Tiziana Tataranni 1 y Claudia Piccoli 1,2
1Laboratoriode Investigación Preclínica y Traslacional, IRCCS-CROB, Centro Oncológico de Referencia de Basilicata, Rionero in Vulture (Pz), 85028, Italia
2Departamento de Medicina Clínica y Experimental, Universidad de Foggia, Foggia 71121, Italia
La correspondencia debe dirigirse a Tiziana Tataranni; [email protected]
Editor invitado: Kanhaiya Singh
Derechos de autor © 2019 Tiziana Tataranni y Claudia Piccoli. Este es un artículo de acceso abierto distribuido bajo la Licencia de Atribución Creative Commons, que permite el uso, distribución y reproducción sin restricciones en cualquier medio, siempre que se cite adecuadamente el trabajo original.
Recibido: 24 de julio de 2019
Revisado: 12 de septiembre de 2019
Aceptado: 11 de octubre de 2019
Publicado en línea: 14 de noviembre de 2019
Un extenso cuerpo de literatura describe la propiedad anticancerígena del dicloroacetato (DCA), pero su administración clínica efectiva en la terapia del cáncer todavía se limita a los ensayos clínicos. La aparición de efectos secundarios como la neurotoxicidad, así como la sospecha de carcinogenicidad del DCA, siguen restringiendo su uso clínico. Sin embargo, en los últimos años, el número de informes que apoyan el empleo del DCA contra el cáncer ha aumentado también debido al gran interés que despierta la acción sobre el metabolismo de las células tumorales. La disección del mecanismo de acción del DCA ayudó a comprender las bases de su eficacia selectiva contra las células cancerosas. En varios modelos de cáncer se ha probado con éxito la coadministración de DCA con quimioterapia convencional, radioterapia, otros fármacos o compuestos naturales. Los nuevos sistemas de administración de fármacos y los compuestos multiacción que contienen DCA y otros fármacos parecen mejorar la biodisponibilidad y parecen más eficaces gracias a la acción sinérgica de múltiples agentes. La difusión de informes que apoyan la eficacia del DCA en la terapia del cáncer ha impulsado estudios adicionales que permiten encontrar otras posibles dianas moleculares del DCA. Curiosamente, el DCA podría afectar significativamente a la fracción de células madre cancerígenas y contribuir a la erradicación del cáncer. En conjunto, estos hallazgos proporcionan una sólida base para nuevos estudios clínicos traslacionales del DCA en la terapia del cáncer.
INTRODUCCIÓN
El cáncer es una de las principales causas de muerte en todo el mundo. A pesar de los importantes avances en los enfoques diagnósticos y terapéuticos, su erradicación sigue representando un reto. Son demasiados los factores responsables del fracaso terapéutico o de la recaída, por lo que urge encontrar nuevos enfoques para tratarlo. Aparte de las típicas propiedades bien conocidas que presentan las células malignas, como la proliferación anormal, la desregulación de la apoptosis y el ciclo celular [1, 2], las células cancerosas también muestran una maquinaria metabólica peculiar que ofrece otro enfoque prometedor para la terapia del cáncer [3-5]. Nuestro grupo ya había sugerido la importancia de una caracterización metabólica de las células cancerosas para predecir la eficacia de un tratamiento metabólico [6]. Ya se están estudiando fármacos capaces de afectar al metabolismo del cáncer, que han mostrado resultados alentadores en términos de eficacia y tolerabilidad [7]. En la última década, la pequeña molécula DCA, ya utilizada para tratar la acidosis láctica aguda y crónica, los errores innatos del metabolismo mitocondrial y la diabetes [8], se ha propuesto en gran medida como fármaco contra el cáncer. El DCA es una molécula ácida soluble en agua de 150 Da, análoga al ácido acético en la que dos de los tres átomos de hidrógeno del grupo metilo han sido sustituidos por átomos de cloro (Figura 1(a)) [9]. La administración de DCA en dosis que oscilan entre 50 y 200 mg/Kg/día se asocia a una disminución del volumen de masa tumoral, de la tasa de proliferación y de la diseminación de metástasis en varios modelos preclínicos [10]. Nuestro grupo ya había observado una correlación inversa entre la capacidad del DCA para destruir células cancerosas y su capacidad respiratoria mitocondrial en carcinomas de células orales [11]. Además, recientemente hemos descrito la capacidad del DCA para afectar a la función mitocondrial y retrasar la progresión del cáncer en un modelo de cáncer de páncreas [12]. Hasta la fecha, se dispone de datos consistentes de ensayos clínicos e informes de casos que describen la administración de DCA en pacientes con cáncer [13-16], pero, a pesar del creciente cuerpo de literatura que sostiene la eficacia del DCA contra el cáncer, todavía no está bajo uso clínico. Esta revisión pretende resumir los informes más recientes que sugieren el empleo del DCA en la terapia del cáncer, en combinación con agentes quimioterápicos, radioterapia y otros compuestos químicos o naturales que muestran propiedades anticancerígenas. Además, describimos datos sobre nuevas formulaciones farmacológicas del DCA capaces de evitar efectos secundarios y mejorar la biodisponibilidad y eficacia del fármaco, lo que fomenta aún más su posible empleo clínico. Por último, revisamos los últimos descubrimientos que sugieren otros posibles mecanismos de acción del DCA, incluyendo nuevos datos sobre su aptitud para afectar a la fracción de células madre cancerígenas.
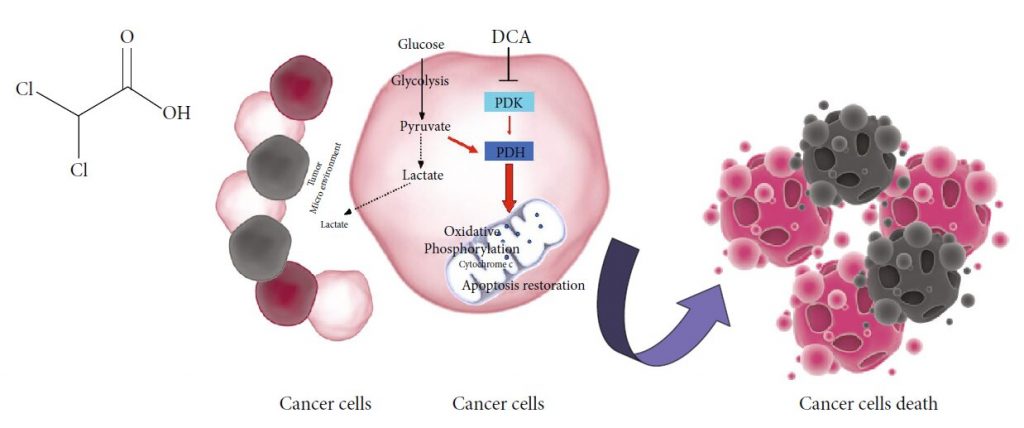
DCA y cáncer: Mecanismo de acción
La eficacia potencial del DCA en la terapia del cáncer proviene de las propiedades metabólicas de las células cancerosas, caracterizadas típicamente por un aumento de la actividad glucolítica y una reducción de la oxidación mitocondrial, independientemente de la disponibilidad de oxígeno, el conocido efecto Warburg [17]. La glucólisis excesiva y la consiguiente sobreproducción de lactato provocan un estado de acidosis metabólica en el microentorno tumoral [18]. El lactato derivado de la glucólisis es absorbido por las células circundantes para favorecer el crecimiento tumoral e inhibe los mecanismos de muerte celular apoptótica [19, 20]. Varias enzimas implicadas en la glucólisis regulan la apoptosis, y su sobreexpresión en células cancerosas contribuye a la supresión de la apoptosis [21]. En este contexto, las sales de DCA se dirigen selectivamente a las células cancerosas cambiando su metabolismo de la glucólisis a la fosforilación oxidativa mediante la inhibición de la piruvato deshidrogenasa quinasa (PDK), el inhibidor de la piruvato deshidrogenasa (PDH) [10]. La activación de la PDH fomenta la oxidación mitocondrial del piruvato y altera la ventaja metabólica de las células cancerosas. Las mutaciones del ADN mitocondrial, que a menudo se producen en la tumorigénesis y dan lugar a una disfunción de la cadena respiratoria [22, 23], hacen que las células malignas sean incapaces de sostener la demanda de energía celular. Además, al reducir la producción de lactato, el DCA contrarresta el estado de acidosis del microambiente tumoral, contribuyendo a inhibir el crecimiento y la diseminación del tumor [24]. La entrada de piruvato en las mitocondrias provoca la remodelación de los orgánulos, lo que se traduce en un aumento del flujo de citocromo c y otros factores apoptóticos, así como en un aumento de los niveles de ROS, con la consiguiente reducción de la viabilidad de las células cancerosas [9] (Figura 1(b)).
Efectos secundarios y limitaciones del empleo del DCA
El empleo clínico del DCA está disponible tanto en formulaciones orales como parenterales, y las dosis oscilan entre 10 y 50 mg/Kg/día [25]. Ninguna evidencia de toxicidad hematológica, hepática, renal o cardiaca grave confirma la seguridad del DCA [26]. Los efectos secundarios gastrointestinales comunes suelen aparecer en un porcentaje de pacientes tratados con DCA [15]. La limitación más conocida de la administración de DCA, observada tanto en estudios preclínicos como clínicos, es la neuropatía periférica [27]. La selectividad del daño inducido por el DCA para el sistema nervioso puede deberse a la falta de maquinaria bien equipada capaz de manejar una fosforilación oxidativa más sostenida en las células que producen ATP principalmente a través de la glucólisis [28]. La sobrecarga mitocondrial resultante compromete la eficacia de los sistemas antioxidantes, incapaces de hacer frente a la cantidad excesiva de ROS. En este contexto, la administración contemporánea de antioxidantes debería representar una estrategia adicional para minimizar la neuropatía inducida por el DCA [27]. La expresión y la actividad de la glutatión transferasa zeta1 (GSTZ1), la primera enzima responsable de la eliminación del DCA, pueden influir en la entidad del daño. Los polimorfismos de un solo nucleótido (SNP) funcionales no sinónimos en el gen humano GSTZ1 dan lugar a diferentes haplotipos que son responsables de una cinética y una dinámica del DCA diferentes. Se ha demostrado una clara asociación entre el haplotipo GSTZ1 y el aclaramiento de DCA. Sobre esta base, una dosificación personalizada de DCA, no sólo basada en el peso corporal, puede minimizar o prevenir los efectos adversos en pacientes tratados crónicamente con este fármaco [29]. La aparición de neuropatía está asociada a la administración oral crónica de DCA y es un efecto reversible, limitado al tiempo de tratamiento [30]. La vía intravenosa reduce, OH Cl Cl O (a) Células cancerígenas Células cancerígenas muerte Lactato Microambiente tumoral Lactato Piruvato Glicólisis PDK DCA PDH Fosforilación oxidativa Apoptosis restauración Citocromo c Glucosa (b) Figura 1: (a) Estructura química del DCA. (b) Mecanismo de acción del DCA: PDK: piruvato deshidrogenasa cinasa; PDH: piruvato deshidrogenasa. Líneas negras discontinuas, procesos bioquímicos inhibidos por el DCA; flechas rojas, vías metabólicas activadas por el DCA. 2 Medicina Oxidativa y Longevidad Celular tanto, el potencial de neurotoxicidad y dejar que la consecución de mayores concentraciones de drogas eludir el sistema digestivo [13].
Dado que el DCA es uno de los subproductos de la desinfección del agua que se encuentra en bajas concentraciones en el agua potable, se está evaluando su potencial carcinogenicidad. Los estudios realizados en modelos de ratón asocian la exposición al DCA en los primeros años de vida a una mayor incidencia de tumores hepatocelulares [31]. Es concebible que los cambios persistentes en el metabolismo celular inducidos por el DCA puedan producir efectos epigenéticos. La inducción a largo plazo de la PDH y otras vías oxidativas relacionadas con el metabolismo de la glucosa podría contribuir a aumentar las especies reactivas del oxígeno y el estrés mitocondrial [27]. Sin embargo, no se ha informado de ninguna evidencia de efecto carcinogénico en estudios clínicos, cuando el DCA se administra en la terapia del cáncer.
Efecto sinérgico del DCA y los agentes quimioterapéuticos
La combinación de diferentes fármacos es una estrategia bien aceptada para producir un efecto beneficioso sinérgico en la terapia del cáncer, reduciendo la dosis de fármacos, minimizando los riesgos de toxicidad y superando la resistencia a los fármacos. La coadministración de DCA y agentes quimioterapéuticos tradicionales se ha propuesto y probado en varios modelos de cáncer (Tabla 1). El tratamiento con DCA parece mejorar la eficacia de la quimioterapia al inducir alteraciones bioquímicas y metabólicas, lo que provoca cambios significativos en el equilibrio energético de las células cancerosas. Un estudio realizado en cáncer de pulmón no microcítico (CPNM) demostró tanto in vitro como in vivo que la coadministración de DCA con paclitaxel aumentaba la eficacia de la muerte celular mediante la inhibición de la autofagia [32]. En células HepG2 se ensayó una combinación eficaz de DCA y doxorrubicina (DOX) que demostró la capacidad del DCA para alterar las defensas antioxidantes celulares, favoreciendo así el daño oxidativo desencadenado a su vez por el tratamiento con DOX [33]. Existe una fuerte asociación entre la sobreexpresión de PDK y la quimiorresistencia; por tanto, es concebible que la inhibición de PDK pueda ayudar a resensibilizar las células cancerosas a los fármacos. La sobreexpresión de la isoforma PDK2 se asoció a la resistencia al paclitaxel en el CPNM. Curiosamente, la combinación de DCA con paclitaxel fue más eficaz para eliminar las células resistentes que el tratamiento con paclitaxel o DCA solos [34]. De forma similar al CPNM, un interesante estudio in vivo realizado en cáncer de vejiga avanzado mostró un aumento de la expresión de la isoforma PDK4 en cánceres de alto grado en comparación con los de bajo grado, y el cotratamiento de DCA y cisplatino redujo drásticamente los volúmenes tumorales en comparación con DCA o cisplatino solos [35]. Un estudio reciente confirmó la capacidad del DCA para revertir la quimiorresistencia relacionada con la PDK4 también en el carcinoma hepatocelular humano (CHC) [36].
| Entidad tumoral | Sistema modelo | Fármaco quimioterapéutico coadministrado con DCA | Mecanismo de acción | Resultado | Referencias |
| Cáncer de pulmón | Líneas celulares A549-H1975/modelo de xenoinjerto | Paclitaxel | Inhibición de la autofagia | Sensibilización eficaz a la quimioterapia del cáncer | [32] |
| Hepatocarcinoma | Línea celular HepG2 | Doxorrubicina | Alteración de la defensa antioxidante | Aumento del daño celular por inducción de estrés oxidativo | [33] |
| Cáncer de pulmón | Línea celular A549 | Paclitaxel | Aumento de la quimiosensibilidad mediante la inhibición de PDK2 | Resistencia al paclitaxel superada | [34] |
| Cáncer de vejiga | Líneas celulares HTB-9, HT-1376, HTB-5, HTB-4/modelo de xenoinjerto | Cisplatino | Aumento de la quimiosensibilidad mediante la inhibición de PDK4 | Aumento de la muerte celular de las células cancerosas y posible ventaja terapéutica | [35] |
| Hepatocarcinoma | Cultivos de esferas de líneas celulares HepaRG y BC2 | Cisplatino, sorafenib | Aumento de la quimiosensibilidad mediante la inhibición de PDK4 | Mejora del efecto terapéutico de la quimioterapia mediante la restauración de la actividad mitocondrial | [36] |
Efecto sinérgico del DCA y otros fármacos anticancerígenos potenciales
Un cuerpo consistente de literatura sugiere efectos positivos de la coadministración de DCA con compuestos actualmente empleados para tratar otras enfermedades pero que muestran propiedades anticancerígenas en varios modelos de cáncer (Tabla 2). La administración simultánea de DCA y el antibiótico salinomicina, redescubierto recientemente por sus propiedades citotóxicas como posible fármaco anticanceroso, se ha probado en líneas celulares de cáncer colorrectal. Su tratamiento parece ejercer un efecto citotóxico sinérgico al inhibir la expresión de proteínas relacionadas con la resistencia a múltiples fármacos [37]. Las células cancerosas que carecen de enzimas metabólicas implicadas en el metabolismo de la arginina pueden resultar sensibles al tratamiento con arginasa. Curiosamente, la administración combinada de arginasa recombinante y DCA produce efectos antiproliferativos en el cáncer de mama triple negativo, debido a la activación de p53 y a la inducción de la detención del ciclo celular [38]. Los inhibidores de la COX2, utilizados principalmente como antiinflamatorios, se han sugerido recientemente como fármacos antitumorales debido a su actividad antiproliferativa. Un interesante estudio realizado en células de cáncer de cuello de útero demostró la incapacidad del DCA para destruir las células de cáncer de cuello de útero que sobreexpresan COX2 y demostró que la inhibición de COX2 mediante celecoxib hace que las células de cáncer de cuello de útero sean más sensibles al DCA tanto en experimentos in vitro como in vivo [39]. Dado que el DCA fomenta la fosforilación oxidativa al disminuir la actividad glucolítica, la combinación de DCA con otros fármacos que potencian un estado de dependencia de la glucosa puede ser una estrategia prometedora. Este enfoque se ha probado en cáncer de cabeza y cuello, en el que la administración de propranolol, un betabloqueante no selectivo capaz de afectar al metabolismo mitocondrial de las células tumorales, produjo dependencia glucolítica y estrés energético, haciendo a las células más vulnerables al tratamiento con DCA [40]. Resultados similares se obtuvieron en células de melanoma en las que la administración de inhibidores del receptor β del ácido retinoico (RARβ) confiere sensibilización al DCA [41]. Se demostró un efecto positivo de la coadministración de DCA con metformina, un fármaco hipoglucemiante ampliamente utilizado para tratar la diabetes, en un modelo preclínico de glioma [ 42], así como en una variante poco metastásica del carcinoma pulmonar de Lewis (LLC) [43]. Jiang y sus colegas investigaron los efectos de la fenformina, un análogo de la metformina, y del DCA en el glioblastoma, demostrando que la inhibición contemporánea del complejo I y de la PDK por la fenformina y el DCA, respectivamente, disminuía la autorrenovación y la viabilidad de las células madre del glioma (GSC), sugiriendo así su posible empleo para afectar a la fracción de células madre del cáncer [44].
| Fármaco | Función principal | Entidad tumoral | Sistema modelo | Resultado | Referencias |
| Salinomicina | Antibiótico | Cáncer colorrectal | Líneas celulares DLD-1 y HCT116 | Inhibición de proteínas relacionadas con la resistencia a múltiples fármacos | [37] |
| Arginasa | Metabolismo de la arginina | Cáncer de mama | MDA-MB231 y MCF-7/ modelo de xenoinjerto | Efecto antiproliferativo debido a la activación de p53 y la detención del ciclo celular | [38] |
| Inhibidores de COX2 | Inflamación | Cáncer cervical | Líneas celulares HeLa y SiHa/modelo de xenoinjerto | Supresión del crecimiento de células cancerosas | [39] |
| Propranolol | Betabloqueante | Cáncer de cabeza y cuello | líneas celulares mEERL y MLM3/C57Bl/6 m | Promoción de la dependencia de la glucosa y potenciación del efecto de la quimiorradiación | [40] |
| Inhibidores de RARβ | Metabolismo de la vitamina A | Melanoma | Líneas celulares ED-007, ED-027, ED-117 y ED196 | Promoción de la dependencia de la glucosa y sensibilización al DCA | [41] |
| Metformina | Diabetes | Glioma, carcinoma pulmonar de Lewis | Modelo de xenoinjerto; células LLC/R9 | Prolongación de la vida útil de ratones con glioma; dependencia grave de la glucosa en el microentorno tumoral | [42, 43] |
| Fenformina | Diabetes | Glioblastoma | Células madre de glioma/modelo de xenoinjerto | Inhibición de la autorrenovación de células madre cancerosas | [44] |
Uso combinado de DCA y compuestos naturales
El empleo clínico de compuestos naturales representa un enfoque novedoso y prometedor para tratar varias enfermedades [45]. Un número creciente de publicaciones apoya la detección, entre los compuestos naturales, de sustancias biológicamente activas aisladas por plantas, hongos y bacterias u organismos marinos que muestran efectos beneficiosos para la salud humana [46-48]. La asunción de compuestos naturales o sus derivados parece representar un enfoque alentador para prevenir el inicio o la recurrencia del cáncer, y se denomina generalmente quimioprevención [49]. Además, las sustancias naturales producen efectos beneficiosos en la terapia del cáncer cuando se coadministran con otros fármacos, demostrando su capacidad para superar la resistencia a los medicamentos, aumentar el potencial anticancerígeno y reducir las dosis y la toxicidad de los fármacos [50, 51]. Curiosamente, recientemente se ha propuesto la coadministración de DCA y compuestos naturales. Un estudio investigó el efecto combinado del DCA con la curcumina mezclada con aceite esencial, un compuesto con propiedades beneficiosas tanto en la prevención como en el tratamiento del cáncer [52], demostrando un potencial anticancerígeno contra el CHC [53]. En particular, la combinación de ambos compuestos redujo sinérgicamente la supervivencia celular, promoviendo la apoptosis celular e induciendo la generación intracelular de ROS. La betulina, un compuesto natural aislado de la corteza de abedul, ya es conocida por sus efectos antiproliferativos y citotóxicos contra varias líneas celulares cancerosas [54-56]. Una investigación in vitro de la actividad antitumoral de derivados de betulina en CPNM confirmó su capacidad para inhibir el crecimiento in vivo e in vitro de células de cáncer de pulmón, bloqueando la fase G2/M del ciclo celular e induciendo la activación de caspasas y la fragmentación del ADN. Curiosamente, el derivado de la betulina Bi-L-RhamBet fue capaz de perturbar la cadena de transporte de electrones (ETC) mitocondrial, induciendo la producción de ROS. Dada la propiedad del DCA de aumentar la oxidación total de la glucosa en las mitocondrias a través del ciclo de Krebs y la ETC, los autores combinaron Bi-L-RhamBet con DCA, demostrando su citotoxicidad significativamente potenciada [57].
DCA y Radiosensibilización
La radioterapia representa otra estrategia para tratar el cáncer y proporciona un enfoque local mediante la administración de rayos de alta energía [58]. El principal efecto de la radiación es la inducción de ROS con el consiguiente daño en el ADN, inestabilidad cromosómica y muerte celular por apoptosis [59]. Sin embargo, varios tumores muestran o desarrollan radioresistencia que es responsable del fracaso de la radioterapia y del alto riesgo de recurrencia tumoral o metástasis [60]. Varios factores pueden ser responsables de la radioresistencia [61]. Entre ellos, la hipoxia, una condición común del microambiente tumoral caracterizada por bajos niveles de oxígeno y reducida generación de especies ROS, puede bloquear la eficacia de las radiaciones ionizantes [62]. Por tanto, aumentar la oxigenación tumoral para favorecer una cantidad considerable de ROS [63] o inducir directamente la producción de ROS puede representar una estrategia para aumentar la radiosensibilización [64, 65]. En este contexto, la administración de DCA, conocido por inducir la producción de ROS [11, 66], podría representar una estrategia para superar la radiorresistencia tumoral. Además, se sabe que las alteraciones metabólicas que caracterizan el desarrollo del cáncer afectan a la radiosensibilidad [67, 68]. Por lo tanto, dirigirse a los intermediarios metabólicos del cáncer puede representar una estrategia para mejorar la respuesta selectiva del cáncer a la irradiación [69]. La eficacia del DCA para aumentar la sensibilidad a la radiación ya se ha demostrado tanto en células de glioblastoma [70] como en carcinoma de células escamosas de esófago [71]. Más recientemente, se ha demostrado que el DCA aumenta la radiosensibilidad en un modelo celular de meduloblastoma, un tumor cerebral mortal en niños, induciendo alteraciones del metabolismo de ROS y de la función mitocondrial y suprimiendo la capacidad de reparación del ADN [72]. Dado que el papel de la inmunoterapia en la restauración de las defensas inmunitarias contra la progresión tumoral y la metástasis está suscitando gran atención en los últimos años [73], Gupta y Dwarakanath proporcionaron un estado de la cuestión sobre los posibles efectos de los inhibidores glucolíticos, incluido el DCA, en la radiosensibilización tumoral, centrando su atención en la interacción entre los modificadores metabólicos y la modulación inmunitaria en los procesos de radiosensibilización [74]. Curiosamente, informaron de la capacidad del DCA para promover la estimulación inmune a través de la inhibición de la acumulación de lactato, sustentando aún más su utilización como adyuvante de la radioterapia.
El DCA y las nuevas formulaciones de fármacos
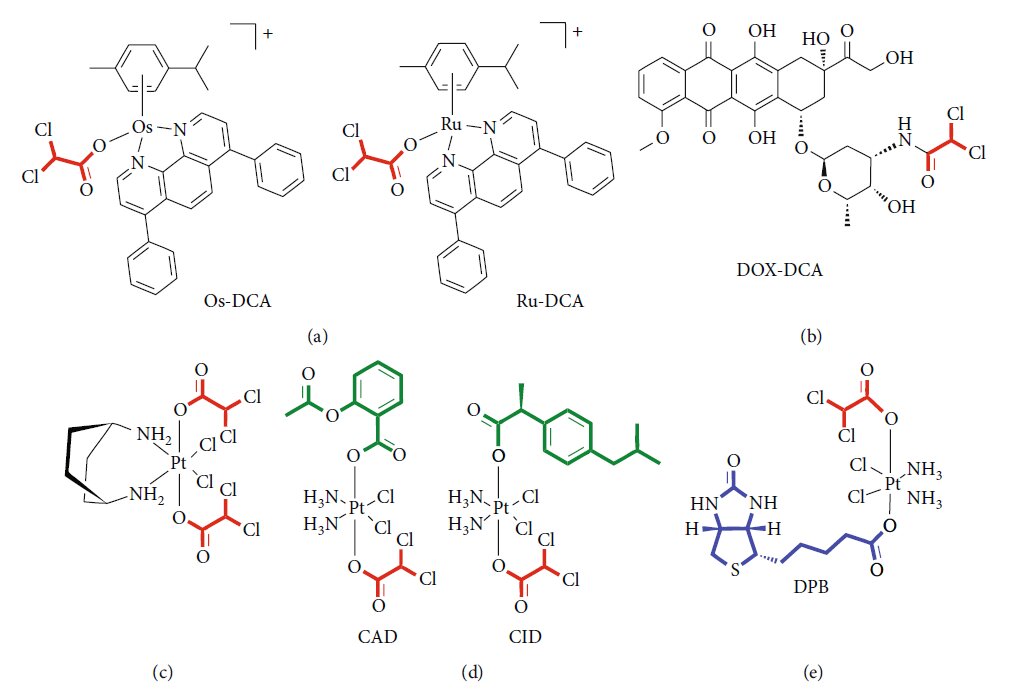
Existe un interés creciente por diseñar nuevas formulaciones de fármacos con el fin de mejorar su administración, aumentar la eficacia y reducir las dosis y, en consecuencia, los efectos indeseables. En este contexto, los sistemas de administración de fármacos (DDS) representan una nueva frontera en la medicina moderna [75]. Los DDSs ofrecen la posibilidad de crear un híbrido de marcos metal-orgánicos (MOFs), combinando la biocompatibilidad del sistema orgánico con las altas cargas de la fracción inorgánica [76]. Varias líneas de evidencia sugieren una funcionalización eficiente de nanopartículas con DCA. Lazaro y colaboradores [77 ] exploraron diferentes protocolos para la funcionalización con DCA de las nanopartículas de tereftalato de circonio (Zr) (UiO-66). Demostraron la citotoxicidad y selectividad de las mismas DDS frente a diferentes líneas celulares de cáncer. Además, excluyeron una posible respuesta del sistema inmunitario al DCA-MOF in vitro. Posteriormente, el mismo grupo demostró la posibilidad de cargar los MOF de Zr con un segundo fármaco anticancerígeno, como el 5-fluorouracilo (5-FU), para reproducir el efecto sinérgico de los dos fármacos [78]. El MOF basado en circonio cargado con DCA también se propuso como una alternativa atractiva al UiO-66, mostrando citotoxicidad selectiva in vitro frente a varias líneas celulares de cáncer y una buena tolerancia por el sistema inmune de varias especies [79]. Recientemente, Štarha et al. [80] sintetizaron y caracterizaron, por primera vez, complejos semisándwich que contienen rutenio u osmio y DCA (Figura 2(a)). Tanto los complejos Ru-dca como Os-DCA se ensayaron en líneas celulares de carcinoma de ovario, demostrando ser más citotóxicos que el cisplatino solo. Ambos complejos fueron capaces de inducir la liberación de citocromo c (Cytc) de las mitocondrias, un índice indirecto de activación del apoptosoma, y parecían ser menos tóxicos para los hepatocitos humanos primarios sanos, lo que indica selectividad para el cáncer frente a las células no cancerosas. También se obtuvieron resultados prometedores en células de cáncer de mama triple negativo [81]. El conjugado de renio (I)-DCA ha demostrado una penetración eficaz en las células cancerosas y una acumulación selectiva en las mitocondrias, induciendo disfunción mitocondrial y trastornos metabólicos [82]. En los últimos años, se han diseñado varios fármacos multiactivos para atacar de forma contemporánea diferentes vías intracelulares utilizando una única formulación. Una nanoformulación segura, sencilla y reproducible del complejo doxorrubicinaDCA (Figura 2(b)) se ensayó con éxito en un modelo de melanoma murino, mostrando un aumento de la capacidad de carga del fármaco, menores efectos secundarios y un efecto terapéutico mejorado [83]. Se han sintetizado profármacos antitumorales de Pt (IV) de doble acción de kiteplatina con ligandos axiales de DCA (Figura 2(c)), se han caracterizado y se han ensayado en diferentes líneas celulares tumorales e in vivo [84]. Para superar la resistencia al cáncer, se han propuesto derivados Pt (IV) de cisplatino de triple acción como nuevos y potentes agentes anticancerígenos, capaces de conjugar la acción del cisplatino, los inhibidores de la ciclooxigenasa y el DCA (Figura 2(d) ) [85]. Se ha ensayado con éxito un nuevo complejo que contiene DCA, platino y biotina (DPB), que presenta propiedades antitumorales multifacéticas (Figura 2(e)). Los autores demostraron la capacidad de este profármaco para afectar al metabolismo energético, promover la apoptosis e interaccionar con el ADN. La alta selectividad de la biotina para las células cancerosas minimiza los efectos perjudiciales sobre las células normales y mejora el efecto curativo sobre los tumores [86]. En la Tabla 3 se resumen las características y pruebas experimentales de las principales clases de compuestos.
| Clase de formulación farmacológica | Características | Pruebas in vitro | Pruebas in vivo | Pruebas experimentales | Referencias |
| Estructuras metal-DCA (sin platino) | Iones metálicos unidos a ligandos orgánicos en andamios porosos | MCF-7/MDA-MB-231 (mama) HeLa/LO2 (cuello uterino) A2780 (ovario) A549/NCl-H1229 (pulmón) | Modelos de ratón para mama | Biocompatibilidad Citotoxicidad selectiva Compatibilidad con el sistema inmunitario Baja mutagenicidad | [77-82] |
| Conjugado de doxorrubicina y DCA | Complejos de DCA y fármacos quimioterapéuticos | B16F10 (melanoma) | Modelos de ratón de sarcoma y melanoma | Citotoxicidad selectiva seguridad Eficacia antitumoral in vivo | [83] |
| Profármacos de platino con DCA | Núcleo de platino asociado a DCA y otros fármacos | MCF-7 (mama) LoVo/HCT-15/HCT116 (colon) A549 (pulmón) BxPC3/PSN-1 (páncreas) A375 (melanoma) BCPAP (tiroides) HeLa (cuello uterino) HepG2 (hepatocarcinoma) | Modelos de ratón de carcinoma de pulmón | Citotoxicidad selectiva acción múltiple Mayor captación celular | [84-86] |
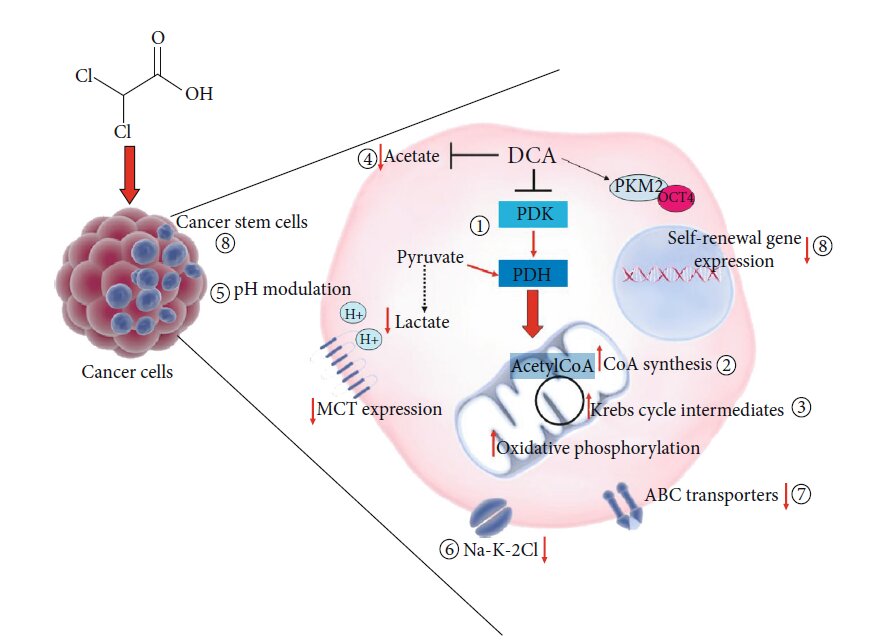
Otros mecanismos de acción propuestos para el DCA
El cambio metabólico de la glucólisis a la oxidación de la glucosa debido a la inhibición de la PDK y la consiguiente activación de la PDH es el efecto molecular más conocido y aceptado de la administración de DCA. Las consiguientes alteraciones bioquímicas, incluyendo el aumento de ROS y la variación del potencial de membrana mitocondrial, pueden ser responsables de la detención de la proliferación y la muerte de las células cancerosas, explicando así el potencial beneficioso del DCA en el tratamiento del cáncer [9]. Sin embargo, aún se desconocen los intermediarios moleculares activados tras la administración de DCA. Es concebible que una molécula tan pequeña pueda afectar directa o indirectamente a otras dianas celulares y moleculares (Figura 3), mostrando otros mecanismos de acción, para explicar así su eficacia también en modelos celulares en los que no produce el cambio metabólico esperado [12]. Un enfoque proteómico aplicado a células de cáncer de pulmón demostró la capacidad del DCA para aumentar la concentración de cada intermediario TCA mientras que no afectó a la captación de glucosa o al proceso glucolítico de glucosa a piruvato [87]. En un intento de arrojar luz sobre el modo de acción del DCA, Dubuis y sus colegas utilizaron un enfoque basado en la metabolómica en varias líneas celulares de cáncer de ovario tratadas con DCA y encontraron una marcada disminución común de pantotenato intracelular, un precursor de CoA, así como un aumento concomitante de CoA, lo que sugiere la capacidad del DCA para aumentar la biosíntesis de novo de CoA. Dado que las altas concentraciones de CoA resultaron ser tóxicas para las células, este efecto metabólico podría ser responsable de la toxicidad de las células cancerosas mediada por el DCA [88]. Un trabajo muy reciente de El Sayed et al. introdujo una nueva hipótesis basada en evidencias, sugiriendo que la eficacia del DCA contra el cáncer puede derivar de su capacidad para antagonizar el acetato [89], conocido por ser un sustrato energético para el glioblastoma y las metástasis cerebrales, capaz de potenciar la síntesis de ADN, ARN y proteínas y las modificaciones postraduccionales, favoreciendo así la proliferación celular y la progresión del cáncer. Además, los niveles elevados de acetato están asociados a la resistencia a los fármacos contra el cáncer [90]. Se ha demostrado que el DCA es capaz de revertir las alteraciones metabólicas inducidas por el acetato restaurando los niveles séricos fisiológicos de lactato y ácido graso libre y la concentración de potasio y fósforo. Según los autores, gracias a una similitud estructural con el acetato, el DCA podría inhibir los efectos metabólicos impulsados por el acetato, responsables del crecimiento de las células cancerosas y de la quimiorresistencia [89]. Otro posible efecto adicional del DCA podría ser la modulación del pH. Se sabe que la modulación del nivel de pH afecta a los procesos de proliferación y apoptosis [91], así como a la sensibilidad a la quimioterapia [92]. El tratamiento con DCA puede tanto aumentar como reducir el pH intracelular. Un efecto secundario de la redirección del piruvato a la mitocondria por el DCA sería la reducción del lactato y el consiguiente aumento del pH intracelular. Por otro lado, el DCA es capaz de disminuir la expresión de transportadores de monocarboxilato y V-ATPasa con la consiguiente reducción del pH, y esto ocurre especialmente en células tumorales, que expresan mayor cantidad de estos transportadores, en comparación con sus homólogas normales [93]. Dada la capacidad de inducir una rápida acidificación intracelular tumoral, Albatany et al. [94] especularon sobre un posible empleo del DCA como rastreador en la obtención de imágenes in vivo de un modelo murino de glioblastoma y apoyaron un uso terapéutico del DCA, ya que se sabe que la acidificación intracelular induce la activación de la caspasa y la fragmentación del ADN de las células cancerosas [95]. Los modelos animales permiten identificar una posible diana molecular adicional del DCA. Los experimentos realizados en ratas pusieron de manifiesto la capacidad del DCA para inhibir la expresión del cotransportador renal Na-K-2Cl (NKCC) en el riñón de las ratas [96]. Dado que el NKCC es un importante biomarcador de la regulación de la homeostasis iónica extracelular e intracelular y participa en la progresión del ciclo celular, desempeña un papel importante en la proliferación, apoptosis e invasión de las células cancerosas. Belkahla et al. [97] investigaron la interacción entre la orientación del metabolismo y la expresión de los transportadores ABC, responsables de la exportación de fármacos de las células y de la consiguiente resistencia a múltiples fármacos, y descubrieron que el tratamiento con DCA es capaz de reducir la expresión génica y proteica de los transportadores ABC en varias células tumorales que expresan p53 de tipo salvaje, tanto in vitro como in vivo [98]. Ya se ha demostrado la capacidad del DCA para inducir la diferenciación a través de la modulación de la interacción PKM2/Oct4 en células de glioma [99]. La reducción resultante de los niveles de transcripción de Oct4 se asoció a una reducción del fenotipo stemness y a un aumento significativo de la sensibilidad al estrés celular. Esta observación permite hipotetizar un papel potencial del DCA contra las células madre cancerígenas (CSCs).
DCA y células madre cancerígenas
Existe un interés creciente por atacar las células madre cancerosas (CSC), que parecen ser las principales responsables de la recaída tumoral [100]. Las CSC comparten la capacidad de autorrenovación con las células madre normales y pueden dar lugar a células diferenciadas, responsables del inicio del tumor así como de la progresión maligna [101]. Su baja tasa de proliferación y su perfil metabólico específico contribuyen a que las CSC sean resistentes a la quimioterapia convencional [102]. Ha surgido una necesidad urgente de desarrollar nuevos agentes terapéuticos capaces de afectar a la viabilidad de las células madre cancerosas [103 ] con el fin de erradicar por completo la masa tumoral. Una extensa literatura está centrando la atención en el fenotipo metabólico de las CSCs, que parecen diferir de las células cancerosas diferenciadas y podrían representar una diana terapéutica [104-108]. En este contexto, se ha planteado la hipótesis de la posible sensibilidad de la fracción de CSC al DCA y se ha probado en diferentes modelos de cáncer. Las células madre embrionarias de carcinoma representan uno de los modelos más apropiados para el estudio del mantenimiento y diferenciación de las CSC y la identificación de fármacos y moléculas capaces de modular estos procesos [109]. Los estudios realizados en células madre embrionarias (ESC) constituyen pruebas preliminares importantes que apoyan una posible eficacia del DCA [110]. Curiosamente, el tratamiento con DCA de ESCs promueve la pérdida de pluripotencia y cambia hacia un metabolismo oxidativo más activo, acompañado de una disminución significativa de la expresión de HIF1a y p53 [111]. Vega-Naredo et al. [112] describieron la importancia del metabolismo mitocondrial en la dirección de la pluripotencia y la diferenciación en un modelo de este tipo. Caracterizaron el perfil metabólico de la fracción de células madre y adivinaron la menor susceptibilidad del fenotipo madre a las terapias dirigidas a las mitocondrias. Forzar las CSC hacia un metabolismo oxidativo mediante el tratamiento con DCA permitió pasar de la condición de célula madre a la de diferenciación. Varios informes apoyan la existencia de CSCs en el glioma [113, 114], y la eficacia del DCA para atacar las CSCs ha sido ampliamente evaluada en este tipo de cáncer, tan difícil de tratar con terapias convencionales y caracterizado por bajas tasas de supervivencia. Ya en 2010, Michelakis y sus colegas habían sugerido, tanto in vitro como in vivo, la capacidad del DCA para inducir la apoptosis de la fracción de células madre cancerosas [26]. Un modelo de glioma en rata, que recapitulaba varias características del glioblastoma humano, confirmó la eficacia del DCA para potenciar la apoptosis de las CSC de glioma, caracterizadas por una sobreestimulación significativa de la vía glucolítica, en comparación con las células madre normales [115]. Asimismo, Jiang et al. investigaron el efecto del DCA sobre la pequeña población de células madre de glioma (GSCs) aisladas de glioblastoma, demostrando una reducción de las propiedades de autorrenovación y un aumento del porcentaje de muerte celular [44]. Además, un ensayo in vivo en ratones portadores de xenoinjertos derivados de GSC tratados con DCA mostró un aumento significativo de la supervivencia global. También se ensayó el tratamiento con DCA en una fracción de células madre de melanoma, y la modulación bioenergética derivada fue capaz de contrarrestar la acción protumorigénica de un inhibidor de c-Met [116]. Un trabajo muy reciente realizado en carcinoma hepatocelular humano identificó la sobreexpresión de PDK4 en esferas originadas a partir de células cancerosas, que presentaban un fenotipo stem-like definido. Curiosamente, el tratamiento con DCA fue capaz de reducir la viabilidad celular tanto de las células diferenciadas del cáncer como de las células madre cancerosas e invirtió la quimiorresistencia a la terapia convencional [36]. Nuestro grupo ha experimentado recientemente la capacidad del DCA para reducir la expresión de los marcadores de células madre cancerosas CD24/CD44/EPCAM en una línea celular de cáncer de páncreas, así como para comprometer la formación de esferoides y la viabilidad [12], corroborando además los datos obtenidos en otros modelos de cáncer. Junto con la quimiorresistencia, también la radioresistencia representa un límite para un tratamiento eficaz del cáncer, y las CSC parecen ser responsables de dicha refractariedad [117]. Sun et al. demostraron la capacidad del DCA para aumentar la radiosensibilidad de las células de meduloblastoma afectando a los clones stem-like, reduciendo el porcentaje de expresión de células CD133-positivas y reduciendo la formación de esferas [72]. Además, en el mismo modelo celular, mostraron un mecanismo alterado de reparación del ADN inducido por el DCA capaz de explicar la mayor eficacia de la radioterapia.
Conclusiones
Atacar el metabolismo de las células cancerosas representa un nuevo enfoque farmacológico para tratar el cáncer. La capacidad del DCA para cambiar el metabolismo de la glucólisis a la fosforilación oxidativa ha aumentado el interés por este fármaco ya conocido por sus propiedades anticancerígenas. Las pruebas acumuladas en los últimos años confirman la capacidad del DCA para superar la quimioresistencia y la radiorresistencia en varios tipos de cáncer y permiten formular hipótesis sobre dianas celulares adicionales capaces de explicar su capacidad para destruir células cancerosas. Es necesario diseñar nuevos estudios clínicos, ahora limitados a pacientes de mal pronóstico con neoplasias avanzadas y recurrentes, ya refractarias a otras terapias convencionales. Su posible eficacia contra las células madre cancerosas, así como el desarrollo de nuevas formulaciones farmacológicas, nos acercan al empleo clínico eficaz del DCA.
Conflictos de intereses
Los autores declaran no tener ningún conflicto de intereses.
Agradecimientos
Este trabajo ha contado con el apoyo de los Fondos de Investigación en Curso, Ministerio de Sanidad italiano, al IRCCS-CROB, Rionero in Vulture, Potenza, Italia.
REFERENCIAS
1 [1] T. G. Lee, E. H. Jeong, I. J. Min, S. Y. Kim, H. R. Kim, y C. H. Kim, «Altered expression of cellular proliferation, apoptosis and the cell cycle-related genes in lung cancer cells with acquired resistance to Egfr tyrosine kinase inhibitors,» Oncology Letters, vol. 14, no. 2, pp. 2191-2197, 2017.
2 J. Chen, «The cell-cycle arrest and apoptotic functions of P53 in tumor initiation and progression,» Cold Spring Harbor Perspectives in Medicine, vol. 6, no. 3, p. a026104, 2016.
3 C. Thakur y F. Chen, «Conexiones entre el metabolismo y la epigenética en los cánceres», Seminars in Cancer Biology, vol. 57, pp. 52-58, 2019.
4 S. Subramaniam, V. Jeet, J. A. Clements, J. H. Gunter y J. Batra, «Emergence of micrornas as key players in cancer cell metabolism», Clinical Chemistry, vol. 65, n.º 9, pp. 1090- 1101, 2019.
5 D. Williams y B. Fingleton, «Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis,» Clinical & Experimental Metastasis, vol. 36, no. 3, pp. 211-224, 2019.
6 T. Tataranni, F. Agriesti, V. Ruggieri y otros, «Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles», Oncotarget, vol. 8, n.º 25, pp. 41265-41281, 2017.
7 A. Luengo, D. Y. Gui y M. G. Vander Heiden, «Targeting metabolism for cancer therapy», Cell Chemical Biology, vol. 24, n.º 9, pp. 1161-1180, 2017.
8 M. O. James, S. C. Jahn, G. Zhong, M. G. Smeltz, Z. Hu y P. W. Stacpoole, «Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1», Pharmacology & Therapeutics, vol. 170, pp. 166-180, 2017.
9 E. D. Michelakis, L. Webster y J. R. Mackey, «Dichloroacetate (Dca) as a potential metabolic-targeting therapy for cancer», British Journal of Cancer, vol. 99, nº 7, pp. 989-994, 2008.
10 S. Kankotia y P. W. Stacpoole, «Dichloroacetate and cancer: new home for an orphan drug?», Biochimica et Biophysica Acta, vol. 1846, nº 2, pp. 617-629, 2014.
11 V. Ruggieri, F. Agriesti, R. Scrima y otros, «Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment», Oncotarget, vol. 6, n.º 2, pp. 1217-1230, 2015.
12 T. Tataranni, F. Agriesti, C. Pacelli et al., «Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines», Cells, vol. 8, no. 5, p. 478, 2019.
13 A. Khan, D. Marier, E. Marsden, D. Andrews e I. Eliaz, «A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases», Alternative Therapies in Health and Medicine, vol. 20, Suplemento 2, pp. 21-28, 2014.
14 E. M. Dunbar, B. S. Coats, A. L. Shroads et al., «Phase 1 trial of dichloroacetate (Dca) in adults with recurrent malignant brain tumors,» Investigational New Drugs, vol. 32, no. 3, pp. 452-464, 2014.
15Q. S.-C. Chu, R. Sangha, J. Spratlin et al., «A phase I openlabeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors,» Investigational New Drugs, vol. 33, no. 3, pp. 603-610, 2015.
16 A. Khan, D. Andrews y A. C. Blackburn, «Long-term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy», World Journal of Clinical Cases, vol. 4, n.º 10, pp. 336-343, 2016.
17 G. Sutendra y E. D. Michelakis, «Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology», Frontiers in Oncology, vol. 3, p. 38, 2013.
18 S. R. Pillai, M. Damaghi, Y. Marunaka, E. P. Spugnini, S. Fais y R. J. Gillies, «Causes, consequences, and therapy of tumors acidosis», Cancer Metastasis Reviews, vol. 38, n.º 1- 2, pp. 205-222, 2019.
19 R. J. DeBerardinis, J. J. Lum, G. Hatzivassiliou y C. B. Thompson, «The biology of cancer: metabolic reprogramming fuels cell growth and proliferation», Cell Metabolism, vol. 7, nº 1, pp. 11-20, 2008.
20 N. Zamzami y G. Kroemer, «The mitochondrion in apoptosis: how Pandora’s box opens», Nature Reviews Molecular Cell Biology, vol. 2, nº 1, pp. 67-71, 2001.
21 J. W. Kim y C. V. Dang, «Multifaceted roles of glycolytic enzymes,» Trends in Biochemical Sciences, vol. 30, no. 3, pp. 142-150, 2005. P. A. Gammage y C. Frezza, «Mitochondrial DNA: the overlooked oncogenome?», BMC Biology, vol. 17, nº 1, p. 53, 2019.
22 L. H. Stockwin, S. X. Yu, S. Borgel et al., «Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC,» International Journal of Cancer, vol. 127, no. 11, pp. 2510-2519, 2010. [
23 P. W. Stacpoole, N. V. Nagaraja y A. D. Hutson, «Efficacy of dichloroacetate as a lactate-lowering drug», Journal of Clinical Pharmacology, vol. 43, nº 7, pp. 683-691, 2003.
24 P. W. Stacpoole, «Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer», JNCI: Journal of the National Cancer Institute, vol. 109, n.º 11, 2017.
25 E. D. Michelakis, G. Sutendra, P. Dromparis et al., «Metabolic modulation of glioblastoma with dichloroacetate,» Science Translational Medicine, vol. 2, no. 31, artículo 31ra34, 2010.
26 P. W. Stacpoole, C. J. Martyniuk, M. O. James y N. A. Calcutt, «Dichloroacetate-induced peripheral neuropathy», International Review of Neurobiology, vol. 145, pp. 211-238, 2019.
27 N. Felitsyn, P. W. Stacpoole, y L. Notterpek, «Dichloroacetate causes reversible demyelination in vitro: potential mechanism for its neuropathic effect,» Journal of Neurochemistry, vol. 100, no. 2, pp. 429-436, 2007.
28 T. Langaee, R. Wagner, L. P. Horne et al., «Personalized dosing of dichloroacetate using Gstz1 clinical genotyping assay,» Genetic Testing and Molecular Biomarkers, vol. 22, no. 4, pp. 266-269, 2018.
29 D. Brandsma, T. P. Dorlo, J. H. Haanen, J. H. Beijnen y W. Boogerd, «Encefalopatía y polineuropatía graves inducidas por dicloroacetato», Journal of Neurology, vol. 257, n.º 12, pp. 2099-2100, 2010.
30 Agencia de Protección del Medio Ambiente de los Estados Unidos, EPA, Toxicological Review of Dichloroacetic Acid, CAS 79-43-6, 2003.
31 X. Lu, D. Zhou, B. Hou et al., «Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer», Cancer Management and Research, vol. 10, pp. 1231-1241, 2018.
32 A. Korga, M. Ostrowska, M. Iwan, M. Herbet y J. Dudka, «Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes Hepg2 cells to doxorubicin treatment», FEBS Open Bio, vol. 9, no. 5, pp. 959-972, 2019.
33 H. Sun, A. Zhu, X. Zhou, y F. Wang, «Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel,» Oncotarget, vol. 8, no. 32, pp. 52642-52650, 2017.
34 B. L. Woolbright, D. Choudhary, A. Mikhalyuk y otros, «The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance», Molecular Cancer Therapeutics, vol. 17, n.º 9, pp. 2004-2012, 2018.
35 K. Fekir, H. Dubois-Pot-Schneider, R. Désert y otros, «La retrodiferenciación de hepatocitos tumorales humanos a células madre conduce a la reprogramación metabólica y la quimiorresistencia», Cancer Research, vol. 79, n.º 8, pp. 1869-1883, 2019.
36A. Skeberdytė, I. Sarapinienė, J. Aleksander-Krasko, V. Stankevičius, K.
37Sužiedėlis, y S. Jarmalaitė, «Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines», Scientific Reports, vol. 8, n.º 1, p. 17744, 2018.
38 A. Verma, Y. M. Lam, Y. C. Leung et al., «Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells,» The Journal of Pharmacy and Pharmacology, vol. 71, no. 3, pp. 306-315, 2019.
39 B. Li, X. Li, H. Xiong et al., «Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells,» Oncotarget, vol. 8, no. 31, pp. 51748-51757, 2017.
40 C. Lucido, W. Miskimins, and P. Vermeer, «Propranolol promotes glucose dependence and synergizes with dichloroacetate for anti-cancer activity in HNSCC,» Cancers, vol. 10, no. 12, p. 476, 2018.
41 C. Abildgaard, C. Dahl, A. Abdul-Al, A. Christensen y P. Guldberg, «Inhibition of retinoic acid receptor Β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells», Oncotarget, vol. 8, no. 48, pp. 84210-84223, 2017.
42 I. V. Prokhorova, O. N. Pyaskovskaya, D. L. Kolesnik y G. I. Solyanik, «Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6», Experimental Oncology, vol. 40, no. 3, pp. 205-210, 2018.
43 D. L. Kolesnik, O. N. Pyaskovskaya, Y. R. Yakshibaeva y G. I. Solyanik, «Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma», Experimental Oncology, vol. 41, n.º 1, pp. 14-19, 2019.
44 W. Jiang, S. Finniss, S. Cazacu et al., «Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma,» Oncotarget, vol. 7, no. 35, pp. 56456- 56470, 2016.
45 B. Waltenberger, A. Mocan, K. Šmejkal, E. Heiss, A. Atanasov y A. G. Atanasov, «Natural products to counteract the epidemic of cardiovascular and metabolic disorders», Molecules, vol. 21, n.º 6, p. 807, 2016.
46 M. Zadorozhna, T. Tataranni y D. Mangieri, «Piperine: role in prevention and progression of cancer», Molecular Biology Reports, vol. 46, no. 5, pp. 5617-5629, 2019.
47G. Della Sala, F. Agriesti, C. Mazzoccoli, T. Tataranni, V. Costantino y C. Piccoli, «Clogging the ubiquitinproteasome machinery with marine natural products: last decade update», Marine Drugs, vol. 16, n.º 12, p. 467, 2018.
48 A. G. Atanasov, B. Waltenberger, E. M. Pferschy-Wenzig et al., «Discovery and resupply of pharmacologically active plant-derived natural products: a review», Biotechnology Advances, vol. 33, n.º 8, pp. 1582-1614, 2015.
49 M. B. Sporn y N. Suh, «Chemoprevention: an essential approach to controlling cancer», Nature Reviews Cancer, vol. 2, nº 7, pp. 537-543, 2002.
50 C. K. Singh, J. George y N. Ahmad, «Resveratrol-based combinatorial strategies for cancer management», Annals of the New York Academy of Sciences, vol. 1290, pp. 113-121, 2013.
51 S. Redondo-Blanco, J. Fernández, I. Gutiérrez-del-Río, C. J. Villar y F. Lombó, «New insights toward colorectal cancer chemotherapy using natural bioactive compounds», Frontiers in Pharmacology, vol. 8, p. 109, 2017.
52 B. B. Aggarwal, A. Kumar y A. C. Bharti, «Anticancer potential of curcumin: preclinical and clinical studies», Anticancer Research, vol. 23, n.º 1A, pp. 363-398, 2003.
53P. C. Kan, Y. J. Chang, C. S. Chien, C. Y. Su y H. W. Fang, «Coupling dichloroacetate treatment with curcumin significantly enhances anticancer potential», Anticancer Research, vol. 38, n.º 11, pp. 6253-6261, 2018.
54 K. Hata, K. Hori, H. Ogasawara y S. Takahashi, «Antileukemia activities of Lup-28-Al-20(29)-En-3-one, a lupane triterpene», Toxicology Letters, vol. 143, n.º 1, pp. 1-7, 2003.
55C. A. Dehelean, S. Feflea, J. Molnár, I. Zupko, y C. Soica, «Betulin as an antitumor agent tested in vitro on A431, Hela and MCF7, and as an angiogenic inhibitor in vivo in the cam assay,» Natural Product Communications, vol. 7, no. 8, pp. 981-985, 2012.
56 M. Drag, P. Surowiak, M. Drag-Zalesinska, M. Dietel, H. Lage y J. Oleksyszyn, «Comparision of the cytotoxic effects of birch bark extract, betulin and betulinic acid towards human gastric carcinoma and pancreatic carcinoma drug-sensitive and drug-resistant cell lines», Molecules, vol. 14, no. 4, pp. 1639-1651, 2009.
57 M. Mihoub, A. Pichette, B. Sylla, C. Gauthier y J. Legault, «Bidesmosidic betulin saponin bearing L-rhamnopyranoside moieties induces apoptosis and inhibition of lung cancer cells growth in vitro and in vivo», PLoS One, vol. 13, no. 3, artículo e0193386, 2018.
58 H. Wang, H. Jiang, M. Van De Gucht y M. De Ridder, «Radioresistencia hipóxica: ¿pueden ser las ERO la clave para superarla?», Cancers, vol. 11, n.º 1, p. 112, 2019.
59 J. P. Pouget, S. Frelon, J. L. Ravanat, I. Testard, F. Odin, y J. Cadet, «Formation of modified DNA bases in cells exposed either to gamma radiation or to high-let particles,» Radiation Research, vol. 157, no. 5, pp. 589-595, 2002.
60 K. Rycaj y D. G. Tang, «Cancer stem cells and radioresistance», International Journal of Radiation Biology, vol. 90, nº 8, pp. 615-621, 2014.
61 L. Tang, F. Wei, Y. Wu y otros, «Role of metabolism in cancer cell radioresistance and radiosensitization methods», Journal of Experimental & Clinical Cancer Research, vol. 37, n.º 1, p. 87, 2018.
62 G. Xie, Y. Liu, Q. Yao y otros, «Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells», Scientific Reports, vol. 7, n.º 1, artículo 42396, 2017.
63 J. Overgaard, «Hypoxic radiosensitization: adored and ignored», Journal of Clinical Oncology, vol. 25, n.º 26, pp. 4066-4074, 2007.
64 Y. Zhang y S. G. Martin, «Redox proteins and radiotherapy», Clinical Oncology, vol. 26, no. 5, pp. 289-300, 2014.
65 H. Jiang, H. Wang y M. De Ridder, «Targeting antioxidant enzymes as a radiosensitizing strategy», Cancer Letters, vol. 438, pp. 154-164, 2018.
66 M. R. Niewisch, Z. Kuçi, H. Wolburg et al., «Influence of dichloroacetate (DCA) on lactate production and oxygen consumption in neuroblastoma cells: is DCA a suitable drug for neuroblastoma therapy?», Cellular Physiology and Biochemistry, vol. 29, no. 3-4, pp. 373-380, 2012.
67 S. P. Pitroda, B. T. Wakim, R. F. Sood y otros, «Stat1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect», BMC Medicine, vol. 7, n.º 1, p. 68, 2009.
68 T. Shimura, N. Noma, Y. Sano et al., «Akt-mediated enhanced aerobic glycolysis
69 V. Bol, A. Bol, C. Bouzin et al., «Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation,» Acta Oncologica, vol. 54, no. 2, pp. 266-274, 2015.
70 H. Shen, E. Hau, S. Joshi, P. J. Dilda y K. L. McDonald, «Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism», Molecular Cancer Therapeutics, vol. 14, n.º 8, pp. 1794-1804, 2015.
71 G. Dong, Q. Chen, F. Jiang y otros, «Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels,» Oncotarget,
72 L. Sun, T. Moritake, K. Ito y otros, «Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets,» PLoS One, vol. 12, no. 4, artículo e0176162, 2017.
73 L. Zitvogel, L. Apetoh, F. Ghiringhelli, F. André, A. Tesniere y G. Kroemer, «The anticancer immune response:indispensable for therapeutic success?», The Journal of Clinical Investigation, vol. 118, n.º 6, pp. 1991-2001, 2008.
74 S. Gupta y B. Dwarakanath, «Modulation of Immunobiome during radio-sensitization of tumors by glycolytic inhibitors», Current Medicinal Chemistry, vol. 25, 2018.
75 D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit, y R. Langer, «Nanocarriers as an emerging platform for cancer therapy,» Nature Nanotechnology, vol. 2, no. 12, pp. 751-760, 2007.
76 R. C. Huxford, J. Della Rocca y W. Lin, «Metal-organic frameworks as potential drug carriers», Current Opinion in Chemical Biology, vol. 14, nº 2, pp. 262-268, 2010. [
77 I. A. Lázaro, S. A. Lázaro y R. S. Forgan, «Enhancing anticancer cytotoxicity through bimodal drug delivery from ultrasmall Zr MOF nanoparticles», Chemical Communications, vol. 54, n.º 22, pp. 2792-2795, 2018.
78 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al., «Surface-functionalization of Zr-fumarate MOF for selective cytotoxicity and immune system compatibility in nanoscale drug delivery,» ACS Applied Materials & Interfaces, vol. 10, no. 37, pp. 31146-31157, 2018.
79 I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz et al., «Mechanistic investigation into the selective anticancer cytotoxicity and immune system response of surface-functionalized, dichloroacetate-loaded, UiO-66 nanoparticles,» ACS Applied Materials & Interfaces, vol. 10, n.º 6, pp. 5255- 5268, 2018.
80 P. Štarha, Z. Trávníček, J. Vančo, y Z. Dvořák, «Half-sandwich Ru(II) and Os(II) bathophenanthroline complexes containing a releasable dichloroacetato ligand,» Molecules, vol. 23, n.º 2, p. 420, 2018.
81 J. Pracharova, V. Novohradsky, H. Kostrhunova et al., «Halfsandwich Os(ii) and Ru(ii) bathophenanthroline complexes: anticancer drug candidates with unusual potency and a cellular activity profile in highly invasive triple-negative breast cancer cells», Dalton Transactions, vol. 47, no. 35, pp. 12197-12208, 2018.
82J. Yang, Q. Cao, H. Zhang y otros, «Inversión dirigida e imágenes de fosforescencia de por vida del metabolismo de las células cancerosas _via_ un conjugado teranóstico de renio(I)-DCA», Biomaterials, vol. 176, pp. 94-105, 2018.
83C. Yang, T. Wu, Y. Qin y otros, «A facile doxorubicindichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery,» International Journal of Nanomedicine, vol. 13, pp. 1281-1293, 2018.
84 S. Savino, V. Gandin, J. D. Hoeschele, C. Marzano, G. Natile y N. Margiotta, «Dual-acting antitumor Pt(IV) prodrugs of kiteplatin with dichloroacetate axial ligands», Dalton Transactions, vol. 47, n.º 21, pp. 7144-7158, 2018.
85 E. Petruzzella, R. Sirota, I. Solazzo, V. Gandin, y D. Gibson, «Triple action Pt(iv) derivatives of cisplatin: a new class of potent anticancer agents that overcome resistance,» Chemical Science, vol. 9, n.º 18, pp. 4299-4307, 2018.
86 S. Jin, Y. Guo, D. Song y otros, «Targeting energy metabolism by a platinum(IV) prodrug as an alternative pathway for cancer suppression», Inorganic Chemistry, vol. 58, n.º 9, pp. 6507-6516, 2019. [
87W. Zhang, X. Hu, W. Zhou y K. Y. Tam, «el método de cromatografía líquida-espectrometría de masas en tándem reveló que las células de cáncer de pulmón exhibían perfiles de metabolitos distintos tras el tratamiento con diferentes inhibidores de la piruvato deshidrogenasa quinasa», Journal of Proteome Research, vol. 17, n.º 9, pp. 3012-3021, 2018.
88S. Dubuis, K. Ortmayr y M. Zampieri, «A framework for large-scale metabolome drug profiling links coenzyme a metabolism to the toxicity of anti-cancer drug dichloroacetate», Communications Biology, vol. 1, n.º 1, pág. 101, 2018.
89S. M. El Sayed, H. Baghdadi, N. S. Ahmed y otros, «El dicloroacetato es un antimetabolito que antagoniza el acetato y priva a las células cancerosas de sus beneficios: una nueva hipótesis médica basada en pruebas», Medical Hypotheses, vol. 122, pp. 206-209, 2019.
90 D. M. Jaworski, A. M. Namboodiri y J. R. Moffett, «Acetate as a metabolic and epigenetic modifier of cancer therapy», Journal of Cellular Biochemistry, vol. 117, no. 3, pp. 574- 588, 2016.
91 B. A. Webb, M. Chimenti, M. P. Jacobson y D. L. Barber, «Dysregulated pH: a perfect storm for cancer progression», Nature Reviews Cancer, vol. 11, n.º 9, pp. 671-677, 2011. [
92 D. Neri y C. T. Supuran, «Interfering with pH regulation in tumours as a therapeutic strategy», Nature Reviews Drug Discovery, vol. 10, n.º 10, pp. 767-777, 2011.
93 A. Kumar, S. Kant y S. M. Singh, «Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation», Toxicology and Applied Pharmacology, vol. 273, n.º 1, pp. 196-208, 2013.
94 M. Albatany, A. Li, S. Meakin y R. Bartha, «Dichloroacetate induced intracellular acidification in glioblastoma: in vivo detection using AACID-CEST MRI at 9.4 Tesla», Journal of Neuro-Oncology, vol. 136, n.º 2, pp. 255-262, 2018.
95 H. J. Park, J. C. Lyons, T. Ohtsubo y C. W. Song, «Acidic environment causes apoptosis by increasing caspase activity», British Journal of Cancer, vol. 80, n.º 12, pp. 1892-1897, 1999.
96 J. Stanevičiūtė, M. Juknevičienė, J. Palubinskienė et al., «Sodium dichloroacetate pharmacological effect as related to Na-K-2Cl cotransporter inhibition in rats,» Dose Response, vol. 16, no. 4, artículo 155932581881152, 2018.
97S. Belkahla, A. U. Haq Khan, D. Gitenay y otros, «Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status», Oncotarget, vol. 9, n.º 1, pp. 1114-1129, 2018.
98 J. A. Bush y G. Li, «Cancer chemoresistance: the relationship between P53 and multidrug transporters», International Journal of Cancer, vol. 98, no. 3, pp. 323-330, 2002.
99 M. Morfouace, L. Lalier, L. Oliver et al., «Control of glioma cell death and differentiation by PKM2-Oct4 interaction,» Cell Death & Disease, vol. 5, no. 1, pp. e1036-e1036, 2014.
100 A. Turdo, V. Veschi, M. Gaggianesi y otros, «Meeting the challenge of targeting cancer stem cells», Frontiers in Cell and Development Biology, vol. 7, pág. 16, 2019.
101 P. Zhu y Z. Fan, «Cancer stem cells and tumorigenesis», Biophysics Reports, vol. 4, no. 4, pp. 178-188, 2018.
102 S. Prasad, S. Ramachandran, N. Gupta, I. Kaushik y S. K. Srivastava, «Cancer cells stemness: a doortep to targeted therapy», Biochimica et Biophysica Acta – Molecular Basis of Disease, p. 165424, 2019.
103 M. Yang, P. Liu y P. Huang, «Cancer stem cells, metabolism, and therapeutic significance», Tumour Biology, vol. 37, no. 5, pp. 5735-5742, 2016.
104 P. Sancho, D. Barneda y C. Heeschen, «Hallmarks of cancer stem cell metabolism», British Journal of Cancer, vol. 114, n.º 12, pp. 1305-1312, 2016.
105 F. Sotgia, M. Fiorillo y M. P. Lisanti, «Hallmarks of the cancer cell of origin: comparisons with «energetic» cancer stem cells (e-CSCs)», Aging, vol. 11, no. 3, pp. 1065-1068, 2019.
106 S. Skvortsov, I. I. Skvortsova, D. G. Tang y A. Dubrovska, «Revisión concisa: células madre del cáncer de próstata: comprensión actual», Stem Cells, vol. 36, n.º 10, pp. 1457-1474, 2018.
107 S. Bordel, «Constraint based modeling of metabolism allows finding metabolic cancer hallmarks and identifying personalized therapeutic windows», Oncotarget, vol. 9, n.º 28, pp. 19716-19729, 2018.
108 Y. Y. Wang, J. Chen, X. M. Liu, R. Zhao y H. Zhe, «Reprogramación metabólica mediada por Nrf2 en el cáncer», Medicina oxidativa y longevidad celular, vol. 2018, ID del artículo 9304091, 7 páginas, 2018.
109 M. W. McBurney, «P19 embryonal carcinoma cells,» The International Journal of Developmental Biology, vol. 37, no. 1, pp. 135-140, 1993.
110 R. Loureiro, S. Magalhães-Novais, K. A. Mesquita et al., «Melatonin antiproliferative effects require active mitochondrial function in embryonal carcinoma cells,» Oncotarget, vol. 6, no. 19, pp. 17081-17096, 2015.
111 A. S. Rodrigues, M. Correia, A. Gomes et al., «Dichloroacetate, the pyruvate dehydrogenase complex and the modulation of mESC pluripotency,» PLoS One, vol. 10, no. 7, article e0131663, 2015.
112 I. Vega-Naredo, R. Loureiro, K. A. Mesquita et al., «Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells,» Cell Death and Differentiation, vol. 21, no. 10, pp. 1560-1574, 2014.
113 S. K. Singh, C. Hawkins, I. D. Clarke y otros, «Identificación de células iniciadoras de tumores cerebrales humanos», Nature, vol. 432, n.º 7015, pp. 396-401, 2004.
114 X. Yuan, J. Curtin, Y. Xiong et al., «Isolation of cancer stem cells from adult glioblastoma multiforme,» Oncogene, vol. 23, no. 58, pp. 9392-9400, 2004.
115 M. Morfouace, L. Lalier, M. Bahut et al., «Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications,» The Journal of Biological Chemistry, vol. 287, no. 40, pp. 33664-33674, 2012.
116 L. Kucerova, L. Demkova, S. Skolekova, R. Bohovic, and M. Matuskova, «Tyrosine kinase inhibitor SU11274 increased tumorigenicity and enriched for melanoma-initiating cells by bioenergetic modulation,» BMC Cancer, vol. 16, no. 1, p. 308, 2016.
117 Z. Zhao, K. Zhang, Z. Wang y otros, «A comprehensive review of available omics data resources and molecular profiling for precision glioma studies», Biomedical Reports, vol. 10, n.º 1, pp. 3-9, 2019.
Contenido relacionado: