Tiziana Tataranni 1 and Claudia Piccoli 1,2
1Laboratory of Pre-Clinical and Translational Research, IRCCS-CROB, Referral Cancer Center of Basilicata, Rionero in Vulture (Pz), 85028, Italy
2Department of Clinical and Experimental Medicine, University of Foggia, Foggia 71121, Italy
Correspondence should be addressed to Tiziana Tataranni; [email protected]
Guest Editor: Kanhaiya Singh
Copyright © 2019 Tiziana Tataranni and Claudia Piccoli. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 24 July 2019
Revised: 12 September 2019
Accepted: 11 October 2019
Published online: 14 November 2019
An extensive body of literature describes anticancer property of dichloroacetate (DCA), but its effective clinical administration in cancer therapy is still limited to clinical trials. The occurrence of side effects such as neurotoxicity as well as the suspicion of DCA carcinogenicity still restricts the clinical use of DCA. However, in the last years, the number of reports supporting DCA employment against cancer increased also because of the great interest in targeting metabolism of tumour cells. Dissecting DCA mechanism of action helped to understand the bases of its selective efficacy against cancer cells. A successful coadministration of DCA with conventional chemotherapy, radiotherapy, other drugs, or natural compounds has been tested in several cancer models. New drug delivery systems and multiaction compounds containing DCA and other drugs seem to ameliorate bioavailability and appear more efficient thanks to a synergistic action of multiple agents. The spread of reports supporting the efficiency of DCA in cancer therapy has prompted additional studies that let to find other potential molecular targets of DCA. Interestingly, DCA could significantly affect cancer stem cell fraction and contribute to cancer eradication. Collectively, these findings provide a strong rationale towards novel clinical translational studies of DCA in cancer therapy.
INTRODUCTION
Cancer is one of the leading causes of death worldwide. Despite the significant progression in diagnostic and therapeutic approaches, its eradication still represents a challenge. Too many factors are responsible for therapy failure or relapse, so there is an urgent need to find new approaches to treat it. Apart from the typical well-known properties featuring malignant cells, including abnormal proliferation, deregulation of apoptosis, and cell cycle [1, 2], cancer cells also display a peculiar metabolic machine that offers a further promising approach for cancer therapy [3–5]. Our group had already suggested the importance of a metabolic characterization of cancer cells to predict the efficacy of a metabolic treatment [6]. Drugs able to affect cancer metabolism are already under consideration, showing encouraging results in terms of efficacy and tolerability [7]. In the last decade, the small molecule DCA, already used to treat acute and chronic lactic acidosis, inborn errors of mitochondrial metabolism, and diabetes [8], has been largely purposed as an anticancer drug. DCA is a 150 Da water-soluble acid molecule, analog of acetic acid in which two of the three hydrogen atoms of the methyl group have been replaced by chlorine atoms (Figure 1(a)) [9]. DCA administration in doses ranging from 50 to 200 mg/Kg/die is associated to a decrease of tumour mass volume, proliferation rate, and metastasis dissemination in several preclinical models [10]. Our group had already observed an inverse correlation between DCA ability to kill cancer cells and their mitochondrial respiratory capacity in oral cell carcinomas [11]. Moreover, we recently described DCA ability to affect mitochondrial function and retarding cancer progression in a pancreatic cancer model [12]. To date, consistent data from clinical trials and case reports describing DCA administration in cancer patients are available [13–16], but, despite the growing body of literature sustaining the efficacy of DCA against cancer, it is not under clinical use yet. This review is aimed at summarizing the very recent reports suggesting the employment of DCA in cancer therapy, in combination with chemotherapy agents, radiotherapy, and other chemical or natural compounds showing anticancer properties. Moreover, we described data about new purposed pharmacological formulations of DCA able to avoid side effects and ameliorate drug bioavailability and efficacy, further encouraging its possible clinical employment. Finally, we reviewed latest findings suggesting other potential mechanisms of action of DCA, including new data about its aptitude to affect cancer stem cell fraction.
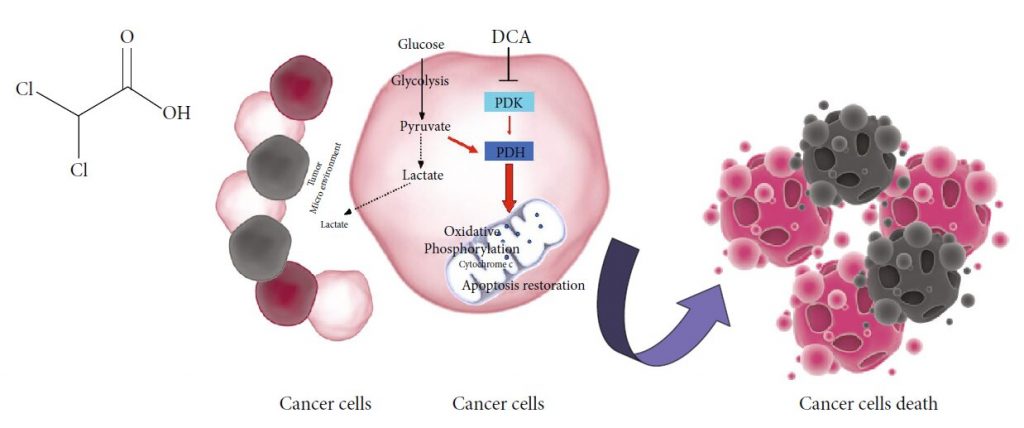
DCA and Cancer: Mechanism of Action
The potential efficacy of DCA in cancer therapy comes from metabolic properties of cancer cells, typically characterized by increased glycolytic activity and reduced mitochondrial oxidation, regardless of oxygen availability, the well-known Warburg effect [17]. The excessive glycolysis and the resulting lactate overproduction provoke a state of metabolic acidosis in tumour microenvironment [18]. Glycolysisderived lactate is taken up by surrounding cells to support tumour growth and inhibits apoptotic cell death mechanisms [19, 20]. Several enzymes involved in glycolysis regulate apoptosis, and their overexpression in cancer cells contributes to apoptosis suppression [21]. In this setting, salts of DCA selectively target cancer cells shifting their metabolism from glycolysis to oxidative phosphorylation by inhibition of pyruvate dehydrogenase kinase (PDK), the inhibitor of pyruvate dehydrogenase (PDH) [10]. PDH activation fosters mitochondrial oxidation of pyruvate and disrupts the metabolic advantage of cancer cells. Mitochondrial DNA mutations, often occurring in tumorigenesis and resulting in respiratory chain dysfunction [22, 23], make malignant cells unable to sustain cellular energy demand. Furthermore, reducing lactate production, DCA counteracts the acidosis state of tumour microenvironment, contributing to the inhibition of tumour growth and dissemination [24]. The delivery of pyruvate into mitochondria causes organelles remodelling resulting in an increased efflux of cytochrome c and other apoptotic-inducing factors and upregulation of ROS levels with a consequent reduction of cancer cell viability [9] (Figure 1(b)).
Side Effects and Limitations to DCA Employment
Clinical employment of DCA is available in both oral and parenteral formulations, and doses range from 10 to 50 mg/Kg/die [25]. No evidence of severe hematologic, hepatic, renal, or cardiac toxicity confirms DCA safety [26]. Common gastrointestinal side effects often occur in a percentage of patients treated with DCA [15]. The best-known limitation to DCA administration, observed both in preclinical and in clinical studies, is peripheral neuropathy [27]. The selectivity of DCA-induced damage for the nervous system may be due to the lack of well-equipped machinery able to handle a more sustained oxidative phosphorylation in cells producing ATP mostly via glycolysis [28]. The resulting mitochondrial overload compromises the antioxidant systems’ efficiency, unable to face the excessive amount of ROS. In this setting, the contemporary administration of antioxidants should represent a further strategy to minimize DCA-induced neuropathy [27]. The expression and the activity of glutathione transferase zeta1 (GSTZ1), the first enzyme responsible for DCA clearance, may influence the entity of damage. Nonsynonymous functional singlenucleotide polymorphisms (SNPs) in human GSTZ1 gene give rise to different haplotypes that are responsible for a different DCA kinetic and dynamics. A clear association between GSTZ1 haplotype and DCA clearance has been demonstrated. On this basis, a personalized DCA dosage, not only based on body weight, may minimize or prevent adverse effects in patients chronically treated with this drug [29]. The occurrence of neuropathy is associated to DCA chronic oral administration and is a reversible effect, limited to the time of treatment [30]. The intravenous route reduces, OH Cl Cl O (a) Cancer cells Cancer cells Cancer cells death Lactate Tumor Micro environment Lactate Pyruvate Glycolysis PDK DCA PDH Oxidative Phosphorylation Apoptosis restoration Cytochrome c Glucose (b) Figure 1: (a) Chemical structure of DCA. (b) Mechanism of action of DCA: PDK: pyruvate dehydrogenase kinase; PDH: pyruvate dehydrogenase. Black dotted lines, biochemical processes inhibited by DCA; Red arrows, metabolic pathways activated by DCA. 2 Oxidative Medicine and Cellular Longevity therefore, the potential for neurotoxicity and let the achievement of higher drug concentrations bypass the digestive system [13].
Since DCA is among water disinfection by-products found in low concentrations in drinking water, its potential carcinogenicity is under evaluation. Studies performed in mouse models associate DCA early-life exposure to an increased incidence of hepatocellular tumours [31]. It is conceivable that persistent changes in cell metabolism induced by DCA may produce epigenetic effects. Long-term induction of PDH and other oxidative pathways related to glucose metabolism could contribute to increase reactive oxygen species and mitochondrial stress [27]. However, no evidence of carcinogenetic effect is reported in clinical studies, when DCA is administered in cancer therapy.
Synergistic Effect of DCA and Chemotherapeutic Agents
Combining different drugs is a well-accepted strategy to produce a synergistic beneficial effect in cancer therapy, reducing drug dosage, minimizing toxicity risks, and overcoming drug resistance. Coadministration of DCA and traditional chemotherapeutic agents has been purposed and tested in several cancer models (Table 1). DCA treatment seems to improve the efficacy of chemotherapy by inducing biochemical and metabolic alterations, resulting in significant changes of cancer cells’ energetic balance. A study performed in non-smallcell lung cancer (NSCLC) showed both in vitro and in vivo that coadministration of DCA with paclitaxel increased the efficiency of cell death through autophagy inhibition [32]. An effective combination of DCA and doxorubicin (DOX) was tested in HepG2 cells, demonstrating the ability of DCA to disrupt cellular antioxidant defences, thus favouring oxidative damage in turn triggered by DOX treatment [33]. There is a strong association between PDK overexpression and chemoresistance; thus, it is conceivable that PDK inhibition might help to resensitize cancer cells to drugs. PDK2 isoform overexpression was associated to paclitaxel resistance in NSCLC. Interestingly, DCA combination with paclitaxel was more effective in killing resistant cells than either paclitaxel or DCA treatment alone [34]. Similarly to NSCLC, an interesting in vivo study performed in advanced bladder cancer showed an increased expression of PDK4 isoform in high grade compared to lower-grade cancers and cotreatment of DCA and cisplatin dramatically reduced tumour volumes as compared to either DCA or cisplatin alone [35]. A recent study confirmed the ability of DCA to revert PDK4-related chemoresistance also in human hepatocellular carcinoma (HCC) [36].
| Tumour entity | Model system | Chemotherapy drug coadministered with DCA | Mechanism of action | Outcome | References |
| Lung cancer | A549-H1975 cell lines/ xenograft model | Paclitaxel | Autophagy inhibition | Efficacious cancer chemotherapy sensitization | [32] |
| Hepatocarcinoma | HepG2 cell line | Doxorubicin | Antioxidant defence disruption | Increased cellular damage by oxidative stress induction | [33] |
| Lung cancer | A549 cell line | Paclitaxel | Increased chemosensitivity through PDK2 inhibition | Paclitaxel resistance overcome | [34] |
| Bladder cancer | HTB-9, HT-1376, HTB-5, HTB-4 cell lines/xenograft model | Cisplatin | Increased chemosensitivity through PDK4 inhibition | Increased cell death of cancer cells and potential therapeutic advantage | [35] |
| Hepatocarcinoma | Sphere cultures from HepaRG and BC2 cell lines | Cisplatin, sorafenib | Increased chemosensitivity through PDK4 inhibition | Improved therapeutic effect of chemotherapy by mitochondrial activity restoration | [36] |
Synergistic Effect of DCA and Other Potential Anticancer Drugs
A consistent body of literature suggests positive effects of DCA coadministration with compounds currently employed to treat other diseases but showing anticancer properties in several cancer models (Table 2). Contemporary administration of DCA and the antibiotic salinomycin, recently rediscovered for its cytotoxic properties as a potential anticancer drug, has been tested in colorectal cancer cell lines. Their treatment seems to exert a synergistic cytotoxic effect by inhibiting the expression of proteins related to multidrug resistance [37]. Cancer cells lacking metabolic enzymes involved in arginine metabolism may result to sensitivity to arginase treatment. Interestingly, a combined administration of recombinant arginase and DCA produces antiproliferative effects in triple-negative breast cancer, due to the activation of p53 and the induction of cell cycle arrest [38]. COX2 inhibitors, primarily used as anti-inflammatory drugs, have been recently suggested as antitumor drugs because of their antiproliferative activity. An intriguing study performed in cervical cancer cells showed the inability of DCA to kill cervical cancer cells overexpressing COX2 and demonstrated that COX2 inhibition by celecoxib makes cervical cancer cells more sensitive to DCA both in vitro and in vivo experiments [39]. Since DCA fosters oxidative phosphorylation by decreasing glycolytic activity, the combination of DCA with other drugs enhancing a state of glucose dependence may be a promising strategy. Such an approach has been tested in head and neck cancer in which the administration of propranolol, a nonselective beta-blocker able to affect tumour cells’ mitochondrial metabolism, produced glycolytic dependence and energetic stress, making cells more vulnerable to DCA treatment [40]. Similar results were obtained in melanoma cells in which the administration of retinoic acid receptor β (RARβ) inhibitors confer sensitization to DCA [41]. A positive effect of DCA coadministration with metformin, a hypoglycaemic drug widely used to treat diabetes was demonstrated in a preclinical model of glioma [42] as well as in a low metastatic variant of Lewis lung carcinoma (LLC) [43]. Jiang and colleagues investigated the effects of phenformin, a metformin analog, and DCA in glioblastoma, demonstrating that contemporary inhibition of complex I and PDK by phenformin and DCA, respectively, decreased self-renewal and viability of glioma stem cells (GSCs), thus suggesting their possible employment to affect cancer stem cell fraction [44].
| Drug | Main function | Tumour entity | Model system | Outcome | References |
| Salinomycin | Antibiotic | Colorectal cancer | DLD-1 and HCT116 cell lines | Inhibition of multidrug resistance-related proteins | [37] |
| Arginase | Arginine metabolism | Breast cancer | MDA-MB231 and MCF-7/ xenograft model | Antiproliferative effect due to p53 activation and cell cycle arrest | [38] |
| COX2 inhibitors | Inflammation | Cervical cancer | HeLa and SiHa cell lines/ xenograft model | Cancer cell growth suppression | [39] |
| Propranolol | Beta-blocker | Head and neck cancer | mEERL and MLM3 cell lines/C57Bl/6 m | Glucose dependence promotion and enhancement of chemoradiation effect | [40] |
| RARβ inhibitors | Vitamin A metabolism | Melanoma | ED-007, ED-027, ED-117, and ED196 cell lines | Glucose dependence promotion and sensitization to DCA | [41] |
| Metformin | Diabetes | Glioma, Lewis lung carcinoma | Xenograft model; LLC/R9 cells | Prolonged lifespan of mice with glioma; severe glucose dependency in tumour microenvironment | [42, 43] |
| Phenformin | Diabetes | Glioblastoma | Glioma stem cells/xenograft model | Self-renewal inhibition of cancer stem cells | [44] |
Combined Use of DCA and Natural Compounds
The clinical employment of natural compounds represents a promising novel approach to treat several diseases [45]. An increasing body of literature supports the detection, among natural compounds, of biologically active substances isolated by plants, mushrooms, and bacteria or marine organism that show beneficial effects for human health [46–48]. The assumption of natural compounds or their derivatives seems to represent an encouraging approach to prevent cancer initiation or recurrence, and it is generally called chemoprevention [49]. Moreover, natural substances produce beneficial effects in cancer therapy when coadministered with other drugs, showing their ability to overcome drug resistance, to increase anticancer potential, and to reduce drug doses and toxicity [50, 51]. Interestingly, the coadministration of DCA and natural compounds has been recently purposed. A study investigated the combined effect of DCA with essential oil-blended curcumin, a compound with beneficial properties both in prevention and treatment of cancer [52], demonstrating an anticancer potential against HCC [53]. In particular, the combination of both compounds synergistically reduced cell survival, promoting cell apoptosis and inducing intracellular ROS generation. Betulin, a natural compound isolated from birch bark, is already known for its antiproliferative and cytotoxic effects against several cancer cell lines [54–56]. An in vitro investigation of the antitumor activity of betulin derivatives in NSCLC confirmed its ability to inhibit in vivo and in vitro growth of lung cancer cells, blocking G2/M phase of the cell cycle and inducing caspase activation and DNA fragmentation. Interestingly, betulin derivative Bi-L-RhamBet was able to perturb mitochondrial electron transport chain (ETC), inducing ROS production. Given the property of DCA to increase the total oxidation of glucose in mitochondria via the Krebs cycle and ETC, the authors combined Bi-L-RhamBet with DCA, demonstrating its significant potentiated cytotoxicity [57].
DCA and Radiosensitization
Radiotherapy represents a further strategy to treat cancer and provides a local approach by the administration of highenergy rays [58]. The main effect of radiation is the induction of ROS with a consequent DNA damage, chromosomal instability, and cell death by apoptosis [59]. However, several tumours show or develop radioresistance that is responsible for radiotherapy failure and high risk of tumour recurrence or metastasis [60]. Several factors may be responsible of radioresistance [61]. Among these, hypoxia, a common condition of tumour microenvironment characterized by low oxygen levels and reduced ROS species generation, can block the efficacy of ionizing radiations [62]. Increasing tumour oxygenation so to favour a considerable amount of ROS [63] or directly induce ROS production may therefore represent a strategy to increase radiosensitization [64, 65]. In this setting, DCA administration, known to induce ROS production [11, 66], could represent a strategy to overcome tumour radioresistance. Moreover, metabolic alterations featuring cancer development are known to affect radiosensitivity [67, 68]. Therefore, targeting cancer metabolic intermediates may represent a strategy to improve a selective cancer response to irradiation [69]. The efficacy of DCA to increase radiation sensitivity has been already demonstrated both in glioblastoma cells [70] and in oesophageal squamous cell carcinoma [71]. More recently, it was demonstrated that DCA increases radiosensitivity in a cellular model of medulloblastoma, a fatal brain tumour in children, inducing alterations of ROS metabolism and mitochondrial function and suppressing DNA repair capacity [72]. Since the role of immunotherapy in the restoration of the immune defences against tumour progression and metastasis is arousing great attention in the last years [73], Gupta and Dwarakanath provided a state of the art of the possible effects of glycolytic inhibitors, including DCA, on tumour radiosensitization, focusing their attention on the interplay between metabolic modifiers and immune modulation in the radiosensitization processes [74]. Interestingly, they reported the ability of DCA to promote immune stimulation through the inhibition of lactate accumulation, further sustaining its utilization as adjuvant of radiotherapy.
DCA and New Drug Formulations
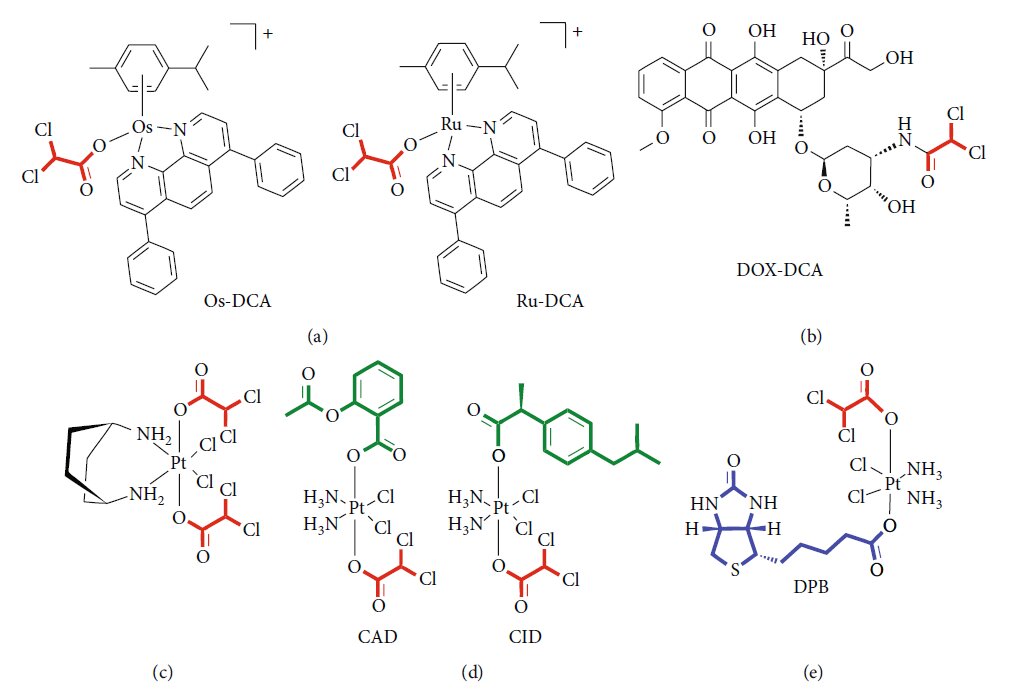
There is a growing interest in designing new drug formulations so to improve drug delivery, increasing the efficacy and reducing the doses and consequently undesirable effects. In this setting, drug delivery systems (DDSs) represent a new frontier in the modern medicine [75]. DDSs offer the possibility to create a hybrid of metal-organic frameworks (MOFs), combining the biocompatibility of organic system to the high loadings of inorganic fraction [76]. Several lines of evidence suggest an efficient functionalization of nanoparticles with DCA. Lazaro and colleagues [77] explored different protocols for DCA functionalization of the zirconium (Zr) terephthalate (UiO-66) nanoparticles. They demonstrated the cytotoxicity and selectivity of the same DDSs against different cancer cell lines. Moreover, they excluded a possible response of the immune system to DCA-MOF in vitro. The same group later showed the possibility to load Zr MOFs with a second anticancer drug, such as 5- fluorouracil (5-FU), so to reproduce the synergistic effect of the two drugs [78]. Zirconium-based MOF loaded with DCA was also purposed as an attractive alternative to UiO-66, showing selective in vitro cytotoxicity towards several cancer cell lines and a good toleration by the immune system of several species [79]. Recently, Štarha et al. [80] synthesized and characterized, for the first time, half-sandwich complexes containing ruthenium or osmium and DCA (Figure 2(a)). Both Ru-dca and Os-DCA complexes were screened in ovarian carcinoma cell lines, demonstrating to be more cytotoxic than cisplatin alone. Both complexes were able to induce cytochrome c (Cytc) release from mitochondria, an indirect index of apoptosome activation and seemed to be less toxic towards healthy primary human hepatocytes, thus indicating selectivity for cancer over noncancerous cells. Promising results were also obtained in triple-negative breast cancer cells [81]. Rhenium (I)-DCA conjugate has demonstrated an efficient penetration into cancer cells and a selective accumulation into mitochondria, inducing mitochondrial dysfunction and metabolic disorders [82]. In the recent years, several multiactive drugs have been designed to contemporary target different intracellular pathways using a single formulation. A safe, simple, reproducible nanoformulation of the complex doxorubicinDCA (Figure 2(b)) was successfully tested in a murine melanoma model, showing an increase in drug-loading capability, lower side effects, and enhanced therapeutic effect [83]. Dualacting antitumor Pt (IV) prodrugs of kiteplatin with DCA axial ligands have been synthesized (Figure 2(c)), characterized, and tested in different tumour cell lines and in vivo [84]. To overcome cancer resistance, triple action Pt (IV) derivatives of cisplatin have been proposed as new potent anticancer agents, able to conjugate the action of cisplatin, cyclooxygenase inhibitors, and DCA (Figure 2(d)) [85]. A novel complex containing DCA, Platinum, and Biotin (DPB) has been successfully tested, exhibiting multifacet antitumor properties (Figure 2(e)). Authors demonstrated the ability of such a prodrug to affect energy metabolism, to promote apoptosis, and to interact with DNA. The high selectivity of biotin for cancer cells minimizes the detrimental effects on normal cells and improves the curative effect on tumours [86]. Features and experimental evidence of the main classes of compounds are summarized in Table 3.
| Class of drug formulation | Features | In vitro tests | In vivo tests | Experimental evidence | References |
| Metal-DCA frameworks (no platinum) | Metal ions linked to organic ligands into porous scaffolds | MCF-7/MDA-MB-231 (breast) HeLa/LO2 (cervix) A2780 (ovary) A549/NCl-H1229 (lung) | Breast mouse models | Biocompatibility selective cytotoxicity Immune system compatibility Low mutagenicity | [77–82] |
| Doxorubicin-DCA conjugate | Complexes of DCA and chemotherapy drugs | B16F10 (melanoma) | Sarcoma and melanoma mouse models | Selective cytotoxicity safety In vivo antitumour efficiency | [83] |
| Platinum prodrugs with DCA | Platinum core associated to DCA and others drugs | MCF-7 (breast) LoVo/HCT-15/HCT116 (colon) A549 (lung) BxPC3/PSN-1 (pancreas) A375 (melanoma) BCPAP (thyroid) HeLa (cervix) HepG2 (hepatocarcinoma) | Lung carcinoma mouse models | Selective cytotoxicity multiple action Increased cellular uptake | [84–86] |
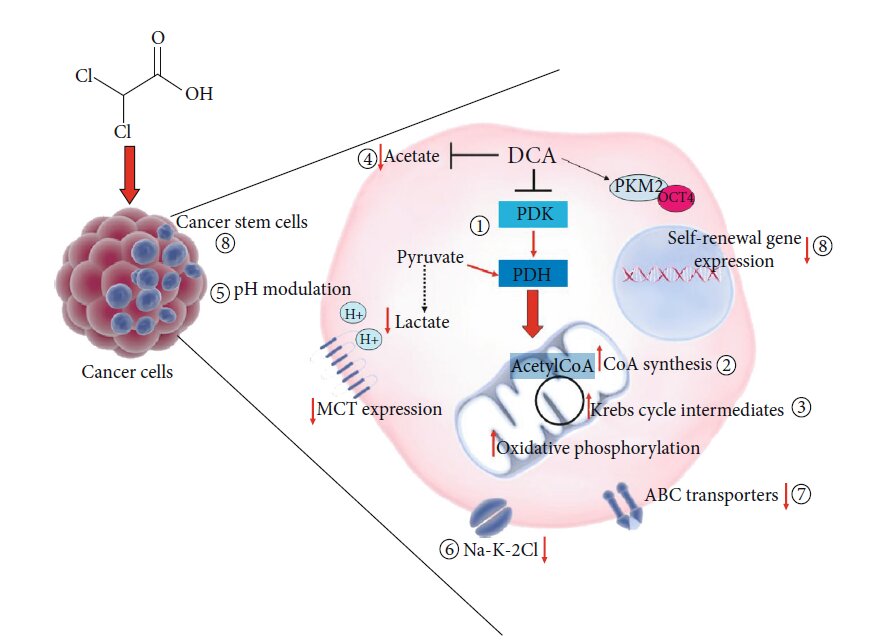
Other Proposed Mechanisms of Action of DCA
The metabolic shift from glycolysis to glucose oxidation due to the inhibition of PDK and the consequent activation of PDH is the best-known and well-accepted molecular effect of DCA administration. The consequent biochemical alterations, including ROS increase and mitochondrial membrane potential variation, may be responsible for proliferation arrest and cancer cell death, thus explaining DCA beneficial potential in cancer treatment [9]. However, the molecular intermediates activated after DCA administration are still unknown. It is conceivable that such a small molecule might directly or indirectly affect other cellular and molecular targets (Figure 3), displaying other mechanisms of action, so to explain its efficacy also in cellular models where it does not produce the expected metabolic shift [12]. A proteomic approach applied to cells of lung cancer demonstrated the ability of DCA to increase the concentration of every TCA intermediate while it did not affect glucose uptake or the glycolytic process from glucose to pyruvate [87]. In the attempt to shed light to DCA mode of action, Dubuis and colleagues used a metabolomics-based approach on several ovarian cancer cell lines treated with DCA and found a common marked depletion of intracellular pantothenate, a CoA precursor, as well as a concomitant increase of CoA, thus suggesting DCA ability to increase CoA de novo biosynthesis. Since high concentrations of CoA resulted to be toxic for cells, this metabolic effect could be responsible of cancer cell toxicity mediated by DCA [88]. A very recent work by El Sayed et al. introduced a novel evidence-based hypothesis, suggesting that DCA efficiency against cancer may derive from its ability to antagonize acetate [89], known to be an energetic substrate for glioblastoma and brain metastases, able to enhance DNA, RNA, and protein synthesis and posttranslational modifications, thus favouring cell proliferation and cancer progression. Moreover, high acetate levels are associated to anticancer drug resistance [90]. It has been shown that DCA is able to revert metabolic alterations induced by acetate by restoring physiological serum levels of lactate and free fatty acid and potassium and phosphorus concentration. According to the authors, thanks to a structural similarity to acetate, DCA could inhibit metabolic effects driven by acetate, responsible for cancer cell growth and chemoresistance [89]. Another possible additional effect of DCA could be pH modulation. pH level modulation is known to affect proliferation and apoptosis processes [91] as well as chemotherapy sensitivity [92]. DCA treatment may both increase and reduce intracellular pH. A secondary effect of pyruvate redirecting into the mitochondria by DCA would be lactate reduction and a consequent increase in intracellular pH. On the other side, DCA is able to decrease the expression of monocarboxilate transporters and V-ATPase with a consequent reduction of pH, and this especially occurs in tumour cells, expressing higher amount of these carriers, compared to normal counterparts [93]. Given the ability to induce rapid tumour intracellular acidification, Albatany et al. [94] speculated about a possible employment of DCA as a tracker in in vivo imaging of a glioblastoma murine model and supported a therapeutic use of DCA since intracellular acidification is known to induce caspase activation and DNA fragmentation of cancer cells [95]. Animal models allow to identify a possible further molecular target of DCA. Experiments performed in rats highlighted the ability of DCA to inhibit the expression of the renal cotransporter Na-K-2Cl (NKCC) in the kidney of rats [96]. As NKCC is an important biomarker of extracellular and intracellular ion homeostasis regulation and participates in cell cycle progression, it plays an important role in cancer cell proliferation, apoptosis, and invasion. Belkahla et al. [97] investigated the interplay between metabolism targeting and the expression of ABC transporters, responsible for drug export from cells and a consequent multidrug resistance, and found that DCA treatment is able to reduce gene and protein expression of ABC transporters in several tumour cells expressing wild type p53, both in vitro and in vivo [98]. It has been already demonstrated the ability of DCA to induce differentiation through the modulation of PKM2/Oct4 interaction in glioma cells [99]. The resulting reduction of Oct4 transcription levels was associated to a reduction of stemness phenotype and a significant increased sensitivity to cell stress. This observation lets to hypothesize a potential role of DCA against cancer stem cells (CSCs).
DCA and Cancer Stem Cells
There is a growing interest in targeting cancer stem cells (CSCs) which seem to be the main responsible for tumour relapse [100]. CSCs share the ability of self-renewal with normal stem cells and can give rise to differentiating cells, responsible for tumour initiation as well as malignant progression [101]. A low proliferation rate and specific metabolic profile contribute to make CSCs resistant to conventional chemotherapy [102]. An urgent need emerged in the developing of new therapeutic agents able to affect cancer stem cell viability [103] in order to completely eradicate the tumour mass. An extensive body of literature is focusing the attention on the metabolic phenotype of CSCs, which seem to differ from differentiated cancer cells and could represent a therapeutic target [104–108]. In this setting, the possible sensitivity of CSC fraction to DCA has been hypothesized and tested in different cancer models. Embryonal carcinoma stem cells represent one of the more appropriate models for the study of CSC maintenance and differentiation and the identification of drugs and molecules able to modulate these processes [109]. Studies performed on embryonic stem cells (ESCs) constitute preliminary important proofs supporting a possible efficacy of DCA [110]. Interestingly, DCA treatment of ESCs promotes loss of pluripotency and shifts towards a more active oxidative metabolism, accompanied by a significant decrease in HIF1a and p53 expression [111]. Vega-Naredo et al. [112] described the importance of mitochondrial metabolism in directing stemness and differentiation in such a model. They characterized the metabolic profile of stem cell fraction and guessed the less susceptibility of stem phenotype to mitochondrial-directed therapies. Forcing CSCs towards an oxidative metabolism by DCA treatment enabled departure from stemness to differentiation. Several reports support the existence of CSCs in glioma [113, 114], and the efficiency of DCA to hit CSCs has been extensively evaluated in such a cancer type, so difficult to treat with conventional therapies and characterized by low rates of survival. Already in 2010, Michelakis and colleagues had suggested, both in vitro and in vivo, DCA ability to induce apoptosis of cancer stem cell fraction [26]. A rat model of glioma, recapitulating several features of human glioblastoma, confirmed the efficacy of DCA to potentiate apoptosis of glioma CSCs, characterized by a significant glycolytic pathway overstimulation, compared to normal stem cells [115]. Also, Jiang et al. investigated the effect of DCA on the small population of glioma stem cells (GSCs) isolated from glioblastoma, demonstrating a reduction of self-renewal properties and an increase in cell death percentage [44]. Moreover, an in vivo test on mice bearing DCA-treated GSC-derived xenografts showed a significant increase in overall survival. DCA treatment was also tested in melanoma stem cell fraction, and the derived bioenergetics modulation was able to counteract protumorigenic action of a c-Met inhibitor [116]. A very recent work performed on human hepatocellular carcinoma identified PDK4 overexpression in spheres originated from cancer cells, featuring a defined stem-like phenotype. Interestingly, DCA treatment was able to reduce cell viability both of cancer-differentiated cells and cancer stem cells and reversed chemoresistance to conventional therapy [36]. Our group has recently experienced the ability of DCA to reduce the expression of cancer stem cell markers CD24/CD44/EPCAM in a pancreatic cancer cell line as well as to compromise spheroid formation and viability [12], further corroborating data obtained in other cancer models. Together with chemoresistance, also radioresistance represents a limit to an efficient cancer treatment, and CSCs seem to be responsible for such refractoriness [117]. Sun et al. demonstrated the ability of DCA to increase radiosensitivity of medulloblastoma cells by affecting stem-like clones, reducing the expression percentage of CD133-positive cells and reducing sphere formation [72]. Moreover, in the same cellular model, they showed an altered mechanism of DNA repair induced by DCA able to explain the increased effectiveness of radiotherapy.
Conclusions
Targeting cancer cell metabolism represents a new pharmacological approach to treat cancer. DCA ability to shift metabolism from glycolysis to oxidative phosphorylation has increased the interest towards this drug already known for its anticancer properties. The evidence accumulated in the last years confirms the capability of DCA to overcome chemo, radioresistance in several cancer types and lets to hypothesize additional cellular targets able to explain its skill to kill cancer cells. There is a need to design further clinical studies now limited to poor-prognosis patients with advanced, recurrent neoplasms, already refractory to other conventional therapies. Its potential efficacy against cancer stem cells as well as the development of new drug formulations takes us closer to reach an effective clinical employment of DCA.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by Current Research Funds, Italian Ministry of Health, to IRCCS-CROB, Rionero in Vulture, Potenza, Italy.
REFERENCES
1 [1] T. G. Lee, E. H. Jeong, I. J. Min, S. Y. Kim, H. R. Kim, and C. H. Kim, “Altered expression of cellular proliferation, apoptosis and the cell cycle-related genes in lung cancer cells with acquired resistance to Egfr tyrosine kinase inhibitors,” Oncology Letters, vol. 14, no. 2, pp. 2191–2197, 2017.
2 J. Chen, “The cell-cycle arrest and apoptotic functions of P53 in tumor initiation and progression,” Cold Spring Harbor Perspectives in Medicine, vol. 6, no. 3, p. a026104, 2016.
3 C. Thakur and F. Chen, “Connections between metabolism and epigenetics in cancers,” Seminars in Cancer Biology, vol. 57, pp. 52–58, 2019.
4 S. Subramaniam, V. Jeet, J. A. Clements, J. H. Gunter, and J. Batra,“Emergence of micrornas as key players in cancer cell metabolism,” Clinical Chemistry, vol. 65, no. 9, pp. 1090– 1101, 2019.
5 D. Williams and B. Fingleton, “Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis,” Clinical & Experimental Metastasis, vol. 36, no. 3, pp. 211–224, 2019.
6 T. Tataranni, F. Agriesti, V. Ruggieri et al., “Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles,” Oncotarget, vol. 8, no. 25, pp. 41265–41281, 2017.
7 A. Luengo, D. Y. Gui, and M. G. Vander Heiden, “Targeting metabolism for cancer therapy,” Cell Chemical Biology, vol. 24, no. 9, pp. 1161–1180, 2017.
8 M. O. James, S. C. Jahn, G. Zhong, M. G. Smeltz, Z. Hu, and P. W. Stacpoole, “Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1,” Pharmacology & Therapeutics, vol. 170, pp. 166–180, 2017.
9 E. D. Michelakis, L. Webster, and J. R. Mackey, “Dichloroacetate (Dca) as a potential metabolic-targeting therapy for cancer,” British Journal of Cancer, vol. 99, no. 7, pp. 989–994, 2008.
10 S. Kankotia and P. W. Stacpoole, “Dichloroacetate and cancer: new home for an orphan drug?,” Biochimica et Biophysica Acta, vol. 1846, no. 2, pp. 617–629, 2014.
11 V. Ruggieri, F. Agriesti, R. Scrima et al., “Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment,” Oncotarget, vol. 6, no. 2, pp. 1217–1230, 2015.
12 T. Tataranni, F. Agriesti, C. Pacelli et al., “Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines,” Cells, vol. 8, no. 5, p. 478, 2019.
13 A. Khan, D. Marier, E. Marsden, D. Andrews, and I. Eliaz, “A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases,” Alternative Therapies in Health and Medicine, vol. 20, Supplement 2, pp. 21–28, 2014.
14 E. M. Dunbar, B. S. Coats, A. L. Shroads et al.,“Phase 1 trial of dichloroacetate (Dca) in adults with recurrent malignant brain tumors,” Investigational New Drugs, vol. 32, no. 3, pp. 452–464, 2014.
15Q. S.-C. Chu, R. Sangha, J. Spratlin et al., “A phase I openlabeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors,” Investigational New Drugs, vol. 33, no. 3, pp. 603–610, 2015.
16 A. Khan, D. Andrews, and A. C. Blackburn, “Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy,” World Journal of Clinical Cases, vol. 4, no. 10, pp. 336–343, 2016.
17 G. Sutendra and E. D. Michelakis, “Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology,” Frontiers in Oncology, vol. 3, p. 38, 2013.
18 S. R. Pillai, M. Damaghi, Y. Marunaka, E. P. Spugnini, S. Fais, and R. J. Gillies, “Causes, consequences, and therapy of tumors acidosis,” Cancer Metastasis Reviews, vol. 38, no. 1- 2, pp. 205–222, 2019.
19 R. J. DeBerardinis, J. J. Lum, G. Hatzivassiliou, and C. B. Thompson, “The biology of cancer: metabolic reprogramming fuels cell growth and proliferation,” Cell Metabolism, vol. 7, no. 1, pp. 11–20, 2008.
20 N. Zamzami and G. Kroemer, “The mitochondrion in apoptosis: how Pandora’s box opens,” Nature Reviews Molecular Cell Biology, vol. 2, no. 1, pp. 67–71, 2001.
21 J. W. Kim and C. V. Dang, “Multifaceted roles of glycolytic enzymes,” Trends in Biochemical Sciences, vol. 30, no. 3, pp. 142–150, 2005. P. A. Gammage and C. Frezza,“Mitochondrial DNA: the overlooked oncogenome?,” BMC Biology, vol. 17, no. 1, p. 53, 2019.
22 L. H. Stockwin, S. X. Yu, S. Borgel et al., “Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC,” International Journal of Cancer, vol. 127, no. 11, pp. 2510–2519, 2010. [
23 P. W. Stacpoole, N. V. Nagaraja, and A. D. Hutson, “Efficacy of dichloroacetate as a lactate-lowering drug,”Journal of Clinical Pharmacology, vol. 43, no. 7, pp. 683–691, 2003.
24 P. W. Stacpoole, “Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer,” JNCI: Journal of the National Cancer Institute, vol. 109, no. 11, 2017.
25 E. D. Michelakis, G. Sutendra, P. Dromparis et al., “Metabolic modulation of glioblastoma with dichloroacetate,” Science Translational Medicine, vol. 2, no. 31, article 31ra34, 2010.
26 P. W. Stacpoole, C. J. Martyniuk, M. O. James, and N. A. Calcutt, “Dichloroacetate-induced peripheral neuropathy,” International Review of Neurobiology, vol. 145, pp. 211–238, 2019.
27 N. Felitsyn, P. W. Stacpoole, and L. Notterpek, “Dichloroacetate causes reversible demyelination in vitro: potential mechanism for its neuropathic effect,” Journal of Neurochemistry, vol. 100, no. 2, pp. 429–436, 2007.
28 T. Langaee, R. Wagner, L. P. Horne et al., “Personalized dosing of dichloroacetate using Gstz1 clinical genotyping assay,” Genetic Testing and Molecular Biomarkers, vol. 22, no. 4, pp. 266–269, 2018.
29 D. Brandsma, T. P. Dorlo, J. H. Haanen, J. H. Beijnen, and W. Boogerd, “Severe encephalopathy and polyneuropathy induced by dichloroacetate,” Journal of Neurology, vol. 257, no. 12, pp. 2099-2100, 2010.
30 United States Environmental Protection Agency, EPA, Toxicological Review of Dichloroacetic Acid, CAS 79-43-6, 2003.
31 X. Lu, D. Zhou, B. Hou et al., “Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer,” Cancer Management and Research, vol. 10, pp. 1231–1241, 2018.
32 A. Korga, M. Ostrowska, M. Iwan, M. Herbet, and J. Dudka, “Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes Hepg2 cells to doxorubicin treatment,” FEBS Open Bio, vol. 9, no. 5, pp. 959–972, 2019.
33 H. Sun, A. Zhu, X. Zhou, and F. Wang, “Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel,” Oncotarget, vol. 8, no. 32, pp. 52642–52650, 2017.
34 B. L. Woolbright, D. Choudhary, A. Mikhalyuk et al., “The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance,” Molecular Cancer Therapeutics, vol. 17, no. 9, pp. 2004–2012, 2018.
35 K. Fekir, H. Dubois-Pot-Schneider, R. Désert et al., “Retrodifferentiation of human tumor hepatocytes to stem cells leads to metabolic reprogramming and chemoresistance,” Cancer Research, vol. 79, no. 8, pp. 1869–1883, 2019.
36A. Skeberdytė, I. Sarapinienė, J. Aleksander-Krasko, V. Stankevičius, K.
37Sužiedėlis, and S. Jarmalaitė, “Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines,” Scientific Reports, vol. 8, no. 1, p. 17744, 2018.
38 A. Verma, Y. M. Lam, Y. C. Leung et al., “Combined use of arginase and dichloroacetate exhibits anti‐proliferative effects in triple negative breast cancer cells,” The Journal of Pharmacy and Pharmacology, vol. 71, no. 3, pp. 306–315, 2019.
39 B. Li, X. Li, H. Xiong et al., “Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells,” Oncotarget, vol. 8, no. 31, pp. 51748–51757, 2017.
40 C. Lucido, W. Miskimins, and P. Vermeer, “Propranolol promotes glucose dependence and synergizes with dichloroacetate for anti-cancer activity in HNSCC,” Cancers, vol. 10, no. 12, p. 476, 2018.
41 C. Abildgaard, C. Dahl, A. Abdul-Al, A. Christensen, and P. Guldberg, “Inhibition of retinoic acid receptor Β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells,” Oncotarget, vol. 8, no. 48, pp. 84210–84223, 2017.
42 I. V. Prokhorova, O. N. Pyaskovskaya, D. L. Kolesnik, and G. I. Solyanik, “Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6,” Experimental Oncology, vol. 40, no. 3, pp. 205–210, 2018.
43 D. L. Kolesnik, O. N. Pyaskovskaya, Y. R. Yakshibaeva, and G. I. Solyanik, “Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma,” Experimental Oncology, vol. 41, no. 1, pp. 14–19, 2019.
44 W. Jiang, S. Finniss, S. Cazacu et al., “Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma,” Oncotarget, vol. 7, no. 35, pp. 56456– 56470, 2016.
45 B. Waltenberger, A. Mocan, K. Šmejkal, E. Heiss, A. Atanasov, and A. G. Atanasov, “Natural products to counteract the epidemic of cardiovascular and metabolic disorders,” Molecules, vol. 21, no. 6, p. 807, 2016.
46 M. Zadorozhna, T. Tataranni, and D. Mangieri, “Piperine: role in prevention and progression of cancer,” Molecular Biology Reports, vol. 46, no. 5, pp. 5617–5629, 2019.
47G. Della Sala, F. Agriesti, C. Mazzoccoli, T. Tataranni, V. Costantino, and C. Piccoli, “Clogging the ubiquitinproteasome machinery with marine natural products: last decade update,” Marine Drugs, vol. 16, no. 12, p. 467, 2018.
48 A. G. Atanasov, B. Waltenberger, E. M. Pferschy-Wenzig et al., “Discovery and resupply of pharmacologically active plant-derived natural products: a review,” Biotechnology Advances, vol. 33, no. 8, pp. 1582–1614, 2015.
49 M. B. Sporn and N. Suh, “Chemoprevention: an essential approach to controlling cancer,” Nature Reviews Cancer, vol. 2, no. 7, pp. 537–543, 2002.
50 C. K. Singh, J. George, and N. Ahmad, “Resveratrol‐based combinatorial strategies for cancer management,” Annals of the New York Academy of Sciences, vol. 1290, pp. 113–121, 2013.
51 S. Redondo-Blanco, J. Fernández, I. Gutiérrez-del-Río, C. J. Villar, and F. Lombó, “New insights toward colorectal cancer chemotherapy using natural bioactive compounds,” Frontiers in Pharmacology, vol. 8, p. 109, 2017.
52 B. B. Aggarwal, A. Kumar, and A. C. Bharti, “Anticancer potential of curcumin: preclinical and clinical studies,” Anticancer Research, vol. 23, no. 1A, pp. 363–398, 2003.
53P. C. Kan, Y. J. Chang, C. S. Chien, C. Y. Su, and H. W. Fang, “Coupling dichloroacetate treatment with curcumin significantly enhances anticancer potential,” Anticancer Research, vol. 38, no. 11, pp. 6253–6261, 2018.
54 K. Hata, K. Hori, H. Ogasawara, and S. Takahashi, “Antileukemia activities of Lup-28-Al-20(29)-En-3-one, a lupane triterpene,” Toxicology Letters, vol. 143, no. 1, pp. 1–7, 2003.
55C. A. Dehelean, S. Feflea, J. Molnár, I. Zupko, and C. Soica, “Betulin as an antitumor agent tested in vitro on A431, Hela and MCF7, and as an angiogenic inhibitor in vivo in the cam assay,” Natural Product Communications, vol. 7, no. 8, pp. 981–985, 2012.
56 M. Drag, P. Surowiak, M. Drag-Zalesinska, M. Dietel, H. Lage, and J. Oleksyszyn, “Comparision of the cytotoxic effects of birch bark extract, betulin and betulinic acid towards human gastric carcinoma and pancreatic carcinoma drug-sensitive and drug-resistant cell lines,” Molecules, vol. 14, no. 4, pp. 1639–1651, 2009.
57 M. Mihoub, A. Pichette, B. Sylla, C. Gauthier, and J. Legault, “Bidesmosidic betulin saponin bearing L-rhamnopyranoside moieties induces apoptosis and inhibition of lung cancer cells growth in vitro and in vivo,” PLoS One, vol. 13, no. 3, article e0193386, 2018.
58 H. Wang, H. Jiang, M. Van De Gucht, and M. De Ridder, “Hypoxic radioresistance: can ROS be the key to overcome it?,” Cancers, vol. 11, no. 1, p. 112, 2019.
59 J. P. Pouget, S. Frelon, J. L. Ravanat, I. Testard, F. Odin, and J. Cadet, “Formation of modified DNA bases in cells exposed either to gamma radiation or to high-let particles,” Radiation Research, vol. 157, no. 5, pp. 589–595, 2002.
60 K. Rycaj and D. G. Tang, “Cancer stem cells and radioresistance,” International Journal of Radiation Biology, vol. 90, no. 8, pp. 615–621, 2014.
61 L. Tang, F. Wei, Y. Wu et al., “Role of metabolism in cancer cell radioresistance and radiosensitization methods,” Journal of Experimental & Clinical Cancer Research, vol. 37, no. 1, p. 87, 2018.
62 G. Xie, Y. Liu, Q. Yao et al., “Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells,” Scientific Reports, vol. 7, no. 1, article 42396, 2017.
63 J. Overgaard, “Hypoxic radiosensitization: adored and ignored,” Journal of Clinical Oncology, vol. 25, no. 26, pp. 4066–4074, 2007.
64 Y. Zhang and S. G. Martin, “Redox proteins and radiotherapy,” Clinical Oncology, vol. 26, no. 5, pp. 289–300, 2014.
65 H. Jiang, H. Wang, and M. De Ridder, “Targeting antioxidant enzymes as a radiosensitizing strategy,” Cancer Letters, vol. 438, pp. 154–164, 2018.
66 M. R. Niewisch, Z. Kuçi, H. Wolburg et al., “Influence of dichloroacetate (DCA) on lactate production and oxygen consumption in neuroblastoma cells: is DCA a suitable drug for neuroblastoma therapy?,” Cellular Physiology and Biochemistry, vol. 29, no. 3-4, pp. 373–380, 2012.
67 S. P. Pitroda, B. T. Wakim, R. F. Sood et al., “Stat1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect,” BMC Medicine, vol. 7, no. 1, p. 68, 2009.
68 T. Shimura, N. Noma, Y. Sano et al., “Akt-mediated enhanced aerobic glycolysis
69 V. Bol, A. Bol, C. Bouzin et al., “Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation,” Acta Oncologica, vol. 54, no. 2, pp. 266–274, 2015.
70 H. Shen, E. Hau, S. Joshi, P. J. Dilda, and K. L. McDonald, “Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism,” Molecular Cancer Therapeutics, vol. 14, no. 8, pp. 1794–1804, 2015.
71 G. Dong, Q. Chen, F. Jiang et al., “Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels,” Oncotarget,
72 L. Sun, T. Moritake, K. Ito et al., “Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets,” PLoS One, vol. 12, no. 4, article e0176162, 2017.
73 L. Zitvogel, L. Apetoh, F. Ghiringhelli, F. André, A. Tesniere, and G. Kroemer, “The anticancer immune response:indispensable for therapeutic success?,” The Journal of Clinical Investigation, vol. 118, no. 6, pp. 1991–2001, 2008.
74 S. Gupta and B. Dwarakanath, “Modulation of Immunobiome during radio-sensitization of tumors by glycolytic inhibitors,” Current Medicinal Chemistry, vol. 25, 2018.
75 D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit, and R. Langer, “Nanocarriers as an emerging platform for cancer therapy,” Nature Nanotechnology, vol. 2, no. 12, pp. 751–760, 2007.
76 R. C. Huxford, J. Della Rocca, and W. Lin, “Metal-organic frameworks as potential drug carriers,” Current Opinion in Chemical Biology, vol. 14, no. 2, pp. 262–268, 2010. [
77 I. A. Lázaro, S. A. Lázaro, and R. S. Forgan, “Enhancing anticancer cytotoxicity through bimodal drug delivery from ultrasmall Zr MOF nanoparticles,” Chemical Communications, vol. 54, no. 22, pp. 2792–2795, 2018.
78 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al., “Surface-functionalization of Zr-fumarate MOF for selective cytotoxicity and immune system compatibility in nanoscale drug delivery,” ACS Applied Materials & Interfaces, vol. 10, no. 37, pp. 31146–31157, 2018.
79 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al., “Mechanistic investigation into the selective anticancer cytotoxicity and immune system response of surface-functionalized, dichloroacetate-loaded, UiO-66 nanoparticles,” ACS Applied Materials & Interfaces, vol. 10, no. 6, pp. 5255– 5268, 2018.
80 P. Štarha, Z. Trávníček, J. Vančo, and Z. Dvořák, “Half-sandwich Ru(II) and Os(II) bathophenanthroline complexes containing a releasable dichloroacetato ligand,” Molecules, vol. 23, no. 2, p. 420, 2018.
81 J. Pracharova, V. Novohradsky, H. Kostrhunova et al., “Halfsandwich Os(ii) and Ru(ii) bathophenanthroline complexes: anticancer drug candidates with unusual potency and a cellular activity profile in highly invasive triple-negative breast cancer cells,” Dalton Transactions, vol. 47, no. 35, pp. 12197–12208, 2018.
82 J. Yang, Q. Cao, H. Zhang et al., “Targeted reversal and phosphorescence lifetime imaging of cancer cell metabolism _via_ a theranostic rhenium(I)-DCA conjugate,” Biomaterials, vol. 176, pp. 94–105, 2018.
83C. Yang, T. Wu, Y. Qin et al., “A facile doxorubicindichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery,” International Journal of Nanomedicine, vol. 13, pp. 1281–1293, 2018.
84 S. Savino, V. Gandin, J. D. Hoeschele, C. Marzano, G. Natile, and N. Margiotta, “Dual-acting antitumor Pt(IV) prodrugs of kiteplatin with dichloroacetate axial ligands,” Dalton Transactions, vol. 47, no. 21, pp. 7144–7158, 2018.
85 E. Petruzzella, R. Sirota, I. Solazzo, V. Gandin, and D. Gibson, “Triple action Pt(iv) derivatives of cisplatin: a new class of potent anticancer agents that overcome resistance,” Chemical Science, vol. 9, no. 18, pp. 4299–4307, 2018.
86 S. Jin, Y. Guo, D. Song et al., “Targeting energy metabolism by a platinum(IV) prodrug as an alternative pathway for cancer suppression,” Inorganic Chemistry, vol. 58, no. 9, pp. 6507–6516, 2019. [
87W. Zhang, X. Hu, W. Zhou, and K. Y. Tam, “liquid chromatography-tandem mass spectrometry method revealed that lung cancer cells exhibited distinct metabolite profiles upon the treatment with different pyruvate dehydrogenase kinase inhibitors,” Journal of Proteome Research, vol. 17, no. 9, pp. 3012–3021, 2018.
88 S. Dubuis, K. Ortmayr, and M. Zampieri, “A framework for large-scale metabolome drug profiling links coenzyme a metabolism to the toxicity of anti-cancer drug dichloroacetate,” Communications Biology, vol. 1, no. 1, p. 101, 2018.
89S. M. El Sayed, H. Baghdadi, N. S. Ahmed et al., “Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: a novel evidencebased medical hypothesis,” Medical Hypotheses, vol. 122, pp. 206–209, 2019.
90 D. M. Jaworski, A. M. Namboodiri, and J. R. Moffett,“Acetate as a metabolic and epigenetic modifier of cancer therapy,” Journal of Cellular Biochemistry, vol. 117, no. 3, pp. 574– 588, 2016.
91 B. A. Webb, M. Chimenti, M. P. Jacobson, and D. L. Barber, “Dysregulated pH: a perfect storm for cancer progression,” Nature Reviews Cancer, vol. 11, no. 9, pp. 671–677, 2011. [
92 D. Neri and C. T. Supuran, “Interfering with pH regulation in tumours as a therapeutic strategy,” Nature Reviews Drug Discovery, vol. 10, no. 10, pp. 767–777, 2011.
93 A. Kumar, S. Kant, and S. M. Singh, “Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation,” Toxicology and Applied Pharmacology, vol. 273, no. 1, pp. 196–208, 2013.
94 M. Albatany, A. Li, S. Meakin, and R. Bartha, “Dichloroacetate induced intracellular acidification in glioblastoma: in vivo detection using AACID-CEST MRI at 9.4 Tesla,” Journal of Neuro-Oncology, vol. 136, no. 2, pp. 255–262, 2018.
95 H. J. Park, J. C. Lyons, T. Ohtsubo, and C. W. Song, “Acidic environment causes apoptosis by increasing caspase activity,” British Journal of Cancer, vol. 80, no. 12, pp. 1892–1897, 1999.
96 J. Stanevičiūtė, M. Juknevičienė, J. Palubinskienė et al., “Sodium dichloroacetate pharmacological effect as related to Na-K-2Cl cotransporter inhibition in rats,” Dose Response, vol. 16, no. 4, article 155932581881152, 2018.
97S. Belkahla, A. U. Haq Khan, D. Gitenay et al., “Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status,” Oncotarget, vol. 9, no. 1, pp. 1114–1129, 2018.
98 J. A. Bush and G. Li, “Cancer chemoresistance: the relationship between P53 and multidrug transporters,” International Journal of Cancer, vol. 98, no. 3, pp. 323–330, 2002.
99 M. Morfouace, L. Lalier, L. Oliver et al., “Control of glioma cell death and differentiation by PKM2-Oct4 interaction,” Cell Death & Disease, vol. 5, no. 1, pp. e1036–e1036, 2014.
100 A. Turdo, V. Veschi, M. Gaggianesi et al., “Meeting the challenge of targeting cancer stem cells,” Frontiers in Cell and Development Biology, vol. 7, p. 16, 2019.
101 P. Zhu and Z. Fan, “Cancer stem cells and tumorigenesis,” Biophysics Reports, vol. 4, no. 4, pp. 178–188, 2018.
102 S. Prasad, S. Ramachandran, N. Gupta, I. Kaushik, and S. K. Srivastava, “Cancer cells stemness: a doorstep to targeted therapy,” Biochimica et Biophysica Acta – Molecular Basis of Disease, p. 165424, 2019.
103 M. Yang, P. Liu, and P. Huang, “Cancer stem cells, metabolism, and therapeutic significance,” Tumour Biology, vol. 37, no. 5, pp. 5735–5742, 2016.
104 P. Sancho, D. Barneda, and C. Heeschen, “Hallmarks of cancer stem cell metabolism,” British Journal of Cancer, vol. 114, no. 12, pp. 1305–1312, 2016.
105 F. Sotgia, M. Fiorillo, and M. P. Lisanti, “Hallmarks of the cancer cell of origin: comparisons with “energetic” cancer stem cells (e-CSCs),” Aging, vol. 11, no. 3, pp. 1065–1068, 2019.
106 S. Skvortsov, I. I. Skvortsova, D. G. Tang, and A. Dubrovska, “Concise review: prostate cancer stem cells: current understanding,” Stem Cells, vol. 36, no. 10, pp. 1457–1474, 2018.
107 S. Bordel, “Constraint based modeling of metabolism allows finding metabolic cancer hallmarks and identifying personalized therapeutic windows,” Oncotarget, vol. 9, no. 28, pp. 19716–19729, 2018.
108 Y. Y. Wang, J. Chen, X. M. Liu, R. Zhao, and H. Zhe, “Nrf2- mediated metabolic reprogramming in cancer,” Oxidative Medicine and Cellular Longevity, vol. 2018, Article ID 9304091, 7 pages, 2018.
109 M. W. McBurney, “P19 embryonal carcinoma cells,” The International Journal of Developmental Biology, vol. 37, no. 1, pp. 135–140, 1993.
110 R. Loureiro, S. Magalhães-Novais, K. A. Mesquita et al., “Melatonin antiproliferative effects require active mitochondrial function in embryonal carcinoma cells,” Oncotarget, vol. 6, no. 19, pp. 17081–17096, 2015.
111 A. S. Rodrigues, M. Correia, A. Gomes et al., “Dichloroacetate, the pyruvate dehydrogenase complex and the modulation of mESC pluripotency,” PLoS One, vol. 10, no. 7, article e0131663, 2015.
112 I. Vega-Naredo, R. Loureiro, K. A. Mesquita et al., “Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells,” Cell Death and Differentiation, vol. 21, no. 10, pp. 1560–1574, 2014.
113 S. K. Singh, C. Hawkins, I. D. Clarke et al., “Identification of human brain tumour initiating cells,” Nature, vol. 432, no. 7015, pp. 396–401, 2004.
114 X. Yuan, J. Curtin, Y. Xiong et al., “Isolation of cancer stem cells from adult glioblastoma multiforme,” Oncogene, vol. 23, no. 58, pp. 9392–9400, 2004.
115 M. Morfouace, L. Lalier, M. Bahut et al., “Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications,” The Journal of Biological Chemistry, vol. 287, no. 40, pp. 33664–33674, 2012.
116 L. Kucerova, L. Demkova, S. Skolekova, R. Bohovic, and M. Matuskova,“Tyrosine kinase inhibitor SU11274 increased tumorigenicity and enriched for melanoma-initiating cells by bioenergetic modulation,” BMC Cancer, vol. 16, no. 1, p. 308, 2016.
117 Z. Zhao, K. Zhang, Z. Wang et al., “A comprehensive review of available omics data resources and molecular profiling for precision glioma studies,” Biomedical Reports, vol. 10, no. 1, pp. 3–9, 2019.
Related content: