Ioanna Papandreou, Tereza Goliasova, und Nicholas C. Denko
Abteilung für Strahlenonkologie, Abteilung für Strahlen- und Krebsbiologie, Stanford University School of Medicine, Stanford, CA
Schlüsselwörter: Tumorstoffwechsel, Pyruvatdehydrogenase, Warburg-Effekt, Stoffwechselinhibitoren
Abkürzungen: DCA: Dichloracetat; HIF1: Hypoxie-induzierbarer Faktor 1; LDH: Laktatdehydrogenase; PDC: Pyruvatdehydrogenase-Komplex; PDH: Pyruvatdehydrogenase; PDK: Pyruvatdehydrogenase-Kinase; PDP: Pyruvatdehydrogenase-Phosphatase
Korrespondenz mit: Nicholas C. Denko, Abteilung für Strahlenonkologie, Abteilung für Strahlen- und Krebsbiologie, Stanford University School of Medicine, Stanford, CA 94305, USATel
.: 650-724-5066, Fax: 650-723-7382,
E-mail: [email protected]
Eingereicht am: 22 Jul 2010Akzeptiert
: Angenommen: 30 Sep 2010Online
: 18 Oct 2010DOI
: 10.1002/ijc.25728
In den letzten 20 Jahren ist die Zahl der Artikel, in denen der Begriff „Tumorstoffwechsel“ vorkommt, von 3 auf 28 pro Jahr gestiegen, und die Zahl der Zitierungen dieser Artikel hat sich von 23 auf 929 pro Jahr erhöht (ISI, Thompson Reuters statistics). Das erneute Interesse am Verständnis der Mechanismen und Folgen eines veränderten Tumorstoffwechsels hat eindeutig die Phantasie der wissenschaftlichen Gemeinschaft beflügelt. Die Idee, dass Tumore einen veränderten Stoffwechsel haben, wurde erstmals von dem Biochemiker und Nobelpreisträger Otto Warburg bei der Beschreibung des Glukosestoffwechsels erkannt.1 In jüngerer Zeit hat sich das Konzept, dass Tumore einen anderen Stoffwechsel haben, auf andere Merkmale ausgeweitet, wie z. B. Glutaminolyse, Fettsäureoxidation und Lipidbiogenese. Es gibt eindeutig einen anderen Stoffwechselbedarf, der diese Veränderungen in sich ständig teilenden Zellen antreibt, als in Zellen, die sich im Endstadium befinden. Die Entdeckung dieser Veränderungen hat die Möglichkeit eröffnet, dass sie aufgrund ihrer einzigartigen Bedeutung für Krebszellen therapeutisch angegangen werden können.2
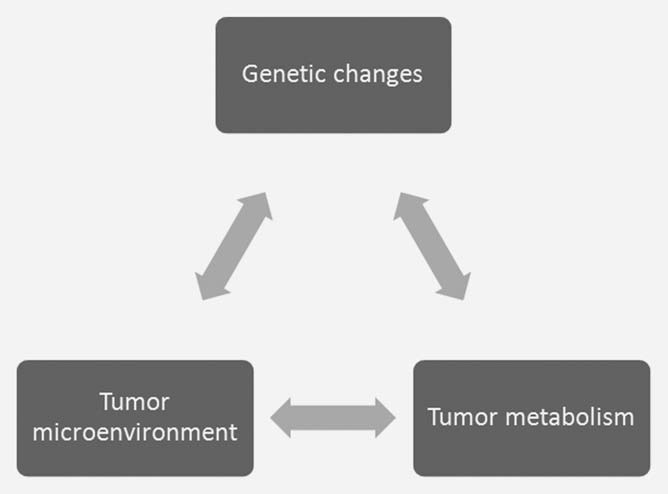
Das Konzept, dass Stoffwechselveränderungen eine Reaktion auf einzigartige Anforderungen innerhalb des Tumors sind, wurdevorgeschlagen3 , auch wenn es schwierig ist, diese Anforderungen zu quantifizieren. Es besteht eine Wechselwirkung zwischen den onkogenen Veränderungen in der Tumorzelle und den besonderen Aspekten der Mikroumgebung des Tumors, die sich auf den Zellstoffwechsel auswirken, und umgekehrt (Abb. 1). Daher ist es schwierig, die genauen Stoffwechselanforderungen innerhalb des Tumors zu bestimmen, indem man die Zellen des ex vivo gezüchteten Tumors untersucht. Die Umweltbedingungen, unter denen die Zellen in der Kultur gezüchtet werden, unterscheiden sich stark von den Umweltbedingungen in vivo. Dulbeccos modifiziertes Eagles-Medium mit hohem Glukosegehalt und einer Atmosphäre von 21 % Sauerstoff unterscheidet sich stark von den hypoxischen und/oder hypoglykämischen Bedingungen, die im Tumor herrschen.4,5 Die Glukosekonzentration von 25 mM ist etwa fünfmal so hoch wie der normale Blutspiegel, und die Sauerstoffspannung ist mindestens viermal so hoch wie in vivo. Die Tatsache, dass die Zellen mit diesen Stoffwechselsubstraten überflutet werden, verändert ihr eigenes Stoffwechselprogramm erheblich.4,6 Erhöhte Glukosekonzentrationen begünstigen die Glykolyse (Crabtree-Effekt7 ), während eine erhöhte Sauerstoffzufuhr zu vermehrten Sauerstoffnebenprodukten führt und die Lebensdauer der Zellen verkürzt.8 Der Glukosestoffwechsel veranschaulicht das Zusammenspiel dieser drei Faktoren im Tumor. Die onkogene Transformation treibt die Proliferation der Tumorzellen stärker an als die Gefäßkapazität, was zu Hypoxie führt. Die Hypoxie in der Mikroumgebung des Tumors steigert den glykolytischen Stoffwechsel, vor allem durch die Aktivierung des Transkriptionsfaktors Hypoxiainducible Factor 1 (HIF1).9 Eine erhöhte Glykolyse führt zu einer vermehrten Produktion von Laktat, was zu einem sauren extrazellulären pH-Wert und weiteren Veränderungen der Genexpression beiträgt.10 Sowohl Hypoxie als auch Azidose können zu einem erhöhten Maß an somatischen Mutationen beitragen, die die Tumorprogression weiter vorantreiben können.11,12 Es ist eindeutig schwierig, diese komplexen Wechselwirkungen in in vitro gezüchteten Zellen zu reproduzieren.
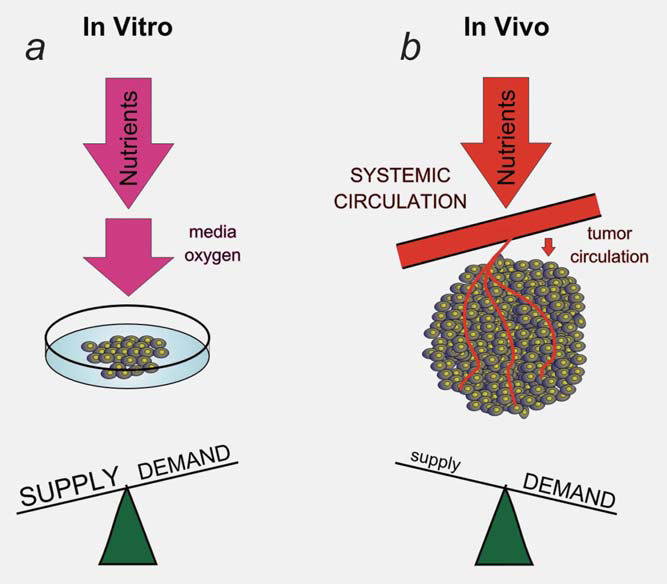
Ein Teil der Interaktion zwischen der Mikroumgebung und dem Stoffwechsel der Tumorzellen wird durch eine adaptive Reaktion auf dynamische Veränderungen des zellulären Angebots und der Nachfrage nach Metaboliten erzeugt. Allein die Tatsache, dass wir innerhalb von Tumoren Regionen mit Hypoxie und Azidose messen können, weist darauf hin, dass die Tumorgefäße keine konstante Umgebung für das Wachstum der Tumorzellen bieten.13 Das Tumorgefäßsystem stellt einen Engpass für die Zufuhr von Nährstoffen und den Abtransport von Abfallprodukten aus dem Tumor dar.4 Die unzureichende Versorgung durch die Tumorgefäße löst eine adaptive Reaktion der Tumorzellen aus, die darauf abzielt, die Nachfrage nach den begrenzten Stoffwechselprodukten zu verringern. Dieser dynamische Prozess ist in vitro nur schwer zu modellieren (Abb. 2). Zum Beispiel induziert ein niedriger Sauerstoffgehalt im Tumor den Transkriptionsfaktor HIF1 und sein Stoffwechselprogramm.9 Ein Teil des von HIF1 initiierten Stoffwechselprogramms besteht darin, den Sauerstoffbedarf durch eine Verringerung der Mitochondrienfunktion zu senken. Ein Teil dieser Reaktion wird durch die HIF1-abhängige Induktion von PDK1 und PDK3 innerhalb der Tumorzellen und eine Verringerung der Pyruvat-Oxidation in den Mitochondrien vermittelt.14-17 Diese adaptive Reaktion ist dafür verantwortlich, dass sich der Sauerstoffbedarf dem begrenzten Angebot annähert.
Dichloracetat (DCA) ist in der Lage, diese Anpassung an die Tumorhypoxie zu stören, indem es die Funktion der PDKs hemmt (Abb. 3). Die Blockierung einer adaptiven Reaktion auf Hypoxie ist am deutlichsten zu beobachten, wenn der Tumor als Ganzes ein Sauerstoffdefizit aufweist und das Tumorgefäßsystem nicht auf diesen erhöhten Bedarf reagieren kann.18 Selbst wenn die Tumorzellen in Hypoxie versetzt werden, ist genügend Sauerstoff in der Umgebung vorhanden (1-2 %), um einen stabilen, wenn auch niedrigen Sauerstoffzustand innerhalb der Zelle aufrechtzuerhalten, selbst wenn DCA hinzugefügt wird. Die Diffusionsrate in die Zellen ist schneller als die Verbrauchsrate, so dass der intrazelluläre Sauerstoffgehalt nicht von der Verbrauchsrate abhängt (es sei denn, es werden sehr große Zellzahlen in Glasschalen verwendet, die die Diffusion von Sauerstoff durch den Kunststoff verhindern14). In diesem Artikel stellen wir eine Analyse der veröffentlichten Daten zu DCA als Beispiel für ein Medikament vor, das den Tumorstoffwechsel beeinflussen soll. Dies stützt unsere Hypothese, dass diese Medikamentenklasse eine ganz andere Wirkung auf Tumorzellen hat, die in vivo mit dem durch die Blutgefäße verursachten metabolischen Engpass wachsen, als auf Zellen, die in vitro wachsen.
Der Pyruvat-Dehydrogenase-Komplex
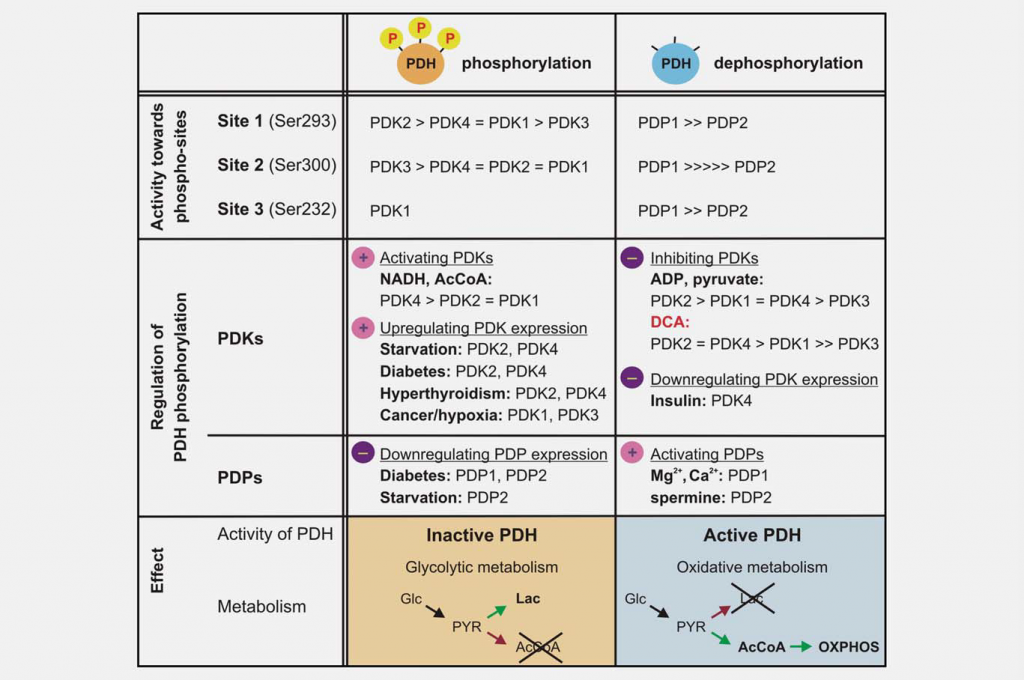
Die Hemmung der Mitochondrienfunktion ist für die Entstehung des Warburg-Effekts ebenso wichtig wie eine erhöhte Glykolyse. Ein wichtiger Regulator der Mitochondrienfunktion ist der Pyruvatdehydrogenase-Komplex (PDC), der die irreversible Decarboxylierung von Pyruvat zu Acetyl-CoA, CO2 und NADH katalysiert. Die Kontrolle des PDC steuert den Eintritt von Kohlenstoffen aus Kohlenhydraten in die Mitochondrien. Diese Reaktion spielt eine zentrale Rolle bei der Regulierung sowohl der mitochondrialen Energieerzeugungswege (Tricarbonsäurezyklus (TCA) und oxidative Phosphorylierung (OXPHOS)) als auch der Erzeugung biosynthetischer Zwischenprodukte, wie z. B. Citrat. PDC besteht aus drei katalytischen Komponenten, der Pyruvat-Dehydrogenase (E1), der Dihydrolipoamid-Transacetylase (E2) und der Dihydrolipoamid-Dehydrogenase (E3), die zusammen mit der strukturellen Untereinheit E3-Bindungsprotein (E3BP) in großen multimeren Komplexen organisiert sind. Der grundlegende Kern der E1-Pyruvatdehydrogenase-Komponente ist ein Heterotetramer aus zwei Alpha- und zwei Beta-Untereinheiten (α2β2) und katalysiert den ersten Schritt der Pyruvat-Decarboxylierung. Die Aktivität des Komplexes wird weitgehend durch die reversible Phosphorylierung von drei Serinresten von E1α reguliert. Pyruvat-Dehydrogenase-Kinasen (PDK1-4) inaktivieren PDC und Pyruvat-Dehydrogenase-Phosphatasen (PDP1-2) aktivieren/reaktivieren es (Abb. 3).
Die verschiedenen PDK-Isoformen unterscheiden sich in ihren regulatorischen Eigenschaften, ihrer Gewebeverteilung und ihrer Modulation durch vorgeschaltete Stoffwechselsignale. Dies führt zu einer dynamischen, organspezifischen Kontrolle der mitochondrialen Funktion und Energieproduktion. Enzymatische Untersuchungen haben gezeigt, dass sich die PDKs in ihrer Spezifität für die drei E1α-Zielstellen und in den kinetischen Parametern der Phosphorylierung unterscheiden. PDK1 ist die einzige Isoform, die in der Lage ist, alle drei Stellen zu phosphorylieren, während PDK2-4 die Stellen 1 und 2 in vitro mit unterschiedlicher Geschwindigkeit phosphorylieren. 19,20 Die Phosphorylierung auch nur einer der sechs E1a-Stellen im Heterotetramer reicht aus, um PDC zu inaktivieren, und man geht davon aus, dass maximal drei der möglichen sechs Stellen des E1-Tetramers zu einem bestimmten Zeitpunkt phosphoryliert werden können.21,22 Auch die räumlich-zeitlichen Veränderungen in den Mengen und der Aktivität der PDC-Phosphatasen (PDP1-2) sind regulierend. Die Dephosphorylierung in vitro scheint zufällig zu sein; beide Isoenzyme sind in der Lage, die drei E1-Stellen von rekombinanten mutierten Substraten zu dephosphorylieren.23
Dichloracetat: Ein PDK-Inhibitor
DCA wurde als Pyruvatdehydrogenase-Aktivator identifiziert, da es die enzymatische Aktivität von PDC in einem perfundierten Rattenherzmodell stimulieren kann.24 Inzwischen ist bekannt, dass dieses Pyruvat-Mimetikum die Wirkung der PDKs hemmt. Die Kristallstruktur von PDK2 im Komplex mit DCA wurde erhalten und zeigt, dass DCA die Pyruvat-Bindungsstelle in der N-terminalen regulatorischen (R) Domäne besetzt.25 Die vier Isoenzyme unterscheiden sich erheblich in ihrer Empfindlichkeit gegenüber der Hemmung durch DCA. PDK2 ist am empfindlichsten, PDK3 am widerstandsfähigsten, während PDK1 und PDK4 relativ empfindlich sind.26,27
Behandlung der Laktatazidose mit DCA
Die Sicherheit und Wirksamkeit von DCA wurde in Fällen von angeborener und erworbener Laktatazidose untersucht. Die DCA-Behandlung senkt den Laktatspiegel im Blutkreislauf wirksam, indem sie die Oxidation von Pyruvat stimuliert; es ist jedoch nicht bekannt, ob DCA die Prognose von Patienten mit diesen Syndromen verbessern kann.28,29 Es wurde vorgeschlagen, dass kleine Kinder mit PDH-Mangel am meisten von einer chronischen DCA-Behandlung profitieren könnten.30 Die wichtigste unerwünschte Wirkung einer langfristigen DCA-Verabreichung ist eine reversible periphere Neuropathie.31,32 Der Schweregrad der Toxizität scheint altersabhängig zu sein, wobei erwachsene Patienten anfälliger sind als Kinder.31,33 Die Gründe für diese Diskrepanz sind nicht ganz klar, hängen aber möglicherweise mit der unterschiedlichen Pharmakokinetik und dem Metabolismus von DCA in den beiden Altersgruppen zusammen.34 DCA wurde auch in klinischen Studien bei Herzerkrankungen, einschließlich kongestiver Herzinsuffizienz und ischämischer Herzkrankheiten, eingesetzt, wobei positive Ergebnisse und eine Verbesserung der Herzmuskelleistung erzielt wurden.35,36
DCA als potenzielles Krebstherapeutikum
In den letzten Jahren hat DCA als potenziell einfaches und kostengünstiges Mittel zur Bekämpfung von glykolytischen Tumoren bei gleichzeitig begrenzten Nebenwirkungen in den oxidativen gesunden Organen Aufmerksamkeit erregt. Das Interesse der wissenschaftlichen Gemeinschaft, der Krebspatienten und der Medien an diesem Medikament wurde 2007 geweckt, nachdem eine Gruppe der University of Alberta berichtet hatte, dass DCA in einzigartiger Weise toxisch für menschliche Krebszelllinien ist und das Wachstum von A549-Lungentumor-Xenograft-Tumoren bei Ratten hemmt.37 Seitdem zeigen die neuen Berichte über die Wirksamkeit von DCA in vitro und in vivo einige interessante und auch rätselhafte Merkmale, die DCA von den meisten als Krebsmittel entwickelten Medikamenten unterscheiden (Tabelle 1). Die Anzahl der verschiedenen Krebsarten und experimentellen Strategien, die bisher getestet wurden, ist zu gering, um verallgemeinerte Schlussfolgerungen über die Wirksamkeit von DCA gegen alle Arten von Tumoren zuzulassen. Ein qualitativer Vergleich der Literatur deutet jedoch darauf hin, dass DCA in vivo eine stärkere krebshemmende Wirkung als in vitro gegen Krebszellen zeigt.
| Krebsart | Referenzen | Wirkung auf Überleben und Wachstum |
| In-vitro-Studien | ||
| Lunge, Glioblastom, Brustkrebs | 37 | Apoptose in vitro, Wachstumshemmung bei Xenotransplantaten |
| Prostata | 38 | Wachstumshemmung in vitro, moderate Radiosensibilisierung |
| Gebärmutterschleimhaut | 39 | Wachstumshemmung |
| Gebärmutterhals | 40 | Wachstumsvorteil unter Hypoxie in vitro |
| Kopf und Hals | 41 | In-vitro-Wachstumshemmung nur bei mutND2 überexprimierenden Zellen |
| Pädiatrie | 42 | Apoptose bei hohen Konzentrationen in vitro, gewisser Einfluss auf das Ansprechen auf eine Chemotherapie |
| Kolorektal | 43 | Apoptose bei sehr hohen Konzentrationen |
| Präklinische Modelle | ||
| Kolorektal | 44 | Geringe Auswirkungen auf das Wachstum, erhöhte Hypoxie durch HRE-Luciferase, sensibilisiert für hypoxische Zytotoxine |
| Kolorektal | 18 | Geringe Auswirkung auf das Wachstum, erhöhte Hypoxie durch 18F-FAZA PET, sensibilisiert für hypoxisches Zytotoxin |
| Präklinische Modelle | 17 | Hemmung des Wachstums von Xenotransplantaten |
| Kolorektal | 45 | Xenotransplantat-Wachstumshemmung |
| Kolorektal | 46 | Schützte in vitro vor Anoxie, förderte das Xenotransplantatwachstum von SW480 |
| Kolorektal, Brust, PML, Prostata | 47 | Nur aktiv gegen Zellen mit defekter Elektronentransportkette |
| Brust | 48 | Hemmung von Wachstum und Metastasierung von Xenotransplantaten |
| Daten von menschlichen Patienten | ||
| Glioblastom | 49 | Klinisch stabile Erkrankung in vivo, vermindertes HIF1, erhöhtes p53 ex vivo |
In-vitro-Studien. Es wurde berichtet, dass DCA in vitro zytotoxische Wirkungen hat, 37-40 einige reagieren bereits bei klinisch relevanten Konzentrationen (0,5-1 mM), andere benötigen suprapharmakologische Konzentrationen (10-100 mM) und wieder andere Gruppen haben in vitro keine direkte Toxizität festgestellt. 18,41,42,47 Eine Bedingung, die Zellen für DCA sensibilisiert, sind Mutationen, die die mitochondriale Atmungsfunktion stören,41,47 was darauf hindeutet, dass die erzwungene Nutzung einer gestörten OXPHOS toxisch ist. Da Sauerstoffmangel auch die mitochondriale Funktion herunterreguliert, lag die Hypothese nahe, dass hypoxische Zellen empfindlicher auf DCA reagieren würden. Diese Hypothese hat sich jedoch nicht bestätigt, zumindest nicht bei der begrenzten Anzahl der bisher getesteten Zelllinien. Eine mäßige In-vitro-Hypoxie hatte keinen Einfluss auf das Zellzyklusprofil der mit DCA behandelten kolorektalen Zellen43 oder die Fortpflanzungsfähigkeit von DCA-behandelten Bauchspeicheldrüsenkrebszellen (unsere unveröffentlichten Beobachtungen). Interessanterweise wurde in einer anderen Studie festgestellt, dass schwere Hypoxie (Anoxie?) vor der DCA-induzierten Apoptose in Darmkrebszellen schützen kann.46 Der Grund für diese Diskrepanzen ist nicht klar.
Insgesamt unterstützen die meisten Daten die Idee, dass klinisch relevante Konzentrationen von DCA (weniger als 1 mM) in vitro nicht direkt zytotoxisch sind. Der Grund für diese offensichtliche zelluläre Resistenz ist nicht die Inaktivierung von DCA unter Gewebekulturbedingungen oder die Unfähigkeit, PDKs zu inaktivieren, da DCA nachweislich die mitochondriale Aktivität vorübergehend erhöht und das mitochondriale Membranpotenzial kollabiert.18,37,50 Die Grundlage für die begrenzte krebshemmende Wirkung von DCA in Kulturen liegt daher wahrscheinlich in der komplexen Zellphysiologie und dem enormen Überschuss an Metaboliten in den Kulturmedien.
Präklinische Modelle. Die Berichte über die Wirksamkeit von DCA bei Modelltumoren, die in Nagetieren gezüchtet wurden, sind ermutigend, obwohl es einige Fälle von Tumorlinien gibt, die nicht auf die Behandlung ansprechen, und sogar ein Beispiel für beschleunigtes Tumorwachstum als Reaktion auf DCA.46 Der erste Bericht über die Antitumoraktivität von DCA stammt von Bonnet et al. 37 Die Autoren berichteten, dass in Nacktratten gezüchtete A549-Lungenadenokarzinom-Xenografts nach der Behandlung mit DCA eine signifikante Verzögerung des Tumorwachstums aufwiesen, wobei bei einigen Versuchsgruppen sogar eine Rückbildung des Tumors zu beobachten war. Diese Effekte wurden mit einer erhöhten Apoptose und einer verringerten Proliferation in Verbindung gebracht. Unter Verwendung der gleichen A549-Zellen bestätigten Stockwin et al. 47 kürzlich, dass DCA das Wachstum von Modelltumoren unterdrückt, obwohl sie in vitro nur eine geringe Toxizität feststellten.
Unsere Gruppe hat berichtet, dass die tägliche DCA-Behandlung von Mäusen mit SU86.86 Xenotransplantaten der Bauchspeicheldrüse zu einer deutlichen Verzögerung des Tumorwachstums sowie zu einem erhöhten hypoxischen Anteil der Tumoren führte. Wir stellten die Hypothese auf, dass ein erhöhter mitochondrialer Sauerstoffverbrauch zu einer größeren Hypoxie führt, die das Wachstum des Tumors hemmt.17 Zur Unterstützung dieses Modells erhöhte DCA das Ausmaß der Tumorhypoxie in kolorektalen Xenotransplantaten von RKO, was entweder durch HRE (Hypoxia Response Element)-gesteuerte Luziferase-Reporter44 oder 18F-Fluoroazomycin-Arabinosid-Positronenemissionstomographieermittelt wurde.18 Dieses RKO-Modell zeigte eine sehr bescheidene Verringerung des Wachstums der mit DCA behandelten Tumoren, aber die akuten Veränderungen der Sauerstoffversorgung nach DCA sensibilisierten sie für die Behandlung mit Hypoxie-aktivierten Zytotoxinen wie Tirapazamin44 oder PR-104.18 Weitere Arbeiten anderer Gruppen an Darmkrebsmodellen haben erhebliche Heterogenität in der Reaktion auf DCA gezeigt. Bei einigen Zelllinien verringerten sich die Tumorwachstumsraten,45 während andere entweder gar nicht reagierten oder sogar schneller wuchsen.46 Darüber hinaus konnte in einem Rattenmodell für Adenokarzinome der Brust durch eine intensive DCA-Behandlung die Zahl der makroskopischen Lungenmetastasen reduziert werden.48 Diese präklinischen Modelle stützen das Konzept, dass DCA in der Lage ist, den Tumormetabolismus in vivo zu modulieren, was je nach dem getesteten Modell zu einer stärkeren oder schwächeren Antitumorwirkung führt.
Daten aus klinischen Studien. Vor kurzem wurden die ersten Daten zur Bewertung von DCA für die Behandlung von Krebs beim Menschen veröffentlicht.49 In dieser Studie wurde DCA in Kombination mit Operation, Temozolomid und Bestrahlung zur Behandlung von fünf Patienten mit Glioblastoma multiforme eingesetzt. Während die Autoren von vielversprechenden klinischen Ergebnissen bei vier der fünf Patienten berichten, lag der Schwerpunkt des Berichts auf der Ex-vivo-Analyse der Tumorzellen vor und nach der Behandlung mit DCA. Sie berichten über Veränderungen des mitochondrialen Membranpotenzials, erhöhte Mengen an mitochondrial erzeugten Sauerstoffradikalen und eine erhöhte Apoptose der Tumorzellen. Mechanistische Studien ergaben veränderte Werte der HIF1-Signalübertragung, eine Aktivierung von p53 und eine verringerte Angiogenese. Diese Daten deuten darauf hin, dass DCA viele Wirkmechanismen hat, die der Hemmung der PDKs nachgelagert sind. Offensichtlich muss eine größere Anzahl von Patienten, vorzugsweise an verschiedenen Orten, behandelt werden, um diese aufregenden Ergebnisse verallgemeinern zu können.
Organisatorische Regulierung des Stoffwechsels
Humorale Wachstums- und Ernährungsfaktoren könnten ebenfalls die Reaktion des Tumors auf die metabolische Umprogrammierung beeinflussen. Die Regulierung der Tumor-PDK-Aktivität in vivo ist noch nicht systematisch untersucht worden, aber die Erkenntnisse, die wir aus dem normalen Gewebestoffwechsel und aus den Bereichen Endokrinologie und Adipositasforschung gewonnen haben, liefern einige interessante Hypothesen. Hungern erhöht die Expression von PDKs und verringert die PDH-Aktivität in peripheren Organen als Strategie zur Aufrechterhaltung einer stabilen Versorgung des Gehirns und anderer neuronaler Gewebe mit Kohlenhydraten. So werden beispielsweise PDK4 und PDK2 in Leber, Niere und anderen Geweben durch Hunger transkriptionell aktiviert.51,52 Glucocorticoide, T3-Schilddrüsenhormone und freie Fettsäuren erhöhen ebenfalls den PDK4-Spiegel.53 Eine erneute Fütterung und/oder ein Anstieg des Insulinspiegels verringern die PDK4-Transkription und reaktivieren PDC. Ebenso wurde gezeigt, dass Hunger und Diabetes die mRNA- und Proteinkonzentration von PDP2 herabsetzen, was durch eine erneute Nahrungsaufnahme oder eine Behandlung mit Insulin wieder rückgängig gemacht werden konnte.54
Daher könnten die vielen Eingangssignale, die die PDH-Aktivität in vivo beeinflussen, die Wirksamkeit von DCA und anderen auf den Stoffwechsel ausgerichteten Medikamenten beeinflussen. Die Verabreichung des Medikaments während des Fastens könnte eine ganz andere Wirkung haben als im nüchternen Zustand. Die Reaktionen auf humorale Bedingungen sind offensichtlich bei verschiedenen Geweben und Tumorarten unterschiedlich und daher in vitro nur schwer nachzuahmen. Ist es also möglich, die antitumorale Wirkung von Molekülen wie DCA in vitro zu bewerten? Viele Jahre der Untersuchung einer großen Anzahl von Tumorzelllinien, die in Kultur gezüchtet wurden, haben gezeigt, dass sie in vitro die abnormen Merkmale der aeroben Glykolyse aufweisen und daher ein wertvolles Instrument für die Untersuchung des Krebsstoffwechsels darstellen.55 Diese Systeme weisen jedoch auch inhärente Einschränkungen auf, wie die uneinheitlichen Daten in der Literatur beim Vergleich der Wirkung von DCA in Zellkulturen mit präklinischen und klinischen Ergebnissen zeigen.
In der vorangegangenen Analyse wurde das Modell vorgestellt, dass ein Krebsmedikament, das auf den Stoffwechsel abzielt und in vitro wenig Toxizität aufweist, in vivo ein erhebliches Potenzial haben kann. Wir haben die begrenzten Stoffwechselsubstrate im Tumor und die systemische Regulierung des Stoffwechsels durch humorale Faktoren hervorgehoben, die die Wirksamkeit des Medikaments in vivo erhöhen könnten. Es ist möglich, dass umgekehrt ein in vitro gut wirksames Medikament in vivo wenig Erfolg haben kann. Auswirkungen auf die normale Gewebetoxizität oder die metabolische Zusammenarbeit zwischen Zelltypen könnten die Wirksamkeit eines Arzneimittels in vivo einschränken. Der vielfach getestete Stoffwechselhemmer 2-Desoxyglukose hat in vitro eine angemessene krebshemmende Wirkung, kann aber wegen seiner negativen Auswirkungen auf normales Gewebe, das auf den Glukoseverbrauch angewiesen ist, nicht bei Patienten eingesetzt werden. Die dosisbegrenzende neurologische Toxizität tritt bei einem Wirkstoffspiegel auf, der weit unter dem liegt, der für eine krebshemmende Wirkung bei Nagetiertumoren erforderlich ist.56,57 Alternativ ist es möglich, dass eine metabolische Kooperation zwischen Zelltypen oder normalen und Tumorzellen die medikamenteninduzierte Stoffwechselblockade umgeht. Während Laktat beispielsweise häufig als Stoffwechselabfallprodukt betrachtet wird, kann es in einigen Zellen als Brennstoff für die Mitochondrienfunktion verwendet werden.58,59 Was
die Kombination von DCA mit bestehenden Therapien betrifft, so zeigen die präklinischen Daten bisher kein offensichtliches Interaktionsmuster, das eine einfache und rationale Auswahl von Therapieschemata ermöglichen würde. Tiermodelle werden auch weiterhin das beste Mittel sein, um empirisch die vielversprechendsten Kombinationen zu ermitteln. Wir haben gezeigt, dass DCA aufgrund seiner Fähigkeit, den Sauerstoffverbrauch zu steigern, die Tumorhypoxie erhöht und xenotransplantierte Bauchspeicheldrüsen- und Dickdarmtumore für hypoxische Zytotoxine sensibilisiert,18,44 so dass es faszinierend ist, sich einen Behandlungsplan vorzustellen, der diese beiden Medikamentenklassen umfasst, die beide darauf ausgelegt sind, die einzigartige hypoxische Mikroumgebung des Tumors zu nutzen.
Über die Wechselwirkung von DCA mit anderen Stoffwechselmodulatoren wurde noch nicht berichtet. Ein mögliches Ziel für eine Kombinationstherapie ist die Laktatdehydrogenase A (LDHA). Es hat sich gezeigt, dass die genetische oder pharmakologische Hemmung von LDHA die mitochondriale Funktion erhöht und die Entstehung und das Fortschreiten von Modelltumoren hemmt.60,61 In diesem Kombinationsschema würde der LDHA-Inhibitor die Umwandlung von Pyruvat in Laktat blockieren, und DCA würde das angesammelte Pyruvat zur mitochondrialen Oxidation umleiten. Wenn die antitumorale Wirkung von DCA auf einer gesteigerten mitochondrialen Funktion beruht, ist es möglich, dass die Kombination von PDK- und LDHA-Inhibitoren eine noch höhere mitochondriale Oxidationsrate erzwingen und das Tumorwachstum noch effizienter beeinträchtigen würde.
Knotenpunkte wichtiger Überlebenswege, wie der PI3KAkt-mTOR-Weg, sind ebenfalls Gegenstand intensiver Bemühungen in der Arzneimittelentwicklung.62 Ein Teil der wachstumsfördernden Eigenschaften dieses Weges beruht auf seiner Fähigkeit, den Stoffwechsel und die Energieproduktion durch direkte oder indirekte Mechanismen zu regulieren. So stimuliert beispielsweise die onkogene Aktivierung von PI3K-Akt die Glukoseaufnahme und die aerobe Glykolyse,63,64 während die Aktivierung von Akt und mTORC1 die Translation von Hif-1a mRNA unter Hypoxie erhöht.65 Vielversprechende Akt-Inhibitoren und mTOR-Inhibitoren der neuen Generation werden derzeit in klinischen Studien getestet66,67 und sind mögliche Kandidaten für eine Kombinationstherapie mit DCA, um sowohl den aeroben als auch den hypoxischen Stoffwechsel zu modulieren.
Schlussfolgerungen
Zusammenfassend lässt sich sagen, dass die jüngsten Erkenntnisse über den einzigartigen Stoffwechsel von soliden Tumoren mehrere neuartige, arzneimittelwirksame Stoffwechselwege identifiziert haben, die in Tumorzellen im Vergleich zu normalen Zellen bevorzugt genutzt werden können. Die Analyse von Kandidaten für Krebsmedikamente, die auf diese Stoffwechselwege abzielen, erfordert eine sorgfältige Versuchsplanung sowohl in vitro als auch in vivo. Die Analyse der veröffentlichten Berichte zur Untersuchung von DCA zeigt eine verwirrende und manchmal widersprüchliche Bandbreite von In-vitro- und In-vivo-Wirkungen. Genetische Studien an Modelltumoren liefern überzeugende Beweise dafür, dass dieser Stoffwechselweg ein guter Kandidat für eine gezielte Therapie ist.68 Für die Analyse des potenziellen Nutzens von DCA wäre es sehr hilfreich, wenn es eine molekulare Signatur gäbe, die sowohl bei Modelltumoren als auch bei Patienten eine Vorhersage über die Empfindlichkeit des Medikaments ermöglichen würde. Vielleicht könnte eine sorgfältige Analyse des mutmaßlichen Ziels von DCA, der Phosphorylierung der Pyruvatdehydrogenase E1α-Untereinheit, diese Signatur liefern.
REFERENZEN
1 1. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927;8:519-30.
2 Pan JG, Mak TW. Metabolic Targeting als Krebsbekämpfungsstrategie: Anbruch einer neuen Ära? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Baustein für Baustein: Stoffwechsel und Tumorzellwachstum. Curr Opin Genet Dev 2008;18:54-61.
4 Vaupel P. Tumor-Mikroumwelt-Physiologie und ihre Bedeutung für die Strahlenonkologie. Semin Radiat Oncol 2004;14:198-206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas: a novel guideline for clinical oncologists? J Cancer Res Clin Oncol 2003;129:321-6.
6 Gstraunthaler G, Seppi T, Pfaller W. Einfluss von Kulturbedingungen, Kulturmedienvolumen und Glukosegehalt auf die metabolischen Eigenschaften von Nierenepithelzellkulturen. Sind Nierenzellen in Gewebekultur hypoxisch? Zellphysiol Biochem 1999;9:150-72.
7 Bloch-Frankenthal L, Ram D. The relationship between the Crabtree effect and the oxidative metabolism of glucose and carbohydrate intermediates in ascites tumor cells. Cancer Res 1959;19:835-42.
8ParrinelloS, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Sauerstoffempfindlichkeit schränkt die replikative Lebensspanne von Mäusefibroblasten stark ein. Nat Cell Biol 2003;5:741-7.
9 Denko NC. Hypoxie, HIF1 und Glukosestoffwechsel in soliden Tumoren. Nat Rev Cancer 2008;8:705-13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. Die genomische Analyse von Laktatazidose und Azidose-Reaktion in menschlichen Krebsarten. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Hypoxie-induzierte Herunterregulierung der BRCA1-Expression durch E2Fs. Cancer Res 2005;65: 11597-604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Zelluläre Anpassungen an Hypoxie und Azidose während der somatischen Entwicklung von Brustkrebs. Br J Cancer 2007;97:646-53.
13 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008;8:425-37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 vermittelt die Anpassung an Hypoxie, indem es den mitochondrialen Sauerstoffverbrauch aktiv herunterreguliert. Cell Metab 2006;3:187-97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Die Induktion der Pyruvat-Dehydrogenase-Kinase-3 durch den Hypoxie-induzierbaren Faktor-1 fördert die Stoffwechselumstellung und die Arzneimittelresistenz. J Biol Chem 2008;283:28106-14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-vermittelte Expression von Pyruvatdehydrogenase-Kinase: ein metabolischer Schalter, der für die zelluläre Anpassung an Hypoxie erforderlich ist. Cell Metab 2006;3:177-85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Der Sauerstoffverbrauch kann das Wachstum von Tumoren regulieren – eine neue Perspektive auf den Warburg-Effekt. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. Pharmakologisch erhöhte Tumorhypoxie kann durch 18F-Fluoroazomycin-Arabinosid-Positronenemissionstomographie gemessen werden und verstärkt die Reaktion des Tumors auf das hypoxische Zytotoxin PR-104. Clin Cancer Res 2009;15: 7170-4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulierung der Pyruvatdehydrogenase-Aktivität durch Phosphorylierung an mehreren Stellen. Biochem J 2001;358:69-77.
20 Korotchkina LG, Patel MS. Ortsspezifität von vier Pyruvatdehydrogenase-Kinase-Isoenzymen gegenüber den drei Phosphorylierungsstellen der menschlichen Pyruvatdehydrogenase. J Biol Chem 2001;276: 37223-9.
21 Sugden PH, Randle PJ. Regulation der Pyruvatdehydrogenase im Schweineherz durch Phosphorylierung. Studies on the subunit and phosphorylation stoicheiometries. Biochem J 1978;173:659-68.
22 Korotchkina LG, Patel MS. Mutagenese-Studien der Phosphorylierungsstellen rekombinanter menschlicher Pyruvatdehydrogenase. Site-specific regulation. J Biol Chem 1995;270:14297-304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Charakterisierung der Isoenzyme der Pyruvat-Dehydrogenase-Phosphatase: Implikationen für die Regulierung der Pyruvat-Dehydrogenase-Aktivität. Biochim Biophys Acta 2003;1652:126-35.
24 Whitehouse S, Randle PJ. Aktivierung der Pyruvat-Dehydrogenase im perfundierten Rattenherz durch Dichloracetat (Short Communication). Biochem J 1973;134: 651-3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatorische Funktionen der N-terminalen Domäne auf der Grundlage von Kristallstrukturen der menschlichen Pyruvat-Dehydrogenase-Kinase 2 mit physiologischen und synthetischen Liganden. Biochemistry 2006;45:402-15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Beweise für die Existenz einer gewebespezifischen Regulierung des Pyruvatdehydrogenase-Komplexes bei Säugetieren. Biochem J 1998;329(Pt 1):191-6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Markante Unterschiede zwischen zwei Isoformen der menschlichen Pyruvatdehydrogenase-Kinase. J Biol Chem 2000; 275:15773-81.
28 Stacpoole PW, Greene YJ. Dichloroacetat. Diabetes Care 1992;15:785-91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Behandlung der Laktatazidose mit Dichloracetat. N Engl J Med 1983;309:390-6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 2008;60:1478-87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurologie 2006;66: 324-30.
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Pharmakokinetik, Metabolismus und Toxikologie von Dichloroacetat. Drug Metab Rev 1998;30: 499-539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Bewertung der Langzeitbehandlung von Kindern mit kongenitaler Laktatazidose mit Dichloracetat. Pediatrics 2008;121:e1223-8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Altersabhängige Kinetik und Metabolismus von Dichloracetat: mögliche Bedeutung für die Toxizität. J Pharmacol Exp Ther 2008;324:1163-71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myokardiale metabolische und hämodynamische Effekte von Dichloracetat bei koronarer Herzkrankheit. Am J Cardiol 1988;61:65-70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994;23:1617-24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2-Prostatakrebszellen in vitro für Strahlung. Prostate 2008;68:1223-31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol 2008;109: 394-402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 2009;29:4579-88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondriale Mutationen tragen über erhöhte reaktive Sauerstoffspezies und hochregulierte Pyruvat-Dehydrogenease-Kinase 2 in Plattenepithelkarzinomen des Kopfes und Halses zur Akkumulation von HIF1alpha bei. Clin Cancer Res 2009;15:476-84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetat metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in Darmkrebszellen. Br J Cancer 2010;102:1746-52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolisches Targeting von Hypoxie und HIF1 in soliden Tumoren kann zytotoxische Chemotherapie verbessern. Proc Natl Acad Sci USA 2007;104:9445-50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. Die Selektion von Krebszellen mit unterdrückten Mitochondrien löst das Fortschreiten von Dickdarmkrebs aus. Carcinogenesis 2010;31:567-76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Natriumdichloracetat (DCA) reduziert die Apoptose bei Hypoxie in kolorektalen Tumoren. Cancer Lett 2010;287:75-83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Natriumdichloracetat (DCA) wirkt selektiv auf Zellen mit Defekten im mitochondrialen ETC. Int J Cancer 2010;127:2510-19.
48SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Die Umkehrung des glykolytischen Phänotyps durch Dichloracetat hemmt das Wachstum metastasierender Brustkrebszellen in vitro und in vivo. Breast Cancer Res Treat 2010;120:253-60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2: 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Überwachung der Phosphorylierung des Pyruvatdehydrogenase-Komplexes. Anal Biochem 2009;389:157-64.
51 Holness MJ, Sugden MC. Pyruvat-Dehydrogenase-Aktivitäten während des Übergangs von der Fütterung zum Hungern und bei erneuter Fütterung nach akuter oder längerer Hungersnot. Biochem J 1989;258:529-33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation der Pyruvatdehydrogenase-Kinase-Expression durch Peroxisom-Proliferator-aktivierte Rezeptor-alpha-Liganden, Glukokortikoide und Insulin. Diabetes 2002;51:276-83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation der Pyruvat-Dehydrogenase-Kinase 4 (PDK4) durch Schilddrüsenhormon: Rolle des Peroxisom-Proliferator-aktivierten Rezeptor-Gamma-Coaktivators (PGC-1 alpha). J Biol Chem 2010;285:2375-85.
54 Huang B, Wu P, Popov KM, Harris RA. Hunger und Diabetes reduzieren die Menge an Pyruvat-Dehydrogenase-Phosphatase in Rattenherz und -niere. Diabetes 2003;52: 1371-6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter-Stoffwechselanalyse zeigt einen engen Zusammenhang zwischen abgeschwächter mitochondrialer bioenergetischer Funktion und erhöhter Glykolyse-Abhängigkeit in menschlichen Tumorzellen. Am J Physiol Cell Physiol 2007;292:C125-36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507-14.
57 Maher JC, Krishan A, Lampidis TJ. Stärkere Zellzyklushemmung und Zytotoxizität durch 2-Desoxy-D-Glucose in Tumorzellen, die unter hypoxischen gegenüber aeroben Bedingungen behandelt wurden. Krebs Chemother Pharmacol 2004;53:116-22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. Targeting lactate-fueled respiration tötet selektiv hypoxische Tumorzellen in Mäusen. J Clin Invest 2008;118:3930-42.
59KoukourakisMI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res 2006;66:632-7.
60 Fantin VR, St-Pierre J, Leder P. Attenuation of LDHA expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006;9:425-34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Die Hemmung der Laktatdehydrogenase A induziert oxidativen Stress und hemmt die Tumorprogression. Proc Natl Acad Sci USA 2010;107:2037-42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235-56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimuliert die aerobe Glykolyse in Krebszellen. Cancer Res 2004; 64:3892-9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. Die Glukoseabhängigkeit von Akt-transformierten Zellen kann durch pharmakologische Aktivierung der Beta-Oxidation von Fettsäuren umgekehrt werden. Oncogene 2005; 24:4165-73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu)-Signalisierung erhöht die Syntheserate des Hypoxie-induzierbaren Faktors 1alpha (HIF-1alpha): neuer Mechanismus für die HIF-1-vermittelte Expression des vaskulären endothelialen Wachstumsfaktors. Mol Cell Biol 2001;21:3995-4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Phase-II-Studie mit dem oralen Mammalian Target of Rapamycin-Inhibitor Everolimus bei rezidivierter oder refraktärer Waldenstrom-Makroglobulinämie. J Clin Oncol 2010;28:1408-14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Targeting des mTOR-Signalnetzwerks für die Krebstherapie. J Clin Oncol 2009;27:2278-87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008;283:22700-8.
Verwandte Inhalte: