Minghao Wang a, Cuiwei Liao a,b, Ying Hu a , Wenqin Pan a , Jun Jiang a,#
a Center of Breast Disease, Southwest Hospital, The Third Military Medical University, Chongqing, 400038, China
b Department of Radiology, Xinqiao Hospital, The Third Military Medical University, Chongqing, 400038, China
Received: 14 May 2017
Accepted: Accepted 17 May 2017
Available online: 19 May 2017
Abstract
Chemotherapy is still the main adjuvant strategy in the treatment of cancer, however, chemoresistance is also frequently encountered. Autophagy inhibition has been widely accepted as a promising therapeutic strategy in cancer, while the lack of effective and specific autophagy inhibitors hinders its application. Here we found that dichloroacetate (DCA), a small molecule compound, could significantly inhibit the autophagy induced by Doxorubicin in breast cancer cells. And DCA markedly enhances Doxorubicin-induced breast cancer cell death and anti-proliferation in vitro. But the sensitization to Dox of DCA was significantly reduced through induction of autophagy by rapamycin. Moreover, the combined therapy of Dox and DCA could significantly inhibit tumor growth in vivo and prolong mouse survival time. Taken together, we demonstrate that DCA could inhibit doxorubicin-inducing autophagy and provide a novel strategy for improving the anti-cancer efficacy of chemotherapy.
Keywords: Dichloroacetate; Breast cancer; Autophagy; Chemotherapy
Highlights:
•Doxorubicin (Dox) induces autophagy in breast cancer cell.
•DCA sensibilizes MDA-MB-231 cells to Dox through inhibiting autophagy.
•Induction autophagy by rapamycin impaired the sensitizing effects of DCA to Dox.
•Synergistic inhibition of tumor growth in mice treated with Dox and DCA.
INTRODUCTION
Breast cancer is a major public health problem worldwide and its universalism and occurrence rate have markedly risen over the past decades. Currently, chemotherapy is still the main aid to breast cancer treatment. Meanwhile, breast cancer is insensitive to regular chemotherapy and radiationtherapy [1], [2].
Autophagy involves the sequestration of portions of the cytoplasmin double-membraned vesicles, the autophagosomes, which then fuse with lysosomes to generate autolysosomes, in which the autophagic cargo is degraded by catabolichydrolases. Autophagy allows cells to degrade their own proteins and organelles to maintain cellular homoeostasis required for normal growth and development, the short-term adaptation to stress as well as for the long-term survival of optimally fit cells [3], [4], [5]. Deregulations of autophagy have been involved in multiple degenerative diseases, aging, including cancer [6], [7], [8], [9]. Notably, autophagy is often a pro-survival response to chemotherapeutic treatment in cancer cells, and suppression of autophagy during chemotherapy has been proposed as anovel therapeutic strategy [10].
Dichloroacetate (DCA) is a small inhibitor of pyruvate dehydrogenase kinase (PDK), which activates pyruvate dehydrogenase (PDH), and increases glucose oxidation by promoting influx of pyruvate into the Krebs cycle. It has been recently demonstrated as a promising nontoxic antineoplastic agent to promote apoptosis in carcinoma cell [11], [12]. But the correlation of DCA and autophagy in breast cancer cell was unknown. In the present study, we investigate the effect of DCA on modulation of autophagy in human breast cancer cells. We found for the first time that DCA potently inhibited autophagy in MDB-231 cell. Furthermore, we investigated the effects of autophagy inhibition by DCA on anticancer potency of chemotherapeutic drugs. Co-treatment of DCA markedly decreased the viability and increased apoptosis in cells treated with Dox, and the sensitizing efficacy of DCA was significantly reduced by rapamycin treatment. The synergistic effect of DCA and doxorubicin was further confirmed in vivo using a mouse xenograft model. Our findings thus demonstrate that inhibition of autophagy with DCA potently enhances the efficacy of Dox, and that such a combination may represent a novel therapeutic strategy for the treatment of breast cancer.
Materials and methods
Cell culture
The human breast cancer cell line MDA-MB-231 was obtained from the American Type Culture Collection (ATCC, HTB-26) and maintained in RPMI1640 medium (Hyclone). The media were supplemented with 10% fetal bovine serum (Gibco) and 100 mg/ml streptomycin (Gibco). Cells were cultured in a humidified incubator with 95% air and 5% CO2 at 37 °C.
Reagents
The primary rabbit monoclonal antibodies against LC3, P62 and β-actin were purchased from Cell Signaling. The goat anti-rabbit IgG secondary antibodies were purchased from Santa Cruz Biotechnology. The FITC-Annexin V and PE-PI was obtained from Biolegend. For dynamic monitoring of autophagy related protein LC3 by immunofluorescence, the adenovirus with tandem EGFP-RFP-LC3 construction was used. The cell viability (MTT) assay kit was purchased from Sangon Biotech (Shanghai). Rapamycin was purchased from Sigma-Aldrich and the doxorubicin was purchased fromSanta Cruz Biotechnology.
Western blot
The Proteins were extracted in lysis buffer after medication and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentrations were measured by BioRad™ protein assay kit. Equal amounts of the protein samples were electrophoresed on 7%–12% SDS-PAGE mini-gel. Proteins were transferred onto Immobilon PVDF membrane at 100 V for 2 h at 4 °C. Membranes were probed with the indicated primary antibody overnight at 4 °C and then incubated with a horse radish peroxidase-coupled secondary antibody. Visualization was performed by incubation for 3 min with enhanced chemiluminescence detection buffer (100 mM Tris-HCl pH 8.5, 1.25 mM luminol, 0.2 mM p-coumaric acid, and 0.03% H2O2) and band intensity was quantified by software Image J.
Immunofluorescence microscopy
Cells were seeded on coverslips in a 24-well plate. Cells were infected with GFP-RFP-LC3 adenovirus before experiment. After 24 h, the medium were replaced and cells were treated with DCA for another 24 h. For immunostaining, the cells were fixed in 4% paraformaldehyde for 15 min and washed. Cells were incubated with DAPI for 5 min then washed with PBS. The coverslips were mounted on slides with Vectashield mounting medium. The images were captured using a LSM780NLO confocal microscope (Carl Zeiss MicroImaging), and processed using the software provided by the manufacturer.
Cell viability (MTT) assay
Cells were seeded in 96-well plates and treated as indicated for 24 h. Then 10 μl MTT (5 mg/ml) was added per well and incubated for 4 h. Each well was supplemented with 100 μl DMSO to dissolve the formazan before being measured by microplate reader at 490 nm. The cell viabilities were normalized to the control group.
Flow cytometry
The extent of apoptosis was determined by flow cytometry. Cells were harvested, washed and incubated with Annexin V staining for 45 min. The propidium iodide staining was added before analyzed by flow cytometry.
Statistical analysis
The data were analyzed by SPSS 21.0 statistical software. The measurement data were expressed inmean ± standard deviation. For comparisons of normal distribution, t-test was used. One-Wayanalysis of variance (ANOVA) was used for comparisons among groups. The count data were expressed in percentage and tested by chi-square test. P < 0.05 was considered statistically significant.
Results
Dox treatment induced autophagy in breast cancer cell
Recent studies have shown that autophagy of tumor cell could be induced during chemotherapy [8], [13]. To determine whether Dox affects autophagy in human breast cancer cells, autophagosome accumulation was detected with a confocal laser-scanning microscope after infected EGFP-RFP-LC3 adenovirus. As shown in Fig. 1A–B, treating cells with Dox for 24 h resulted in a marked increase in RFP-LC3 puncta formation in MDA-MB-231 cells (p < 0.05). Then, we examined the effect of Dox on classic autophagy protein LC3B conversion and P62 in MDA-MB-231 cells. Western blot analysis showed that Dox treatment for 24 h resulted in accumulation of LC3B-II and decrease of P62 (ubiquitin bindingreceptor protein) in a dose- (Fig. 1C–E) and time-dependent manner (Fig. 1F–H) (p < 0.05). These results indicated that Dox is able to induce autophagy in MDA-MB-231 cells.
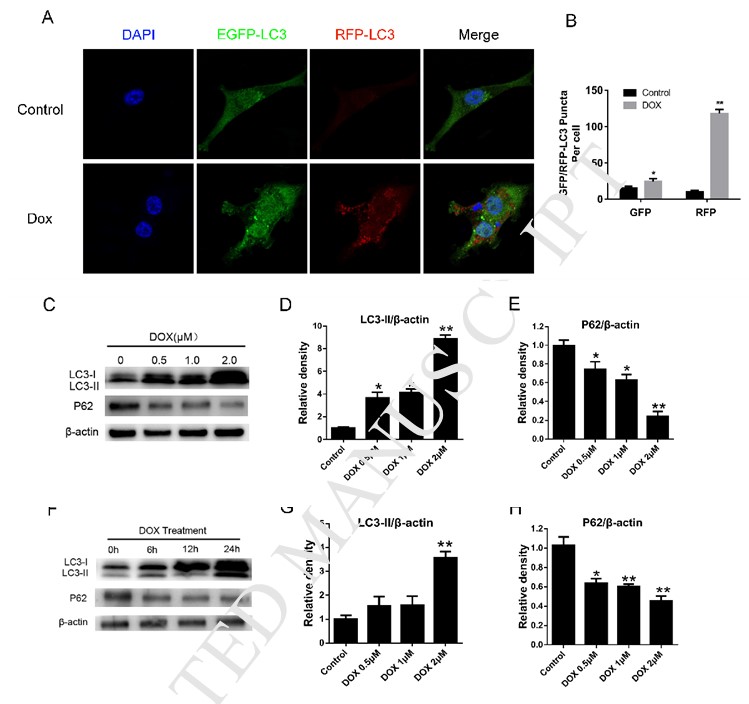
DCA sensitizes doxorubicin-induced cell death through inhibiting autophagy
To explore the influence of DCA treatment on the drug sensitivity of MDA-MB-231 cells to Dox, we therefore detected the apoptosis of MDA-MB-231 cells after treatment of Dox with or without DCA. As shown in Fig. 2A–B, treatment of MDA-MB-231 cells with varied concentration of Dox and DCA for 24 h caused higher levels of apoptosis when compared with Dox treatment alone as shown by FACS (p < 0.05). The results of proliferation experiment showed that DCA had a synergistic effect on the antiproliferative actions of Dox from the 24 h timepoint (Fig. 2C–D). Inhibition of autophagy in tumor cells was considered to enhance the sensitivity of chemotherapy drugs [8], [13], [14]. Next, to determine whether DCA could affect the autophagy induced by Dox in MDA-MB-231 cells, we investigate the change in level of autophagy proteins via indirect immunofluorescent assay and western blotting. Treating cells with Dox or rapamycin resulted in a marked increase in RFP-LC3 puncta formation in MDA-MB-231 cells, which was significantly decreased when co-treatment of DCA (Fig. 1A, Fig. 2E–F). Although the LC3-II level increased after Dox treatment, but the LC3-II level was most weakened after the combined treatment of Dox and DCA, and level of P62 protein was significantly increased after co-treatment (Fig. 2G–I). All these results indicated that DCA enhances the drug sensitivity of MDA-MB-231 cells to Dox through inhibiting autophagy in vitro.
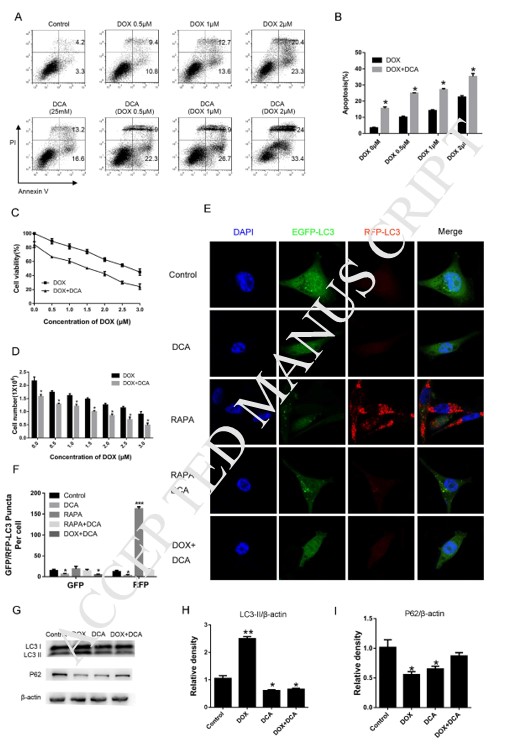
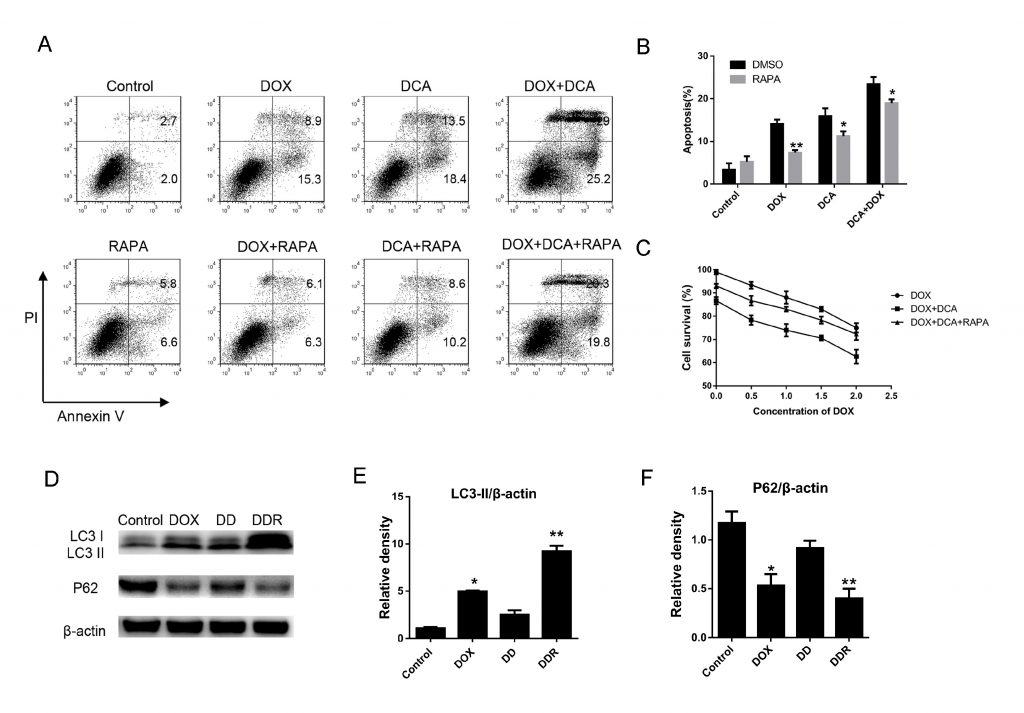
Inducing autophagy of rapamycinreduced the drug sensitivity of DCA and Dox
To further confirm autophagy inhibition by DCA could improve the effect of combination chemotherapy, rapamycin, a classic autophagy accelerant, was used to induce autophagy. As Fig. 3 A–B shown, although the level of apoptosis significantly increased after co-treatment Dox and DCA when compared with Dox treatment alone, but the level of apoptosis was markedly decreased after the combined treatment rapamycin (p < 0.05). The results of MTT experiment showed that rapamycin had anantagonistic effect on the antiproliferative actions of Dox and DCA from the 24 h timepoint (p < 0.05) (Fig. 3C). Furthermore, we detected the change of autophagy related protein LC3-II and P62 after co-treatment of rapamycin.The results showed that the level of LC3-II was markedly increased and the p62 band was significantly reduced in the DDR (Dox, DCA and rapamycin) group when compared to the group treated with DD (Dox and DCA) (Fig. 3D–F). These results further confirmed that DCA sensitizes doxorubicin-induced MDA-MB-231 cells apoptosis through inhibiting autophagy.
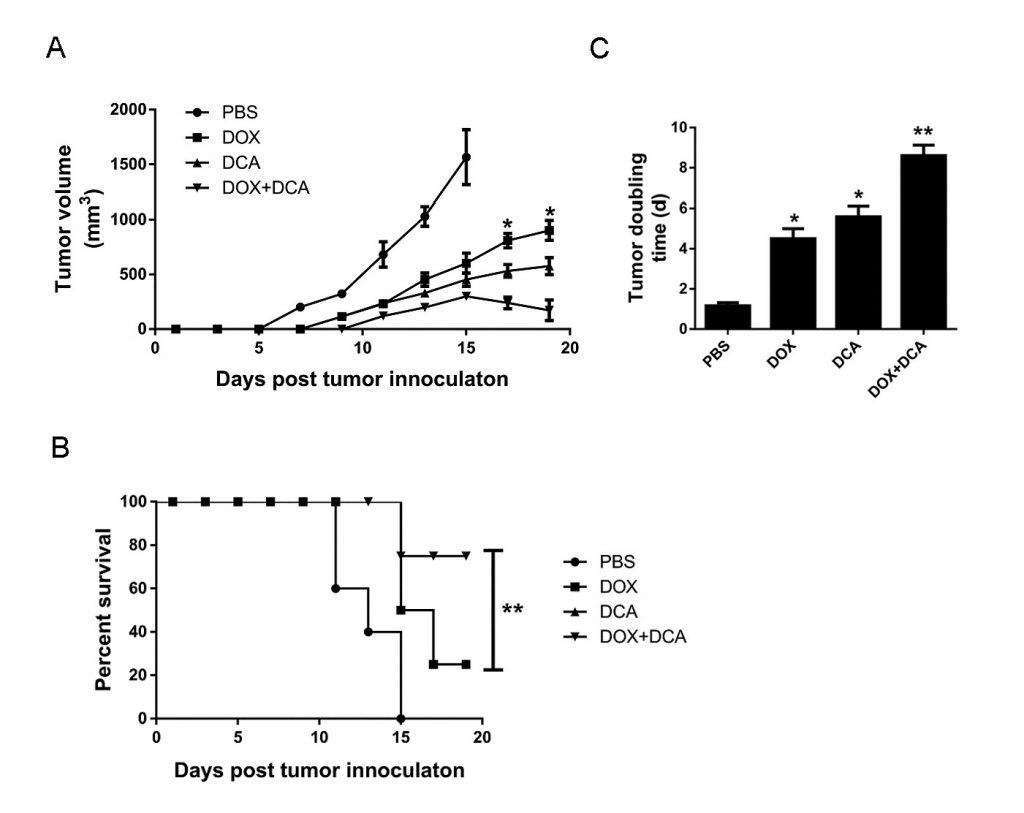
In vivo inhibition of autophagy by DCA potentiates the anticancer capacity of Dox
Considering that Dox induces more cell death after inhibiting autophagy, we assessed the potential of the combined therapy of Dox and DCA in vivo. When the tumor was palpable, the animals were treated with: (1) PBS, (2) Dox, (3) DCA, and (4) Dox plus DCA. As shown in Fig. 4A–B, although Dox or DCA treatment alone showed some inhibitory effect on the growth of tumor when compared to PBS. However, the combined treatment of Dox and DCA significantly inhibited tumor growth and prolonged mouse survival time (p < 0.05). As shown in Fig. 4C, the tumor doubling time significantly increased from 4.65 d (CI, 4.17–5.85 days) in the Dox group to 8.80 d (CI, 7.33–10.3 days) in the Dox plus DCA group (p < 0.05).
Discussion
Chemotherapy is still the main strategy of cancer treatment, however, tolerance always develops for a short period of time. Therefore, current research is focused on searching for new ways to sensitization or delay the drug resistance. Currently the leading view is that cancer cell could tolerate the stress from the outside and hypoxia by regulation of autophagy.
Autophagy is a physiological behavior which is to help maintain cellular homeostasis, senile or defective proteins, organelles, and other cellular components are delivered to the lysosome for degradation and recycling through this process [15], [16]. Lots of studies over the past few years has attempted to link autophagy to cancer and cancer therapy [8], [13], [17]. There were opinions that most cancer drugs as well as ionizing radiation or target-specific drug induce autophagy of tumor cell during the treatment [18], [19], [20]. Inhibition of autophagy in tumor cell was considered to be a strategy to sensibilize chemotherapy and radiotherapy [8], [21], [22]. Although inhibition of autophagy in tumor cell has been widely accepted as a promising therapeutic project in cancer, while the lack ofeffective and specific autophagy inhibitors hinders its application. Chloroquine and hydroxychloroquineare, are the only two FDA-approved autophagy inhibitors. Recently several phase I/II clinical studies involving autophagy inhibition using chloroquine or hydroxychloroquinein combination with chemotherapy for the treatment of different cancers, including breast cancer [13], [23]. Therefore, development of a new type of autophagy inhibitor for the treatment of cancer has important clinical significance. DCA is which could inhibit the critical enzyme pyruvate dehydrogenase kinase (PDK) leads to change mitochondrial respiration of tumor cell and apoptosis [11]. To date, there is no evidence whether DCA was involved in autophagy regulation in breast cancer cell. In thepresent study, we provide evidence that DCA exhibits the ability of inhibition of autophagy and markedly enhances chemotherapeutic drugs-induced breast cancer cell death. However, the sensitization to Dox of DCA was significantly reduced by treating with rapamycin. To the best of our knowledge, this is the first report to demonstrate the effectiveness of autophagy inhibition by DCA in combination with doxorubicin in breast cancer cells in vitro and invivo. However, we did not know whether other chemotherapy drug-induced autophagy could be inhibited by DCA, more research involved in the universality of DCA need to be proceeded.
In summary, the present findings demonstrate that inhibition of autophagy by DCA enhanced doxorubicin-mediated apoptosis and provide a novel research perspective to finding autophagy inhibitors for combination chemotherapy. Our findings suggest that DCA could potentially be further developed as anovel autophagy inhibitor, and a combination of DCA with classical chemotherapeutic drugs could represent a novel therapeutic strategy for treatment of breast cancer.
Conflict of interest
The authors declare that there is no conflict of interests.
Acknowledgement
This work was supported by the Project belongs to the Southwest Hospital [cstc2015shms-ztzx10005].
REFERENCES
1 Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359-386.
2 Siegel RL, Miller KD and Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017; 67: 7-30.
3 Jiang X, Overholtzer M and Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest 2015; 125: 47-54.
4 Klionsky DJ and Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717-1721.
5 Mizushima N and Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147: 728-741.
6 Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H and Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885-889.
7 Hardie DG. Cell biology. Why starving cells eat themselves. Science 2011; 331: 410-411.
8 Mathew R, Karantza-Wadsworth V and White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7: 961-967.
9 Rubinsztein DC, Marino G and Kroemer G. Autophagy and aging. Cell 2011; 146: 682-695.
10 Rubinsztein DC, Codogno P and Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11: 709-730.
11 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B and Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37-51.
12 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C and Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008; 68: 1223-1231.
13 Janku F, McConkey DJ, Hong DS and Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8: 528-539.
14 Kondo Y, Kanzawa T, Sawaya R and Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726-734.
15 Mizushima N. Autophagy: process and function. Genes Dev 2007; 21: 2861-2873.
16 Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR and Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90: 1383-1435.
17 Kondo Y and Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2: 85-90.
18 Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S and White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51-64.
19 Kim DG, Jung KH, Lee DG, Yoon JH, Choi KS, Kwon SW, Shen HM, Morgan MJ, Hong SS and Kim YS. 20(S)-Ginsenoside Rg3 is a novel inhibitor of autophagy and sensitizes hepatocellular carcinoma to doxorubicin. Oncotarget 2014; 5: 4438-4451.
20 Manov I, Pollak Y, Broneshter R and Iancu TC. Inhibition of doxorubicin-induced autophagy in hepatocellular carcinoma Hep3B cells by sorafenib–the role of extracellular signal-regulated kinase counteraction. FEBS J 2011; 278: 3494-3507.
21 Chen K and Shi W. Autophagy regulates resistance of non-small cell lung cancer cells to paclitaxel. Tumour Biol 2016; 37: 10539-10544.
22 Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, Huang C, Li P and Gao N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015; 11: 1259-1279.
23 Kimura T, Takabatake Y, Takahashi A and Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73: 3-7.
Related content: