Tiziana Tataranni 1 und Claudia Piccoli 1,2
1Laborfür präklinische und translationale Forschung, IRCCS-CROB, Krebsreferenzzentrum der Basilicata, Rionero in Vulture (Pz), 85028, Italien
2Abteilungfür klinische und experimentelle Medizin, Universität Foggia, Foggia 71121, Italien
Die Korrespondenz sollte an Tiziana Tataranni gerichtet werden; [email protected]
Gastherausgeber: Kanhaiya Singh
Copyright © 2019 Tiziana Tataranni und Claudia Piccoli. Dies ist ein Open-Access-Artikel, der unter der Creative-Commons-Attributionslizenz verbreitet wird, die die uneingeschränkte Nutzung, Verbreitung und Vervielfältigung in jedem Medium erlaubt, sofern das Originalwerk ordnungsgemäß zitiert wird.
Received: 24 July 2019Revised
: 12 September 2019Accepted
: 11. Oktober
2019Online-Veröffentlichung: 14. November 2019
Die krebsbekämpfenden Eigenschaften von Dichloracetat (DCA) sind in der Literatur umfangreich beschrieben, seine effektive klinische Anwendung in der Krebstherapie ist jedoch noch auf klinische Studien beschränkt. Das Auftreten von Nebenwirkungen wie Neurotoxizität sowie der Verdacht der Karzinogenität von DCA schränken den klinischen Einsatz von DCA immer noch ein. In den letzten Jahren hat jedoch die Zahl der Berichte, die den Einsatz von DCA gegen Krebs unterstützen, zugenommen, auch aufgrund des großen Interesses an der Beeinflussung des Stoffwechsels von Tumorzellen. Die Entschlüsselung des Wirkmechanismus von DCA half, die Grundlagen seiner selektiven Wirksamkeit gegen Krebszellen zu verstehen. Die erfolgreiche gleichzeitige Verabreichung von DCA mit konventioneller Chemotherapie, Strahlentherapie, anderen Medikamenten oder Naturstoffen wurde in mehreren Krebsmodellen getestet. Neue Systeme zur Verabreichung von Arzneimitteln und Mehrfachwirkstoffe, die DCA und andere Arzneimittel enthalten, scheinen die Bioverfügbarkeit zu verbessern und dank einer synergistischen Wirkung mehrerer Wirkstoffe effizienter zu sein. Die Verbreitung von Berichten, die die Wirksamkeit von DCA in der Krebstherapie belegen, hat zu weiteren Studien geführt, die andere potenzielle molekulare Angriffspunkte von DCA finden sollen. Interessanterweise könnte DCA den Anteil der Krebsstammzellen erheblich beeinflussen und zur Krebsausrottung beitragen. Zusammengenommen liefern diese Ergebnisse eine gute Grundlage für neue klinische translationale Studien mit DCA in der Krebstherapie.
EINFÜHRUNG
Krebs ist eine der häufigsten Todesursachen weltweit. Trotz erheblicher Fortschritte bei den diagnostischen und therapeutischen Ansätzen stellt seine Ausrottung nach wie vor eine Herausforderung dar. Zu viele Faktoren sind für Therapieversagen oder Rückfälle verantwortlich, so dass dringend neue Behandlungsansätze gesucht werden müssen. Abgesehen von den typischen bekannten Eigenschaften bösartiger Zellen, wie abnorme Vermehrung, Deregulierung der Apoptose und des Zellzyklus [1, 2]weisen Krebszellen auch eine besondere Stoffwechselmaschine auf, die einen weiteren vielversprechenden Ansatz für die Krebstherapie bietet [3-5]. Unsere Gruppe hatte bereits auf die Bedeutung einer metabolischen Charakterisierung von Krebszellen hingewiesen, um die Wirksamkeit einer metabolischen Behandlung vorherzusagen [6]. Medikamente, die den Stoffwechsel von Krebszellen beeinflussen können, werden bereits in Betracht gezogen und zeigen ermutigende Ergebnisse in Bezug auf Wirksamkeit und Verträglichkeit [7]. In den letzten zehn Jahren wurde das kleine Molekül DCA, das bereits zur Behandlung von akuter und chronischer Laktatazidose, angeborenen Fehlern des mitochondrialen Stoffwechsels und Diabetes [8]eingesetzt wird, wurde weitgehend als Krebsmedikament eingesetzt. DCA ist ein 150 Da großes, wasserlösliches Säuremolekül, ein Analogon der Essigsäure, bei dem zwei der drei Wasserstoffatome der Methylgruppe durch Chloratome ersetzt wurden (Abbildung 1(a)) [9]. Die Verabreichung von DCA in Dosen von 50 bis 200 mg/Kg/Tag führt in mehreren präklinischen Modellen zu einer Verringerung des Tumormassenvolumens, der Proliferationsrate und der Metastasenausbreitung [10]. Unsere Gruppe hatte bereits eine umgekehrte Korrelation zwischen der Fähigkeit von DCA, Krebszellen abzutöten, und ihrer mitochondrialen Atmungskapazität in Mundhöhlenkarzinomen beobachtet [11]. Darüber hinaus haben wir vor kurzem die Fähigkeit von DCA beschrieben, die mitochondriale Funktion zu beeinflussen und das Fortschreiten von Krebs in einem Pankreaskrebsmodell zu verzögern [12]. Bislang liegen konsistente Daten aus klinischen Studien und Fallberichten über die Verabreichung von DCA bei Krebspatienten vor [13-16]trotz der zunehmenden Literatur, die die Wirksamkeit von DCA gegen Krebs bestätigt, wird es noch nicht klinisch eingesetzt. In dieser Übersicht sollen die jüngsten Berichte zusammengefasst werden, die den Einsatz von DCA in der Krebstherapie in Kombination mit Chemotherapeutika, Strahlentherapie und anderen chemischen oder natürlichen Substanzen mit krebsbekämpfenden Eigenschaften vorschlagen. Darüber hinaus haben wir Daten über neue pharmakologische Formulierungen von DCA beschrieben, mit denen Nebenwirkungen vermieden und die Bioverfügbarkeit und Wirksamkeit des Medikaments verbessert werden können, was seinen möglichen klinischen Einsatz weiter begünstigt. Schließlich haben wir die neuesten Erkenntnisse zu anderen potenziellen Wirkmechanismen von DCA besprochen, einschließlich neuer Daten zu seiner Fähigkeit, die Krebsstammzellfraktion zu beeinflussen.
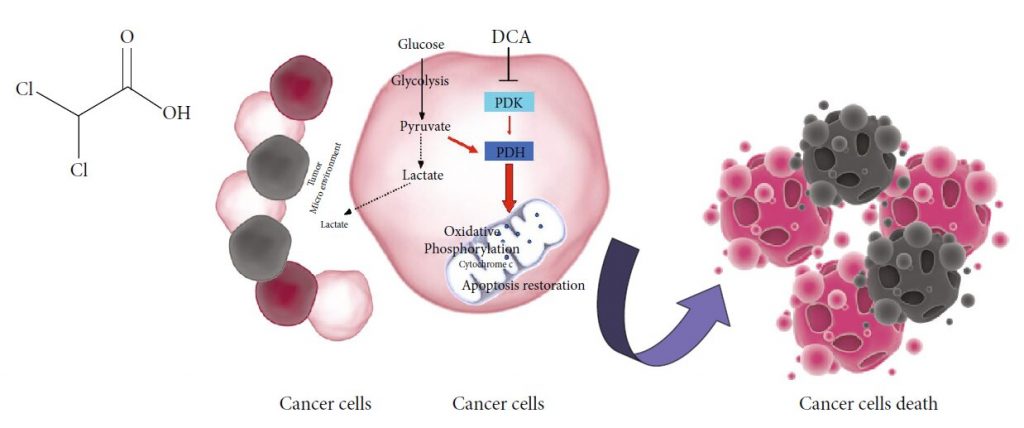
DCA und Krebs: Mechanismus der Wirkung
Die potenzielle Wirksamkeit von DCA in der Krebstherapie ergibt sich aus den metabolischen Eigenschaften von Krebszellen, die typischerweise durch eine erhöhte glykolytische Aktivität und eine reduzierte mitochondriale Oxidation gekennzeichnet sind, unabhängig von der Sauerstoffverfügbarkeit, dem bekannten Warburg-Effekt [17]. Die übermäßige Glykolyse und die daraus resultierende Laktatüberproduktion führen zu einem Zustand der metabolischen Azidose in der Mikroumgebung des Tumors [18]. Das aus der Glykolyse stammende Laktat wird von den umliegenden Zellen aufgenommen, um das Tumorwachstum zu unterstützen, und hemmt die apoptotischen Zelltodmechanismen [19, 20]. Mehrere an der Glykolyse beteiligte Enzyme regulieren die Apoptose, und ihre Überexpression in Krebszellen trägt zur Unterdrückung der Apoptose bei [21]. Vor diesem Hintergrund wirken DCA-Salze selektiv auf Krebszellen, indem sie ihren Stoffwechsel von der Glykolyse auf die oxidative Phosphorylierung verlagern, indem sie die Pyruvatdehydrogenase-Kinase (PDK), den Inhibitor der Pyruvatdehydrogenase (PDH), hemmen [10]. Die Aktivierung der PDH fördert die mitochondriale Oxidation von Pyruvat und stört den metabolischen Vorteil der Krebszellen. Mitochondriale DNA-Mutationen, die häufig bei der Tumorentstehung auftreten und zu einer Dysfunktion der Atmungskette führen [22, 23]führen, machen bösartige Zellen unfähig, den zellulären Energiebedarf zu decken. Durch die Verringerung der Laktatproduktion wirkt DCA außerdem der Übersäuerung der Mikroumgebung des Tumors entgegen und trägt so zur Hemmung des Tumorwachstums und der Ausbreitung bei [24]. Die Zufuhr von Pyruvat in die Mitochondrien bewirkt einen Umbau der Organellen, was zu einem verstärkten Ausfluss von Cytochrom c und anderen apoptotisch wirkenden Faktoren sowie zu einer Erhöhung des ROS-Spiegels führt und damit die Lebensfähigkeit der Krebszellen verringert [9] (Abbildung 1(b)).
Nebenwirkungen und Beschränkungen der DCA-Beschäftigung
Für den klinischen Einsatz von DCA stehen sowohl orale als auch parenterale Formulierungen zur Verfügung, und die Dosis reicht von 10 bis 50 mg/Kg/Tag [25]. Keine Hinweise auf schwere hämatologische, hepatische, renale oder kardiale Toxizität bestätigen die Sicherheit von DCA [26]. Häufige gastrointestinale Nebenwirkungen treten häufig bei einem Prozentsatz der mit DCA behandelten Patienten auf [15]. Die bekannteste Einschränkung bei der Verabreichung von DCA, die sowohl in präklinischen als auch in klinischen Studien beobachtet wurde, ist die periphere Neuropathie [27]. Die Selektivität der DCA-induzierten Schädigung des Nervensystems könnte darauf zurückzuführen sein, dass es keine gut ausgestattete Maschinerie gibt, die in der Lage ist, eine länger andauernde oxidative Phosphorylierung in Zellen zu bewältigen, die ATP hauptsächlich über Glykolyse produzieren [28]. Die daraus resultierende Überlastung der Mitochondrien beeinträchtigt die Effizienz der antioxidativen Systeme, die nicht in der Lage sind, die übermäßige Menge an ROS zu bewältigen. Vor diesem Hintergrund dürfte die gleichzeitige Verabreichung von Antioxidantien eine weitere Strategie zur Minimierung der DCA-induzierten Neuropathie darstellen [27]. Die Expression und die Aktivität der Glutathiontransferase zeta1 (GSTZ1), des ersten Enzyms, das für die DCA-Clearance verantwortlich ist, kann die Entität der Schädigung beeinflussen. Nicht-synonyme funktionelle Singlenukleotid-Polymorphismen (SNPs) im menschlichen GSTZ1-Gen führen zu verschiedenen Haplotypen, die für eine unterschiedliche DCA-Kinetik und -Dynamik verantwortlich sind. Es wurde ein eindeutiger Zusammenhang zwischen dem GSTZ1-Haplotyp und der DCA-Clearance nachgewiesen. Auf dieser Grundlage kann eine personalisierte DCA-Dosierung, die nicht nur auf dem Körpergewicht basiert, unerwünschte Wirkungen bei Patienten, die chronisch mit diesem Medikament behandelt werden, minimieren oder verhindern [29]. Das Auftreten von Neuropathie wird mit der chronischen oralen Verabreichung von DCA in Verbindung gebracht und ist ein reversibler Effekt, der auf die Dauer der Behandlung beschränkt ist [30]. Der intravenöse Weg reduziert, OH Cl Cl O (a) Krebszellen Krebszellen Krebszellen Tod Laktat Tumor Mikroumgebung Laktat Pyruvat Glykolyse PDK DCA PDH Oxidative Phosphorylierung Apoptose Wiederherstellung Cytochrom c Glucose (b) Abbildung 1: (a) Chemische Struktur von DCA. (b) Wirkmechanismus von DCA: PDK: Pyruvatdehydrogenase-Kinase; PDH: Pyruvatdehydrogenase. Schwarze gestrichelte Linien: biochemische Prozesse, die durch DCA gehemmt werden; rote Pfeile: Stoffwechselwege, die durch DCA aktiviert werden. 2 Oxidative Medizin und zelluläre Langlebigkeit daher das Potenzial für Neurotoxizität und lassen das Erreichen höherer Arzneimittelkonzentrationen das Verdauungssystem umgehen [13].
Da DCA zu den Wasserdesinfektionsnebenprodukten gehört, die in geringen Konzentrationen im Trinkwasser vorkommen, wird seine potenzielle Karzinogenität derzeit untersucht. Studien an Mäusen zeigen, dass eine DCA-Exposition in jungen Jahren mit einer erhöhten Inzidenz hepatozellulärer Tumore in Verbindung gebracht wird [31]. Es ist denkbar, dass anhaltende Veränderungen im Zellstoffwechsel, die durch DCA induziert werden, epigenetische Auswirkungen haben. Die langfristige Induktion von PDH und anderen oxidativen Stoffwechselwegen, die mit dem Glukosestoffwechsel zusammenhängen, könnte zu einer Zunahme reaktiver Sauerstoffspezies und mitochondrialem Stress beitragen [27]. In klinischen Studien gibt es jedoch keine Hinweise auf eine karzinogene Wirkung, wenn DCA in der Krebstherapie verabreicht wird.
Synergistische Wirkung von DCA und chemotherapeutischen Wirkstoffen
Die Kombination verschiedener Medikamente ist eine anerkannte Strategie, um in der Krebstherapie eine synergistische Wirkung zu erzielen, die Dosierung der Medikamente zu verringern, die Toxizitätsrisiken zu minimieren und die Arzneimittelresistenz zu überwinden. Die gleichzeitige Verabreichung von DCA und herkömmlichen Chemotherapeutika wurde in verschiedenen Krebsmodellen getestet (Tabelle 1). Die DCA-Behandlung scheint die Wirksamkeit der Chemotherapie zu verbessern, indem sie biochemische und metabolische Veränderungen hervorruft, die zu erheblichen Veränderungen im Energiehaushalt der Krebszellen führen. Eine bei nicht-kleinzelligem Lungenkrebs (NSCLC) durchgeführte Studie zeigte sowohl in vitro als auch in vivo, dass die gleichzeitige Verabreichung von DCA mit Paclitaxel die Effizienz des Zelltods durch Hemmung der Autophagie erhöht [32]. Eine wirksame Kombination von DCA und Doxorubicin (DOX) wurde an HepG2-Zellen getestet, wobei sich zeigte, dass DCA die Fähigkeit besitzt, die zelluläre antioxidative Abwehr zu stören, und damit oxidative Schäden begünstigt, die wiederum durch die DOX-Behandlung ausgelöst werden [33]. Es besteht ein enger Zusammenhang zwischen PDK-Überexpression und Chemoresistenz; daher ist es denkbar, dass eine PDK-Hemmung dazu beitragen könnte, Krebszellen gegen Medikamente unempfindlich zu machen. Die Überexpression der PDK2-Isoform wurde mit einer Paclitaxel-Resistenz bei NSCLC in Verbindung gebracht. Interessanterweise war die Kombination von DCA mit Paclitaxel bei der Abtötung resistenter Zellen wirksamer als die Behandlung mit Paclitaxel oder DCA allein [34]. Ähnlich wie beim NSCLC zeigte eine interessante In-vivo-Studie, die bei fortgeschrittenem Blasenkrebs durchgeführt wurde, eine erhöhte Expression der PDK4-Isoform bei hochgradigem Krebs im Vergleich zu niedriggradigem Krebs, und die gemeinsame Behandlung mit DCA und Cisplatin führte zu einer drastischen Verringerung des Tumorvolumens im Vergleich zu DCA oder Cisplatin allein [35]. Eine kürzlich durchgeführte Studie bestätigte die Fähigkeit von DCA, die PDK4-bedingte Chemoresistenz auch beim menschlichen hepatozellulären Karzinom (HCC) umzukehren [36].
| Tumorentität | Modellsystem | Chemotherapeutikum, das gleichzeitig mit DCA verabreicht wird | Mechanismus der Wirkung | Ergebnis | Referenzen |
| Lungenkrebs | A549-H1975 Zelllinien/ Xenograft-Modell | Paclitaxel | Autophagie-Hemmung | Wirksame Sensibilisierung für Krebs-Chemotherapie | [32] |
| Hepatokarzinom | HepG2-Zelllinie | Doxorubicin | Störung der antioxidativen Abwehr | Erhöhte zelluläre Schäden durch Induktion von oxidativem Stress | [33] |
| Lungenkrebs | A549 Zelllinie | Paclitaxel | Erhöhte Chemosensitivität durch PDK2-Inhibition | Überwindung der Paclitaxel-Resistenz | [34] |
| Harnblasenkrebs | HTB-9, HT-1376, HTB-5, HTB-4 Zelllinien/Xenograft-Modell | Cisplatin | Erhöhte Chemosensitivität durch PDK4-Inhibition | Erhöhter Zelltod von Krebszellen und potenzieller therapeutischer Vorteil | [35] |
| Hepatokarzinom | Sphärenkulturen aus HepaRG- und BC2-Zelllinien | Cisplatin, Sorafenib | Erhöhte Chemosensitivität durch PDK4-Inhibition | Verbesserte therapeutische Wirkung der Chemotherapie durch Wiederherstellung der mitochondrialen Aktivität | [36] |
Synergistische Wirkung von DCA und anderen potenziellen Krebsmedikamenten
In der Literatur finden sich zahlreiche Hinweise auf positive Wirkungen der gleichzeitigen Verabreichung von DCA mit Wirkstoffen, die derzeit zur Behandlung anderer Krankheiten eingesetzt werden, aber in verschiedenen Krebsmodellen krebshemmende Eigenschaften zeigen (Tabelle 2). Die gleichzeitige Verabreichung von DCA und dem Antibiotikum Salinomycin, das vor kurzem wegen seiner zytotoxischen Eigenschaften als potenzielles Krebsmedikament wiederentdeckt wurde, wurde an Darmkrebs-Zelllinien getestet. Ihre Behandlung scheint eine synergistische zytotoxische Wirkung auszuüben, indem sie die Expression von Proteinen hemmt, die mit der Multidrug-Resistenz [37]. Krebszellen, denen Stoffwechselenzyme fehlen, die am Arginin-Stoffwechsel beteiligt sind, reagieren möglicherweise empfindlich auf die Behandlung mit Arginase. Interessanterweise führt eine kombinierte Verabreichung von rekombinanter Arginase und DCA zu einer antiproliferativen Wirkung bei dreifach negativem Brustkrebs, die auf die Aktivierung von p53 und die Induktion eines Zellzyklusarrests zurückzuführen ist [38]. COX2-Inhibitoren, die in erster Linie als entzündungshemmende Medikamente eingesetzt werden, wurden kürzlich aufgrund ihrer antiproliferativen Wirkung als Antitumormittel vorgeschlagen. Eine interessante Studie, die an Gebärmutterhalskrebszellen durchgeführt wurde, zeigte, dass DCA Gebärmutterhalskrebszellen, die COX2 überexprimieren, nicht abtöten kann, und dass die Hemmung von COX2 durch Celecoxib Gebärmutterhalskrebszellen sowohl in vitro als auch in vivo empfindlicher gegenüber DCA macht [39]. Da DCA die oxidative Phosphorylierung durch Verringerung der glykolytischen Aktivität fördert, könnte die Kombination von DCA mit anderen Arzneimitteln, die einen Zustand der Glukoseabhängigkeit fördern, eine vielversprechende Strategie sein. Ein solcher Ansatz wurde bei Kopf- und Halskrebs getestet, bei dem die Verabreichung von Propranolol, einem nichtselektiven Betablocker, der den mitochondrialen Stoffwechsel der Tumorzellen beeinflussen kann, eine glykolytische Abhängigkeit und energetischen Stress erzeugte, wodurch die Zellen anfälliger für die Behandlung mit DCA wurden [40]. Ähnliche Ergebnisse wurden bei Melanomzellen erzielt, bei denen die Verabreichung von Hemmstoffen des Retinsäure-Rezeptors β (RARβ) eine Sensibilisierung für DCA bewirkt [41]. Eine positive Wirkung der gleichzeitigen Verabreichung von DCA mit Metformin, einem zur Behandlung von Diabetes weit verbreiteten Hypoglykämikum, wurde in einem präklinischen Gliom-Modell [42] sowie bei einer schwach metastasierenden Variante des Lewis-Lungenkarzinoms (LLC) [43]. Jiang und Kollegen untersuchten die Wirkungen von Phenformin, einem Metformin-Analogon, und DCA bei Glioblastomen und zeigten, dass die gleichzeitige Hemmung von Komplex I und PDK durch Phenformin bzw. DCA die Selbsterneuerung und Lebensfähigkeit von Gliomstammzellen (GSC) verringerte, was auf ihren möglichen Einsatz zur Beeinflussung der Krebsstammzellenfraktion hindeutet [44].
| Droge | Hauptfunktion | Tumorentität | Modellsystem | Ergebnis | Referenzen |
| Salinomycin | Antibiotikum | Kolorektaler Krebs | DLD-1 und HCT116 Zelllinien | Hemmung von Multidrug-Resistenz-Proteinen | [37] |
| Arginase | Arginin-Stoffwechsel | Brustkrebs | MDA-MB231 und MCF-7/ Xenotransplantationsmodell | Antiproliferative Wirkung durch p53-Aktivierung und Zellzyklus-Stillstand | [38] |
| COX2-Hemmer | Entzündung | Gebärmutterhalskrebs | HeLa- und SiHa-Zelllinien/ Xenograft-Modell | Unterdrückung des Krebszellwachstums | [39] |
| Propranolol | Beta-Blocker | Kopf- und Halskrebs | mEERL und MLM3 Zelllinien/C57Bl/6 m | Förderung der Glukoseabhängigkeit und Verstärkung der Wirkung der Chemostrahlung | [40] |
| RARβ-Hemmer | Vitamin-A-Stoffwechsel | Melanom | ED-007, ED-027, ED-117, und ED196 Zelllinien | Förderung der Glukoseabhängigkeit und Sensibilisierung gegenüber DCA | [41] |
| Metformin | Diabetes | Gliom, Lewis-Lungenkarzinom | Xenotransplantationsmodell; LLC/R9-Zellen | Verlängerte Lebensspanne von Mäusen mit Gliom; starke Glukoseabhängigkeit in der Tumormikroumgebung | [42, 43] |
| Phenformin | Diabetes | Glioblastom | Gliom-Stammzellen/Exenograft-Modell | Selbsterneuerungshemmung von Krebsstammzellen | [44] |
Kombinierte Verwendung von DCA und Naturstoffen
Der klinische Einsatz von Naturstoffen ist ein vielversprechender neuer Ansatz zur Behandlung verschiedener Krankheiten [45]. In der Literatur werden immer mehr biologisch aktive Substanzen, die aus Pflanzen, Pilzen, Bakterien oder Meeresorganismen isoliert werden, nachgewiesen, die sich positiv auf die menschliche Gesundheit auswirken [46-48]. Die Annahme von Naturstoffen oder deren Derivaten scheint ein vielversprechender Ansatz zu sein, um die Entstehung oder das Wiederauftreten von Krebs zu verhindern, und wird allgemein als Chemoprävention bezeichnet [49]. Darüber hinaus haben Naturstoffe positive Auswirkungen auf die Krebstherapie, wenn sie zusammen mit anderen Medikamenten verabreicht werden, da sie in der Lage sind, Arzneimittelresistenzen zu überwinden, das krebsbekämpfende Potenzial zu erhöhen und die Dosierung und Toxizität von Medikamenten zu verringern [50, 51]. Interessanterweise wurde vor kurzem die gleichzeitige Verabreichung von DCA und Naturstoffen erprobt. Eine Studie untersuchte die kombinierte Wirkung von DCA mit einer Mischung aus ätherischem Öl und Curcumin, einer Verbindung mit positiven Eigenschaften sowohl bei der Prävention als auch bei der Behandlung von Krebs [52]und zeigte dabei ein krebshemmendes Potenzial gegen HCC [53]. Insbesondere reduzierte die Kombination beider Verbindungen synergistisch das Überleben der Zellen, indem sie die Apoptose der Zellen förderte und die intrazelluläre ROS-Bildung induzierte. Betulin, eine aus Birkenrinde isolierte natürliche Verbindung, ist bereits für seine antiproliferativen und zytotoxischen Wirkungen gegen mehrere Krebszelllinien bekannt [54-56]. Eine In-vitro-Untersuchung der Antitumoraktivität von Betulinderivaten bei nicht-kleinzelligem Lungenkrebs (NSCLC) bestätigte seine Fähigkeit, das In-vivo- und In-vitro-Wachstum von Lungenkrebszellen zu hemmen, indem es die G2/M-Phase des Zellzyklus blockiert und die Caspase-Aktivierung und DNA-Fragmentierung induziert. Interessanterweise war das Betulinderivat Bi-L-RhamBet in der Lage, die mitochondriale Elektronentransportkette (ETC) zu stören und die ROS-Produktion zu induzieren. Angesichts der Eigenschaft von DCA, die Gesamtoxidation von Glukose in den Mitochondrien über den Krebs-Zyklus und die ETC zu erhöhen, kombinierten die Autoren Bi-L-RhamBet mit DCA und wiesen dessen signifikant verstärkte Zytotoxizität nach [57].
DCA und Radiosensibilisierung
Die Strahlentherapie stellt eine weitere Strategie zur Behandlung von Krebs dar und bietet einen lokalen Ansatz durch die Verabreichung von hochenergetischen Strahlen [58]. Die Hauptwirkung der Bestrahlung ist die Induktion von ROS mit der Folge von DNA-Schäden, chromosomaler Instabilität und Zelltod durch Apoptose [59]. Einige Tumoren zeigen oder entwickeln jedoch eine Strahlenresistenz, die für das Scheitern der Strahlentherapie und das hohe Risiko eines Tumorrezidivs oder einer Metastasierung verantwortlich ist [60]. Mehrere Faktoren können für die Radioresistenz verantwortlich sein [61]. Unter anderem kann Hypoxie, ein häufiger Zustand der Tumormikroumgebung, der durch einen niedrigen Sauerstoffgehalt und eine reduzierte Bildung von ROS-Spezies gekennzeichnet ist, die Wirksamkeit ionisierender Strahlen blockieren [62]. Eine Erhöhung der Sauerstoffversorgung des Tumors, die eine beträchtliche Menge an ROS [63] oder die direkte Induktion der ROS-Produktion kann daher eine Strategie zur Erhöhung der Radiosensibilisierung darstellen [64, 65]. In diesem Zusammenhang könnte die Verabreichung von DCA, von dem bekannt ist, dass es die ROS-Produktion anregt [11, 66]könnte eine Strategie zur Überwindung der Radioresistenz von Tumoren darstellen. Darüber hinaus ist bekannt, dass Stoffwechselveränderungen, die die Krebsentwicklung begünstigen, die Radiosensitivität beeinflussen [67, 68]. Daher könnte die gezielte Beeinflussung von Krebs-Stoffwechselintermediaten eine Strategie zur Verbesserung einer selektiven Krebsreaktion auf Bestrahlung darstellen [69]. Die Wirksamkeit von DCA zur Erhöhung der Strahlenempfindlichkeit wurde bereits sowohl bei Glioblastomzellen [70] und bei Plattenepithelkarzinomen der Speiseröhre [71]. Kürzlich wurde gezeigt, dass DCA die Strahlenempfindlichkeit in einem Zellmodell des Medulloblastoms, eines tödlichen Hirntumors bei Kindern, erhöht, indem es Veränderungen des ROS-Stoffwechsels und der Mitochondrienfunktion bewirkt und die DNA-Reparaturkapazität unterdrückt [72]. Da die Rolle der Immuntherapie bei der Wiederherstellung der Immunabwehr gegen das Fortschreiten und die Metastasierung von Tumoren in den letzten Jahren große Aufmerksamkeit erregt hat [73]gupta und Dwarakanath lieferten einen aktuellen Überblick über die möglichen Auswirkungen von Glykolytik-Inhibitoren, einschließlich DCA, auf die Radiosensibilisierung von Tumoren, wobei sie ihr Augenmerk auf das Zusammenspiel von Stoffwechselmodifikatoren und Immunmodulation bei den Radiosensibilisierungsprozessen richteten [74]. Interessanterweise berichteten sie über die Fähigkeit von DCA, die Immunstimulation durch die Hemmung der Laktatakkumulation zu fördern, was seine Verwendung als Adjuvans der Strahlentherapie weiter unterstützt.
DCA und neue Medikamentenformulierungen
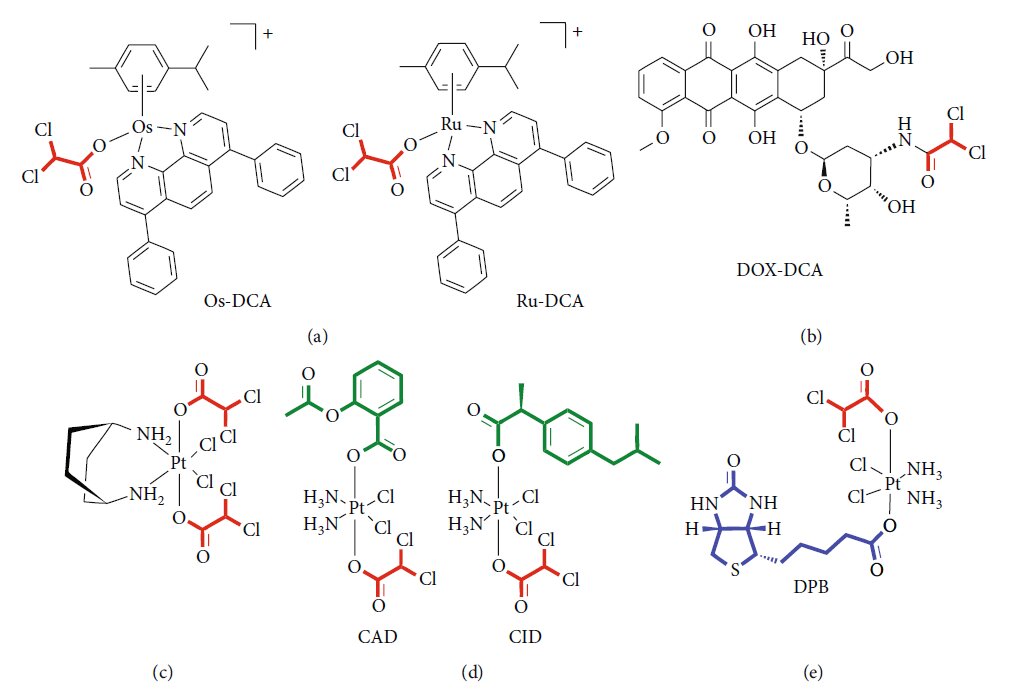
Es besteht ein wachsendes Interesse an der Entwicklung neuer Arzneimittelformulierungen, um die Verabreichung von Arzneimitteln zu verbessern, die Wirksamkeit zu erhöhen und die Dosierung und damit unerwünschte Wirkungen zu verringern. In diesem Zusammenhang stellen Arzneimittelverabreichungssysteme (DDS) eine neue Grenze in der modernen Medizin dar [75]. DDS bieten die Möglichkeit, ein Hybrid aus metallorganischen Gerüsten (MOF) zu schaffen, das die Biokompatibilität eines organischen Systems mit der hohen Beladung einer anorganischen Fraktion kombiniert [76]. Mehrere Hinweise deuten auf eine effiziente Funktionalisierung von Nanopartikeln mit DCA hin. Lazaro und Kollegen [77] untersuchten verschiedene Protokolle für die DCA-Funktionalisierung von Nanopartikeln aus Zirkonium(Zr)terephthalat (UiO-66). Sie wiesen die Zytotoxizität und Selektivität der gleichen DDSs gegen verschiedene Krebszelllinien nach. Außerdem schlossen sie eine mögliche Reaktion des Immunsystems auf DCA-MOF in vitro aus. Dieselbe Gruppe zeigte später, dass es möglich ist, Zr-MOFs mit einem zweiten Krebsmedikament, z. B. 5-Fluorouracil (5-FU), zu beladen, um so die synergistische Wirkung der beiden Medikamente zu reproduzieren [78]. MOF auf Zirkoniumbasis, die mit DCA beladen sind, wurden auch als attraktive Alternative zu UiO-66 betrachtet, da sie eine selektive In-vitro-Zytotoxizität gegenüber mehreren Krebszelllinien und eine gute Verträglichkeit für das Immunsystem verschiedener Spezies zeigten [79]. Vor kurzem haben Štarha et al. [80] zum ersten Mal Halbsandwich-Komplexe synthetisiert und charakterisiert, die Ruthenium oder Osmium und DCA enthalten (Abbildung 2(a)). Sowohl Ru-DCA- als auch Os-DCA-Komplexe wurden in Ovarialkarzinom-Zelllinien getestet und erwiesen sich als zytotoxischer als Cisplatin allein. Beide Komplexe waren in der Lage, die Freisetzung von Cytochrom c (Cytc) aus Mitochondrien zu induzieren, ein indirekter Index für die Apoptosom-Aktivierung, und schienen für gesunde primäre menschliche Hepatozyten weniger toxisch zu sein, was auf eine Selektivität für Krebszellen gegenüber Nicht-Krebszellen hinweist. Vielversprechende Ergebnisse wurden auch bei dreifach negativen Brustkrebszellen erzielt [81]. Das Rhenium(I)-DCA-Konjugat hat ein effizientes Eindringen in Krebszellen und eine selektive Anreicherung in Mitochondrien gezeigt, was zu mitochondrialer Dysfunktion und Stoffwechselstörungen führt [82]. In den letzten Jahren wurden mehrere multiaktive Arzneimittel entwickelt, die mit einer einzigen Formulierung auf verschiedene intrazelluläre Signalwege abzielen. Eine sichere, einfache und reproduzierbare Nanoformulierung des Doxorubicin-DCA-Komplexes (Abbildung 2(b)) wurde erfolgreich in einem Melanommodell der Maus getestet und zeigte eine höhere Wirkstoffbeladung, geringere Nebenwirkungen und eine verbesserte therapeutische Wirkung [83]. Doppelt wirkende antitumorale Pt(IV)-Prodrugs von Kiteplatin mit axialen DCA-Liganden wurden synthetisiert (Abbildung 2(c)), charakterisiert und in verschiedenen Tumorzelllinien und in vivo getestet [84]. Zur Überwindung der Krebsresistenz wurden dreifach wirksame Pt(IV)-Derivate von Cisplatin als neue potente Krebsmittel vorgeschlagen, die die Wirkung von Cisplatin, Cyclooxygenase-Inhibitoren und DCA konjugieren können (Abbildung 2(d)) [85]. Ein neuartiger Komplex, der DCA, Platin und Biotin (DPB) enthält, wurde erfolgreich getestet und zeigte vielseitige Antitumoreigenschaften (Abbildung 2(e)). Die Autoren zeigten die Fähigkeit eines solchen Prodrugs, den Energiestoffwechsel zu beeinflussen, die Apoptose zu fördern und mit der DNA zu interagieren. Die hohe Selektivität von Biotin für Krebszellen minimiert die schädlichen Auswirkungen auf normale Zellen und verbessert die heilende Wirkung auf Tumore [86]. Die Merkmale und experimentellen Nachweise der wichtigsten Verbindungsklassen sind in Tabelle 3 zusammengefasst.
| Klasse der Arzneimittelformulierung | Merkmale | In-vitro-Tests | In-vivo-Tests | Experimenteller Nachweis | Referenzen |
| Metall-DCA-Gerüste (kein Platin) | Metallionen verbunden mit organischen Liganden in porösen Gerüsten | MCF-7/MDA-MB-231 (Brust) HeLa/LO2 (Gebärmutterhals) A2780 (Eierstöcke) A549/NCl-H1229 (Lunge) | Mausmodelle für die Brust | Biokompatibilität selektive Zytotoxizität Verträglichkeit mit dem Immunsystem geringe Mutagenität | [77-82] |
| Doxorubicin-DCA-Konjugat | Komplexe aus DCA und Chemotherapeutika | B16F10 (Melanom) | Sarkom- und Melanom-Mausmodelle | Selektive Zytotoxizität Sicherheit In vivo Antitumoreffizienz | [83] |
| Platin-Prodrugs mit DCA | Platin-Kern assoziiert mit DCA und anderen Wirkstoffen | MCF-7 (Brust) LoVo/HCT-15/HCT116 (Dickdarm) A549 (Lunge) BxPC3/PSN-1 (Bauchspeicheldrüse) A375 (Melanom) BCPAP (Schilddrüse) HeLa (Gebärmutterhals) HepG2 (Hepatokarzinom) | Lungenkarzinom-Mausmodelle | Selektive Zytotoxizität Mehrfachwirkung Erhöhte zelluläre Aufnahme | [84-86] |
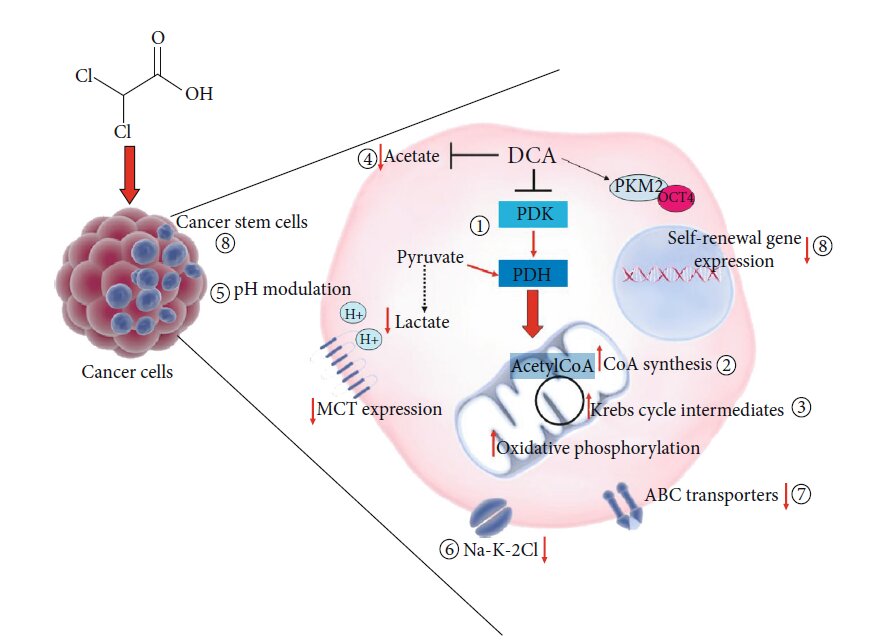
Andere vorgeschlagene Wirkmechanismen von DCA
Die Verlagerung des Stoffwechsels von der Glykolyse zur Glukoseoxidation aufgrund der Hemmung der PDK und der daraus resultierenden Aktivierung der PDH ist die bekannteste und am besten akzeptierte molekulare Wirkung der DCA-Verabreichung. Die sich daraus ergebenden biochemischen Veränderungen, einschließlich des Anstiegs der ROS und der Veränderung des mitochondrialen Membranpotenzials, könnten für den Proliferationsstopp und den Tod der Krebszellen verantwortlich sein und damit das positive Potenzial von DCA bei der Krebsbehandlung erklären [9]. Die molekularen Zwischenstufen, die nach der DCA-Verabreichung aktiviert werden, sind jedoch noch unbekannt. Es ist denkbar, dass ein solches kleines Molekül direkt oder indirekt auf andere zelluläre und molekulare Ziele einwirkt (Abbildung 3) und andere Wirkmechanismen aufweist, um seine Wirksamkeit auch in Zellmodellen zu erklären, in denen es nicht die erwartete Stoffwechselverschiebung bewirkt [12]. Ein proteomischer Ansatz, der bei Lungenkrebszellen angewandt wurde, zeigte die Fähigkeit von DCA, die Konzentration aller TCA-Zwischenprodukte zu erhöhen, während es die Glukoseaufnahme oder den glykolytischen Prozess von Glukose zu Pyruvat nicht beeinflusste [87]. In dem Versuch, die Wirkungsweise von DCA zu erhellen, verwendeten Dubuis und Kollegen einen auf Metabolomik basierenden Ansatz bei mehreren mit DCA behandelten Ovarialkarzinom-Zelllinien und stellten eine gemeinsame deutliche Verarmung an intrazellulärem Pantothenat, einem CoA-Vorläufer, sowie einen gleichzeitigen Anstieg von CoA fest, was auf die Fähigkeit von DCA hinweist, die CoA-De-novo-Biosynthese zu steigern. Da sich hohe CoA-Konzentrationen als zelltoxisch erwiesen haben, könnte dieser metabolische Effekt für die durch DCA vermittelte Toxizität von Krebszellen verantwortlich sein [88]. In einer kürzlich erschienenen Arbeit von El Sayed et al. wurde eine neue, evidenzbasierte Hypothese aufgestellt, die besagt, dass die Wirksamkeit von DCA gegen Krebs möglicherweise auf seiner Fähigkeit beruht, Acetat [89]das als energetisches Substrat für Glioblastome und Hirnmetastasen bekannt ist und die DNA-, RNA- und Proteinsynthese sowie posttranslationale Modifikationen fördern kann, wodurch die Zellproliferation und das Fortschreiten von Krebs begünstigt werden. Außerdem werden hohe Acetatspiegel mit einer Resistenz gegen Krebsmedikamente in Verbindung gebracht [90]. Es hat sich gezeigt, dass DCA in der Lage ist, durch Acetat induzierte Stoffwechselveränderungen rückgängig zu machen, indem es den physiologischen Serumspiegel von Laktat und freien Fettsäuren sowie die Kalium- und Phosphorkonzentration wiederherstellt. Den Autoren zufolge könnte DCA dank seiner strukturellen Ähnlichkeit mit Acetat die durch Acetat ausgelösten metabolischen Effekte hemmen, die für das Wachstum von Krebszellen und die Chemoresistenz verantwortlich sind [89]. Ein weiterer möglicher zusätzlicher Effekt von DCA könnte die Modulation des pH-Werts sein. Es ist bekannt, dass die Modulation des pH-Werts die Proliferations- und Apoptoseprozesse [91] sowie die Empfindlichkeit gegenüber Chemotherapie [92]. Die DCA-Behandlung kann den intrazellulären pH-Wert sowohl erhöhen als auch senken. Ein sekundärer Effekt der Umleitung von Pyruvat in die Mitochondrien durch DCA wäre die Verringerung von Laktat und ein daraus folgender Anstieg des intrazellulären pH-Werts. Andererseits ist DCA in der Lage, die Expression von Monocarboxilat-Transportern und der V-ATPase zu verringern, was zu einer Senkung des pH-Wertes führt, und zwar insbesondere in Tumorzellen, die diese Transporter in größerer Menge exprimieren als normale Zellen [93]. Angesichts der Fähigkeit, eine rasche intrazelluläre Ansäuerung von Tumorzellen zu bewirken, spekulierten Albatany et al. [94] über einen möglichen Einsatz von DCA als Tracker bei der In-vivo-Bildgebung eines Glioblastom-Mausmodells spekuliert und einen therapeutischen Einsatz von DCA befürwortet, da die intrazelluläre Ansäuerung bekanntermaßen die Caspase-Aktivierung und DNA-Fragmentierung von Krebszellen induziert [95]. Tiermodelle ermöglichen es, ein weiteres mögliches molekulares Ziel von DCA zu identifizieren. In Experimenten an Ratten wurde die Fähigkeit von DCA hervorgehoben, die Expression des renalen Kotransporters Na-K-2Cl (NKCC) in der Niere von Ratten zu hemmen [96]. Da NKCC ein wichtiger Biomarker für die Regulierung der extrazellulären und intrazellulären Ionenhomöostase ist und an der Progression des Zellzyklus beteiligt ist, spielt er eine wichtige Rolle bei der Proliferation, Apoptose und Invasion von Krebszellen. Belkahla et al. [97] untersuchten das Zusammenspiel zwischen dem Targeting des Stoffwechsels und der Expression von ABC-Transportern, die für den Arzneimittelexport aus den Zellen und eine daraus resultierende Multidrug-Resistenz verantwortlich sind, und fanden heraus, dass eine DCA-Behandlung in der Lage ist, die Gen- und Proteinexpression von ABC-Transportern in verschiedenen Tumorzellen, die Wildtyp p53 exprimieren, sowohl in vitro als auch in vivo zu verringern [98]. Es wurde bereits nachgewiesen, dass DCA in der Lage ist, durch die Modulation der PKM2/Oct4-Interaktion in Gliomzellen eine Differenzierung zu bewirken [99]. Die sich daraus ergebende Verringerung der Oct4-Transkriptionsmengen war mit einer Verringerung des Stammzellphänotyps und einer deutlich erhöhten Empfindlichkeit gegenüber Zellstress verbunden. Diese Beobachtung lässt auf eine mögliche Rolle von DCA bei der Bekämpfung von Krebsstammzellen (CSCs) schließen.
DCA und Krebsstammzellen
Es besteht ein wachsendes Interesse an der Bekämpfung von Krebsstammzellen (CSCs), die offenbar die Hauptverantwortlichen für Tumorrezidive sind [100]. CSC haben die Fähigkeit zur Selbsterneuerung mit normalen Stammzellen gemeinsam und können differenzierende Zellen hervorbringen, die für die Tumorentstehung und das Fortschreiten des Tumors verantwortlich sind [101]. Eine niedrige Proliferationsrate und ein spezifisches Stoffwechselprofil tragen dazu bei, dass CSCs gegen konventionelle Chemotherapie resistent sind [102]. Es besteht ein dringender Bedarf an der Entwicklung neuer therapeutischer Wirkstoffe, die die Lebensfähigkeit von Krebsstammzellen beeinflussen können [103] um die Tumormasse vollständig zu beseitigen. Eine umfangreiche Literatur konzentriert sich auf den metabolischen Phänotyp von CSCs, der sich von differenzierten Krebszellen zu unterscheiden scheint und ein therapeutisches Ziel darstellen könnte [104-108]. In diesem Zusammenhang wurde die mögliche Empfindlichkeit der CSC-Fraktion gegenüber DCA vermutet und in verschiedenen Krebsmodellen getestet. Embryonale Karzinomstammzellen sind eines der geeignetsten Modelle für die Untersuchung der Aufrechterhaltung und Differenzierung von CSCs und die Identifizierung von Medikamenten und Molekülen, die diese Prozesse beeinflussen können [109]. Studien, die an embryonalen Stammzellen (ESCs) durchgeführt wurden, stellen erste wichtige Beweise für eine mögliche Wirksamkeit von DCA [110]. Interessanterweise fördert die DCA-Behandlung von WSZ den Verlust der Pluripotenz und die Umstellung auf einen aktiveren oxidativen Stoffwechsel, begleitet von einem deutlichen Rückgang der HIF1a- und p53-Expression [111]. Vega-Naredo et al. [112] beschrieben die Bedeutung des mitochondrialen Stoffwechsels bei der Steuerung von Stammzellen und Differenzierung in einem solchen Modell. Sie charakterisierten das metabolische Profil der Stammzellfraktion und vermuteten eine geringere Anfälligkeit des Stammzellphänotyps für mitochondrial gesteuerte Therapien. Durch eine DCA-Behandlung konnten die CSCs zu einem oxidativen Stoffwechsel gezwungen werden, was den Übergang von der Stammzellen- zur Differenzierungsphase ermöglichte. Mehrere Berichte belegen die Existenz von CSCs in Gliomen [113, 114]und die Wirksamkeit von DCA zur Bekämpfung von CSCs wurde bei dieser Krebsart, die so schwer mit konventionellen Therapien zu behandeln ist und sich durch niedrige Überlebensraten auszeichnet, eingehend untersucht. Bereits 2010 hatten Michelakis und Kollegen sowohl in vitro als auch in vivo gezeigt, dass DCA in der Lage ist, die Apoptose der Krebsstammzellfraktion zu induzieren [26]. Ein Gliom-Modell bei der Ratte, das mehrere Merkmale des menschlichen Glioblastoms rekapituliert, bestätigte die Wirksamkeit von DCA bei der Potenzierung der Apoptose von Gliom-Stammzellen, die im Vergleich zu normalen Stammzellen durch eine signifikante Überstimulation des glykolytischen Stoffwechsels gekennzeichnet sind [115]. Außerdem untersuchten Jiang et al. die Wirkung von DCA auf die kleine Population von Gliom-Stammzellen (GSC), die aus Glioblastomen isoliert wurden, und zeigten eine Verringerung der Selbsterneuerungseigenschaften und eine Erhöhung des Prozentsatzes der absterbenden Zellen [44]. Darüber hinaus zeigte ein In-vivo-Test an Mäusen mit DCA-behandelten GSC-Xenotransplantaten eine signifikante Verlängerung der Gesamtüberlebenszeit. Die DCA-Behandlung wurde auch in der Melanom-Stammzellfraktion getestet, und die daraus abgeleitete bioenergetische Modulation konnte der protumorigenen Wirkung eines c-Met-Inhibitors entgegenwirken [116]. In einer kürzlich durchgeführten Arbeit am menschlichen Leberzellkarzinom wurde eine Überexpression von PDK4 in Sphären festgestellt, die aus Krebszellen mit einem bestimmten stammähnlichen Phänotyp entstanden sind. Interessanterweise konnte eine DCA-Behandlung die Lebensfähigkeit sowohl von krebsdifferenzierten Zellen als auch von Krebsstammzellen verringern und die Chemoresistenz gegenüber herkömmlichen Therapien aufheben [36]. Unsere Gruppe hat kürzlich festgestellt, dass DCA die Expression der Krebsstammzellmarker CD24/CD44/EPCAM in einer Zelllinie des Bauchspeicheldrüsenkrebses reduzieren und die Sphäroidbildung und Lebensfähigkeit beeinträchtigen kann [12]und bestätigt damit die in anderen Krebsmodellen gewonnenen Daten. Zusammen mit der Chemoresistenz stellt auch die Strahlenresistenz eine Grenze für eine effiziente Krebsbehandlung dar, und CSCs scheinen für diese Refraktärität verantwortlich zu sein [117]. Sun et al. wiesen nach, dass DCA die Radiosensitivität von Medulloblastomzellen erhöhen kann, indem es stammähnliche Klone beeinflusst, den Prozentsatz der CD133-positiven Zellen reduziert und die Sphärenbildung verringert [72]. Darüber hinaus zeigten sie in demselben Zellmodell einen durch DCA induzierten veränderten Mechanismus der DNA-Reparatur, der die erhöhte Wirksamkeit der Strahlentherapie erklären kann.
Schlussfolgerungen
Der Angriff auf den Stoffwechsel von Krebszellen stellt einen neuen pharmakologischen Ansatz zur Behandlung von Krebs dar. Die Fähigkeit von DCA, den Stoffwechsel von der Glykolyse auf die oxidative Phosphorylierung umzustellen, hat das Interesse an diesem Medikament, das bereits für seine krebsbekämpfenden Eigenschaften bekannt ist, verstärkt. Die in den letzten Jahren gesammelten Beweise bestätigen die Fähigkeit von DCA, die Chemo- und Radioresistenz bei verschiedenen Krebsarten zu überwinden, und lassen Hypothesen über zusätzliche zelluläre Ziele zu, die seine Fähigkeit, Krebszellen zu töten, erklären können. Es ist notwendig, weitere klinische Studien zu konzipieren, die sich auf Patienten mit schlechter Prognose und fortgeschrittenen, rezidivierenden Neoplasien beschränken, die bereits refraktär gegenüber anderen konventionellen Therapien sind. Seine potenzielle Wirksamkeit gegen Krebsstammzellen sowie die Entwicklung neuer Arzneimittelformulierungen bringen uns einem effektiven klinischen Einsatz von DCA näher.
Interessenkonflikte
Die Autoren erklären, dass keine Interessenkonflikte bestehen.
Danksagung
Diese Arbeit wurde von Current Research Funds, Italian Ministry of Health, an IRCCS-CROB, Rionero in Vulture, Potenza, Italien, unterstützt.
REFERENZEN
1 [1] T. G. Lee, E. H. Jeong, I. J. Min, S. Y. Kim, H. R. Kim, and C. H. Kim, „Altered expression of cellular proliferation, apoptosis and the cell cycle-related genes in lung cancer cells with acquired resistance to Egfr tyrosine kinase inhibitors,“ Oncology Letters, vol. 14, no. 2, pp. 2191-2197, 2017.
2 J. Chen, „The cell-cycle arrest and apoptotic functions of P53 in tumor initiation and progression“, Cold Spring Harbor Perspectives in Medicine, vol. 6, no. 3, p. a026104, 2016.
3 C. Thakur und F. Chen, „Connections between metabolism and epigenetics in cancers,“ Seminars in Cancer Biology, vol. 57, pp. 52-58, 2019.
4 S. Subramaniam, V. Jeet, J. A. Clements, J. H. Gunter, and J. Batra, „Emergence of micrornas as key players in cancer cell metabolism,“ Clinical Chemistry, vol. 65, no. 9, pp. 1090- 1101, 2019.
5 D. Williams und B. Fingleton, „Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis“, Clinical & Experimental Metastasis, vol. 36, no. 3, pp. 211-224, 2019.
6 T. Tataranni, F. Agriesti, V. Ruggieri et al. „Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles“, Oncotarget, vol. 8, no. 25, pp. 41265-41281, 2017.
7 A. Luengo, D. Y. Gui, and M. G. Vander Heiden, „Targeting metabolism for cancer therapy“, Cell Chemical Biology, vol. 24, no. 9, pp. 1161-1180, 2017.
8 M. O. James, S. C. Jahn, G. Zhong, M. G. Smeltz, Z. Hu, and P. W. Stacpoole, „Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1,“ Pharmacology & Therapeutics, vol. 170, pp. 166-180, 2017.
9 E. D. Michelakis, L. Webster und J. R. Mackey, „Dichloroacetate (Dca) as a potential metabolic-targeting therapy for cancer,“ British Journal of Cancer, vol. 99, no. 7, pp. 989-994, 2008.
10 S. Kankotia und P. W. Stacpoole, „Dichloroacetate and cancer: new home for an orphan drug?“, Biochimica et Biophysica Acta, Bd. 1846, Nr. 2, S. 617-629, 2014.
11 V. Ruggieri, F. Agriesti, R. Scrima et al. „Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment“, Oncotarget, vol. 6, no. 2, pp. 1217-1230, 2015.
12 T. Tataranni, F. Agriesti, C. Pacelli et al. „Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines“, Cells, vol. 8, no. 5, p. 478, 2019.
13 A. Khan, D. Marier, E. Marsden, D. Andrews, and I. Eliaz, „A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases,“ Alternative Therapies in Health and Medicine, vol. 20, Supplement 2, pp. 21-28, 2014.
14 E. M. Dunbar, B. S. Coats, A. L. Shroads et al., „Phase 1 trial of dichloroacetate (Dca) in adults with recurrent malignant brain tumors,“ Investigational New Drugs, vol. 32, no. 3, pp. 452-464, 2014.
15Q. S.-C. Chu, R. Sangha, J. Spratlin et al., „A phase I openlabeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors,“ Investigational New Drugs, vol. 33, no. 3, pp. 603-610, 2015.
16 A. Khan, D. Andrews, und A. C. Blackburn, „Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy,“ World Journal of Clinical Cases, vol. 4, no. 10, pp. 336-343, 2016.
17 G. Sutendra und E. D. Michelakis, „Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology“ (Pyruvat-Dehydrogenase-Kinase als neues therapeutisches Ziel in der Onkologie), Frontiers in Oncology, Bd. 3, S. 38, 2013.
18 S. R. Pillai, M. Damaghi, Y. Marunaka, E. P. Spugnini, S. Fais, und R. J. Gillies, „Causes, consequences, and therapy of tumors acidosis,“ Cancer Metastasis Reviews, vol. 38, no. 1- 2, pp. 205-222, 2019.
19 R. J. DeBerardinis, J. J. Lum, G. Hatzivassiliou und C. B. Thompson, „The biology of cancer: metabolic reprogramming fuels cell growth and proliferation,“ Cell Metabolism, vol. 7, no. 1, pp. 11-20, 2008.
20 N. Zamzami und G. Kroemer, „The mitochondrion in apoptosis: how Pandora’s box opens“ (Das Mitochondrium in der Apoptose: wie sich die Büchse der Pandora öffnet), Nature Reviews Molecular Cell Biology, Bd. 2, Nr. 1, S. 67-71, 2001.
21 J. W. Kim und C. V. Dang, „Multifaceted roles of glycolytic enzymes“, Trends in Biochemical Sciences, vol. 30, no. 3, S. 142-150, 2005. P. A. Gammage und C. Frezza, „Mitochondrial DNA: the overlooked oncogenome?“, BMC Biology, Bd. 17, Nr. 1, S. 53, 2019.
22 L. H. Stockwin, S. X. Yu, S. Borgel et al., „Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC,“ International Journal of Cancer, vol. 127, no. 11, pp. 2510-2519, 2010. [
23 P. W. Stacpoole, N. V. Nagaraja und A. D. Hutson, „Efficacy of dichloroacetate as a lactate-lowering drug“ (Wirksamkeit von Dichloracetat als laktatsenkendes Medikament), Journal of Clinical Pharmacology, Bd. 43, Nr. 7, S. 683-691, 2003.
24 P. W. Stacpoole, „Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer“, JNCI: Journal of the National Cancer Institute, Bd. 109, Nr. 11, 2017.
25 E. D. Michelakis, G. Sutendra, P. Dromparis et al., „Metabolic modulation of glioblastoma with dichloroacetate,“ Science Translational Medicine, vol. 2, no. 31, article 31ra34, 2010.
26 P. W. Stacpoole, C. J. Martyniuk, M. O. James und N. A. Calcutt, „Dichloroacetate-induced peripheral neuropathy“, International Review of Neurobiology, Bd. 145, S. 211-238, 2019.
27 N. Felitsyn, P. W. Stacpoole und L. Notterpek, „Dichloroacetate causes reversible demyelination in vitro: potential mechanism for its neuropathic effect“, Journal of Neurochemistry, vol. 100, no. 2, pp. 429-436, 2007.
28 T. Langaee, R. Wagner, L. P. Horne et al., „Personalized dosing of dichloroacetate using Gstz1 clinical genotyping assay,“ Genetic Testing and Molecular Biomarkers, vol. 22, no. 4, pp. 266-269, 2018.
29 D. Brandsma, T. P. Dorlo, J. H. Haanen, J. H. Beijnen, and W. Boogerd, „Severe encephalopathy and polyneuropathy induced by dichloroacetate,“ Journal of Neurology, vol. 257, no. 12, pp. 2099-2100, 2010.
30 United States Environmental Protection Agency, EPA, Toxicological Review of Dichloroacetic Acid, CAS 79-43-6, 2003.
31 X. Lu, D. Zhou, B. Hou et al. „Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer“, Cancer Management and Research, vol. 10, pp. 1231-1241, 2018.
32 A. Korga, M. Ostrowska, M. Iwan, M. Herbet, and J. Dudka, „Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes Hepg2 cells to doxorubicin treatment,“ FEBS Open Bio, vol. 9, no. 5, pp. 959-972, 2019.
33 H. Sun, A. Zhu, X. Zhou, and F. Wang, „Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel,“ Oncotarget, vol. 8, no. 32, pp. 52642-52650, 2017.
34 B. L. Woolbright, D. Choudhary, A. Mikhalyuk et al. „The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance“, Molecular Cancer Therapeutics, vol. 17, no. 9, pp. 2004-2012, 2018.
35 K. Fekir, H. Dubois-Pot-Schneider, R. Désert et al. „Retrodifferenzierung von menschlichen Tumorhepatozyten zu Stammzellen führt zu metabolischer Umprogrammierung und Chemoresistenz,“ Cancer Research, vol. 79, no. 8, pp. 1869-1883, 2019.
36A. Skeberdytė, I. Sarapinienė, J. Aleksander-Krasko, V. Stankevičius, K.
37Sužiedėlis, and S. Jarmalaitė, „Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines,“ Scientific Reports, vol. 8, no. 1, p. 17744, 2018.
38 A. Verma, Y. M. Lam, Y. C. Leung et al., „Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells,“ The Journal of Pharmacy and Pharmacology, vol. 71, no. 3, pp. 306-315, 2019.
39 B. Li, X. Li, H. Xiong et al., „Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells,“ Oncotarget, vol. 8, no. 31, pp. 51748-51757, 2017.
40 C. Lucido, W. Miskimins, and P. Vermeer, „Propranolol promotes glucose dependence and synergizes with dichloroacetate for anti-cancer activity in HNSCC,“ Cancers, vol. 10, no. 12, p. 476, 2018.
41 C. Abildgaard, C. Dahl, A. Abdul-Al, A. Christensen, and P. Guldberg, „Inhibition of retinoic acid receptor Β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells,“ Oncotarget, vol. 8, no. 48, pp. 84210-84223, 2017.
42 I. V. Prokhorova, O. N. Pyaskovskaya, D. L. Kolesnik, and G. I. Solyanik, „Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6,“ Experimental Oncology, vol. 40, no. 3, pp. 205-210, 2018.
43 D. L. Kolesnik, O. N. Pyaskovskaya, Y. R. Yakshibaeva, and G. I. Solyanik, „Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma,“ Experimental Oncology, vol. 41, no. 1, pp. 14-19, 2019.
44 W. Jiang, S. Finniss, S. Cazacu et al., „Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma,“ Oncotarget, vol. 7, no. 35, pp. 56456- 56470, 2016.
45 B. Waltenberger, A. Mocan, K. Šmejkal, E. Heiss, A. Atanasov, and A. G. Atanasov, „Natural products to counteract the epidemic of cardiovascular and metabolic disorders,“ Molecules, vol. 21, no. 6, p. 807, 2016.
46 M. Zadorozhna, T. Tataranni, und D. Mangieri, „Piperine: role in prevention and progression of cancer,“ Molecular Biology Reports, vol. 46, no. 5, pp. 5617-5629, 2019.
47G. Della Sala, F. Agriesti, C. Mazzoccoli, T. Tataranni, V. Costantino, and C. Piccoli, „Clogging the ubiquitinproteasome machinery with marine natural products: last decade update,“ Marine Drugs, vol. 16, no. 12, p. 467, 2018.
48 A. G. Atanasov, B. Waltenberger, E. M. Pferschy-Wenzig et al. „Discovery and resupply of pharmacologically active plant-derived natural products: a review,“ Biotechnology Advances, vol. 33, no. 8, pp. 1582-1614, 2015.
49 M. B. Sporn und N. Suh, „Chemoprävention: ein wesentlicher Ansatz zur Kontrolle von Krebs“, Nature Reviews Cancer, Bd. 2, Nr. 7, S. 537-543, 2002.
50 C. K. Singh, J. George und N. Ahmad, „Resveratrol-based combinatorial strategies for cancer management“, Annals of the New York Academy of Sciences, Bd. 1290, S. 113-121, 2013.
51 S. Redondo-Blanco, J. Fernández, I. Gutiérrez-del-Río, C. J. Villar, and F. Lombó, „New insights towards colorectal cancer chemotherapy using natural bioactive compounds,“ Frontiers in Pharmacology, vol. 8, p. 109, 2017.
52 B. B. Aggarwal, A. Kumar, und A. C. Bharti, „Anticancer potential of curcumin: preclinical and clinical studies,“ Anticancer Research, vol. 23, no. 1A, pp. 363-398, 2003.
53P. C. Kan, Y. J. Chang, C. S. Chien, C. Y. Su, and H. W. Fang, „Coupling dichloroacetate treatment with curcumin significantly enhances anticancer potential,“ Anticancer Research, vol. 38, no. 11, pp. 6253-6261, 2018.
54 K. Hata, K. Hori, H. Ogasawara, and S. Takahashi, „Antileukemia activities of Lup-28-Al-20(29)-En-3-one, a lupane triterpene,“ Toxicology Letters, vol. 143, no. 1, pp. 1-7, 2003.
55C. A. Dehelean, S. Feflea, J. Molnár, I. Zupko, and C. Soica, „Betulin as an antitumor agent tested in vitro on A431, Hela and MCF7, and as an angiogenic inhibitor in vivo in the cam assay,“ Natural Product Communications, vol. 7, no. 8, pp. 981-985, 2012.
56 M. Drag, P. Surowiak, M. Drag-Zalesinska, M. Dietel, H. Lage, and J. Oleksyszyn, „Comparision of the cytotoxic effects of birch bark extract, betulin and betulinic acid towards human gastric carcinoma and pancreatic carcinoma drug-sensitive and drug-resistant cell lines,“ Molecules, vol. 14, no. 4, pp. 1639-1651, 2009.
57 M. Mihoub, A. Pichette, B. Sylla, C. Gauthier, and J. Legault, „Bidesmosidic betulin saponin bearing L-rhamnopyranoside moieties induces apoptosis and inhibition of lung cancer cells growth in vitro and in vivo,“ PLoS One, vol. 13, no. 3, article e0193386, 2018.
58 H. Wang, H. Jiang, M. Van De Gucht, and M. De Ridder, „Hypoxic radioresistance: can ROS be the key to overcome it?“, Cancers, vol. 11, no. 1, p. 112, 2019.
59 J. P. Pouget, S. Frelon, J. L. Ravanat, I. Testard, F. Odin, und J. Cadet, „Formation of modified DNA bases in cells exposed either to gamma radiation or to high-let particles,“ Radiation Research, vol. 157, no. 5, S. 589-595, 2002.
60 K. Rycaj und D. G. Tang, „Cancer stem cells and radioresistance“, International Journal of Radiation Biology, Bd. 90, Nr. 8, S. 615-621, 2014.
61 L. Tang, F. Wei, Y. Wu et al., „Role of metabolism in cancer cell radioresistance and radiosensitization methods,“ Journal of Experimental & Clinical Cancer Research, vol. 37, no. 1, p. 87, 2018.
62 G. Xie, Y. Liu, Q. Yao et al. „Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells“, Scientific Reports, vol. 7, no. 1, article 42396, 2017.
63 J. Overgaard, „Hypoxic radiosensitization: adored and ignored,“ Journal of Clinical Oncology, vol. 25, no. 26, pp. 4066-4074, 2007.
64 Y. Zhang und S. G. Martin, „Redox proteins and radiotherapy,“ Clinical Oncology, vol. 26, no. 5, pp. 289-300, 2014.
65 H. Jiang, H. Wang und M. De Ridder, „Targeting antioxidant enzymes as a radiosensitizing strategy“, Cancer Letters, Bd. 438, S. 154-164, 2018.
66 M. R. Niewisch, Z. Kuçi, H. Wolburg et al. „Influence of dichloroacetate (DCA) on lactate production and oxygen consumption in neuroblastoma cells: is DCA a suitable drug for neuroblastoma therapy?“, Cellular Physiology and Biochemistry, vol. 29, no. 3-4, pp. 373-380, 2012.
67 S. P. Pitroda, B. T. Wakim, R. F. Sood et al., „Stat1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect,“ BMC Medicine, vol. 7, no. 1, p. 68, 2009.
68 T. Shimura, N. Noma, Y. Sano et al., „Akt-mediated enhanced aerobic glycolysis
69 V. Bol, A. Bol, C. Bouzin et al., „Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation,“ Acta Oncologica, vol. 54, no. 2, pp. 266-274, 2015.
70 H. Shen, E. Hau, S. Joshi, P. J. Dilda, and K. L. McDonald, „Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism,“ Molecular Cancer Therapeutics, vol. 14, no. 8, pp. 1794-1804, 2015.
71 G. Dong, Q. Chen, F. Jiang et al., „Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels,“ Oncotarget,
72 L. Sun, T. Moritake, K. Ito et al., „Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets,“ PLoS One, vol. 12, no. 4, article e0176162, 2017.
73 L. Zitvogel, L. Apetoh, F. Ghiringhelli, F. André, A. Tesniere, und G. Kroemer, „The anticancer immune response:indispensable for therapeutic success?“, The Journal of Clinical Investigation, vol. 118, no. 6, pp. 1991-2001, 2008.
74 S. Gupta und B. Dwarakanath, „Modulation of Immunobiome during radio-sensitization of tumors by glycolytic inhibitors,“ Current Medicinal Chemistry, vol. 25, 2018.
75 D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit, and R. Langer, „Nanocarriers as an emerging platform for cancer therapy,“ Nature Nanotechnology, vol. 2, no. 12, pp. 751-760, 2007.
76 R. C. Huxford, J. Della Rocca und W. Lin, „Metal-organic frameworks as potential drug carriers,“ Current Opinion in Chemical Biology, Bd. 14, Nr. 2, S. 262-268, 2010. [
77 I. A. Lázaro, S. A. Lázaro, and R. S. Forgan, „Enhancing anticancer cytotoxicity through bimodal drug delivery from ultrasmall Zr MOF nanoparticles,“ Chemical Communications, vol. 54, no. 22, pp. 2792-2795, 2018.
78 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al., „Surface-functionalization of Zr-fumarate MOF for selective cytotoxicity and immune system compatibility in nanoscale drug delivery,“ ACS Applied Materials & Interfaces, vol. 10, no. 37, pp. 31146-31157, 2018.
79 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al. „Mechanistic investigation into the selective anticancer cytotoxicity and immune system response of surface-functionalized, dichloroacetate-loaded, UiO-66 nanoparticles,“ ACS Applied Materials & Interfaces, vol. 10, no. 6, pp. 5255- 5268, 2018.
80 P. Štarha, Z. Trávníček, J. Vančo, and Z. Dvořák, „Half-sandwich Ru(II) and Os(II) bathophenanthroline complexes containing a releasable dichloroacetato ligand,“ Molecules, vol. 23, no. 2, p. 420, 2018.
81 J. Pracharova, V. Novohradsky, H. Kostrhunova et al. „Halfsandwich Os(ii) and Ru(ii) bathophenanthroline complexes: anticancer drug candidates with unusual potency and a cellular activity profile in highly invasive triple-negative breast cancer cells,“ Dalton Transactions, vol. 47, no. 35, pp. 12197-12208, 2018.
82 J. Yang, Q. Cao, H. Zhang et al. „Targeted reversal and phosphorescence lifetime imaging of cancer cell metabolism _via_ a theranostic rhenium(I)-DCA conjugate,“ Biomaterials, vol. 176, pp. 94-105, 2018.
83C. Yang, T. Wu, Y. Qin et al. „A facile doxorubicindichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery,“ International Journal of Nanomedicine, vol. 13, pp. 1281-1293, 2018.
84 S. Savino, V. Gandin, J. D. Hoeschele, C. Marzano, G. Natile, and N. Margiotta, „Dual-acting antitumor Pt(IV) prodrugs of kiteplatin with dichloroacetate axial ligands,“ Dalton Transactions, vol. 47, no. 21, pp. 7144-7158, 2018.
85 E. Petruzzella, R. Sirota, I. Solazzo, V. Gandin, and D. Gibson, „Triple action Pt(iv) derivatives of cisplatin: a new class of potent anticancer agents that overcome resistance,“ Chemical Science, vol. 9, no. 18, pp. 4299-4307, 2018.
86 S. Jin, Y. Guo, D. Song et al. „Targeting energy metabolism by a platinum(IV) prodrug as an alternative pathway for cancer suppression,“ Inorganic Chemistry, vol. 58, no. 9, pp. 6507-6516, 2019. [
87W. Zhang, X. Hu, W. Zhou, and K. Y. Tam, „liquid chromatography-tandem mass spectrometry method revealed that lung cancer cells exhibited distinct metabolite profiles upon the treatment with different pyruvate dehydrogenase kinase inhibitors,“ Journal of Proteome Research, vol. 17, no. 9, pp. 3012-3021, 2018.
88 S. Dubuis, K. Ortmayr, and M. Zampieri, „A framework for large-scale metabolome drug profiling links coenzyme a metabolism to the toxicity of anti-cancer drug dichloroacetate,“ Communications Biology, vol. 1, no. 1, p. 101, 2018.
89S. M. El Sayed, H. Baghdadi, N. S. Ahmed et al. „Dichloracetate is an antimetabolite that antagonizes acetate and deprived cancer cells from its benefits: a novel evidencebased medical hypothesis,“ Medical Hypotheses, vol. 122, pp. 206-209, 2019.
90 D. M. Jaworski, A. M. Namboodiri, and J. R. Moffett, „Acetate as a metabolic and epigenetic modifier of cancer therapy,“ Journal of Cellular Biochemistry, vol. 117, no. 3, pp. 574- 588, 2016.
91 B. A. Webb, M. Chimenti, M. P. Jacobson, und D. L. Barber, „Dysregulated pH: a perfect storm for cancer progression,“ Nature Reviews Cancer, vol. 11, no. 9, pp. 671-677, 2011. [
92 D. Neri und C. T. Supuran, „Interfering with pH regulation in tumours as a therapeutic strategy“, Nature Reviews Drug Discovery, Bd. 10, Nr. 10, S. 767-777, 2011.
93 A. Kumar, S. Kant, and S. M. Singh, „Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation,“ Toxicology and Applied Pharmacology, vol. 273, no. 1, pp. 196-208, 2013.
94 M. Albatany, A. Li, S. Meakin, and R. Bartha, „Dichloroacetate induced intracellular acidification in glioblastoma: in vivo detection using AACID-CEST MRI at 9.4 Tesla,“ Journal of Neuro-Oncology, vol. 136, no. 2, pp. 255-262, 2018.
95 H. J. Park, J. C. Lyons, T. Ohtsubo, and C. W. Song, „Acidic environment causes apoptosis by increasing caspase activity,“ British Journal of Cancer, vol. 80, no. 12, pp. 1892-1897, 1999.
96 J. Stanevičiūtė, M. Juknevičienė, J. Palubinskienė et al., „Sodium dichloroacetate pharmacological effect as related to Na-K-2Cl cotransporter inhibition in rats,“ Dose Response, vol. 16, no. 4, article 155932581881152, 2018.
97S. Belkahla, A. U. Haq Khan, D. Gitenay et al. „Veränderungen im Stoffwechsel beeinflussen die Expression von ABC-Transportern durch ERK5 und in Abhängigkeit vom p53-Status,“ Oncotarget, vol. 9, no. 1, pp. 1114-1129, 2018.
98 J. A. Bush und G. Li, „Cancer chemoresistance: the relationship between P53 and multidrug transporters,“ International Journal of Cancer, vol. 98, no. 3, S. 323-330, 2002.
99 M. Morfouace, L. Lalier, L. Oliver et al., „Control of glioma cell death and differentiation by PKM2-Oct4 interaction“, Cell Death & Disease, vol. 5, no. 1, pp. e1036-e1036, 2014.
100 A. Turdo, V. Veschi, M. Gaggianesi et al., „Meeting the challenge of targeting cancer stem cells“, Frontiers in Cell and Development Biology, Bd. 7, S. 16, 2019.
101 P. Zhu und Z. Fan, „Cancer stem cells and tumorigenesis,“ Biophysics Reports, vol. 4, no. 4, pp. 178-188, 2018.
102 S. Prasad, S. Ramachandran, N. Gupta, I. Kaushik, and S. K. Srivastava, „Cancer cells stemness: a doorstep to targeted therapy,“ Biochimica et Biophysica Acta – Molecular Basis of Disease, p. 165424, 2019.
103 M. Yang, P. Liu, and P. Huang, „Cancer stem cells, metabolism, and therapeutic significance,“ Tumour Biology, vol. 37, no. 5, pp. 5735-5742, 2016.
104 P. Sancho, D. Barneda, and C. Heeschen, „Hallmarks of cancer stem cell metabolism,“ British Journal of Cancer, vol. 114, no. 12, pp. 1305-1312, 2016.
105 F. Sotgia, M. Fiorillo, and M. P. Lisanti, „Hallmarks of the cancer cell of origin: comparisons with „energetic“ cancer stem cells (e-CSCs),“ Aging, vol. 11, no. 3, pp. 1065-1068, 2019.
106 S. Skvortsov, I. I. Skvortsova, D. G. Tang, and A. Dubrovska, „Concise review: prostate cancer stem cells: current understanding,“ Stem Cells, vol. 36, no. 10, pp. 1457-1474, 2018.
107 S. Bordel, „Constraint based modeling of metabolism allows finding metabolic cancer hallmarks and identifying personalized therapeutic windows,“ Oncotarget, vol. 9, no. 28, pp. 19716-19729, 2018.
108 Y. Y. Wang, J. Chen, X. M. Liu, R. Zhao, and H. Zhe, „Nrf2- mediated metabolic reprogramming in cancer,“ Oxidative Medicine and Cellular Longevity, vol. 2018, Article ID 9304091, 7 pages, 2018.
109 M. W. McBurney, „P19 embryonal carcinoma cells,“ The International Journal of Developmental Biology, vol. 37, no. 1, pp. 135-140, 1993.
110 R. Loureiro, S. Magalhães-Novais, K. A. Mesquita et al., „Melatonin antiproliferative effects require active mitochondrial function in embryonal carcinoma cells,“ Oncotarget, vol. 6, no. 19, pp. 17081-17096, 2015.
111 A. S. Rodrigues, M. Correia, A. Gomes et al., „Dichloroacetate, the pyruvate dehydrogenase complex and the modulation of mESC pluripotency,“ PLoS One, vol. 10, no. 7, article e0131663, 2015.
112 I. Vega-Naredo, R. Loureiro, K. A. Mesquita et al., „Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells,“ Cell Death and Differentiation, vol. 21, no. 10, pp. 1560-1574, 2014.
113 S. K. Singh, C. Hawkins, I. D. Clarke et al., „Identification of human brain tumour initiating cells“, Nature, Bd. 432, Nr. 7015, S. 396-401, 2004.
114 X. Yuan, J. Curtin, Y. Xiong et al., „Isolation of cancer stem cells from adult glioblastoma multiforme,“ Oncogene, vol. 23, no. 58, pp. 9392-9400, 2004.
115 M. Morfouace, L. Lalier, M. Bahut et al., „Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications“, The Journal of Biological Chemistry, vol. 287, no. 40, pp. 33664-33674, 2012.
116 L. Kucerova, L. Demkova, S. Skolekova, R. Bohovic, and M. Matuskova, „Tyrosine kinase inhibitor SU11274 increased tumorigenicity and enriched for melanoma-initiating cells by bioenergetic modulation,“ BMC Cancer, vol. 16, no. 1, p. 308, 2016.
117 Z. Zhao, K. Zhang, Z. Wang et al., „A comprehensive review of available omics data resources and molecular profiling for precision glioma studies,“ Biomedical Reports, vol. 10, no. 1, pp. 3-9, 2019.
Verwandte Inhalte: