Ioanna Papandreou, Tereza Goliasova, and Nicholas C. Denko
Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA
Keywords: tumor metabolism, pyruvate dehydrogenase, Warburg effect, metabolic inhibitors
Abbreviations: DCA: dichloroacetate; HIF1: hypoxia-inducible factor 1; LDH: lactate dehydrogenase; PDC: pyruvate dehydrogenase complex; PDH: pyruvate dehydrogenase; PDK: pyruvate dehydrogenase kinase; PDP: pyruvate dehydrogenase phosphatase
Correspondence to: Nicholas C. Denko, Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA 94305, USA
Tel.: 650-724-5066, Fax: 650-723-7382,
E-mail: [email protected]
Received: 22 Jul 2010
Accepted: Accepted 30 Sep 2010
Online: 18 Oct 2010
DOI: 10.1002/ijc.25728
In the past 20 years, the number of articles containing ‘‘tumor metabolism’’ has increased from 3 to 28 per year, and the number of times these articles have been cited has increased from 23 to 929 per year (ISI, Thompson Reuters statistics). The renewed interest in understanding the mechanisms and consequences of altered tumor metabolism has clearly captured the imagination of the scientific community. The idea that tumors have altered metabolism was first recognized by Nobel Prizewinning biochemist Otto Warburg when describing glucose metabolism.1 More recently, the concept that tumors are metabolically different has grown to encompass other characteristics, such as glutaminolysis, fatty acid oxidation and lipid biogenesis. There is clearly a different metabolic demand that drives these changes in cells that are continuously dividing when compared with terminally differentiated cells. The discovery of these alterations has raised the possibility that they may be therapeutically targeted because of their unique importance to cancer cells.2
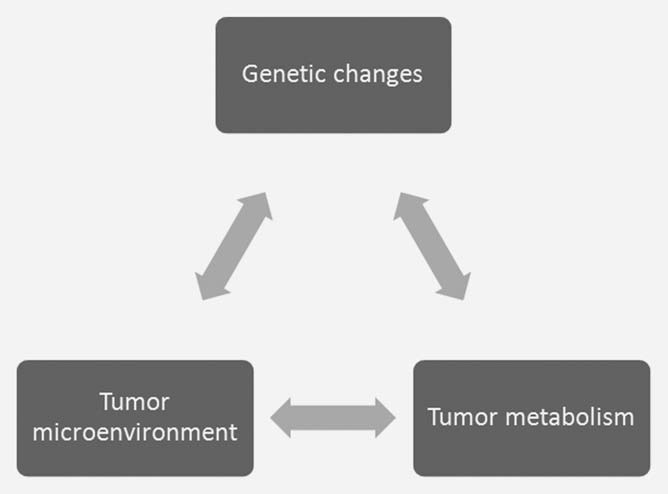
The concept that metabolic changes are a response to unique demands within the tumor has been proposed,3 even when it is hard to quantitate those demands. There is an interplay between oncogenic changes in the tumor cell with the unique aspects of the tumor microenvironment that impact on cellular metabolism and vice versa (Fig. 1). It is therefore difficult to establish the exact metabolic demands within the tumor by studying the cells from the tumor grown ex vivo. The environmental conditions used to grow cells in culture are very different from the environmental conditions in vivo. High glucose Dulbecco’s modified Eagles media and an atmosphere of 21% oxygen is very different from the hypoxic and/or hypoglycemic conditions found in the tumor.4,5 The glucose concentration of 25 mM is approximately five times that of normal blood levels, and the oxygen tension is at least four times greater than that found in vivo. The fact that the cells are bathed in these metabolic substrates significantly alters their inherent metabolic programs.4,6 Elevated glucose concentrations favors glycolysis (the Crabtree effect7 ) while elevated oxygenation produces increased oxygen byproducts and shortens cellular lifespan.8 Glucose metabolism illustrates the interplay of these three factors in the tumor. Oncogenic transformation drives tumor cell proliferation more than vascular capacity, generating hypoxia. Hypoxia within the tumor microenvironment enhances glycolytic metabolism, largely through the activation of the hypoxiainducible factor 1 (HIF1) transcription factor.9 Increased glycolysis leads to increased production of lactate, which contributes to an acidic extracellular pH and further changes in gene expression.10 Both hypoxia and acidosis can contribute to increased levels of somatic mutation that can further drive tumor progression.11,12 It is clearly difficult to reproduce these complex interactions in cells grown in vitro.
Part of the interaction between the microenvironment and tumor cell metabolism is generated through an adaptive response to dynamic changes in cellular supply and demand for metabolites. The simple fact that we can measure regions of hypoxia and acidosis within tumors indicates that the tumor vessels do not maintain a constant environment for the growth of the tumor cells.13 The tumor vasculature represents a bottleneck in the delivery of nutrients and the removal of waste products from the tumor.4 The inadequate supply from the tumor vessels initiates an adaptive response from the tumor cells designed to decrease the demand for the limited metabolites. This dynamic process is difficult to model in vitro (Fig. 2). For example, low levels of oxygen within the tumor induce HIF1 transcription factor and its metabolic program.9 Part of the HIF1-initiated metabolic program is to reduce oxygen demand by decreasing mitochondrial function. Part of this response is mediated through the HIF1-dependent induction of PDK1 and PDK3 within the tumor cells and a reduction in pyruvate oxidation within the mitochondria.14–17 This adaptive response is responsible for bringing the demand for oxygen closer to the limited supply.
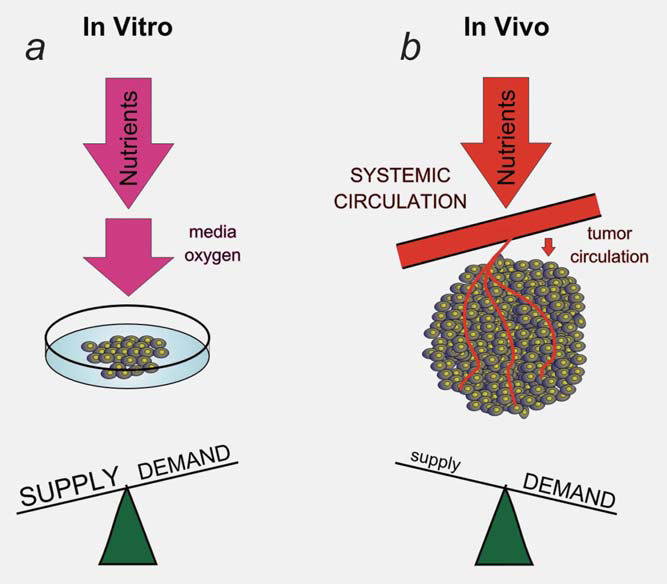
Dichloroacetate (DCA) is capable of interfering with this adaptation to tumor hypoxia by inhibiting the function of the PDKs (Fig. 3). The block of an adaptive response to hypoxia is most clearly observed when the tumor as a whole is in oxygen deficit, and the tumor vasculature cannot respond to this increased demand.18 Even when tumor cells are placed in hypoxia, there is enough oxygen in the environment (1–2%) to maintain a stable, albeit low oxygen condition within the cell even with the addition of DCA. The rate of diffusion into the cells is faster than the rate of consumption, so the intracellular level of oxygen does not depend on the rate of consumption (unless very high cell numbers are used in glass dishes that prevent diffusion of oxygen through the plastic14). In this article, we present an analysis of the published data regarding DCA as an example of a drug designed to impact tumor metabolism that supports our hypothesis that this class of drugs has a very different effect on tumor cells growing in vivo with the vasculature-produced metabolic bottleneck, when compared with their effect on cells grown in vitro.
The Pyruvate Dehydrogenase Complex
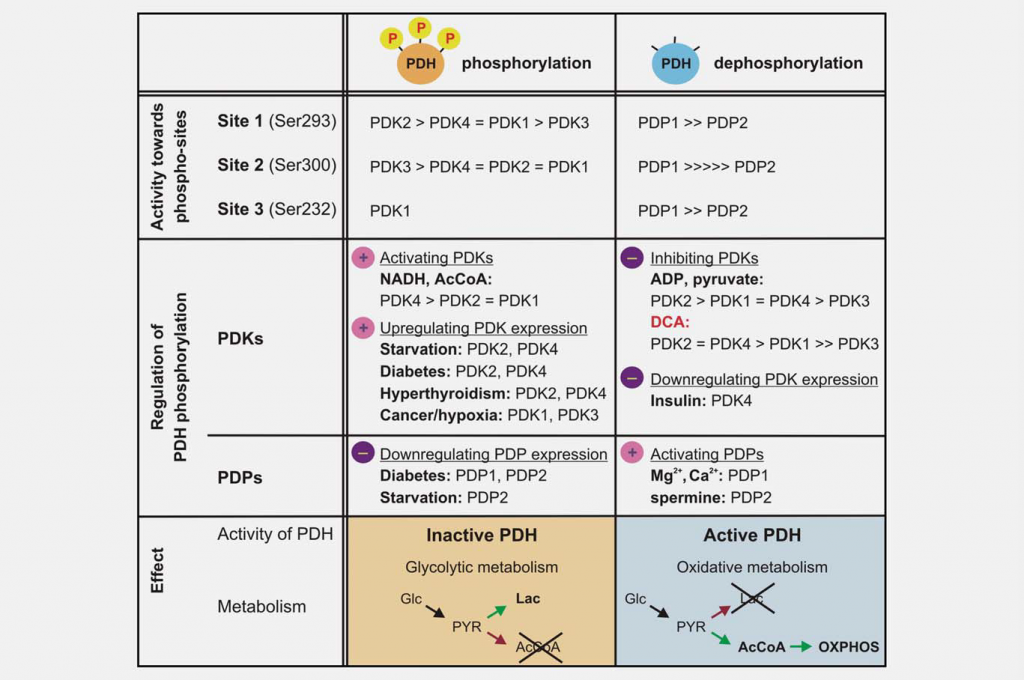
Inhibition of mitochondrial function is as important as increased glycolysis to produce the Warburg effect. One major regulator of mitochondrial function is the pyruvate dehydrogenase complex (PDC), which catalyzes the irreversible decarboxylation of pyruvate to acetyl-CoA, CO2 and NADH. Controlling PDC controls the entry of carbons derived from carbohydrates into the mitochondria. This reaction plays a central role in regulating both mitochondrial energy-producing pathways (tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS)) and the generation of biosynthetic intermediates, such as citrate. PDC consists of three catalytic components, pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2) and dihydrolipoamide dehydrogenase (E3), which are organized into large multimeric complexes together with the structural subunit E3 binding protein (E3BP). The basic core of the E1 pyruvate dehydrogenase component is a heterotetramer of two alpha and two beta subunits (α2β2), and it catalyzes the first step of pyruvate decarboxylation. The activity of the complex is largely regulated by the reversible phosphorylation of three serine residues of E1α. Pyruvate dehydrogenase kinases (PDK1–4) inactivate PDC and pyruvate dehydrogenase phosphatases (PDP1–2) activate/reactivate it (Fig. 3).
The different PDK isoforms vary in their regulatory properties, tissue distribution and modulation by upstream metabolic signals. This results in dynamic, organ-specific control of mitochondrial function and energy production. Enzymatic assays have shown that the PDKs differ in their specificity toward the three E1α target sites and the kinetic parameters of phosphorylation. PDK1 is the only isoform able to phosphorylate all three sites, whereas PDK2–4 phosphorylate sites 1 and 2 with different rates in vitro. 19,20 Phosphorylation of even one of the six E1a sites in the heterotetramer is sufficient to inactivate PDC, and it is believed that maximally three of the possible six sites of the E1 tetramer can be phosphorylated at any given time.21,22 Spatiotemporal changes in the levels and activity of the PDC phosphatases (PDP1–2) are also regulatory. Dephosphorylation in vitro appears to be random; both isozymes are able to dephosphorylate the three E1 sites of recombinant mutated substrates.23
Dichloroacetate: A PDK Inhibitor
DCA was identified as a pyruvate dehydrogenase activator through its ability to stimulate PDC enzymatic activity in a perfused rat heart model.24 It is now known that this pyruvate mimetic acts by inhibiting the action of the PDKs. The crystal structure of PDK2 in complex with DCA has been obtained, and it shows that DCA occupies the pyruvate binding site in the N-terminal regulatory (R) domain.25 The four isozymes vary considerably in their sensitivity to inhibition by DCA. PDK2 is the most sensitive, PDK3 the most resistant, while PDK1 and PDK4 are relatively sensitive.26,27
Treatment of lactic acidosis with DCA
The safety and efficacy of DCA has been evaluated in cases of congenital and acquired lactic acidosis. DCA treatment effectively reduces lactate levels in the circulation by stimulating oxidation of pyruvate; however, it remains not known if DCA can improve the prognosis of patients with these syndromes.28,29 It has been proposed that young children with PDH deficiency may benefit the most from chronic DCA treatment.30 The most significant adverse effect of long-term DCA administration is a reversible peripheral neuropathy.31,32 The severity of the toxicity appears to be age dependent, with adult patients being more susceptible than children.31,33 The reasons for this discrepancy are not entirely clear, but they are possibly related to the different pharmacokinetics and metabolism of DCA in the two age groups.34 DCA has also been used in clinical trials for heart disease, including congestive heart failure and ischemic heart disease, showing positive results and improving myocardial performance.35,36
DCA as a potential cancer therapeutic
In recent years, DCA has attracted attention as a potentially simple and economical means to target glycolytic tumors while producing limited side effects in the oxidative healthy organs. The interest in this drug by the scientific community, cancer patients and the media was kindled in 2007, after a group from the University of Alberta reported that DCA was uniquely toxic to human cancer cell lines and inhibited the growth of A549 lung tumor xenograft tumors in rats.37 Since then, the emerging reports on the efficacy of DCA in vitro and in vivo reveal some interesting and also puzzling characteristics, which distinguish the case of DCA from the majority of drugs developed as anticancer agents (Table 1). The number of different cancer types and experimental strategies tested to date is too limited to allow for generalized conclusions about the efficacy of DCA against all kinds of tumors. With this caveat, a qualitative comparison of the literature suggests that DCA shows more anticancer effect in vivo than anticancer cell effect in vitro.
| Cancer type | References | Effect on survival and growth |
| In vitro studies | ||
| Lung, Glioblastoma, Breast | 37 | Apoptosis in vitro, xenograft growth inhibition |
| Prostate | 38 | Growth inhibition in vitro, moderate radiosensitization |
| Endometrial | 39 | Growth inhibition |
| Cervical | 40 | Growth advantage under hypoxia in vitro |
| Head and Neck | 41 | In vitro growth inhibition only in mutND2 overexpressing cells |
| Pediatric | 42 | Apoptosis at high concentrations in vitro, some influence on response to chemotherapy |
| Colorectal | 43 | Apoptosis at very high concentrations |
| Preclinical models | ||
| Colorectal | 44 | Little effect on growth, increased hypoxia by HRE-luciferase, sensitized to hypoxic cytotoxins |
| Colorectal | 18 | Little effect on growth, increased hypoxia by 18F-FAZA PET, sensitized to hypoxic cytotoxin |
| Preclinical models | 17 | Xenograft growth inhibition |
| Colorectal | 45 | Xenograft growth inhibition |
| Colorectal | 46 | Protected from anoxia in vitro, promoted xenograft growth of SW480 |
| Colorectal, Breast, PML, Prostate | 47 | Active only against cells with defective electron transport chain |
| Breast | 48 | Inhibition of xenograft growth and metastasis |
| Human patient data | ||
| Glioblastoma | 49 | Clinically stable disease in vivo, decreased HIF1, increased p53 ex vivo |
In vitro studies. DCA has been reported to have cytotoxic effects in vitro, 37–40 with some responses at clinically relevant concentrations (0.5–1 mM), while others require suprapharmacologic levels (10–100 mM) and still other groups have found no direct toxicity in vitro. 18,41,42,47 One condition that has been identified that sensitizes cells to DCA is mutations that perturb mitochondrial respiratory function,41,47 suggesting that enforced utilization of defective OXPHOS is toxic. As oxygen deprivation also downregulates mitochondrial function, it seemed reasonable to hypothesize that hypoxic cells would be more sensitive to DCA. However, this hypothesis has not been supported, at least in the limited number of cell lines tested to date. Moderate in vitro hypoxia did not influence the cell cycle profile of DCA-treated colorectal cells43 or the reproductive viability of DCA-treated pancreatic cancer cells (our unpublished observations). Interestingly, another study found severe hypoxia (anoxia?) can be protective against DCA-induced apoptosis in colorectal cancer cells.46 The reason for these discrepancies is not clear.
Overall, the majority of the data support the idea that clinically relevant concentrations of DCA (less than 1 mM) are not directly cytotoxic in vitro. The reason for this apparent cellular resistance is not an inactivation of DCA under tissue culture conditions or an inability to inactivate PDKs, as DCA has been shown to transiently increase mitochondrial activity and collapse the mitochondrial membrane potential.18,37,50 Therefore, the basis for the limited anticancer effect of DCA in culture likely lies in the complex cellular physiology and the enormous excess of metabolites present in culture media.
Preclinical models. Reports of DCA activity against model tumors grown in rodents are encouraging, although there are certain cases of tumor lines that do not respond to treatment, and even one example of accelerated tumor growth in response to DCA.46 The first report of DCA’s antitumor activity was that of Bonnet et al. 37 The authors reported that A549 lung adenocarcinoma xenografts grown in nude rats showed significant tumor growth delay after treatment with DCA, with some experimental groups even showing tumor regression. These effects were associated with increased apoptosis and reduced proliferation. Using the same A549 cells, Stockwin et al. 47 recently confirmed that DCA was growth suppressive in model tumors, even though they found little toxicity in vitro.
Our group has reported that daily DCA treatment of mice with pancreatic SU86.86 xenografts caused a significant tumor growth delay, as well as an increased hypoxic fraction of the tumors. We hypothesized that increased mitochondrial oxygen consumption resulted in greater hypoxia which was growth inhibitory to the tumor.17 In support of this model, DCA increased the extent of tumor hypoxia in RKO colorectal xenografts as assessed by either HRE (hypoxia response element)-driven luciferase reporters44 or 18F-fluoroazomycin arabinoside positron emission tomography.18 This RKO model showed very modest reduction in the growth of the DCA-treated tumors, but the acute changes in oxygenation post DCA sensitized them to treatment with hypoxia-activated cytotoxins such as Tirapazamine44 or PR-104.18 Additional work on colorectal cancer models, by other groups, has revealed significant heterogeneity in the response to DCA. Some cell lines reduce their tumor growth rates,45 whereas others either failed to respond or even grew faster.46 Additionally, in a rat model of mammary adenocarcinoma, an intensive schedule of DCA treatment was able to reduce the number of macroscopic lung metastases.48 These preclinical models support the concept that DCA has the ability to modulate tumor metabolism in vivo, resulting in greater of lesser antitumor effects based on the model tested.
Clinical trial data. The first data on the evaluation of DCA for the treatment of human cancer were reported recently.49 In this study, DCA was used in combination with surgery, temozolomide and radiation for the treatment of five patients with glioblastoma multiforme. While the authors report promising clinical results in four of the five patients, the emphasis of the report was the ex vivo analysis of the tumor cells before and after treatment with DCA. They report changes in mitochondrial membrane potential, increased amounts of mitochondrially generated oxygen radicals and increased tumor cell apoptosis. Mechanistic studies found altered levels of HIF1 signaling, p53 activation and decreased angiogenesis. These data suggest that DCA has many mechanisms of action downstream of its inhibition of PDKs. Obviously, greater numbers of patients, preferably from a number of different sites, need to be treated to be able to generalize these exciting results.
Organismal Regulation of Metabolism
Humoral growth- and nutritional factors could also influence tumor response to metabolic reprograming. The regulation of tumor PDC activity in vivo has not been systematically studied yet, but the knowledge we have gained from normal tissue metabolism and the fields of endocrinology and obesity research provide some interesting hypotheses. Starvation increases the expression of PDKs and decreases PDH activity in peripheral organs as a strategy to maintain a stable supply of carbohydrates to the brain and other neuronal tissues. For example, starvation transcriptionally activates PDK4 and PDK2 in liver, kidney and other tissues.51,52 Glucocorticoids, T3 thyroid hormone and free fatty acids also increase PDK4 levels.53 Refeeding and/or increases in insulin levels decrease PDK4 transcription and reactivate PDC. Likewise, starvation and diabetes were shown to downregulate PDP2 mRNA and protein levels, effects that were reversed by refeeding or treatment with insulin.54
Therefore, the many input signals that impact PDH activity in vivo could influence the efficacy of DCA and other metabolically targeted drugs. Delivery of the drug during fasting could have a very different effect when compared with the fed state. The responses to humoral conditions are obviously different for different tissues and tumor types and so, difficult to mimic in vitro. So, is it possible to evaluate the antitumor effect of molecules like DCA in vitro? Many years of study of a large number of tumor cell lines grown in culture show that they do mantain the abnormal characteristics of aerobic glycolysis in vitro, and therefore provide a valuable tool in the study of cancer metabolism.55 However, there also inherent limitations in these systems, as evidenced by the inconsistent data in the literature when comparing the effect of DCA in cell culture versus preclinical and clinical outcomes.
The preceeding analysis presents the model that an anticancer drug that targets metabolism and has little toxicity in vitro may have significant potential in vivo. We have highlighted the limited metabolic substrates in the tumor, and the systemic regulation of metabolism by humoral factors that could increase in vivo drug efficacy. It is possible that the converse may also be true that a drug with good activity in vitro may have little success in vivo. Effects on normal tissue toxicity or metabolic cooperation between cell types could limit a drug’s effectiveness in vivo. The widely tested metabolic inhibitor 2-deoxyglucose has reasonable anticancer activity in vitro but cannot be used in patients because of its negative effects on normal tissues that rely on glucose consumption. The dose-limiting neurologic toxicity occurs at a drug level well below that needed for anticancer effects in rodent tumors.56,57 Alternatively, it is possible that metabolic cooperation between cell types or normal and tumor cells may bypass the druginduced metabolic block. For example, while lactate is often viewed as a metabolic waste product, it can be used in some cells as a fuel to power mitochondrial function.58,59
With respect to combining DCA with existing therapies, the preclinical data so far do not show an obvious pattern of interaction that would allow for an easy and rational selection of therapeutic regiments. Animal models will continue to be the best means of testing to determine empirically the most promising combinations. We have shown that because of its ability to increase oxygen consumption, DCA increased tumor hypoxia and sensitized xenografted pancreatic and colon tumors to hypoxic cytotoxins,18,44 so it is intriguing to envision a treatment plan including these two classes of drugs that are both designed to exploit the unique hypoxic microenvironment of the tumor.
The interaction of DCA with other metabolic modulators has not been reported. A potential target for combination therapy is lactate dehydrogenase A (LDHA). Genetic or pharmacologic inhibition of LDHA has been shown to increase mitochondrial function and inhibit model tumors’ formation and progression.60,61 In this combination scheme, the LDHA inhibitor would block the conversion of pyruvate to lactate and DCA would divert the accumulated pyruvate toward mitochondrial oxidation. If DCA’s antitumor effect is derived from increased mitochondrial function, it is possible that combining PDK and LDHA inhibitors would force an even greater rate of mitochondrial oxidation and impair tumor growth more efficiently.
Nodal points of key survival pathways, such as the PI3KAkt-mTOR pathway are also the subject of intense drug development efforts.62 Part of the growth-promoting properties of this pathway comes from its ability to regulate metabolism and energy production by direct or indirect mechanisms. For example, oncogenic activation of PI3K-Akt stimulates glucose uptake and aerobic glycolysis,63,64 whereas activation of Akt and mTORC1 increases the translation of Hif-1a mRNA under hypoxia.65 Promising Akt inhibitors and new generation mTOR inhibitors are being tested in clinical trials66,67 and represent viable candidates for combination therapy with DCA to modulate both aerobic and hypoxic metabolism.
Conclusions
In conclusion, recent understanding about the unique metabolism of the solid tumor has identified several novel, druggable pathways that may be preferentially used in tumor cells compared with normal cells. Analysis of candidate anticancer drugs designed to target these metabolic pathways will require careful experimental design, both in vitro and in vivo. Analysis of the published reports studying DCA shows a confusing, and sometime contradictory, range of in vitro and in vivo effects. Genetic studies in model tumors offer compelling evidence that this pathway is a good candidate for therapeutic targeting.68 It would be very helpful in the analysis of the potential utility of DCA if there could be some molecular signature that could predict for drug senstitivity, both in model tumors and eventually in patients. Perhaps a careful analysis of the presumed target of DCA, the phosphorylation of pyruvate dehydrogenase E1α subunit may offer this signature.
REFERENCES
1 1. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927;8:519–30.
2 Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 2008;18:54–61.
4 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004;14:198–206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas: a novel guideline for clinical oncologists? J Cancer Res Clin Oncol 2003;129:321–6.
6 Gstraunthaler G, Seppi T, Pfaller W. Impact of culture conditions, culture media volumes, and glucose content on metabolic properties of renal epithelial cell cultures. Are renal cells in tissue culture hypoxic? Cell Physiol Biochem 1999;9:150–72.
7 Bloch-Frankenthal L, Ram D. The relationship between the Crabtree effect and the oxidative metabolism of glucose and carbohydrate intermediates in ascites tumor cells. Cancer Res 1959;19:835–42.
8Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003;5:741–7.
9 Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 2008;8:705–13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Hypoxiainduced down-regulation of BRCA1 expression by E2Fs. Cancer Res 2005;65: 11597–604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer 2007;97:646–53.
13 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008;8:425–37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187–97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008;283:28106–14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177–85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. Pharmacologically increased tumor hypoxia can be measured by 18F-fluoroazomycin arabinoside positron emission tomography and enhances tumor response to hypoxic cytotoxin PR-104. Clin Cancer Res 2009;15: 7170–4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358:69–77.
20 Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 2001;276: 37223–9.
21 Sugden PH, Randle PJ. Regulation of pig heart pyruvate dehydrogenase by phosphorylation. Studies on the subunit and phosphorylation stoicheiometries. Biochem J 1978;173:659–68.
22 Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem 1995;270:14297–304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Characterization of the isozymes of pyruvate dehydrogenase phosphatase: implications for the regulation of pyruvate dehydrogenase activity. Biochim Biophys Acta 2003;1652:126–35.
24 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 1973;134: 651–3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006;45:402–15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191–6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem 2000; 275:15773–81.
28 Stacpoole PW, Greene YJ. Dichloroacetate. Diabetes Care 1992;15:785–91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med 1983;309:390–6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 2008;60:1478–87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 2006;66: 324–30.
32Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab Rev 1998;30: 499–539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 2008;121:e1223–8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther 2008;324:1163–71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol 1988;61:65–70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994;23:1617–24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37–51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008;68:1223–31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008;109: 394–402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 2009;29:4579–88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin Cancer Res 2009;15:476–84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 2010;102:1746–52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA 2007;104:9445–50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010;31:567–76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;287:75–83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate (DCA) selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 2010;127:2510–19.
48Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010;120:253–60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2: 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 2009;389:157–64.
51 Holness MJ, Sugden MC. Pyruvate dehydrogenase activities during the fed-to-starved transition and on re-feeding after acute or prolonged starvation. Biochem J 1989;258:529–33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptoralpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276–83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J Biol Chem 2010;285:2375–85.
54 Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003;52: 1371–6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 2007;292:C125–36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507–14.
57 Maher JC, Krishan A, Lampidis TJ. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol 2004;53:116–22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930–42.
59Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res 2006;66:632–7.
60 Fantin VR, St-Pierre J, Leder P. Attenuation of LDHA expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006;9:425–34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 2010;107:2037–42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235–56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 2004; 64:3892–9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005; 24:4165–73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 2001;21:3995–4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Phase II trial of the oral mammalian target of rapamycin inhibitor everolimus in relapsed or refractory Waldenstrom macroglobulinemia. J Clin Oncol 2010;28:1408–14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009;27: 2278–87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008;283:22700–8.
Related content: