Ioanna Papandreou, Tereza Goliasova y Nicholas C. Denko
Departamento de Oncología Radioterápica, División de Radiación y Biología del Cáncer, Facultad de Medicina de la Universidad de Stanford, Stanford, CA
Palabras clave: metabolismo tumoral, piruvato deshidrogenasa, efecto Warburg, inhibidores metabólicos
Abreviaturas: DCA: dicloroacetato; HIF1: factor 1 inducible por hipoxia; LDH: lactato deshidrogenasa; PDC: complejo piruvato deshidrogenasa; PDH: piruvato deshidrogenasa; PDK: piruvato deshidrogenasa cinasa; PDP: piruvato deshidrogenasa fosfatasa
Correspondencia a: Nicholas C. Denko, Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA 94305, USA
Tel.: 650-724-5066, Fax: 650-723-7382,
E-mail: [email protected]
Recibido: 22 Jul 2010
Aceptado: Aceptado: 30 sep 2010
En línea: 18 oct 2010
DOI: 10.1002/ijc.25728
En los últimos 20 años, el número de artículos que contienen »metabolismo tumoral» ha aumentado de 3 a 28 al año, y el número de veces que estos artículos han sido citados ha aumentado de 23 a 929 al año (estadísticas ISI, Thompson Reuters). El renovado interés por comprender los mecanismos y consecuencias de la alteración del metabolismo tumoral ha captado claramente la imaginación de la comunidad científica. La idea de que los tumores tienen un metabolismo alterado fue reconocida por primera vez por el bioquímico Otto Warburg, ganador del Premio Nobel, al describir el metabolismo de la glucosa.1 Más recientemente, el concepto de que los tumores son metabólicamente diferentes ha crecido hasta abarcar otras características, como la glutaminolisis, la oxidación de ácidos grasos y la biogénesis lipídica. Es evidente que existe una demanda metabólica diferente que impulsa estos cambios en las células que se dividen continuamente en comparación con las células diferenciadas en fase terminal. El descubrimiento de estas alteraciones ha planteado la posibilidad de que puedan ser un objetivo terapéutico debido a su importancia única para las células cancerosas.2
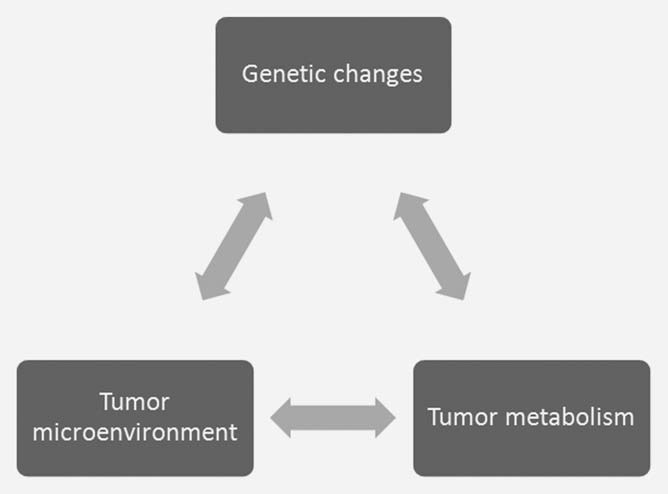
Se ha propuesto el concepto de que los cambios metabólicos son una respuesta a demandas únicas dentro del tumor,3 incluso cuando es difícil cuantificar dichas demandas. Existe una interacción entre los cambios oncogénicos en la célula tumoral y los aspectos únicos del microambiente tumoral que repercuten en el metabolismo celular y viceversa (Fig. 1). Por lo tanto, es difícil establecer las demandas metabólicas exactas dentro del tumor estudiando las células del tumor cultivadas ex vivo. Las condiciones ambientales utilizadas para cultivar células son muy diferentes de las condiciones ambientales in vivo. El medio Dulbecco’s modified Eagles con alto contenido en glucosa y una atmósfera con un 21% de oxígeno es muy diferente de las condiciones hipóxicas y/o hipoglucémicas que se encuentran en el tumor.4,5 La concentración de glucosa de 25 mM es aproximadamente cinco veces superior a los niveles normales en sangre, y la tensión de oxígeno es al menos cuatro veces superior a la que se encuentra in vivo. El hecho de que las células estén bañadas en estos sustratos metabólicos altera significativamente sus programas metabólicos inherentes.4,6 Las concentraciones elevadas de glucosa favorecen la glucólisis (el efecto Crabtree7 ) mientras que la oxigenación elevada produce un aumento de los subproductos del oxígeno y acorta la vida celular.8 El metabolismo de la glucosa ilustra la interacción de estos tres factores en el tumor. La transformación oncogénica impulsa la proliferación de células tumorales más que la capacidad vascular, generando hipoxia. La hipoxia dentro del microambiente tumoral aumenta el metabolismo glucolítico, en gran medida a través de la activación del factor de transcripción HIF1 (factor inducible por hipoxia 1).9 El aumento de la glucólisis conduce a una mayor producción de lactato, lo que contribuye a un pH extracelular ácido y a más cambios en la expresión génica.10 Tanto la hipoxia como la acidosis pueden contribuir a aumentar los niveles de mutación somática que pueden impulsar aún más la progresión tumoral.11, 12 Es claramente difícil reproducir estas complejas interacciones en células cultivadas in vitro.
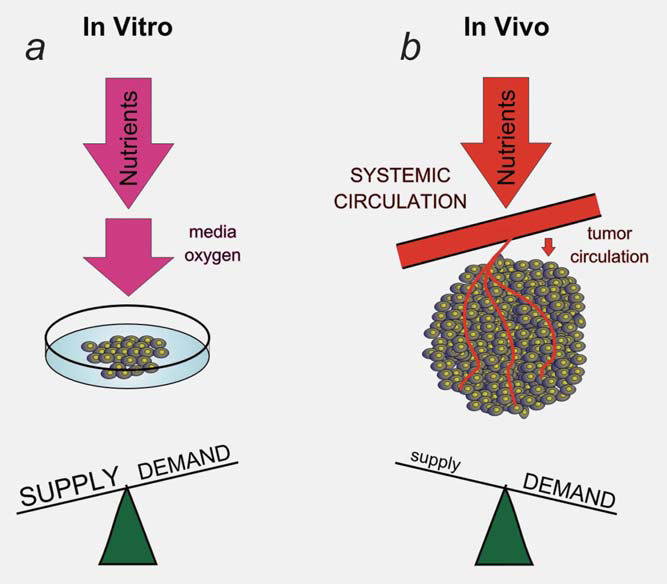
Parte de la interacción entre el microentorno y el metabolismo de las células tumorales se genera a través de una respuesta adaptativa a los cambios dinámicos en la oferta y demanda celular de metabolitos. El simple hecho de que podamos medir regiones de hipoxia y acidosis dentro de los tumores indica que los vasos tumorales no mantienen un entorno constante para el crecimiento de las células tumorales.13 La vasculatura tumoral representa un cuello de botella en el suministro de nutrientes y la eliminación de productos de desecho del tumor.4 El suministro inadecuado de los vasos tumorales inicia una respuesta adaptativa de las células tumorales diseñada para disminuir la demanda de los metabolitos limitados. Este proceso dinámico es difícil de modelizar in vitro (Fig. 2). Por ejemplo, los bajos niveles de oxígeno dentro del tumor inducen el factor de transcripción HIF1 y su programa metabólico.9 Parte del programa metabólico iniciado por HIF1 consiste en reducir la demanda de oxígeno disminuyendo la función mitocondrial. Parte de esta respuesta está mediada por la inducción dependiente de HIF1 de PDK1 y PDK3 dentro de las células tumorales y una reducción de la oxidación de piruvato dentro de las mitocondrias.14-17 Esta respuesta adaptativa es responsable de acercar la demanda de oxígeno al suministro limitado.
El dicloroacetato (DCA) es capaz de interferir en esta adaptación a la hipoxia tumoral inhibiendo la función de las PDKs (Fig. 3). El bloqueo de una respuesta adaptativa a la hipoxia se observa más claramente cuando el tumor en su conjunto está en déficit de oxígeno, y la vasculatura tumoral no puede responder a este aumento de la demanda.18 Incluso cuando las células tumorales se colocan en hipoxia, hay suficiente oxígeno en el medio ambiente (1-2%) para mantener una condición estable, aunque baja de oxígeno dentro de la célula, incluso con la adición de DCA. La velocidad de difusión en las células es más rápida que la velocidad de consumo, por lo que el nivel intracelular de oxígeno no depende de la velocidad de consumo (a menos que se utilice un número muy elevado de células en placas de vidrio que impidan la difusión de oxígeno a través del plástico14). En este artículo, presentamos un análisis de los datos publicados relativos al DCA como ejemplo de fármaco diseñado para incidir en el metabolismo tumoral que apoya nuestra hipótesis de que esta clase de fármacos tiene un efecto muy diferente en las células tumorales que crecen in vivo con el cuello de botella metabólico producido por la vasculatura, en comparación con su efecto en las células cultivadas in vitro.
El complejo piruvato deshidrogenasa
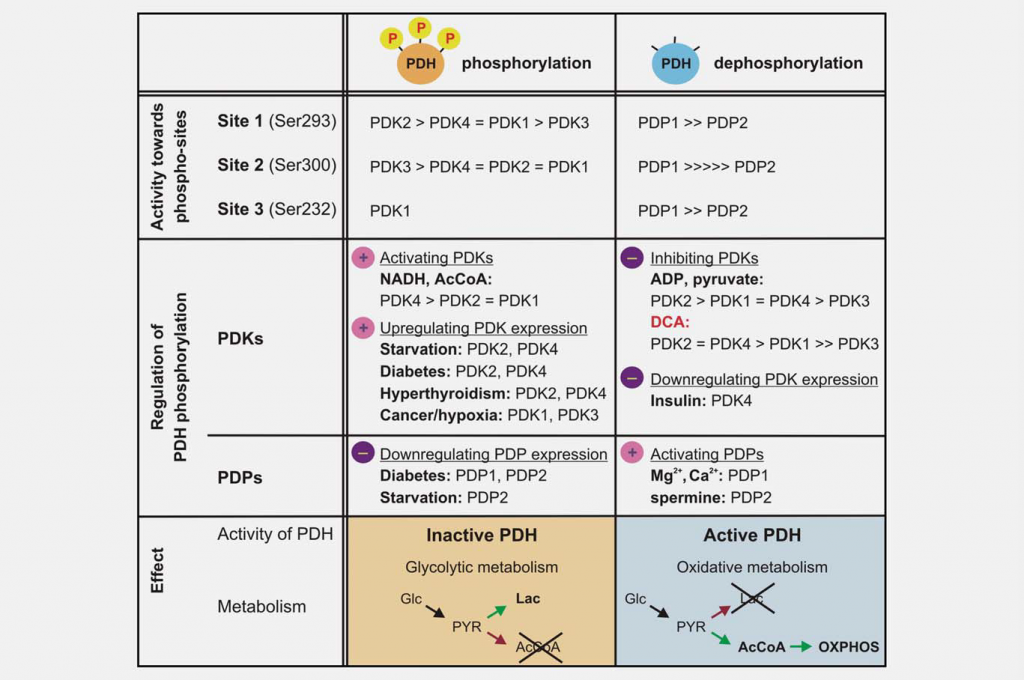
La inhibición de la función mitocondrial es tan importante como el aumento de la glucólisis para producir el efecto Warburg. Uno de los principales reguladores de la función mitocondrial es el complejo piruvato deshidrogenasa (PDC), que cataliza la descarboxilación irreversible del piruvato a acetil-CoA, CO2 y NADH. El control de la PDC controla la entrada de los carbonos derivados de los hidratos de carbono en la mitocondria. Esta reacción desempeña un papel central en la regulación tanto de las vías mitocondriales de producción de energía (ciclo del ácido tricarboxílico [TCA] y fosforilación oxidativa [OXPHOS]) como de la generación de intermediarios biosintéticos, como el citrato. La PDC consta de tres componentes catalíticos, la piruvato deshidrogenasa (E1), la dihidrolipoamida transacetilasa (E2) y la dihidrolipoamida deshidrogenasa (E3), que se organizan en grandes complejos multiméricos junto con la subunidad estructural E3 binding protein (E3BP). El núcleo básico del componente E1 de la piruvato deshidrogenasa es un heterotetrámero de dos subunidades alfa y dos beta (α2β2), y cataliza el primer paso de la descarboxilación del piruvato. La actividad del complejo está regulada en gran medida por la fosforilación reversible de tres residuos de serina de E1α. Las piruvato deshidrogenasa quinasas (PDK1-4) inactivan la PDC y las piruvato deshidrogenasa fosfatasas (PDP1-2) la activan/reactivan (Fig. 3).
Las diferentes isoformas de PDK varían en sus propiedades reguladoras, distribución tisular y modulación por señales metabólicas ascendentes. El resultado es un control dinámico y órgano-específico de la función mitocondrial y de la producción de energía. Los ensayos enzimáticos han demostrado que las PDK difieren en su especificidad hacia los tres sitios diana E1α y los parámetros cinéticos de la fosforilación. La PDK1 es la única isoforma capaz de fosforilar los tres sitios, mientras que las PDK2-4 fosforilan los sitios 1 y 2 con diferentes velocidades in vitro. 19,20 La fosforilación de incluso uno de los seis sitios E1a en el heterotetrámero es suficiente para inactivar la PDC, y se cree que como máximo tres de los seis sitios posibles del tetrámero E1 pueden ser fosforilados en un momento dado.21, 22 Los cambios espaciotemporales en los niveles y la actividad de las fosfatasas de la PDC (PDP1-2) también son reguladores. La desfosforilación in vitro parece ser aleatoria; ambas isozimas son capaces de desfosforilar los tres sitios E1 de sustratos mutados recombinantes.23
Dicloroacetato: Un inhibidor de PDK
El DCA fue identificado como un activador de la piruvato deshidrogenasa por su capacidad de estimular la actividad enzimática de la PDC en un modelo de corazón de rata perfundido.24 Ahora se sabe que este mimético del piruvato actúa inhibiendo la acción de las PDKs. Se ha obtenido la estructura cristalina de PDK2 en complejo con DCA, y muestra que el DCA ocupa el sitio de unión del piruvato en el dominio regulador N-terminal (R).25 Las cuatro isozimas varían considerablemente en su sensibilidad a la inhibición por DCA. La PDK2 es la más sensible, la PDK3 la más resistente, mientras que la PDK1 y la PDK4 son relativamente sensibles.26,27
Tratamiento de la acidosis láctica con D CA
La seguridad y eficacia del DCA se ha evaluado en casos de acidosis láctica congénita y adquirida. El tratamiento con DCA reduce eficazmente los niveles de lactato en la circulación al estimular la oxidación del piruvato; sin embargo, aún no se sabe si el DCA puede mejorar el pronóstico de los pacientes con estos síndromes.28,29 Se ha propuesto que los niños pequeños con deficiencia de PDH pueden ser los que más se beneficien del tratamiento crónico con DCA.30 El efecto adverso más significativo de la administración de DCA a largo plazo es una neuropatía periférica reversible.31,32 La gravedad de la toxicidad parece depender de la edad, siendo los pacientes adultos más susceptibles que los niños.31,33 Las razones de esta discrepancia no están del todo claras, pero posiblemente estén relacionadas con la diferente farmacocinética y metabolismo del DCA en los dos grupos de edad.34 El DCA también se ha utilizado en ensayos clínicos para enfermedades cardiacas, incluyendo la insuficiencia cardiaca congestiva y la cardiopatía isquémica, mostrando resultados positivos y mejorando el rendimiento miocárdico.35,36
DCA como potencial terapéutico contra el cáncer
En los últimos años, el DCA ha atraído la atención como un medio potencialmente sencillo y económico para atacar tumores glucolíticos a la vez que produce efectos secundarios limitados en los órganos oxidativos sanos. El interés por este fármaco por parte de la comunidad científica, los pacientes con cáncer y los medios de comunicación se despertó en 2007, después de que un grupo de la Universidad de Alberta informara de que el DCA era singularmente tóxico para las líneas celulares de cáncer humano e inhibía el crecimiento de tumores de xenoinjerto de tumor de pulmón A549 en ratas.37 Desde entonces, los informes emergentes sobre la eficacia del DCA in vitro e in vivo revelan algunas características interesantes y también desconcertantes, que distinguen el caso del DCA de la mayoría de los fármacos desarrollados como agentes anticancerígenos (Tabla 1). El número de diferentes tipos de cáncer y estrategias experimentales probadas hasta la fecha es demasiado limitado para permitir conclusiones generalizadas sobre la eficacia del DCA contra todo tipo de tumores. Con esta salvedad, una comparación cualitativa de la bibliografía sugiere que el DCA muestra más efecto anticancerígeno in vivo que efecto anticélulas cancerígenas in vitro.
| Tipo de cáncer | Referencias | Efecto sobre la supervivencia y el crecimiento |
| Estudios in vitro | ||
| Pulmón, Glioblastoma, Mama | 37 | Apoptosis in vitro, inhibición del crecimiento de xenoinjertos |
| Próstata | 38 | Inhibición del crecimiento in vitro, radiosensibilización moderada |
| Endometrio | 39 | Inhibición del crecimiento |
| Cervical | 40 | Ventaja de crecimiento bajo hipoxia in vitro |
| Cabeza y cuello | 41 | Inhibición del crecimiento in vitro sólo en células que sobreexpresan mutND2 |
| Pediatría | 42 | Apoptosis a altas concentraciones in vitro, cierta influencia en la respuesta a la quimioterapia |
| Colorrectal | 43 | Apoptosis a concentraciones muy elevadas |
| Modelos preclínicos | ||
| Colorrectal | 44 | Poco efecto sobre el crecimiento, aumento de la hipoxia por HRE-luciferasa, sensibilizado a citotoxinas hipóxicas |
| Colorrectal | 18 | Poco efecto sobre el crecimiento, aumento de la hipoxia por 18F-FAZA PET, sensibilizado a la citotoxina hipóxica |
| Modelos preclínicos | 17 | Inhibición del crecimiento de xenoinjertos |
| Colorrectal | 45 | Inhibición del crecimiento de xenoinjertos |
| Colorrectal | 46 | Protegido de la anoxia in vitro, promueve el crecimiento de xenoinjertos de SW480 |
| Colorrectal, mama, PML, próstata | 47 | Activo sólo contra células con cadena de transporte de electrones defectuosa |
| Mama | 48 | Inhibición del crecimiento de xenoinjertos y metástasis |
| Datos de pacientes humanos | ||
| Glioblastoma | 49 | Enfermedad clínicamente estable in vivo, disminución de HIF1, aumento de p53 ex vivo |
Estudios in vitro. Se ha informado de que el DCA tiene efectos citotóxicos in vitro, 37-40 con algunas respuestas a concentraciones clínicamente relevantes (0,5-1 mM), mientras que otras requieren niveles suprafarmacológicos (10-100 mM) y otros grupos no han encontrado toxicidad directa in vitro. 18,41,42,47 Una condición que se ha identificado que sensibiliza a las células al DCA son las mutaciones que perturban la función respiratoria mitocondrial,41,47 lo que sugiere que la utilización forzada de la OXPHOS defectuosa es tóxica. Como la privación de oxígeno también regula a la baja la función mitocondrial, parecía razonable plantear la hipótesis de que las células hipóxicas serían más sensibles al DCA. Sin embargo, esta hipótesis no se ha confirmado, al menos en el limitado número de líneas celulares analizadas hasta la fecha. La hipoxia moderada in vitro no influyó en el perfil del ciclo celular de las células colorrectales tratadas con DCA43 ni en la viabilidad reproductiva de las células de cáncer de páncreas tratadas con DCA (nuestras observaciones no publicadas). Curiosamente, otro estudio descubrió que la hipoxia grave (¿anoxia?) puede ser protectora frente a la apoptosis inducida por DCA en células de cáncer colorrectal.46 La razón de estas discrepancias no está clara.
En general, la mayoría de los datos apoyan la idea de que las concentraciones clínicamente relevantes de DCA (inferiores a 1 mM) no son directamente citotóxicas in vitro. La razón de esta aparente resistencia celular no es una inactivación del DCA en condiciones de cultivo de tejidos o una incapacidad para inactivar las PDK, ya que se ha demostrado que el DCA aumenta transitoriamente la actividad mitocondrial y colapsa el potencial de membrana mitocondrial.18,37,50 Por lo tanto, la base del limitado efecto anticancerígeno del DCA en cultivo probablemente radique en la compleja fisiología celular y el enorme exceso de metabolitos presentes en los medios de cultivo.
Modelos preclínicos. Los informes sobre la actividad del DCA contra tumores modelo cultivados en roedores son alentadores, aunque hay ciertos casos de líneas tumorales que no responden al tratamiento, e incluso un ejemplo de crecimiento tumoral acelerado en respuesta al DCA.46 El primer informe sobre la actividad antitumoral del DCA fue el de Bonnet et al. 37 Los autores informaron de que los xenoinjertos de adenocarcinoma de pulmón A549 cultivados en ratas desnudas mostraron un retraso significativo del crecimiento tumoral tras el tratamiento con DCA, y algunos grupos experimentales incluso mostraron regresión tumoral. Estos efectos se asociaron con un aumento de la apoptosis y una reducción de la proliferación. Utilizando las mismas células A549, Stockwin et al. 47 confirmaron recientemente que el DCA suprimía el crecimiento en los tumores modelo, aunque encontraron poca toxicidad in vitro.
Nuestro grupo ha informado de que el tratamiento diario con DCA de ratones con xenoinjertos pancreáticos SU86.86 provocó un retraso significativo del crecimiento tumoral, así como un aumento de la fracción hipóxica de los tumores. En apoyo de este modelo, el DCA aumentó el grado de hipoxia tumoral en xenoinjertos colorrectales RKO, evaluado mediante reporteros de luciferasa impulsados por HRE (elemento de respuesta a la hipoxia)44 o tomografía por emisión de positrones conarabinosido de 18F-fluoroazomicina.18 Este modelo RKO mostró una hipoxia muy modesta en los xenoinjertos colorrectales RKO Este modelo RKO mostró una reducción muy modesta en el crecimiento de los tumores tratados con DCA, pero los cambios agudos en la oxigenación tras el DCA los sensibilizaron al tratamiento con citotoxinas activadas por hipoxia como la Tirapazamina44 o la PR-104.18 Trabajos adicionales en modelos de cáncer colorrectal, realizados por otros grupos, han revelado una heterogeneidad significativa en la respuesta al DCA. Algunas líneas celulares reducen sus tasas de crecimiento tumoral,45 mientras que otras no responden o incluso crecen más rápido.46 Además, en un modelo de rata de adenocarcinoma mamario, un programa intensivo de tratamiento con DCA fue capaz de reducir el número de metástasis pulmonares macroscópicas.48 Estos modelos preclínicos apoyan el concepto de que el DCA tiene la capacidad de modular el metabolismo tumoral in vivo, dando lugar a mayores o menores efectos antitumorales en función del modelo probado.
Datos de ensayos clínicos. Los primeros datos sobre la evaluación del DCA para el tratamiento del cáncer humano se comunicaron recientemente.49 En este estudio, el DCA se utilizó en combinación con cirugía, temozolomida y radiación para el tratamiento de cinco pacientes con glioblastoma multiforme. Aunque los autores informan de resultados clínicos prometedores en cuatro de los cinco pacientes, el énfasis del informe fue el análisis ex vivo de las células tumorales antes y después del tratamiento con DCA. Informan de cambios en el potencial de membrana mitocondrial, aumento de las cantidades de radicales de oxígeno generados mitocondrialmente y aumento de la apoptosis de las células tumorales. Los estudios mecanísticos hallaron niveles alterados de señalización HIF1, activación de p53 y disminución de la angiogénesis. Estos datos sugieren que el DCA tiene muchos mecanismos de acción posteriores a su inhibición de las PDK. Obviamente, es necesario tratar a un mayor número de pacientes, preferiblemente de diferentes localizaciones, para poder generalizar estos interesantes resultados.
Regulación organísmica del metabolismo
Los factores humorales de crecimiento y nutricionales también podrían influir en la respuesta tumoral a la reprogramación metabólica. La regulación de la actividad de las PDC tumorales in vivo aún no se ha estudiado sistemáticamente, pero los conocimientos que hemos adquirido del metabolismo de los tejidos normales y de los campos de la endocrinología y la investigación de la obesidad proporcionan algunas hipótesis interesantes. La inanición aumenta la expresión de PDKs y disminuye la actividad de PDH en órganos periféricos como estrategia para mantener un suministro estable de carbohidratos al cerebro y otros tejidos neuronales. Por ejemplo, la inanición activa transcripcionalmente la PDK4 y la PDK2 en el hígado, el riñón y otros tejidos.51, 52 Los glucocorticoides, la hormona tiroidea T3 y los ácidos grasos libres también aumentan los niveles de PDK4.53 La realimentación y/o el aumento de los niveles de insulina disminuyen la transcripción de PDK4 y reactivan la PDC. Del mismo modo, se demostró que la inanición y la diabetes reducen los niveles de ARNm y proteína de PDP2, efectos que se invirtieron con la realimentación o el tratamiento con insulina.54
Por lo tanto, las numerosas señales de entrada que afectan a la actividad de la PDH in vivo podrían influir en la eficacia del DCA y otros fármacos metabólicamente dirigidos. La administración del fármaco durante el ayuno podría tener un efecto muy diferente en comparación con el estado alimentado. Las respuestas a las condiciones humorales son obviamente diferentes para los distintos tejidos y tipos de tumores y, por tanto, difíciles de imitar in vitro. Entonces, ¿es posible evaluar el efecto antitumoral de moléculas como el DCA in vitro? Muchos años de estudio de un gran número de líneas celulares tumorales cultivadas demuestran que sí mantienen las características anormales de la glucólisis aeróbica in vitro y, por lo tanto, proporcionan una herramienta valiosa en el estudio del metabolismo del cáncer.55 Sin embargo, también hay limitaciones inherentes en estos sistemas, como lo demuestran los datos inconsistentes en la literatura al comparar el efecto del DCA en cultivo celular frente a los resultados preclínicos y clínicos.
El análisis precedente presenta el modelo de que un fármaco anticanceroso dirigido al metabolismo y con escasa toxicidad in vitro puede tener un potencial significativo in vivo. Hemos destacado los sustratos metabólicos limitados en el tumor y la regulación sistémica del metabolismo por factores humorales que podrían aumentar la eficacia del fármaco in vivo. Es posible que también ocurra lo contrario, que un fármaco con buena actividad in vitro tenga poco éxito in vivo. Los efectos sobre la toxicidad de los tejidos normales o la cooperación metabólica entre tipos celulares podrían limitar la eficacia de un fármaco in vivo. El inhibidor metabólico 2-deoxiglucosa, ampliamente probado, tiene una actividad anticancerígena razonable in vitro, pero no puede utilizarse en pacientes debido a sus efectos negativos sobre los tejidos normales que dependen del consumo de glucosa. La toxicidad neurológica limitante de la dosis se produce a un nivel de fármaco muy inferior al necesario para obtener efectos anticancerígenos en tumores de roedores.56, 57 Otra posibilidad es que la cooperación metabólica entre tipos celulares o entre células normales y tumorales pueda eludir el bloqueo metabólico inducido por el fármaco. Por ejemplo, aunque el lactato suele considerarse un producto de desecho metabólico, en algunas células puede utilizarse como combustible para potenciar la función mitocondrial.58,59
Con respecto a la combinación del DCA con las terapias existentes, los datos preclínicos hasta la fecha no muestran un patrón obvio de interacción que permita una selección fácil y racional de los regímenes terapéuticos. Los modelos animales seguirán siendo el mejor medio de experimentación para determinar empíricamente las combinaciones más prometedoras. Hemos demostrado que, debido a su capacidad para aumentar el consumo de oxígeno, el DCA incrementa la hipoxia tumoral y sensibiliza a los tumores de páncreas y colon xenotrasplantados a las citotoxinas hipóxicas,18,44 por lo que resulta intrigante imaginar un plan de tratamiento que incluya estas dos clases de fármacos, ambos diseñados para explotar el microambiente hipóxico único del tumor.
No se ha descrito la interacción del DCA con otros moduladores metabólicos. Una diana potencial para la terapia combinada es la lactato deshidrogenasa A (LDHA). Se ha demostrado que la inhibición genética o farmacológica de la LDHA aumenta la función mitocondrial e inhibe la formación y progresión de tumores modelo.60,61 En este esquema de combinación, el inhibidor de la LDHA bloquearía la conversión de piruvato en lactato y el DCA desviaría el piruvato acumulado hacia la oxidación mitocondrial. Si el efecto antitumoral del DCA se deriva del aumento de la función mitocondrial, es posible que la combinación de inhibidores de la PDK y de la LDHA fuerce una tasa de oxidación mitocondrial aún mayor y frene el crecimiento tumoral con mayor eficacia.
Los puntos nodales de vías de supervivencia clave, como la vía PI3KAkt-mTOR, también son objeto de intensos esfuerzos de desarrollo farmacológico.62 Parte de las propiedades promotoras del crecimiento de esta vía proviene de su capacidad para regular el metabolismo y la producción de energía mediante mecanismos directos o indirectos. Por ejemplo, la activación oncogénica de PI3K-Akt estimula la captación de glucosa y la glucólisis aeróbica,63, 64 mientras que la activación de Akt y mTORC1 aumenta la traducción del ARNm de Hif-1a en condiciones de hipoxia.65 Se están probando en ensayos clínicos inhibidores prometedores de Akt e inhibidores de mTOR de nueva generación66,67 que representan candidatos viables para la terapia combinada con DCA para modular tanto el metabolismo aeróbico como el hipóxico.
Conclusiones
En conclusión, los conocimientos recientes sobre el metabolismo único de los tumores sólidos han identificado varias vías novedosas, susceptibles de fármacos, que pueden utilizarse preferentemente en las células tumorales en comparación con las células normales. El análisis de los fármacos candidatos contra el cáncer diseñados para atacar estas vías metabólicas requerirá un cuidadoso diseño experimental, tanto in vitro como in vivo. El análisis de los informes publicados que estudian el DCA muestra una gama confusa, y a veces contradictoria, de efectos in vitro e in vivo. Los estudios genéticos en tumores modelo ofrecen pruebas convincentes de que esta vía es una buena candidata para la orientación terapéutica.68 Sería muy útil en el análisis de la utilidad potencial del DCA si pudiera haber alguna firma molecular que pudiera predecir la sensibilidad al fármaco, tanto en tumores modelo como, eventualmente, en pacientes. Quizás un análisis cuidadoso de la presunta diana del DCA, la fosforilación de la subunidad E1α de la piruvato deshidrogenasa, pueda ofrecer esta firma.
REFERENCIAS
1 1. Warburg O, Wind F, Negelein E. El metabolismo de los tumores en el cuerpo. J Gen Physiol 1927;8:519-30.
2 Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Ladrillo a ladrillo: metabolismo y crecimiento de células tumorales. Curr Opin Genet Dev 2008;18:54-61.
4 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004;14:198-206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas: a novel guideline for clinical oncologists? J Cancer Res Clin Oncol 2003;129:321-6.
6 Gstraunthaler G, Seppi T, Pfaller W. Impact of culture conditions, culture media volumes, and glucose content on metabolic properties of renal epithelial cell cultures. ¿Son hipóxicas las células renales en cultivo tisular? Cell Physiol Biochem 1999;9:150-72.
7 Bloch-Frankenthal L, Ram D. The relationship between the Crabtree effect and the oxidative metabolism of glucose and carbohydrate intermediates in ascites tumor cells. Cancer Res 1959;19:835-42.
8ParrinelloS, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003;5:741-7.
9 Denko NC. Hipoxia, HIF1 y metabolismo de la glucosa en el tumor sólido. Nat Rev Cancer 2008;8:705-13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. El análisis genómico de la acidosis láctica y la respuesta a la acidosis en los cánceres humanos. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Hypoxiainduced down-regulation of BRCA1 expression by E2Fs. Cancer Res 2005;65: 11597-604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Adaptaciones celulares a la hipoxia y acidosis durante la evolución somática del cáncer de mama. Br J Cancer 2007;97:646-53.
13 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008;8:425-37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187-97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. La inducción de la piruvato deshidrogenasa quinasa-3 por el factor inducible por hipoxia-1 promueve el cambio metabólico y la resistencia a los medicamentos. J Biol Chem 2008;283:28106-14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177-85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. El consumo de oxígeno puede regular el crecimiento de los tumores, una nueva perspectiva sobre el efecto Warburg. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. Pharmacologically increased tumor hypoxia can be measured by 18F-fluoroazomycin arabinoside positron emission tomography and enhances tumour response to hypoxic cytotoxin PR-104. Clin Cancer Res 2009;15: 7170-4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358:69-77.
20 Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 2001;276: 37223-9.
21 Sugden PH, Randle PJ. Regulation of pig heart pyruvate dehydrogenase by phosphorylation. Studies on the subunit and phosphorylation stoicheiometries. Biochem J 1978;173:659-68.
22 Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem 1995;270:14297-304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Characterization of the isozymes of pyruvate dehydrogenase phosphatase: implications for the regulation of pyruvate dehydrogenase activity. Biochim Biophys Acta 2003;1652:126-35.
24 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 1973;134: 651-3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006;45:402-15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Diferencias marcadas entre dos isoformas de la piruvato deshidrogenasa cinasa humana. J Biol Chem 2000; 275:15773-81.
28 Stacpoole PW, Greene YJ. Dicloroacetato. Diabetes Care 1992;15:785-91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med 1983;309:390-6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 2008;60:1478-87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 2006;66: 324-30.
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Farmacocinética, metabolismo y toxicología del dicloroacetato. Drug Metab Rev 1998;30: 499-539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluación del tratamiento a largo plazo de niños con acidosis láctica congénita con dicloroacetato. Pediatrics 2008;121:e1223-8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther 2008;324:1163-71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol 1988;61:65-70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994;23:1617-24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008;68:1223-31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008;109: 394-402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 2009;29:4579-88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin Cancer Res 2009;15:476-84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. El dicloroacetato induce la apoptosis y la detención del ciclo celular en células de cáncer colorrectal. Br J Cancer 2010;102:1746-52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA 2007;104:9445-50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. La selección de células cancerosas con mitocondrias reprimidas desencadena la progresión del cáncer de colon. Carcinogenesis 2010;31:567-76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;287:75-83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. El dicloroacetato de sodio (DCA) se dirige selectivamente a las células con defectos en el ETC mitocondrial. Int J Cancer 2010;127:2510-19.
48SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010;120:253-60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Modulación metabólica del glioblastoma con dicloroacetato. Sci Transl Med 2010;2: 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitorización de la fosforilación del complejo piruvato deshidrogenasa. Anal Biochem 2009;389:157-64.
51 Holness MJ, Sugden MC. Pyruvate dehydrogenase activities during the fed-to-starved transition and on re-feeding after acute or prolonged starvation. Biochem J 1989;258:529-33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptoralpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276-83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulación de la piruvato deshidrogenasa quinasa 4 (PDK4) por la hormona tiroidea: papel del coactivador del receptor gamma activado por proliferador de peroxisomas (PGC-1 alfa). J Biol Chem 2010;285:2375-85.
54 Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003;52: 1371-6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 2007;292:C125-36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507-14.
57 Maher JC, Krishan A, Lampidis TJ. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol 2004;53:116-22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930-42.
59KoukourakisMI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res 2006;66:632-7.
60 Fantin VR, St-Pierre J, Leder P. Attenuation of LDHA expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006;9:425-34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. La inhibición de la lactato deshidrogenasa A induce estrés oxidativo e inhibe la progresión tumoral. Proc Natl Acad Sci USA 2010;107:2037-42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235-56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt estimula la glucólisis aeróbica en células cancerosas. Cancer Res 2004; 64:3892-9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. La dependencia de la glucosa de las células transformadas por Akt puede revertirse mediante la activación farmacológica de la betaoxidación de ácidos grasos. Oncogene 2005; 24:4165-73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 2001;21:3995-4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Phase II trial of the oral mammalian target of rapamycin inhibitor everolimus in relapsed or refractory Waldenstrom macroglobulinemia. J Clin Oncol 2010;28:1408-14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009;27: 2278-87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008;283:22700-8.
Contenido relacionado: