, Pirsig S, Bruchelt G and Senekowitsch-Schmidtke R: DCA способствует прогрессированию опухолей нейробластомы у голых мышей. Am J Cancer Res. 5:812-820. 2015.
41 Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC and Altieri DC: Контроль апоптоза и контрольного пункта митотического веретена сурвивином. Nature. 396:580-584. 1998.
42 Ambrosini G, Adida C and Altieri DC: Новый антиапоптозный ген, сурвивин, экспрессируемый в раке и лимфоме. Nat Med. 3:917-921. 1997.
43 Abemayor E, Kovachich GB and Haugaard N: Effects of dichloroacetate on brain pyruvate dehydrogenase. J Neurochem. 42:38-42. 1984.
44 McPherson NO, Zander-Fox D и Lane M: Стимуляция митохондриального метаболизма эмбрионов дихлоруксусной кислотой в модели пожилой мыши улучшает развитие и жизнеспособность эмбрионов. Fertil Steril. 101:1458-1466. 2014.
45 Zhou N, Qu Y, Xu C and Tang Y: Upregulation of microRNA-375 increases the cisplatin-sensitivity of human gastric cancer cells by regulating ERBB2. Exp Ther Med. 11:625-630. 2016.
46 Zhou J, Song S, He S, Zhu X, Zhang Y, Yi B, Zhang B, Qin G and Li D: МикроРНК-375 нацелена на PDK1 в панкреатической карциноме и подавляет рост клеток через сигнальный путь Akt. Int J Mol Med. 33:950-956. 2014.
47 Wang L, Song G, Liu M, Chen B, Chen Y, Shen Y, Zhu J and Zhou X: Сверхэкспрессия микроРНК-375 влияет на пролиферацию, апоптоз и дифференцировку клеток P19 через сигнальный путь Notch. Int J Mol Med. 37:47-55. 2016.
48 Zaharie F, Muresan MS, Petrushev B, Berce C, Gafencu GA, Selicean S, Jurj A, Cojocneanu-Petric R, Lisencu CI, Pop LA, et al: Exosome-carried microRNA-375 inhibits cell progression and dissemination through Bcl-2 blocking in colon cancer. J Gastrointestin Liver Dis. 24:435-443. 2015.
49 Costa-Pinheiro P, Ramalho-Carvalho J, Vieira FQ, Torres-Ferreira J, Oliveira J, Gonçalves CS, Costa BM, Henrique R and Jerónimo C: MicroRNA-375 plays a dual role in prostate carcinogenesis. Clin Epigenetics. 7:422015.
50 Wang Z, Lei H and Sun Q: MicroRNA-141 и связанный с ней ген FUS модулируют пролиферацию, миграцию и химиочувствительность к цисплатину в клеточных линиях нейробластомы. Oncol Rep. 35:2943-2951. 2016.
51 Mahdavinezhad A, Mousavi-Bahar SH, Poorolajal J, Yadegarazari R, Jafari M, Shabab N and Saidijam M: Evaluation of miR-141, miR-200c, miR-30b Expression and Clinicopathological features of bladder cancer. Int J Mol Cell Med. 4:32-39. 2015.
52 Brase JC, Johannes M, Schlomm T, Fälth M, Haese A, Steuber T, Beissbarth T, Kuner R and Sültmann H: Circulating miRNAs are correlated with tumor progression in prostate cancer. Int J Cancer. 128:608-616. 2011.
53 Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang PC, Zhao YL, Xu Y, Jia L, Zhang X, et al: Mitaplatin увеличивает чувствительность опухолевых клеток к цисплатину, вызывая дисфункцию митохондрий. Mol Pharm. 9:634-644. 2012.
54 Porten SP, Leapman MS and Greene KL: Intravesical chemotherapy in non-muscle-invasive bladder cancer. Indian J Urol. 31:297-303. 2015.
55 Song D, Wientjes MG, Gan Y and Au JL: Bladder tissue pharmacokinetics and antitumor effect of intravesical 5-fluorouridine. Clin Cancer Res. 3:901-909. 1997.
56 Abbo AH, Jones DR, Masters AR, Stewart JC, Fourez L и Knapp DW: Фаза I клинического испытания и фармакокинетика внутрипузырного митомицина C у собак с локализованной переходноклеточной карциномой мочевого пузыря. J Vet Intern Med. 24:1124-1130. 2010.