Ioanna Papandreou, Tereza Goliasova en Nicholas C. Denko
Afdeling stralingsoncologie, afdeling stralings- en kankerbiologie, Stanford University School of Medicine, Stanford, CA
Trefwoorden: tumormetabolisme, pyruvaatdehydrogenase, Warburg-effect, metabole remmers
Afkortingen: DCA: dichlooracetaat; HIF1: hypoxia-inducible factor 1; LDH: lactaat dehydrogenase; PDC: pyruvaat dehydrogenase complex; PDH: pyruvaat dehydrogenase; PDK: pyruvaat dehydrogenase kinase; PDP: pyruvaat dehydrogenase fosfatase
Correspondentie aan: Nicholas C. Denko, Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA 94305, USA
Tel.: 650-724-5066, Fax: 650-723-7382,
E-mail: [email protected]
Ontvangen: 22 Jul 2010
Geaccepteerd: Aanvaard 30 sep 2010
Online: 18 okt 2010
DOI: 10.1002/ijc.25728
In de afgelopen 20 jaar is het aantal artikelen met ”tumormetabolisme” gestegen van 3 naar 28 per jaar, en het aantal keren dat deze artikelen zijn geciteerd is gestegen van 23 naar 929 per jaar (ISI, Thompson Reuters statistieken). De hernieuwde belangstelling voor het begrijpen van de mechanismen en gevolgen van een veranderd tumormetabolisme heeft duidelijk tot de verbeelding van de wetenschappelijke gemeenschap gesproken. Het idee dat tumoren een veranderd metabolisme hebben werd voor het eerst erkend door de Nobelprijswinnende biochemicus Otto Warburg toen hij het glucosemetabolisme beschreef.1 Meer recent is het concept dat tumoren een ander metabolisme hebben, uitgebreid met andere kenmerken, zoals glutaminolyse, vetzuuroxidatie en lipidenbiogenese. Er is duidelijk een andere metabole behoefte die deze veranderingen in cellen die zich voortdurend delen aanstuurt dan in terminaal gedifferentieerde cellen. De ontdekking van deze veranderingen heeft de mogelijkheid geopend dat zij therapeutisch kunnen worden aangepakt vanwege hun unieke belang voor kankercellen.2
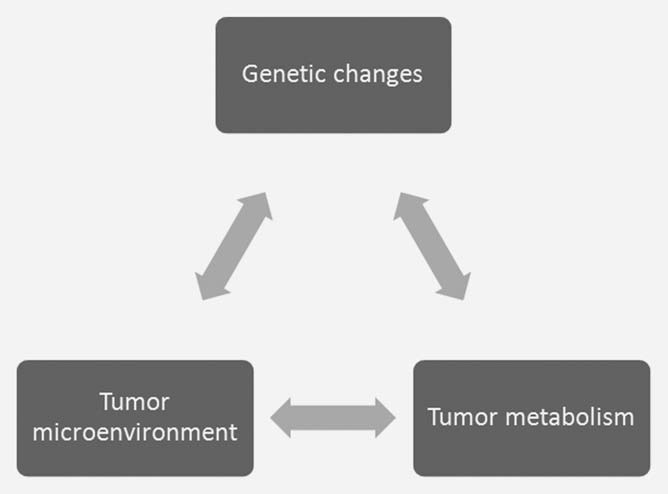
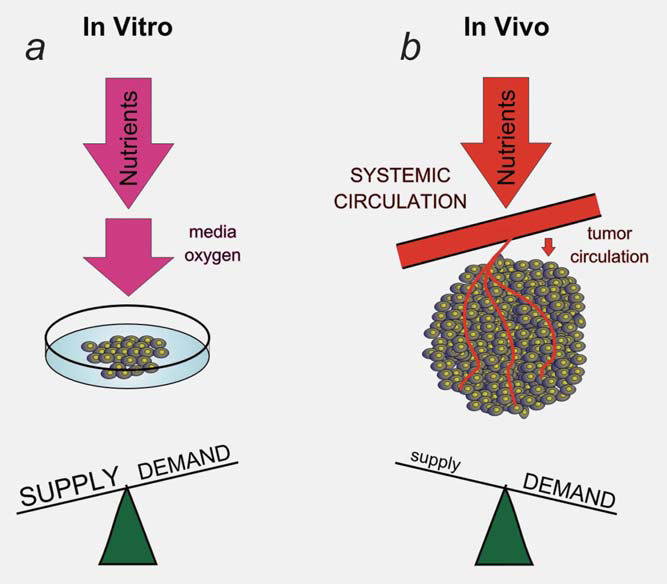
Het concept dat metabole veranderingen een reactie zijn op unieke eisen binnen de tumor is voorgesteld,3 ook al is het moeilijk die eisen te kwantificeren. Er is een wisselwerking tussen oncogene veranderingen in de tumorcel en de unieke aspecten van het tumormilieu die van invloed zijn op het cellulaire metabolisme en vice versa (Fig. 1). Daarom is het moeilijk om de exacte metabolische eisen binnen de tumor vast te stellen door de cellen van de ex vivo gekweekte tumor te bestuderen. De milieuomstandigheden die worden gebruikt om cellen in cultuur te kweken, verschillen sterk van de milieuomstandigheden in vivo. Dulbecco’s gemodificeerde Eagles-media met hoge glucose en een atmosfeer van 21% zuurstof is heel anders dan de hypoxische en/of hypoglycemische omstandigheden die in de tumor worden aangetroffen.4,5 De glucoseconcentratie van 25 mM is ongeveer vijf keer zo hoog als de normale bloedwaarden, en de zuurstofspanning is ten minste vier keer zo hoog als in vivo. Het feit dat de cellen baden in deze metabole substraten verandert hun inherente metabole programma’s aanzienlijk.4,6 Verhoogde glucoseconcentraties begunstigen de glycolyse (het Crabtree-effect7 ), terwijl verhoogde zuurstoftoevoer meer zuurstofbijproducten produceert en de levensduur van de cellen verkort.8 Het glucosemetabolisme illustreert het samenspel van deze drie factoren in de tumor. Oncogene transformatie stimuleert de proliferatie van tumorcellen meer dan de vasculaire capaciteit, waardoor hypoxie ontstaat. Hypoxie in de tumormicro-omgeving bevordert het glycolytisch metabolisme, voornamelijk door de activering van de hypoxia-induceerbare factor 1 (HIF1) transcriptiefactor.9 Verhoogde glycolyse leidt tot verhoogde productie van lactaat, wat bijdraagt tot een zure extracellulaire pH en verdere veranderingen in genexpressie.10 Zowel hypoxie als acidose kunnen bijdragen tot verhoogde niveaus van somatische mutatie die de tumorprogressie verder kunnen aanjagen.11,12 Het is duidelijk moeilijk om deze complexe interacties te reproduceren in cellen die in vitro worden gekweekt.
Een deel van de interactie tussen de micro-omgeving en het metabolisme van de tumorcellen ontstaat door een adaptieve reactie op dynamische veranderingen in de vraag naar en het aanbod van metabolieten door de cellen. Het simpele feit dat we gebieden van hypoxie en acidose kunnen meten binnen tumoren geeft aan dat de tumorvaten geen constante omgeving handhaven voor de groei van de tumorcellen.13 De tumor vasculatuur vormt een knelpunt in de aanvoer van voedingsstoffen en de afvoer van afvalstoffen uit de tumor.4 De ontoereikende aanvoer vanuit de tumorvaten initieert een adaptieve respons van de tumorcellen om de vraag naar de beperkte metabolieten te verminderen. Dit dynamische proces is moeilijk in vitro te modelleren (Fig. 2). Lage zuurstofniveaus in de tumor leiden bijvoorbeeld tot de HIF1-transcriptiefactor en het bijbehorende stofwisselingsprogramma.9 Een deel van het door HIF1 geïnitieerde stofwisselingsprogramma bestaat uit het verminderen van de vraag naar zuurstof door het verminderen van de mitochondriale functie. Een deel van deze respons wordt gemedieerd door de HIF1-afhankelijke inductie van PDK1 en PDK3 in de tumorcellen en een vermindering van de pyruvaatoxidatie in de mitochondriën.14-17 Deze adaptieve respons is verantwoordelijk voor het dichter bij elkaar brengen van de vraag naar zuurstof en het beperkte aanbod.
Dichlooracetaat (DCA) kan deze aanpassing aan tumorhypoxie verstoren door de functie van de PDK’s te remmen (fig. 3). Het blokkeren van een adaptieve respons op hypoxie wordt het duidelijkst waargenomen wanneer de tumor als geheel een zuurstoftekort heeft, en de tumorvasculatuur niet kan reageren op deze verhoogde vraag.18 Zelfs wanneer tumorcellen in hypoxie worden gebracht, is er voldoende zuurstof in de omgeving (1-2%) om een stabiele, zij het lage zuurstofconditie binnen de cel te handhaven, zelfs met toevoeging van DCA. De diffusiesnelheid in de cellen is hoger dan de verbruikssnelheid, zodat het intracellulaire zuurstofniveau niet afhangt van de verbruikssnelheid (tenzij zeer hoge aantallen cellen worden gebruikt in glazen schaaltjes die de diffusie van zuurstof door het plastic verhinderen14). In dit artikel presenteren wij een analyse van de gepubliceerde gegevens over DCA als een voorbeeld van een geneesmiddel dat is ontworpen om het tumormetabolisme te beïnvloeden, die onze hypothese ondersteunt dat deze klasse van geneesmiddelen een heel ander effect heeft op tumorcellen die in vivo groeien met het door de vasculatuur veroorzaakte metabole knelpunt, in vergelijking met hun effect op cellen die in vitro worden gekweekt.
Het Pyruvaat Dehydrogenase Complex
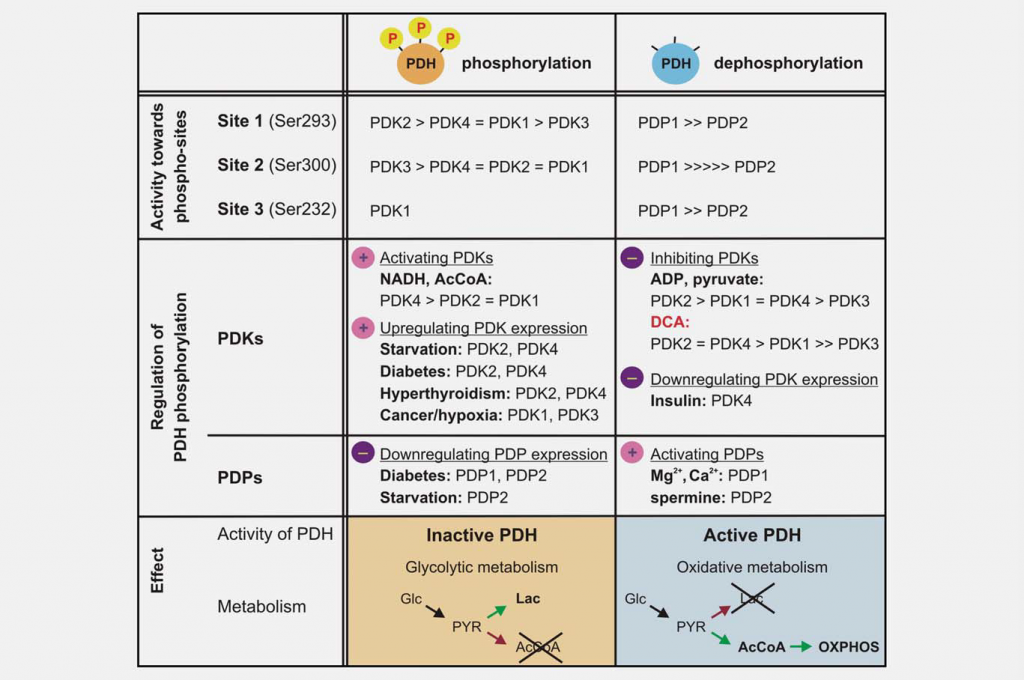
Remming van de mitochondriale functie is even belangrijk als verhoogde glycolyse om het Warburg-effect te veroorzaken. Een belangrijke regulator van de mitochondriale functie is het pyruvaat dehydrogenase complex (PDC), dat de onomkeerbare decarboxylering van pyruvaat tot acetyl-CoA, CO2 en NADH katalyseert. De controle van het PDC regelt de opname van koolhydraten in de mitochondriën. Deze reactie speelt een centrale rol bij de regulering van zowel de mitochondriale energieproducerende routes (tricarbonzuurcyclus (TCA) en oxidatieve fosforylering (OXPHOS)) als de generatie van biosynthetische tussenproducten, zoals citraat. PDC bestaat uit drie katalytische componenten, pyruvaat dehydrogenase (E1), dihydrolipoamide transacetylase (E2) en dihydrolipoamide dehydrogenase (E3), die samen met de structurele subeenheid E3-bindingseiwit (E3BP) georganiseerd zijn in grote multimere complexen. De basiskern van de E1 pyruvaat dehydrogenase component is een heterotetrameer van twee alfa en twee beta subeenheden (α2β2), en het katalyseert de eerste stap van pyruvaat decarboxylering. De activiteit van het complex wordt grotendeels geregeld door de omkeerbare fosforylering van drie serineresten van E1α. Pyruvaatdehydrogenase kinases (PDK1-4) inactiveren PDC en pyruvaatdehydrogenase fosfatases (PDP1-2) activeren/reactiveren het (Fig. 3).
De verschillende PDK-isovormen variëren in hun regulerende eigenschappen, weefseldistributie en modulatie door upstream metabole signalen. Dit resulteert in een dynamische, orgaanspecifieke controle van de mitochondriale functie en energieproductie. Enzymatische tests hebben aangetoond dat de PDK’s verschillen in hun specificiteit ten aanzien van de drie E1α-doelwitlocaties en de kinetische parameters van de fosforylering. PDK1 is de enige isovorm die alle drie de plaatsen kan fosforyleren, terwijl PDK2-4 de plaatsen 1 en 2 met verschillende snelheid in vitro fosforyleren. 19,20 Fosforylering van zelfs maar één van de zes E1a-plaatsen in het heterotetrameer is voldoende om PDC te inactiveren, en aangenomen wordt dat maximaal drie van de mogelijke zes plaatsen van het E1-tetrameer op elk moment kunnen worden gefosforyleerd.21,22 Spatiotemporele veranderingen in de niveaus en activiteit van de PDC-fosfatases (PDP1-2) zijn ook regulerend. Fosforylering in vitro lijkt willekeurig te zijn; beide isozymes zijn in staat de drie E1-plaatsen van recombinant gemuteerde substraten te defosforyleren.23
Dichlooracetaat: Een PDK-remmer
DCA werd geïdentificeerd als een pyruvaatdehydrogenase activator door zijn vermogen om de enzymactiviteit van PDC te stimuleren in een geperforeerd rattenhartmodel.24 Het is nu bekend dat dit pyruvaatnabootsende middel werkt door de werking van de PDK’s te remmen. De kristalstructuur van PDK2 in complex met DCA is verkregen, en daaruit blijkt dat DCA de pyruvaatbindingsplaats in het N-terminale regulerende (R) domein bezet.25 De vier isozymes verschillen aanzienlijk in hun gevoeligheid voor remming door DCA. PDK2 is het meest gevoelig, PDK3 het meest resistent, terwijl PDK1 en PDK4 relatief gevoelig zijn.26,27
Behandeling van melkzuursyndroom met DCA
De veiligheid en werkzaamheid van DCA is geëvalueerd in gevallen van aangeboren en verworven melkzuursyndroom. Behandeling met DCA verlaagt effectief het lactaatgehalte in de circulatie door de oxidatie van pyruvaat te stimuleren; het is echter nog niet bekend of DCA de prognose van patiënten met deze syndromen kan verbeteren.28,29 Er is voorgesteld dat jonge kinderen met PDH-deficiëntie het meeste baat hebben bij chronische DCA-behandeling.30 Het belangrijkste nadelige effect van langdurige toediening van DCA is een omkeerbare perifere neuropathie.31,32 De ernst van de toxiciteit lijkt leeftijdsafhankelijk te zijn, waarbij volwassen patiënten gevoeliger zijn dan kinderen.31,33 De redenen voor deze discrepantie zijn niet helemaal duidelijk, maar ze houden mogelijk verband met de verschillende farmacokinetiek en het metabolisme van DCA in de twee leeftijdsgroepen.34 DCA is ook gebruikt in klinische proeven voor hartaandoeningen, waaronder congestief hartfalen en ischemische hartaandoeningen, waarbij positieve resultaten werden geboekt en de hartspierprestaties verbeterden.35,36
DCA als potentieel kankertherapeuticum
De laatste jaren heeft DCA de aandacht getrokken als een potentieel eenvoudig en economisch middel om glycolytische tumoren aan te pakken en tegelijkertijd beperkte bijwerkingen te veroorzaken in de oxidatieve gezonde organen. De belangstelling van de wetenschappelijke gemeenschap, kankerpatiënten en de media voor dit geneesmiddel werd aangewakkerd in 2007, nadat een groep van de Universiteit van Alberta had gemeld dat DCA uniek toxisch was voor menselijke kankercellijnen en de groei van A549 longtumor xenograft tumoren bij ratten remde.37 Sindsdien onthullen de opkomende rapporten over de werkzaamheid van DCA in vitro en in vivo enkele interessante en ook raadselachtige kenmerken, die het geval van DCA onderscheiden van de meeste geneesmiddelen die als antikankermiddel zijn ontwikkeld (tabel 1). Het aantal verschillende kankertypes en experimentele strategieën dat tot op heden is getest, is te beperkt om algemene conclusies te trekken over de werkzaamheid van DCA tegen alle soorten tumoren. Met dit voorbehoud suggereert een kwalitatieve vergelijking van de literatuur dat DCA meer antikankereffect in vivo vertoont dan antikankerceleffect in vitro.
| Type kanker | Referenties | Effect op overleving en groei |
| In vitro studies | ||
| Long, Glioblastoom, Borst | 37 | Apoptose in vitro, groeiremming bij xenograften |
| Prostaat | 38 | Groeiremming in vitro, matige radiosensitisatie |
| Endometrium | 39 | Groeiremming |
| Baarmoederhals | 40 | Groeivoordeel onder hypoxie in vitro |
| Hoofd en nek | 41 | In vitro groeiremming alleen in mutND2-overexpressiecellen |
| Pediatrisch | 42 | Apoptose bij hoge concentraties in vitro, enige invloed op respons op chemotherapie |
| Colorectaal | 43 | Apoptose bij zeer hoge concentraties |
| Preklinische modellen | ||
| Colorectaal | 44 | Weinig effect op groei, verhoogde hypoxie door HRE-luciferase, gevoelig voor hypoxische cytotoxinen |
| Colorectaal | 18 | Weinig effect op groei, verhoogde hypoxie door 18F-FAZA PET, gevoelig voor hypoxisch cytotoxine |
| Preklinische modellen | 17 | Xenograft groeiremming |
| Colorectaal | 45 | Xenograft groeiremming |
| Colorectaal | 46 | Beschermde tegen anoxie in vitro, bevorderde xenograft-groei van SW480 |
| Colorectaal, Borst, PML, Prostaat | 47 | Alleen actief tegen cellen met defecte elektronentransportketen |
| Borst | 48 | Remming van xenograftgroei en metastase |
| Gegevens van menselijke patiënten | ||
| Glioblastoom | 49 | Klinisch stabiele ziekte in vivo, verlaagd HIF1, verhoogd p53 ex vivo |
In vitro studies. Er is gemeld dat DCA cytotoxische effecten heeft in vitro, 37-40 met sommige reacties bij klinisch relevante concentraties (0,5-1 mM), terwijl andere suprafarmacologische niveaus vereisen (10-100 mM) en weer andere groepen geen directe toxiciteit in vitro hebben gevonden. 18,41,42,47 Een voorwaarde die cellen gevoelig maakt voor DCA zijn mutaties die de mitochondriale ademhalingsfunctie verstoren,41,47 wat suggereert dat gedwongen gebruik van defecte OXPHOS toxisch is. Aangezien zuurstofgebrek ook de mitochondriale functie downreguleert, leek het redelijk te veronderstellen dat hypoxische cellen gevoeliger zouden zijn voor DCA. Deze hypothese is echter niet bevestigd, althans niet in het beperkte aantal cellijnen dat tot dusver is getest. Matige in vitro hypoxie had geen invloed op het celcyclusprofiel van met DCA behandelde colorectale cellen43 of de voortplantingslevensvatbaarheid van met DCA behandelde pancreaskankercellen (onze ongepubliceerde waarnemingen). Interessant is dat in een ander onderzoek ernstige hypoxie (anoxie?) beschermend kan werken tegen door DCA geïnduceerde apoptose in colorectale kankercellen.46 De reden voor deze discrepanties is niet duidelijk.
In het algemeen ondersteunen de meeste gegevens het idee dat klinisch relevante concentraties DCA (minder dan 1 mM) in vitro niet direct cytotoxisch zijn. De reden voor deze schijnbare cellulaire weerstand is niet een inactivatie van DCA onder weefselkweekomstandigheden of een onvermogen om PDK’s te inactiveren, aangezien is aangetoond dat DCA tijdelijk de mitochondriale activiteit verhoogt en de mitochondriale membraanpotentiaal doet instorten.18,37,50 Daarom ligt de basis voor het beperkte antikanker effect van DCA in kweek waarschijnlijk in de complexe cellulaire fysiologie en de enorme overmaat aan metabolieten die in kweekmedia aanwezig zijn.
Preklinische modellen. De rapporten over de activiteit van DCA tegen modeltumoren gekweekt in knaagdieren zijn bemoedigend, hoewel er bepaalde gevallen zijn van tumorlijnen die niet reageren op de behandeling, en zelfs een voorbeeld van versnelde tumorgroei in reactie op DCA.46 Het eerste rapport over de antitumoractiviteit van DCA was dat van Bonnet et al. 37 De auteurs meldden dat A549 longadenocarcinoom xenografts gekweekt in naakte ratten een significante vertraging van de tumorgroei vertoonden na behandeling met DCA, waarbij sommige experimentele groepen zelfs tumorregressie vertoonden. Deze effecten gingen gepaard met verhoogde apoptose en verminderde proliferatie. Met dezelfde A549 cellen hebben Stockwin et al. 47 onlangs bevestigd dat DCA groeionderdrukkend was in modeltumoren, hoewel zij in vitro weinig toxiciteit vonden.
Onze groep heeft gemeld dat dagelijkse behandeling met DCA van muizen met pancreas SU86.86 xenograften een aanzienlijke vertraging van de tumorgroei veroorzaakte, alsmede een verhoogde hypoxische fractie van de tumoren. Wij veronderstelden dat een verhoogd mitochondriaal zuurstofverbruik resulteerde in een grotere hypoxie die groeiremmend was voor de tumor.17 Ter ondersteuning van dit model, verhoogde DCA de omvang van de tumorhypoxie in RKO colorectale xenograften, zoals beoordeeld door ofwel HRE (hypoxia response element)-gestuurde luciferase reporters44 of 18F-fluoroazomycinearabinoside positronemissietomografie.18 Dit RKO model toonde een zeer bescheiden reductie in de groei van de met DCA behandelde tumoren, maar de acute veranderingen in oxygenatie na DCA maakte ze gevoelig voor behandeling met hypoxia-geactiveerde cytotoxines zoals Tirapazamine44 of PR-104.18 Aanvullend werk met colorectale kankermodellen, door andere groepen, heeft significante heterogeniteit in de respons op DCA aangetoond. Bij sommige cellijnen nam de tumorgroei af,45 terwijl andere niet reageerden of zelfs sneller groeiden.46 Bovendien kon in een rattenmodel van mammadenocarcinoom een intensief behandelingsschema met DCA het aantal macroscopische longmetastasen verminderen.48 Deze preklinische modellen ondersteunen het concept dat DCA in staat is het tumormetabolisme in vivo te moduleren, wat resulteert in grotere of kleinere antitumoreffecten, afhankelijk van het geteste model.
Klinische onderzoeksgegevens. De eerste gegevens over de evaluatie van DCA voor de behandeling van menselijke kanker werden onlangs gerapporteerd.49 In deze studie werd DCA gebruikt in combinatie met chirurgie, temozolomide en bestraling voor de behandeling van vijf patiënten met glioblastoma multiforme. Hoewel de auteurs veelbelovende klinische resultaten melden bij vier van de vijf patiënten, lag de nadruk van het verslag op de ex vivo analyse van de tumorcellen voor en na de behandeling met DCA. Zij melden veranderingen in het mitochondriale membraanpotentieel, verhoogde hoeveelheden mitochondriaal gegenereerde zuurstofradicalen en verhoogde tumorcelapoptose. Mechanistische studies vonden gewijzigde niveaus van HIF1 signalering, p53 activering en verminderde angiogenese. Deze gegevens suggereren dat DCA vele werkingsmechanismen heeft na de remming van de PDK’s. Uiteraard moeten grotere aantallen patiënten, bij voorkeur van verschillende locaties, worden behandeld om deze opwindende resultaten te kunnen veralgemenen.
Organische regulering van het metabolisme
Humorale groei- en voedingsfactoren zouden ook de tumorrespons op metabole herprogrammering kunnen beïnvloeden. De regulering van de PDC-activiteit van tumoren in vivo is nog niet systematisch bestudeerd, maar de kennis die we hebben opgedaan met het normale weefselmetabolisme en de gebieden endocrinologie en obesitasonderzoek leveren enkele interessante hypotheses op. Verhongering verhoogt de expressie van PDK’s en verlaagt de PDH-activiteit in perifere organen als strategie om een stabiele toevoer van koolhydraten naar de hersenen en andere neuronale weefsels te handhaven. Zo activeert verhongering transcriptioneel PDK4 en PDK2 in lever, nieren en andere weefsels.51,52 Glucocorticoïden, T3 schildklierhormoon en vrije vetzuren verhogen ook de PDK4-niveaus.53 Bijvoeding en/of verhoging van de insulinespiegel verminderen de PDK4-transcriptie en reactiveren de PDC. Evenzo werd aangetoond dat honger en diabetes het PDP2-mRNA en -eiwitgehalte downreguleren, effecten die ongedaan werden gemaakt door hervoeding of behandeling met insuline.54
Daarom zouden de vele inputsignalen die de PDH-activiteit in vivo beïnvloeden, de werkzaamheid van DCA en andere metabolisch gerichte geneesmiddelen kunnen beïnvloeden. Toediening van het geneesmiddel tijdens het vasten zou een heel ander effect kunnen hebben dan tijdens het voeden. De reacties op humorale omstandigheden zijn uiteraard verschillend voor verschillende weefsels en tumortypes en dus moeilijk na te bootsen in vitro. Is het dus mogelijk het antitumoreffect van moleculen als DCA in vitro te evalueren? Uit jarenlang onderzoek van een groot aantal in cultuur gekweekte tumorcellijnen blijkt dat deze in vitro inderdaad de abnormale kenmerken van aërobe glycolyse vertonen, en dus een waardevol instrument vormen voor de studie van het kankermetabolisme.55 Er zijn echter ook inherente beperkingen aan deze systemen, zoals blijkt uit de inconsistente gegevens in de literatuur bij de vergelijking van het effect van DCA in celkweek met preklinische en klinische resultaten.
De voorgaande analyse presenteert het model dat een middel tegen kanker dat zich richt op het metabolisme en in vitro weinig toxisch is, in vivo een aanzienlijk potentieel kan hebben. Wij hebben gewezen op de beperkte metabole substraten in de tumor en de systemische regulering van het metabolisme door humorale factoren die de werkzaamheid van het geneesmiddel in vivo zouden kunnen verhogen. Het is mogelijk dat het omgekeerde ook waar is, namelijk dat een geneesmiddel met een goede activiteit in vitro weinig succes heeft in vivo. Effecten op normale weefseltoxiciteit of metabole samenwerking tussen celtypes zouden de doeltreffendheid van een geneesmiddel in vivo kunnen beperken. De veel geteste stofwisselingsremmer 2-deoxyglucose heeft een redelijke antikankeractiviteit in vitro, maar kan niet bij patiënten worden gebruikt vanwege de negatieve effecten op normaal weefsel dat afhankelijk is van glucoseverbruik. De dosislimiterende neurologische toxiciteit treedt op bij een medicijnniveau dat veel lager is dan het niveau dat nodig is voor antikankereffecten in tumoren bij knaagdieren.56,57 Het is ook mogelijk dat metabole samenwerking tussen celtypen of normale en tumorcellen de door het medicijn veroorzaakte metabole blokkade omzeilt. Hoewel lactaat bijvoorbeeld vaak wordt beschouwd als een metabool afvalproduct, kan het in sommige cellen worden gebruikt als brandstof voor de mitochondriale functie.58,59
Wat betreft het combineren van DCA met bestaande therapieën, laten de preklinische gegevens tot nu toe geen duidelijk interactiepatroon zien dat een eenvoudige en rationele selectie van therapeutische behandelingen mogelijk maakt. Diermodellen zullen het beste middel blijven om de meest veelbelovende combinaties empirisch te bepalen. Wij hebben aangetoond dat DCA door zijn vermogen om het zuurstofverbruik te verhogen, de hypoxie van de tumor verhoogt en xenografisch getransplanteerde pancreatische en colontumoren gevoelig maakt voor hypoxische cytotoxinen,18,44 dus het is intrigerend om een behandelingsplan voor te stellen dat deze twee klassen van geneesmiddelen omvat, die beide zijn ontworpen om de unieke hypoxische micro-omgeving van de tumor te benutten.
De interactie van DCA met andere metabole modulatoren is niet gerapporteerd. Een mogelijk doelwit voor combinatietherapie is lactaatdehydrogenase A (LDHA). Genetische of farmacologische remming van LDHA heeft aangetoond de mitochondriale functie te verhogen en de vorming en progressie van modeltumoren te remmen.60,61 In dit combinatieschema zou de LDHA-remmer de omzetting van pyruvaat in lactaat blokkeren en zou DCA het geaccumuleerde pyruvaat afleiden naar mitochondriale oxidatie. Als het antitumoreffect van DCA voortkomt uit een verhoogde mitochondriale functie, is het mogelijk dat een combinatie van PDK- en LDHA-remmers een nog grotere mitochondriale oxidatie zou afdwingen en de tumorgroei efficiënter zou belemmeren.
Nodale punten van belangrijke overlevingsroutes, zoals de PI3KAkt-mTOR-route, zijn ook het onderwerp van intensieve inspanningen om geneesmiddelen te ontwikkelen.62 Een deel van de groeibevorderende eigenschappen van deze route komt voort uit het vermogen om het metabolisme en de energieproductie te reguleren door directe of indirecte mechanismen. Zo stimuleert oncogene activering van PI3K-Akt de glucose-opname en aërobe glycolyse,63,64 terwijl activering van Akt en mTORC1 de translatie van Hif-1a mRNA onder hypoxie verhoogt.65 Veelbelovende Akt-remmers en mTOR-remmers van de nieuwe generatie worden momenteel getest in klinische proeven66,67 en zijn geschikte kandidaten voor combinatietherapie met DCA om zowel het aërobe als het hypoxische metabolisme te moduleren.
Conclusies
De conclusie is dat recente inzichten in het unieke metabolisme van vaste tumoren verschillende nieuwe, geneesbare pathways hebben opgeleverd die bij voorkeur worden gebruikt in tumorcellen in vergelijking met normale cellen. De analyse van kandidaat-antikankergeneesmiddelen die gericht zijn op deze metabolische routes vereist een zorgvuldig experimenteel ontwerp, zowel in vitro als in vivo. Analyse van de gepubliceerde rapporten over DCA laat een verwarrende en soms tegenstrijdige reeks in vitro en in vivo effecten zien. Genetische studies in modeltumoren leveren overtuigend bewijs dat deze pathway een goede kandidaat is voor therapeutische targeting.68 Het zou zeer nuttig zijn bij de analyse van het potentiële nut van DCA als er een moleculaire handtekening zou zijn die de gevoeligheid voor geneesmiddelen zou kunnen voorspellen, zowel in modeltumoren als uiteindelijk in patiënten. Misschien kan een zorgvuldige analyse van het vermoedelijke doelwit van DCA, de fosforylering van de pyruvaatdehydrogenase E1α-subeenheid, deze handtekening bieden.
VERWIJZINGEN
1 1. Warburg O, Wind F, Negelein E. Het metabolisme van tumoren in het lichaam. J Gen Physiol 1927;8:519-30.
2 Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Steen voor steen: metabolisme en tumorcelgroei. Curr Opin Genet Dev 2008;18:54-61.
4 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Seminar Radiat Oncol 2004;14:198-206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas: a novel guideline for clinical oncologists? J Cancer Res Clin Oncol 2003;129:321-6.
6 Gstraunthaler G, Seppi T, Pfaller W. Impact of culture conditions, culture media volumes, and glucose content on metabolic properties of renal epithelial cell cultures. Zijn niercellen in weefselkweek hypoxisch? Cell Physiol Biochem 1999;9:150-72.
7 Bloch-Frankenthal L, Ram D. The relationship between the Crabtree effect and the oxidative metabolism of glucose and carbohydrate intermediates in ascites tumor cells. Cancer Res 1959;19:835-42.
8ParrinelloS, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003;5:741-7.
9 Denko NC. Hypoxie, HIF1 en glucosemetabolisme in de vaste tumor. Nat Rev Cancer 2008;8:705-13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. De genomische analyse van verzuring en acidose respons in menselijke kankers. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Hypoxiainduced down-regulation of BRCA1 expression by E2Fs. Cancer Res 2005;65: 11597-604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Cellulaire aanpassingen aan hypoxie en acidose tijdens de somatische evolutie van borstkanker. Br J Cancer 2007;97:646-53.
13 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008;8:425-37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 medieert aanpassing aan hypoxie door actieve downregulatie van mitochondriaal zuurstofverbruik. Cell Metab 2006;3:187-97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008;283:28106-14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177-85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Zuurstofverbruik kan de groei van tumoren reguleren, een nieuw perspectief op het Warburg-effect. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. Pharmacologically increased tumor hypoxia can be measured by 18F-fluoroazomycin arabinoside positron emission tomography and enhances tumor response to hypoxic cytotoxin PR-104. Clin Cancer Res 2009;15: 7170-4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358:69-77.
20 Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 2001;276: 37223-9.
21 Sugden PH, Randle PJ. Regulation of pig heart pyruvate dehydrogenase by phosphorylation. Studies over de stoicheiometrieën van de subunit en fosforylering. Biochem J 1978;173:659-68.
22 Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specifieke regulatie. J Biol Chem 1995;270:14297-304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Characterization of the isozymes of pyruvate dehydrogenase phosphatase: implications for the regulation of pyruvate dehydrogenase activity. Biochim Biophys Acta 2003;1652:126-35.
24 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 1973;134: 651-3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemie 2006;45:402-15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem 2000; 275:15773-81.
28 Stacpoole PW, Greene YJ. Dichlooracetaat. Diabetes Care 1992;15:785-91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med 1983;309:390-6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 2008;60:1478-87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurologie 2006;66: 324-30.
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Farmacokinetiek, metabolisme en toxicologie van dichlooracetaat. Drug Metab Rev 1998;30: 499-539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluatie van langdurige behandeling van kinderen met congenitale lactose met dichlooracetaat. Pediatrics 2008;121:e1223-8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther 2008;324:1163-71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol 1988;61:65-70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994;23:1617-24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostaat 2008;68:1223-31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induceert apoptose in endometriumkankercellen. Gynecol Oncol 2008;109: 394-402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 2009;29:4579-88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondriale mutaties dragen bij aan HIF1alpha accumulatie via verhoogde reactieve zuurstofsoorten en upgereguleerde pyruvaat dehydrogenease kinase 2 in hoofd-hals plaveiselcelcarcinoom. Clin Cancer Res 2009;15:476-84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichlooracetaat induceert apoptose en celcyclusstilstand in colorectale kankercellen. Br J Cancer 2010;102:1746-52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA 2007;104:9445-50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010;31:567-76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Natriumdichlooracetaat (DCA) vermindert apoptose in colorectale tumorhypoxie. Cancer Lett 2010;287:75-83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Natriumdichlooracetaat (DCA) richt zich selectief op cellen met defecten in de mitochondriale ETC. Int J Cancer 2010;127:2510-19.
48SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010;120:253-60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2: 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 2009;389:157-64.
51 Holness MJ, Sugden MC. Pyruvate dehydrogenase activities during the fed-to-starved transition and on re-feeding after acute or prolonged starvation. Biochem J 1989;258:529-33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptoralpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276-83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J Biol Chem 2010;285:2375-85.
54 Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003;52: 1371-6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 2007;292:C125-36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507-14.
57 Maher JC, Krishan A, Lampidis TJ. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol 2004;53:116-22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930-42.
59KoukourakisMI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res 2006;66:632-7.
60 Fantin VR, St-Pierre J, Leder P. Attenuation of LDHA expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006;9:425-34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibitie van lactaat dehydrogenase A induceert oxidatieve stress en remt tumorprogressie. Proc Natl Acad Sci USA 2010;107:2037-42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235-56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimuleert aërobe glycolyse in kankercellen. Cancer Res 2004; 64:3892-9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005; 24:4165-73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signalering verhoogt de snelheid van hypoxia-inducible factor 1alpha (HIF-1alpha) synthese: nieuw mechanisme voor HIF-1-gemedieerde vasculaire endotheliale groeifactor expressie. Mol Cell Biol 2001;21:3995-4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Phase II trial of the oral mammalian target of rapamycin inhibitor everolimus in relapsed or refractory Waldenstrom macroglobulinemia. J Clin Oncol 2010;28:1408-14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009;27: 2278-87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008;283:22700-8.
Gerelateerde inhoud: