Tatjana Harting1, Mandy Stubbendorff 2, Saskia Willenbrock1, Siegfried Wagner1, Patrik Schadzek3, Anaclet Ngezahayo3, Hugo Murua Escobar1, Ingo Nolte1
1 Clinica dei piccoli animali, Università di Medicina Veterinaria di Hannover, Fondazione, D-30559 Hannover
2 Divisione di Medicina Clinica III, Ematologia, Oncologia e Medicina Palliativa, Università di Rostock, D-18057 Rostock
3 Evotec AG, D-22419 Amburgo
4 Istituto di Biofisica, Università Leibniz, D-30419 Hannover, Germania
Corrispondenza: Professor Ingo Nolte, Small Animal Clinic, University of Veterinary Medicine Hannover, Foundation, Bünteweg 9, D-30559 Hannover, Germania; E-mail: [email protected]
Ricevuto: 12 luglio 2016
Accettato: 5 settembre 2016
Pubblicato online: 5 ottobre 2016
Abstract
L’effetto Warburg descrive la capacità delle cellule tumorali di produrre energia attraverso la glicolisi aerobica invece della fosforilazione ossidativa del piruvato. Questa deviazione del metabolismo mitocondriale inibisce l’apoptosi, consentendo una maggiore proliferazione in condizioni di ridotti livelli di ossigeno. Il dicloroacetato (DCA) è stato utilizzato con successo in diverse linee cellulari tumorali umane per riattivare la fosforilazione ossidativa nei mitocondri. Lo scopo di questo studio era la caratterizzazione e la risposta delle linee cellulari cancerose canine dopo l’esposizione al DCA. L’effetto di 10 mM di DCA è stato caratterizzato in vitro su una serie di sei linee cellulari derivate dall’adenocarcinoma prostatico e dal carcinoma a cellule transizionali (TCC) canino. Sono stati analizzati il numero di cellule, i livelli di lattato, l’apoptosi, l’espressione di proteine apoptotiche, fattori di sopravvivenza e diversi miRNA. Inoltre, sono state analizzate l’attività metabolica, l’attività mitocondriale e la proliferazione. Il DCA ha ridotto significativamente il numero di cellule di tutte le linee cellulari utilizzate, tranne una, e ha portato a una riduzione significativa del rilascio di lattato. I livelli di survivin sono diminuiti in tutte le linee cellulari, due delle quali hanno presentato una riduzione significativa dell’attività metabolica. In tutte le linee cellulari TCC è stato misurato un aumento dei livelli di miR-375. La riattivazione della piruvato deidrogenasi e un’elevata attività mitocondriale sembrano indurre la transizione dalla glicolisi aerobica alla fosforilazione ossidativa. Inoltre, questi risultati dimostrano che il trattamento con DCA ha un effetto soppressivo sulla proliferazione delle cellule tumorali canine.
Parole chiave: dicloroacetato, adenocarcinoma prostatico canino, carcinoma a cellule transizionali canino, effetto Warburg
DOI: 10.3892/ijo.2016.3720
INTRODUZIONE
Negli ultimi anni i trattamenti antitumorali come la chemioterapia, la radioterapia e la chirurgia, utilizzati nel trattamento del cancro umano, hanno acquisito maggiore importanza in medicina veterinaria. Gli agenti chemioterapici convenzionali colpiscono le cellule in divisione, sia quelle cancerose che quelle non neoplastiche, causando diversi effetti collaterali come mielosoppressione, diarrea, vomito e anoressia [1]. Inoltre, a causa dello stadio avanzato della malattia e della resistenza del cancro alla prostata e alla vescica, il trattamento è difficile e spesso associato a una prognosi infausta [2,3]. Pertanto, è necessario studiare nuove alternative più efficaci.
Il dicloroacetato (DCA), una molecola piccola ed economica, agisce su diverse vie metaboliche inibendo la piruvato deidrogenasi chinasi (PDK) [4,5]. Ciò implica che la piruvato deidrogenasi (PDH) è potenzialmente attivata indirettamente dal DCA, il che produce uno spostamento metabolico a favore dell’ossidazione del piruvato ad acetil-coenzima-A nei mitocondri [5]. Nonostante ciò, negli ultimi decenni il DCA è stato utilizzato nel trattamento di una moltitudine di disturbi come l’acidosi lattatica congenita [6,7], l’ipercolesterolemia [8], l’iperglicemia [9], l’insufficienza cardiaca congestizia [10] e solo recentemente nella ricerca sul cancro [11-16]. Il DCA è stato testato in diversi approcci in vitro nel campo dell’oncologia umana, tra cui il cancro del colon-retto [17,18], il cancro dell’endometrio [14], il carcinoma orale a cellule squamose [19] e il cancro al seno [20]. In uno studio clinico su pazienti affetti da glioblastoma e altri tumori solidi, il DCA ha ridotto la crescita tumorale e l’angiogenesi [15]. Ad eccezione di diversi studi che hanno analizzato gli effetti farmacocinetici del DCA nel cane [21-23], attualmente non esistono pubblicazioni sull’effetto del DCA nel cancro canino. Tuttavia, il DCA è stato utilizzato con successo in cani con acidosi lattica [24] e risulta essere ben tollerato nei cani [22]. Effetti collaterali gravi, come morte e paralisi, sono stati osservati solo in uno studio a lungo termine ad alte dosi [21].
In condizioni aerobiche le cellule non neoplastiche fanno riferimento all’ossidazione del glucosio attraverso i mitocondri che ossidano il piruvato ad acetil-coenzima-A [25]. La PDH permette al piruvato di entrare nei mitocondri. La produzione di energia delle cellule tumorali si sposta principalmente dall’ossidazione del glucosio alla glicolisi aerobica, che porta a un aumento della produzione di lattato citosolico nonostante la disponibilità di ossigeno sufficiente [26]. Questo comportamento viene definito effetto Warburg. Il biochimico Otto Warburg segnalò per la prima volta queste caratteristiche nel 1926 e ipotizzò che la causa potesse essere un’insufficienza mitocondriale [26]. La carcinogenesi si instaura di preferenza nei tessuti ipossici, dove il consumo di glucosio è basso. Di conseguenza, il fattore inducibile 1α dell’ipossia viene attivato e porta a un’upregolazione dei trasportatori di glucosio e della PDK. L’attivazione della PDK determina l’inibizione della PDH e quindi della glicolisi [27,28]. Grazie a questo cambiamento metabolico e alla diminuzione della depolarizzazione mitocondriale, le cellule tumorali hanno un vantaggio in termini di sopravvivenza e non vengono colpite dalle vie di apoptosi intrinseca [29,30].
Per la valutazione preclinica dei farmaci antitumorali, gli esperimenti in vitro con linee cellulari sono approcci importanti sia nella ricerca umana che in quella veterinaria. Le indagini in vitro offrono la possibilità di ricevere maggiori informazioni sull’efficacia e sulla sensibilità di diverse entità tumorali [31-33]. In questo studio sono state utilizzate diverse linee cellulari consolidate [34] e nuove.
A nostra conoscenza, questo è il primo studio in cui è stato analizzato l’effetto del DCA sulle cellule di adenocarcinoma prostatico canino e di carcinoma a cellule transizionali (TCC). È stata determinata l’influenza del DCA sulla conta delle cellule, sui livelli di lattato, sull’attività mitocondriale, sull’apoptosi e sull’attività metabolica. Inoltre, è stato valutato l’effetto del DCA sulla PDH e sulle proteine coinvolte nell’apoptosi. L’effetto del DCA su diversi microRNA (miR) non è stato determinato in precedenza. Inoltre, non esiste letteratura sull’influenza del DCA sul cancro della vescica nell’uomo o nella medicina veterinaria.
Materiali e metodi
Linee cellulari e coltura cellulare
Per gli esperimenti sono stati utilizzati tre adenocarcinomi prostatici canini (DT08/46, CT1258, DT15/08) e tre TCC canini (DT08/40, DT15/06, DT15/09). Due linee cellulari TCC (DT08/40 e DT15/09) derivavano da tessuto prostatico e una TCC (DT15/06) da tessuto vescicale femminile. Le linee cellulari sono state classificate come adenocarcinoma prostatico o TCC dopo l’esame patoistologico dei tessuti iniziali. Tutte le linee cellulari sono state create nella Clinica dei piccoli animali dell’Università di Medicina Veterinaria di Hannover, in Germania. Le linee cellulari sono state coltivate in fiasche da 75 cm2 (TPP, Faust Lab Science, Klettgau, Germania) con 10 ml di terreno 199 (Gibco™, Thermo Fisher Scientific, Darmstadt, Germania), 10% di siero fetale di vitello (Hyclone®, Thermo Fisher Scientific), 2% di penicillina-streptomicina (Biochrom, Berlino, Germania); un cambio di terreno è stato effettuato ogni 48 ore. Le cellule sono state lasciate crescere fino a una densità del 90% prima di essere divise 1:3. Le cellule sono state incubate a 37°C e 5% diCO2 in aria umidificata. Per gli esperimenti le cellule sono state trattate con 10 mM DCA (Sigma-Aldrich GmbH, Taufkirchen, Germania) per 48 h. Per la divisione delle cellule o dopo il periodo di trattamento le cellule sono state tripsinizzate con TrypLE™ Express (Gibco™, Thermo Fisher Scientific) e il numero di cellule è stato contato con un Cellometer™ Auto T4 (Nexcelom Bioscience, Lawrence, MA, USA) e confrontato con il controllo negativo. Le cellule sono state lavate con PBS (Biochrom) e conservate a -80°C per ulteriori esami (RT-PCR quantitativa e analisi delle proteine). Il DCA è stato disciolto in acqua deionizzata, filtrato e il pH è stato regolato a 7,4 con NaOH. La dose di 10 mM è stata scelta in base a studi precedenti con cellule umane HeLa [14]. Anche se questa concentrazione potrebbe non essere raggiunta con sicurezza in vivo, è stata scelta per consentire la comparabilità con altri studi umani in vitro.
Livelli di lattato
Per le misurazioni del livello di lattato, il surnatante della coltura cellulare è stato centrifugato a 1.000 giri/min per 10 minuti per rimuovere le cellule fluttuanti e i detriti e 1,3 ml sono stati trasferiti in un contenitore di fluoruro di sodio (Sarstedt, Nümbrecht, Germania). Per eliminare le alterazioni dovute al rosso fenolo e al lattato nativo del siero fetale di vitello, il terreno è stato utilizzato come controllo negativo e sottratto dalle misurazioni. La determinazione colorimetrica del lattato è stata eseguita con Cobas® C311 (Hitachi, Tokyo, Giappone). Per mettere in relazione i livelli di lattato valutati con il numero e il volume delle cellule, il contenuto totale di lattato è stato normalizzato alla concentrazione di proteine intracellulari, determinata con il saggio Pierce™ BCA (Thermo Fisher Scientific) secondo le istruzioni del produttore.
Attività metabolica
Le cellule sono state seminate in una piastra a 96 pozzetti (Falcon, Corning, Amsterdam, Paesi Bassi) con 200 μl di terreno 199, 10% di siero fetale di vitello, 2% di penicillina-streptomicina e incubate a 37°C e 5% diCO2 umidificata. Il terreno è stato cambiato ogni 24 ore e per la misurazione sono stati aggiunti 20 μl di MTT (CellTiter96® Aqueous One Solution assay, Promega, Mannheim, Germania) in ogni pozzetto. L’assorbanza è stata determinata dopo 2 ore con un lettore di piastre Synergy2 (BioTek, Bad Friedrichshall, Germania). Le misurazioni sono state eseguite ogni 24 ore per un periodo di quattro giorni. I dati sono stati analizzati con il software Gen5™ 1.11 (BioTek) e normalizzati rispetto al controllo negativo del terreno.
Citometria a flusso
Per la determinazione dell’apoptosi,105 cellule sono state coltivate in una piastra a 6 pozzetti (TPP, Faust Lab Science) con 4 ml di terreno e trattate con 10 mM DCA come descritto sopra per 48 h. Dopo questo periodo, le cellule sono state tripsinizzate e centrifugate insieme al terreno contenente cellule non aderenti e morte a 1.000 rpm per 6 min. Il sovranatante è stato scartato e le cellule sono state risospese in 500 μl di tampone di saggio. La colorazione è stata eseguita con 5 μl di Annexin-FITC e 1 μl di Sytox (Annexin V-FITC Detection Kit Plus, PromoCell, Heidelberg, Germania). Dopo 5 minuti di incubazione a temperatura ambiente,104 cellule sono state analizzate con BD FACScalibur™ (BD Biosciences, Heidelberg, Germania) e il software CellQuest™ Pro 6.0 (BD Biosciences). L’Annexina e la Sytox sono state rilevate in FL-1. L’analisi dei dati è stata eseguita con FlowJo versione 10.0.8r1 (FlowJo, Ashland, OR, USA). Le porte sono state impostate in base alla media dei controlli positivi (cellule permeabilizzate con saponina) e dei controlli negativi di ciascuna linea cellulare (cellule vitali non trattate).
Isolamento dell’RNA e RT-PCR quantitativa
L’RNA totale è stato isolato da106 cellule utilizzando il kit NucleoSpin Small RNA (Macherey Nagel, Düren, Germania) come descritto nel protocollo del produttore. Il cDNA è stato preparato con 35 ng di RNA totale mediante trascrizione inversa utilizzando il kit TaqMan® MicroRNA Reverse Transkription (Applied Biosystems™, Thermo Fisher Scientific) secondo le istruzioni del produttore. La quantificazione relativa dell’espressione dei microRNA delle cellule trattate rispetto al controllo negativo è stata eseguita con il ciclatore Eppendorf realplex4 (Eppendorf, Wesseling-Berzdorf, Germania) utilizzando 1.33 μl di cDNA in un volume totale di 20 μl contenente TaqMan® Universal Master Mix NoAmpErase® UNG (Applied Biosystems, Thermo Fisher Scientific) e i saggi TaqMan® MicroRNA per Mir-141 (ID 245445_mat), Mir-145 (ID 002278), Mir-375 (ID 000564) acquistati da Thermo Fisher Scientific. La procedura è stata mantenuta come descritto nel protocollo del produttore. I dati sono stati normalizzati rispetto al gene housekeeping RNU6B (ID 001093) e l’analisi è stata eseguita con Rest2009 (Qiagen, Hilden, Germania).
Analisi delle microsfere magnetiche Luminex
L’analisi dell’espressione proteica è stata eseguita con la tecnologia xMAP® Luminex Bead utilizzando uno strumento Luminex 200™ (Luminex Corp., Hertogenbosch, Paesi Bassi). I dati sono indicati come quantità totale (survivin pg/ml) o MFI netto (altri target) e sono stati mostrati con il software xPONENT 3.1 (Luminex Corp.). I risultati dei campioni con valori inferiori come MFI < background MFI + 2× deviazione standard sono stati esclusi dall’analisi. La quantificazione della survivin è stata eseguita con il kit ProcartaPlex Human Survivin Simplex (eBioscience, Francoforte sul Meno, Germania) utilizzando il surnatante della coltura cellulare come descritto nel protocollo del produttore. La quantità di Survivin è stata normalizzata alla concentrazione di proteine, stabilita con il saggio Pierce BCA (Thermo Fisher Scientific). Le proteine PDH e apoptotiche (BAD e JNK) sono state rilevate con test multiplex di Merck Millipore (Multi-species PDH Complex Magnetic Bead Panel e 7-Plex Early Apoptosis Magnetic Bead kit, Darmstadt, Germania). I campioni sono stati trattati come descritto nelle istruzioni del produttore. Inoltre, i campioni per le misurazioni della PDH sono stati filtrati con unità filtranti centrifughe ultrafree con una dimensione dei pori di 0,65 μm (Merck Millipore) a 7.000 rpm per 4 minuti.
Attività mitocondriale
Le cellule cresciute su piastre μ a 8 pozzetti (Ibidi, Martinsried, Germania) trattate con e senza 10 mM DCA sono state fissate con paraformaldeide al 4%, lavate tre volte con HBSS contenente calcio e magnesio e colorate con 4 μM MitoSox (Invitrogen, Thermo Fisher Scientific) per 15 minuti. Dopo la colorazione, le cellule sono state lavate tre volte con HBSS (Gibco, Thermo Fisher Scientific) e il nucleo cellulare è stato controtrattato con DAPI (diluizione 1:1.000, Sigma-Aldrich GmbH) per 5 minuti. L’imaging in fluorescenza è stato eseguito con un microscopio confocale a scansione laser invertito (Eclipse TE2000-E, Nikon, Düsseldorf, Germania) utilizzando un obiettivo a immersione in acqua 60× (Nikon). Le immagini sono state acquisite con il software EZ-C1 1.80 (Nikon). L’eccitazione è avvenuta con un laser a diodi a 408 nm (DAPI) e con un laser elio/neon a 543 nm (MitoSox). La fluorescenza cellulare totale di MitoSox, dedotto lo sfondo, è stata analizzata con ImageJ e normalizzata al numero di cellule.
Colorazione in immunofluorescenza di Ki67 e TUNEL
Le cellule sono state seminate, fissate e lavate come descritto sopra. Dopo il lavaggio con HBSS, le cellule sono state permeabilizzate con Triton X-100 allo 0,2% per 20 minuti, lavate e incubate per una notte con un anticorpo policlonale di coniglio specifico per il canino Ki67 con una diluizione di 1:150 (Life Technologies, Thermo Fisher Scientific). Per la colorazione, un anticorpo monoclonale anti-rabbit Alexa Fluor® 555 (Cell Signaling Technology, Leiden, Paesi Bassi) è stato incubato per 1 ora con una diluizione di 1:250 e le cellule sono state controcolorate con DAPI (1:1.000) per 5 minuti. Il protocollo di imaging in fluorescenza è stato lo stesso descritto sopra. La fluorescenza totale delle cellule è stata stabilita come descritto sopra. Per la colorazione TUNEL è stato utilizzato il kit Apoptag Fluorescein Direct (Merck Millipore) secondo le istruzioni del produttore. L’eccitazione è avvenuta con un argonlaser a 488 nm e l’imaging è stato eseguito come descritto sopra. È stata valutata la percentuale di cellule TUNEL positive.
Analisi statistica
L’analisi statistica dei dati è stata eseguita con il software SAS 7.1 (SAS Institute Inc., Cary, NC, USA). Per il confronto tra due medie è stato utilizzato il t-test a due code. Il valore di confidenza è stato fissato al 5% (P<0,05) ed è stato considerato statisticamente significativo.
Risultati
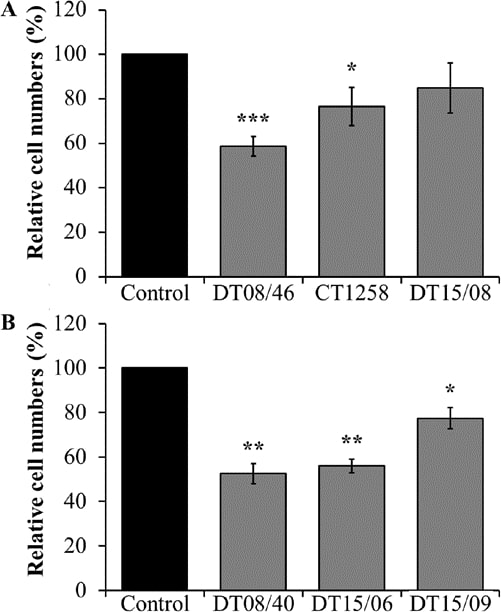
Conta delle cellule dopo il trattamento con DCA
Rispetto ai controlli negativi non trattati, le linee cellulari di adenocarcinoma prostatico DT08/46 (P<0,0001) e CT1258 (P=0,0122) hanno mostrato un numero di cellule significativamente inferiore dopo il trattamento con 10 mM DCA per 48 ore (Fig. 1A). La terza linea cellulare prostatica DT15/08 DCA non ha mostrato una riduzione significativa (P=0,0748), ma è stata osservata una tendenza a una quantità di cellule inferiore, rispettivamente un tasso di proliferazione inferiore nella coltura cellulare nativa (Fig. 1A ). Come mostrato nella Fig. 1B, lo stesso effetto di riduzione del DCA è stato osservato in tutte le linee cellulari TCC esaminate DT08/40 (P=0,0031), DT15/06 (P=0,0016) e DT15/09 (P=0,0143).
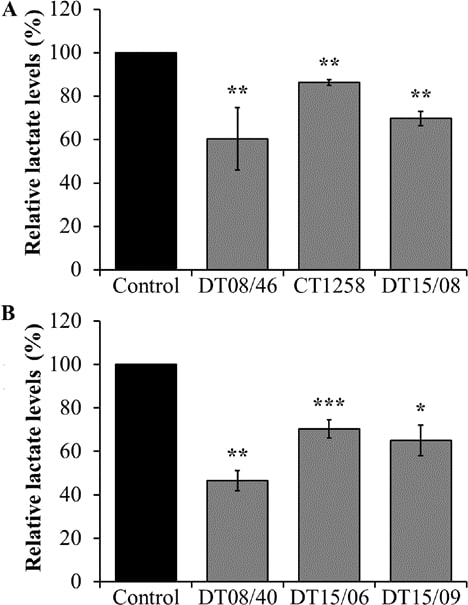
Livelli di lattato nel surnatante della coltura cellulare dopo esposizione al DCA
Per valutare l’effetto di riduzione del rilascio di lattato del trattamento con DCA dopo 48 ore, è stata misurata la quantità di lattato nel surnatante della coltura cellulare. In tutte le linee cellulari di entrambe le entità tumorali canine, il DCA 10 mM ha avuto un effetto significativo di riduzione del lattato (Fig. 2).
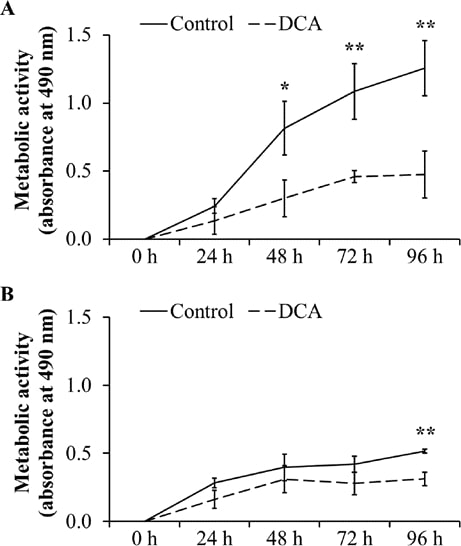
Attività metabolica dopo trattamento con DCA per un periodo di 96 ore
Per confermare l’effetto negativo sulla proliferazione cellulare, l’attività metabolica come indicatore di proliferazione e vitalità cellulare è stata analizzata mediante saggi MTT. Come mostrato nella Fig. 3A, l’attività metabolica di DT15/08 è risultata significativamente ridotta rispetto al rispettivo controllo negativo dopo 48 ore di trattamento con DCA (P=0,0202). Dopo 96 ore di trattamento continuo con DCA l’attività metabolica è risultata significativa (P=0,007). Un effetto simile è stato osservato nella DT08/40 (Fig. 3B) dopo 96 ore (P=0,0025) e nella DT15/06 (P=0,0419) dopo 96 ore (dati non mostrati) di trattamento con DCA. Le linee cellulari DT08/46, CT1258 e DT15/09 non hanno mostrato alcun effetto statisticamente significativo del DCA sull’attività metabolica (dati non mostrati).
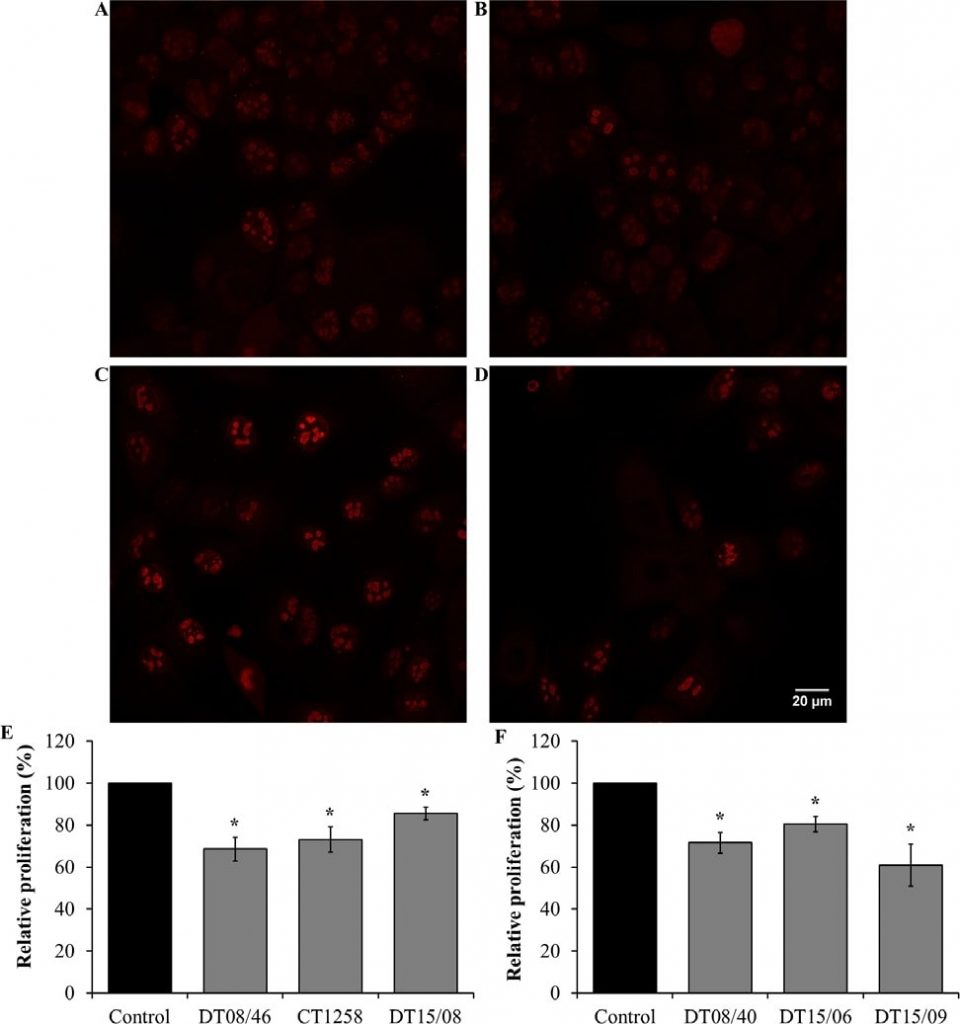
Proliferazione dopo trattamento con DCA per 48 h
La proliferazione cellulare nelle linee cellulari esposte a DCA 10 mM è stata analizzata mediante colorazione Ki67 e visualizzata con microscopia confocale a fluorescenza. Il DCA ha diminuito la quantità di Ki67, indicando una riduzione della proliferazione cellulare dopo 48 ore in tutte le linee cellulari valutate con significatività (P<0,05) (Fig. 4).
Effetto del DCA sull’apoptosi
L’effetto del DCA 10 mM sull’apoptosi e sulla vitalità è stato valutato con Annexin e Sytox utilizzando la citometria a flusso. Per quanto riguarda l’apoptosi e le cellule morte, non sono stati rilevati effetti statisticamente significativi in nessuna linea cellulare (dati non mostrati). La conferma dei tassi di apoptosi al FACS è stata effettuata mediante colorazione TUNEL per eliminare gli effetti negativi della tripsinizzazione sulle cellule coltivate. L’imaging è stato eseguito con microscopia confocale a fluorescenza. I risultati confermano il rapporto di apoptosi in tutte le linee cellulari trattate con DCA, eccetto la DT15/06. Livelli di apoptosi significativamente ridotti sono stati osservati in DT15/08 (P=0,022) e DT15/09 (P=0,0265). DT15/06 ha mostrato valori di apoptosi significativamente aumentati (P=0,041) (dati non mostrati). Le altre linee cellulari non hanno mostrato valori significativi di apoptosi.
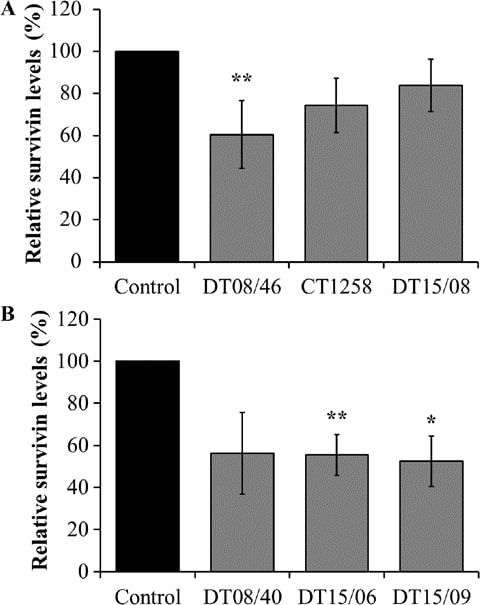
Espressione di Survivin analizzata con la tecnologia delle microsfere magnetiche xMAP
Il DCA ha ridotto significativamente la produzione di survivin nella linea cellulare di adenocarcinoma prostatico DT08/46 (P=0,0053) e nelle linee cellulari TCC DT15/06 (P=0,0027) e DT15/09 (P=0,0206). Le linee cellulari CT1258 (P=0,0761), DT15/08 (P=0,1551) e DT08/40 (P=0,0598) non hanno mostrato effetti significativi nella produzione di survivin (Fig. 5).
Espressione di Bcl-2-antagonista della morte cellulare (BAD) e c-jun N-terminal kinases (JNK) analizzata con la tecnologia delle microsfere magnetiche xMAP
Dopo 48 ore di incubazione con DCA, la forma fosforilata e quindi inattiva di BAD è aumentata in modo significativo solo in DT08/46 (P=0,0285), CT1258 (P=0,0039) e DT15/06 (P=0,0235). Inoltre, non è stato osservato alcun effetto del DCA sulla JNK fosforilata attiva, ad eccezione del DT08/46 (P=0,0044) che ha mostrato valori ridotti (dati non mostrati).
Espressione della piruvato-deidrogenasi (PDH) analizzata con la tecnologia delle microsfere magnetiche xMAP
Per confermare la diminuzione dei livelli di lattato come conseguenza della riduzione della glicolisi, la quantità di PDH fosforilata (PDH-P) (Ser232, Ser293, Ser300) è stata misurata con la Luminex Magnetic Bead Technology. La diminuzione dei livelli di PDH-P e quindi l’aumento degli enzimi attivi sono associati a una maggiore ossidazione del piruvato e al metabolismo dell’acetil coenzima A, che porta a un aumento dell’attività del ciclo TCA [35]. In PDH-P (Ser232) è stata osservata una diminuzione statisticamente significativa rispetto ai controlli non trattati in tutte le linee cellulari, ad eccezione di DT08/46. Questa linea cellulare ha mostrato un livello di PDH-P (Ser232) più alto ma non significativo. La PDH-P al residuo Ser293 è stata significativamente influenzata dal DCA solo nelle linee cellulari DT15/08 (P=0,0082) e DT15/06 (P=0,0122). In tutte le altre linee cellulari non è stato osservato alcun effetto dopo il trattamento con DCA. La PDH-P a Ser300 è stata compromessa dal DCA in modo significativo in DT08/46 (P=0,0060), CT1258 (P=0,0215), DT15/08 (P=0,0002) e DT15/06 (P=0,0097) (Tabella I).
Tabella I
Espressione di PDH-P dopo esposizione a DCA con valori medi relativi ± SD (%).
| Linea cellulare | PDH-P Ser232 | Valore P | PDH-P Ser293 | Valore P | PDH-P Ser300 | Valore P |
|---|---|---|---|---|---|---|
| Controllo | 100 | 100 | 100 | |||
| Adenocarcinoma prostatico | ||||||
| dT08/46 | 121.2±31.5 | 0.2715 | 127.7±27.8 | 0.0900 | 47.7±28.0 | 0.0060b |
| cT1258 | 11.0±5.1 | 0.0011b | 73.7±30.5 | 0.2740 | 24.3±19.5 | 0.0215a |
| dT15/08 | 18.0±17.6 | 0.0026b | 39.7±19.2 | 0.0082b | 18.4±6.8 | 0.0002c |
| Carcinoma a cellule transizionali | ||||||
| dT08/40 | 14.3±10.2 | 0.0047b | 84.5±22.8 | 0.3594 | 48.6±32.1 | 0.1090 |
| dT15/06 | 28.2±17.7 | 0.0039b | 72.3±5.4 | 0.0122a | 39.1±10.4 | 0.0097b |
| dT15/09 | 20.5±24.8 | 0.0308a | 99.8±71.3 | 0.9967 | 34.1±41.0 | 0.1085 |
PDH-P, piruvato deidrogenasi fosforilato; Ser, serina; DCA, dicloroacetato; ±, più meno.
a P<0,05;
b P<0,01;
c P<0,001;
SD, deviazione standard; %, percentuale.
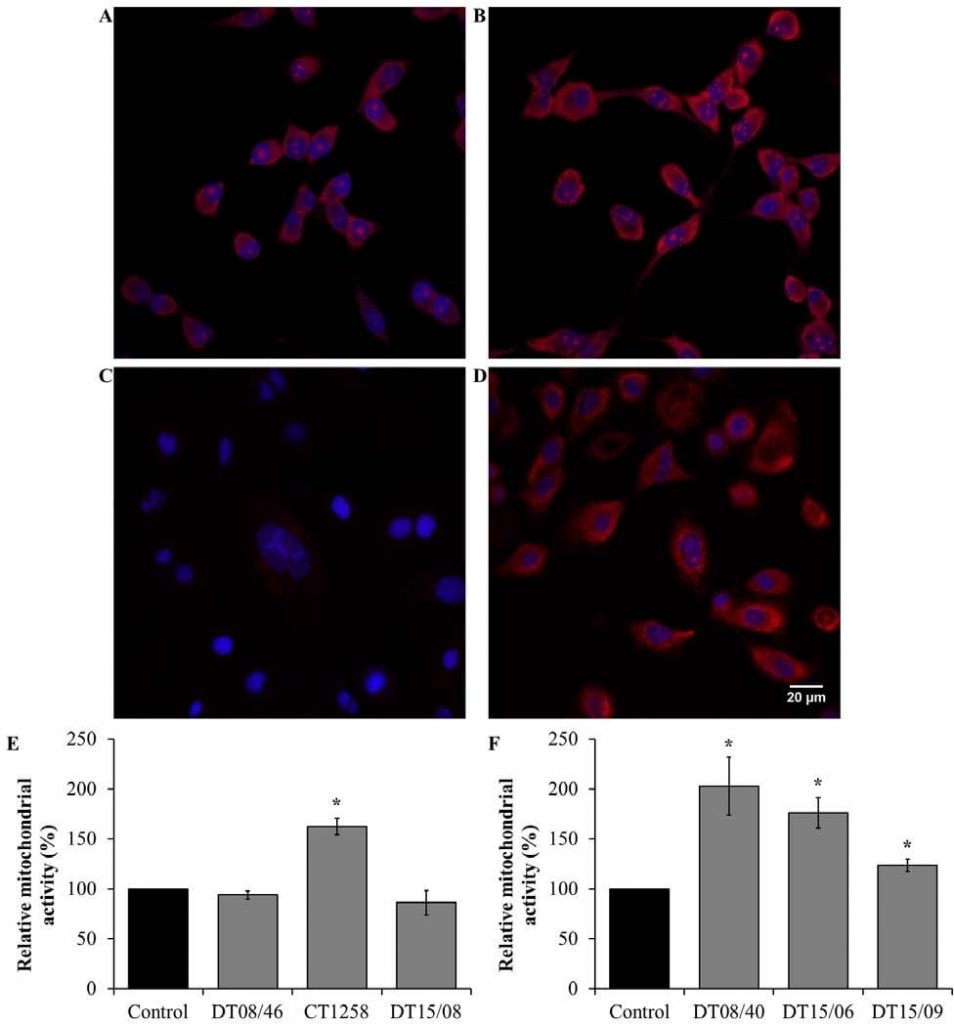
Attività mitocondriale dopo il trattamento con DCA
La verifica dell’alterazione metabolica dell’attività mitocondriale di ossidazione del glucosio è stata effettuata mediante la rilevazione delle specie reattive dell’ossigeno (ROS) derivate dai mitocondri. Nelle linee cellulari di adenocarcinoma prostatico (Fig. 6E) è stato osservato un aumento significativo dell’attività mitocondriale in CT1258 (P=0,0153). Le linee cellulari DT08/46 (P=0,2829) e DT15/08 (P=0,3082) non hanno mostrato alcun effetto di diminuzione dopo il trattamento con DCA. Al contrario, il DCA è stato in grado di aumentare significativamente l’attività mitocondriale (P<0,05) in tutte le linee cellulari TCC (Fig. 6F).
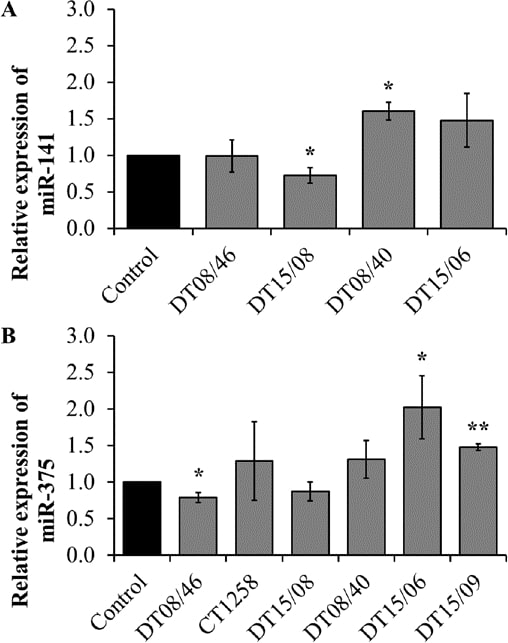
RT-PCR quantitativa dei miRNA
Per valutare se i tassi di proliferazione più bassi osservati siano correlati a cambiamenti nei microRNA, sono stati valutati tre miR coinvolti nella proliferazione o nell’apoptosi. L’espressione del miR-141 ha rivelato cambiamenti significativi con livelli ridotti nel DT15/08 (P=0,0451) e aumentati nel DT08/40 (P=0,0135) rispetto ai controlli non trattati, mentre il DT08/46 (P=0,9285) e il DT15/06 (P=0,1520) non hanno mostrato differenze rispetto ai controlli. In CT1258 e DT15/09 il miR-141 è stato downregolato ed escluso dall’analisi. DT08/46 (P=0,0322) ha mostrato una minore e DT15/06 (P=0,0267) e DT15/09 (P=0,0031) una maggiore espressione del miR-375. La linea cellulare di adenocarcinoma prostatico CT1258 non ha mostrato cambiamenti significativi in tutti i microRNA esaminati, rispettivamente. L’espressione del miR-145 non è stata influenzata dal trattamento con DCA in nessuna delle linee cellulari (escluse dall’analisi a causa di valori Ct >30). In sintesi, le linee cellulari di adenocarcinoma prostatico tendono a mostrare una downregulation di miR-141 (DT15/08) e miR-375 (DT08/46, DT15/08), mentre le linee cellulari di TCC hanno mostrato una upregulation di entrambi (Fig. 7).
Discussione
Il DCA ha ridotto il numero di cellule nell’adenocarcinoma prostatico e nelle linee cellulari di TCC. Ciò può avvenire sulla base di una ridotta proliferazione o di un aumento dell’apoptosi. In questo studio è stato osservato un effetto di riduzione della proliferazione, indicato dalla diminuzione del Ki67 in tutte le linee cellulari e dalla riduzione delle attività metaboliche in DT15/08 e DT08/40. Questo dato è in accordo con i risultati precedenti presentati da Bonnet et al in diverse linee cellulari umane [35] e da Sun et al nel cancro al seno [12]. Inoltre, una diminuzione della proliferazione cellulare dopo l’esposizione al DCA è stata osservata anche nel carcinoma prostatico umano [36], nel carcinoma del colon [37], nel colon [11] e nel carcinoma polmonare [38]. Tuttavia, in questo studio non è stata osservata l’induzione dell’apoptosi dipendente dai mitocondri da parte del DCA, come precedentemente riportato da diversi gruppi di ricerca [14,35,39]. Tuttavia, questo fenomeno è stato osservato anche da Feuerecker et al. in cellule di neuroblastoma murino e umano (40). Stockwin et al. hanno riportato che per l’induzione dell’apoptosi sono necessarie alte concentrazioni di DCA [38]. I risultati incoerenti in una linea cellulare (DT15/06) tra citometria a flusso e colorazione TUNEL non consentono di valutare se il DCA influisce sull’apoptosi. I risultati dell’espressione delle proteine apoptotiche JNK e BAD sono coerenti con gli effetti osservati in tutte le altre linee cellulari e supportano l’ipotesi che il DCA non abbia mostrato alcuna influenza sull’apoptosi nelle cellule tumorali della prostata e del TCC canino.
La Survivin, un inibitore dell’apoptosi e promotore del tumore [41,42], è risultata significativamente ridotta in tutte le linee cellulari che hanno mostrato un aumento dell’attività mitocondriale (DT15/06, DT15/09). È stato riferito che la diminuzione dei livelli di survivin induce l’apoptosi attraverso vie intrinseche mediante l’attivazione della caspasi-3 [41] ed è stata osservata in linee cellulari di cancro endometriale dopo l’esposizione al DCA [14]. Inaspettatamente, non è stato possibile osservare un aumento dell’apoptosi in queste linee cellulari, ad eccezione di DT15/06 (P=0,041), che ha mostrato un lieve ma inconsistente aumento della morte cellulare. Ciò porta a concludere che una diminuzione dei livelli di survivin potrebbe comportare una diminuzione della proliferazione. Inoltre, è ipotizzabile una deregolazione di altri geni coinvolti nell’induzione dell’apoptosi, che spiegherebbe perché la diminuzione dei livelli di survivin non ha portato all’apoptosi.
Il DCA è un inibitore della PDK che attiva indirettamente la PDH. A causa della diminuzione dei valori di PDH-P, il piruvato può essere ossidato dai mitocondri [4,5]. Una diminuzione della fosforilazione della PDH è stata osservata in diverse linee cellulari tumorali umane [16,37,43]. In accordo con i risultati pubblicati nelle linee cellulari umane, questo studio ha confermato la diminuzione della fosforilazione della PDH in tutte le cellule trattate con DCA, confermata dalla riduzione del rilascio di lattato in tutte le linee cellulari. Ciò indica che il DCA promuove l’ossidazione del glucosio nelle cellule tumorali canine e in quelle umane. Inoltre, in questo studio è stato dimostrato un aumento dei livelli di ROS nei mitocondri di tutte le linee cellulari, tranne due linee cellulari di adenocarcinoma prostatico. L’aumento delle specie ROS nei mitocondri, dovuto alla respirazione cellulare, conferma la diminuzione dei valori di PDH-P e la riduzione del lattato. È possibile che l’aumento dell’attività mitocondriale non si traduca in apoptosi, ma in un aumento della respirazione cellulare che impedisce la sopravvivenza delle cellule cancerose con un metabolismo modificato. Questo potrebbe spiegare l’aumento della vitalità delle cellule dopo l’esposizione al DCA. L’aumento della vitalità dopo l’esposizione al DCA è stato osservato anche da McPherson et al. che hanno riportato un aumento dell’ossidazione del piruvato e della vitalità degli embrioni in un modello di topo invecchiato [44].
è stato riportato che l’upregolazione del miR-375 ha effetti antiproliferativi in molte cellule come il cancro gastrico [45], il cancro del pancreas [46], le cellule simil-cardiomiocitarie fetali [47] e il cancro del colon [48]. Nella carcinogenesi della prostata il microRNA-375 ha effetti variabili a seconda del fenotipo tumorale. Costa-Pinheiro et al. hanno dimostrato effetti antiproliferativi nelle cellule PC-3 dovuti all’upregolazione e all’aumento dell’apoptosi nelle cellule 22Rv1 in seguito al knockdown del miR-375 [49]. I nostri risultati dimostrano che l’up- o downregulation del miR-375 dopo il trattamento con DCA non è coerente nelle linee cellulari di adenocarcinoma prostatico, il che è in accordo con i risultati sopra descritti. Rispetto alle cellule di carcinoma prostatico, tutte le linee cellulari di TCC hanno mostrato un aumento dei livelli di miR-375. Non esiste letteratura che descriva gli effetti del miR-375 nel cancro della vescica. È possibile che questi risultati siano in accordo con gli effetti antiproliferativi descritti in molte altre cellule. Gli stessi risultati sono stati osservati nel microRNA-141. Un’upregolazione del miR-141 ha inibito la proliferazione del cancro e la progressione del ciclo cellulare nelle cellule di neuroblastoma [50]. Inoltre, il miR-141 è downregolato nel cancro della vescica con invasione muscolare [51]. Nel nostro studio i miR-141 e miR-375 sono stati trovati upregolati in tutte le linee cellulari di TCC dopo il trattamento con DCA, indicando che questi cambiamenti causano tassi di proliferazione più bassi. Nel cancro alla prostata è stata segnalata l’upregolazione del miR-141 [52]. In questo studio i livelli di miR-141 sono leggermente diminuiti, ma riteniamo che questo effetto sia troppo lieve per influenzare le cellule tumorali. Tuttavia, un’analisi che consenta di determinare se un legame diretto tra DCA e cambiamenti in diversi microRNA sia causale per le risposte biologiche osservate richiederebbe un approccio trascrittomico completo.
In conclusione, questo studio illustra che le linee cellulari tumorali canine rispondono al trattamento con DCA e l’effetto può essere ridotto alla diminuzione del tasso di proliferazione, all’aumento dell’ossidazione del piruvato e dell’attività mitocondriale. I risultati mostrano anche differenze tra le entità tumorali esaminate. Le linee cellulari di TCC sembrano rispondere in modo coerente e sono più sensibili al trattamento con DCA rispetto alle linee cellulari di adenocarcinoma prostatico. Rispetto alla maggior parte delle linee cellulari umane, il DCA non ha influenzato l’apoptosi, il che potrebbe indicare che il DCA potrebbe essere utile per limitare la crescita del tumore nel cancro canino, ma non per ridurne le dimensioni.
Inoltre, il DCA può essere vantaggioso nel sensibilizzare le cellule tumorali canine ad altri farmaci antitumorali e quindi potrebbe essere appropriato per le terapie di combinazione [53]. Per garantire concentrazioni più elevate di DCA nei tessuti cancerosi ed evitare gravi effetti collaterali generalizzati, una terapia intralesionale, paragonabile alla chemioterapia intravescicale nell’uomo [54] e nei cani con TCC [55,56], potrebbe essere un’ulteriore possibilità. La concentrazione di 10 mM di DCA è stata scelta per consentire la comparabilità con altri studi umani in vitro [14,18,37]. Per gli studi clinici, la concentrazione di DCA deve essere rivalutata per quanto riguarda la compatibilità e gli effetti collaterali negativi. Pertanto, dovrebbero essere esaminati ulteriori studi con concentrazioni di DCA inferiori.
Abbreviazioni:
| DCA | dicloroacetato |
| PDH | piruvato deidrogenasi |
| PDK | piruvato deidrogenasi chinasi |
| TCC | carcinoma a cellule transizionali |
| RNA | acido ribonucleico |
| PCR | reazione a catena della polimerasi |
| JNK | c-jun N-terminale chinasi |
| BAD | Bcl-2-antagonista della morte cellulare |
| ROS | specie reattive dell’ossigeno |
| PBS | soluzione salina tamponata con fosfato |
RIFERIMENTI
1 Vail DM: Supporto al paziente oncologico veterinario in chemioterapia: Neutropenia e tossicità gastrointestinale. Top Companion Anim Med. 24:122-129. 2009.2 Cornell KK, Bostwick DG, Cooley DM, Hall G, Harvey HJ, Hendrick MJ, Pauli BU, Render JA, Stoica G, Sweet DC, et al: Clinical and pathologic aspects of spontaneous canine prostate carcinoma: A retrospective analysis of 76 cases. Prostata. 45:173-183. 2000.
3 Mutsaers AJ, Widmer WR e Knapp DW: Carcinoma a cellule transizionali canino. J Vet Intern Med. 17:136-144. 2003.
4 Sutendra G e Michelakis ED: La piruvato deidrogenasi chinasi come nuovo bersaglio terapeutico in oncologia. Front Oncol. 3:382013.
5 Stacpoole PW: La farmacologia del dicloroacetato. Metabolismo. 38:1124-1144. 1989.
6 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW e Shuster JJ: Valutazione del trattamento a lungo termine di bambini con acidosi lattica congenita con dicloroacetato. Pediatria. 121:e1223-e1228. 2008.
7 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, et al: Studio clinico controllato del dicloroacetato per il trattamento dell’acidosi lattica congenita nei bambini. Pediatria. 117:1519-1531. 2006.
8 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA e Stacpoole PW: riduzione del colesterolo sierico in due pazienti con ipercolesterolemia familiare omozigote mediante dicloroacetato. Aterosclerosi. 33:285-293. 1979.
9 Stacpoole PW, Moore GW e Kornhauser DM: Effetti metabolici del dicloroacetato in pazienti con diabete mellito e iperlipoproteinemia. N Engl J Med. 298:526-530. 1978.
10 Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, et al: Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 3:420-430. 2010.
11 Sánchez-Aragó M, Chamorro M e Cuezva JM: La selezione di cellule tumorali con mitocondri repressi innesca la progressione del cancro del colon. Carcinogenesi. 31:567-576. 2010.
12 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG e Blackburn AC: L’inversione del fenotipo glicolitico da parte del dicloroacetato inibisce la crescita delle cellule metastatiche del cancro al seno in vitro e in vivo. Breast Cancer Res Treat. 120:253-260. 2010.
13 Saed GM, Fletcher NM, Jiang ZL, Abu-Soud HM e Diamond MP: Il dicloroacetato induce l’apoptosi delle cellule epiteliali di cancro ovarico attraverso un meccanismo che coinvolge la modulazione dello stress ossidativo. Reprod Sci. 18:1253-1261. 2011.
14 Wong JY, Huggins GS, Debidda M, Munshi NC e De Vivo I: Il dicloroacetato induce apoptosi nelle cellule del cancro endometriale. Gynecol Oncol. 109:394-402. 2008.
15 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2:31ra342010.
16 Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al: Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 69:734-744. 2016.
17 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD e Coomber BL: Il dicloroacetato influisce sulla proliferazione ma non sulla sopravvivenza delle cellule di cancro colorettale umano. Apoptosi. 20:63-74. 2015.
18 Madhok BM, Yeluri S, Perry SL, Hughes TA e Jayne DG: Il dicloroacetato induce apoptosi e arresto del ciclo cellulare in cellule di cancro del colon-retto. Br J Cancer. 102:1746-1752. 2010.
19RuggieriV, Agriesti F, Scrima R, Laurenzana I, Perrone D, Tataranni T, Mazzoccoli C, Lo Muzio L, Capitanio N e Piccoli C: Il dicloroacetato, un farmaco mirato ai mitocondri selettivo per il carcinoma orale a cellule squamose: una prospettiva metabolica del trattamento. Oncotarget. 6:1217-1230. 2015.
20 Xintaropoulou C, Ward C, Wise A, Marston H, Turnbull A e Langdon SP: Un’analisi comparativa degli inibitori della via della glicolisi in modelli di linee cellulari di cancro al seno e all’ovaio. Oncotarget. 6:25677-25695. 2015.
21 Cicmanec JL, Condie LW, Olson GR e Wang SR: Studio di tossicità a 90 giorni del dicloroacetato nei cani. Fundam Appl Toxicol. 17:376-389. 1991.
22 Maisenbacher HW III, Shroads AL III, Zhong G, Daigle AD, Abdelmalak MM, Samper IS, Mincey BD, James MO e Stacpoole PW: Farmacocinetica del dicloroacetato orale nei cani. J Biochem Mol Toxicol. 27:522-525. 2013.
23 Lukas G, Vyas KH, Brindle SD, Le Sher AR e Wagner WE Jr: Disposizione biologica del dicloroacetato di sodio negli animali e nell’uomo dopo somministrazione endovenosa. J Pharm Sci. 69:419-421. 1980.
24 Park R, Arieff AI, Leach W e Lazarowitz VC: Trattamento dell’acidosi lattica con dicloroacetato nei cani. J Clin Invest. 70:853-862. 1982.
25 Racker E: Storia dell’effetto Pasteur e della sua patobiologia. Mol Cell Biochem. 5:17-23. 1974.
26 Warburg O, Wind F e Negelein E: Über den Stoffwechsel von Tumoren im Körper. Klin Wochenschr. 5:829-832. 1926.
27 Kim JW, Tchernyshyov I, Semenza GL e Dang CV: Espressione mediata da HIF-1 della piruvato deidrogenasi chinasi: Un interruttore metabolico necessario per l’adattamento cellulare all’ipossia. Cell Metab. 3:177-185. 2006.
28 Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC e Thompson CB: Il fattore di trascrizione HIF-1alfa svolge un ruolo critico nella regolazione dipendente dai fattori di crescita della glicolisi aerobica e anaerobica. Genes Dev. 21:1037-1049. 2007.
29 Zhao Y, Butler EB e Tan M: Prendere di mira il metabolismo cellulare per migliorare la terapia del cancro. Cell Death Dis. 4:e5322013.
30 Plas DR e Thompson CB: Il metabolismo cellulare nella regolazione della morte cellulare programmata. Trends Endocrinol Metab. 13:75-78. 2002.
31 Simon D, Knebel JW, Baumgartner W, Aufderheide M, Meyer-Lindenberg A e Nolte I: Efficacia in vitro dei chemioterapici determinata dalle concentrazioni inibitorie del 50% in colture cellulari di tumori della ghiandola mammaria ottenuti da cani. Am J Vet Res. 62:1825-1830. 2001.
32 Knapp DW, Chan TC, Kuczek T, Reagan WJ e Park B: Valutazione della citotossicità in vitro di farmaci antinfiammatori non steroidei contro cellule tumorali canine. Am J Vet Res. 56:801-805. 1995.
33 Sartin EA, Barnes S, Toivio-Kinnucan M, Wright JC e Wolfe LG: Proprietà eterogenee di linee cellulari clonali derivate da carcinomi mammari canini e sensibilità al tamoxifene e alla doxorubicina. Anticancer Res. 13:229-236. 1993.
34 Winkler S, Murua Escobar H, Eberle N, Reimann-Berg N, Nolte I e Bullerdiek J: Costituzione di una linea cellulare derivata da un carcinoma prostatico canino con cariotipo altamente riarrangiato. J Hered. 96:782-785. 2005.
35 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 11:37-51. 2007.
36 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C e Rosser CJ: Il dicloroacetato (DCA) sensibilizza alle radiazioni in vitro sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata. 68:1223-1231. 2008.
37 Ho N e Coomber BL: Espressione della piruvato deidrogenasi chinasi e cambiamenti metabolici in seguito all’esposizione al dicloroacetato in cellule umane anossiche di cancro colorettale. Exp Cell Res. 331:73-81. 2015.
38 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG e Newton DL: Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer. 127:2510-2519. 2010.
39 Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C e Chen HZ: Il dicloroacetato sposta il metabolismo dalla glicolisi all’ossidazione del glucosio e mostra un’inibizione sinergica della crescita con il cisplatino nelle cellule HeLa. Int J Oncol. 38:409-417. 2011.
40 Feuerecker B, Seidl C, Pirsig S, Bruchelt G e Senekowitsch-Schmidtke R: Il DCA promuove la progressione dei tumori del neuroblastoma in topi nudi. Am J Cancer Res. 5:812-820. 2015.
41 Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC e Altieri DC: Controllo dell’apoptosi e del checkpoint del fuso mitotico da parte della survivin. Nature. 396:580-584. 1998.
42 Ambrosini G, Adida C e Altieri DC: Un nuovo gene anti-apoptosi, la survivin, espresso nel cancro e nel linfoma. Nat Med. 3:917-921. 1997.
43 Abemayor E, Kovachich GB e Haugaard N: Effetti del dicloroacetato sulla piruvato deidrogenasi cerebrale. J Neurochem. 42:38-42. 1984.
44 McPherson NO, Zander-Fox D e Lane M: La stimolazione del metabolismo mitocondriale dell’embrione con l’acido dicloroacetico in un modello di topo invecchiato migliora lo sviluppo e la vitalità dell’embrione. Fertil Steril. 101:1458-1466. 2014.
45 Zhou N, Qu Y, Xu C e Tang Y: L’up-regolazione del microRNA-375 aumenta la sensibilità al cisplatino delle cellule di cancro gastrico umano regolando ERBB2. Exp Ther Med. 11:625-630. 2016.
46 Zhou J, Song S, He S, Zhu X, Zhang Y, Yi B, Zhang B, Qin G e Li D: Il microRNA-375 ha come bersaglio PDK1 nel carcinoma pancreatico e sopprime la crescita cellulare attraverso la via di segnalazione Akt. Int J Mol Med. 33:950-956. 2014.
47 Wang L, Song G, Liu M, Chen B, Chen Y, Shen Y, Zhu J e Zhou X: La sovraespressione del microRNA-375 influenza la proliferazione, l’apoptosi e la differenziazione delle cellule P19 attraverso la via di segnalazione Notch. Int J Mol Med. 37:47-55. 2016.
48 Zaharie F, Muresan MS, Petrushev B, Berce C, Gafencu GA, Selicean S, Jurj A, Cojocneanu-Petric R, Lisencu CI, Pop LA, et al: il microRNA-375 trasportato dagli esosomi inibisce la progressione e la diffusione cellulare attraverso il blocco di Bcl-2 nel cancro del colon. J Gastrointestin Liver Dis. 24:435-443. 2015.
49 Costa-Pinheiro P, Ramalho-Carvalho J, Vieira FQ, Torres-Ferreira J, Oliveira J, Gonçalves CS, Costa BM, Henrique R e Jerónimo C: Il microRNA-375 svolge un doppio ruolo nella carcinogenesi della prostata. Clin Epigenetics. 7:422015.
50 Wang Z, Lei H e Sun Q: Il microRNA-141 e il suo gene associato FUS modulano la proliferazione, la migrazione e la chemiosensibilità al cisplatino in linee cellulari di neuroblastoma. Oncol Rep. 35:2943-2951. 2016.
51 Mahdavinezhad A, Mousavi-Bahar SH, Poorolajal J, Yadegarazari R, Jafari M, Shabab N e Saidijam M: Valutazione dell’espressione di miR-141, miR-200c, miR-30b e delle caratteristiche clinicopatologiche del cancro della vescica. Int J Mol Cell Med. 4:32-39. 2015.
52 Brase JC, Johannes M, Schlomm T, Fälth M, Haese A, Steuber T, Beissbarth T, Kuner R e Sültmann H: I miRNA circolanti sono correlati alla progressione del tumore nel cancro alla prostata. Int J Cancer. 128:608-616. 2011.
53 Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang PC, Zhao YL, Xu Y, Jia L, Zhang X, et al: Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction. Mol Pharm. 9:634-644. 2012.
54 Porten SP, Leapman MS e Greene KL: Chemioterapia intravescicale nel cancro della vescica non muscolo-invasivo. Indian J Urol. 31:297-303. 2015.
55 Song D, Wientjes MG, Gan Y e Au JL: Farmacocinetica del tessuto vescicale ed effetto antitumorale della 5-fluorouridina intravescicale. Clin Cancer Res. 3:901-909. 1997.
56 Abbo AH, Jones DR, Masters AR, Stewart JC, Fourez L e Knapp DW: Studio clinico di fase I e farmacocinetica della mitomicina C intravescicale in cani con carcinoma a cellule transizionali localizzato della vescica urinaria. J Vet Intern Med. 24:1124-1130. 2010.