Ioanna Papandreou, Tereza Goliasova e Nicholas C. Denko
Dipartimento di radioterapia oncologica, Divisione di radioterapia e biologia del cancro, Stanford University School of Medicine, Stanford, CA
Parole chiave: metabolismo tumorale, piruvato deidrogenasi, effetto Warburg, inibitori metabolici
Abbreviazioni: DCA: dicloroacetato; HIF1: hypoxia-inducible factor 1; LDH: lattato deidrogenasi; PDC: complesso piruvato deidrogenasi; PDH: piruvato deidrogenasi; PDK: piruvato deidrogenasi chinasi; PDP: piruvato deidrogenasi fosfatasi
Corrispondenza a: Nicholas C. Denko, Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA 94305, USA
Tel.: 650-724-5066, Fax: 650-723-7382,
E-mail: [email protected]
Ricevuto: 22 Jul 2010
Accettato: Accettato il 30 settembre 2010
Online: 18 ottobre 2010
DOI: 10.1002/ijc.25728
Negli ultimi 20 anni, il numero di articoli contenenti ”metabolismo tumorale” è aumentato da 3 a 28 all’anno e il numero di volte in cui questi articoli sono stati citati è aumentato da 23 a 929 all’anno (statistiche ISI, Thompson Reuters). Il rinnovato interesse per la comprensione dei meccanismi e delle conseguenze delle alterazioni del metabolismo tumorale ha chiaramente catturato l’immaginazione della comunità scientifica. L’idea che i tumori abbiano un metabolismo alterato è stata riconosciuta per la prima volta dal biochimico premio Nobel Otto Warburg quando ha descritto il metabolismo del glucosio.1 Più recentemente, il concetto che i tumori sono metabolicamente diversi è cresciuto fino a comprendere altre caratteristiche, come la glutaminolisi, l’ossidazione degli acidi grassi e la biogenesi dei lipidi. È evidente che la domanda metabolica che guida questi cambiamenti è diversa nelle cellule in continua divisione rispetto alle cellule differenziate in modo terminale. La scoperta di queste alterazioni ha sollevato la possibilità che possano essere oggetto di un intervento terapeutico, data la loro importanza unica per le cellule tumorali.2
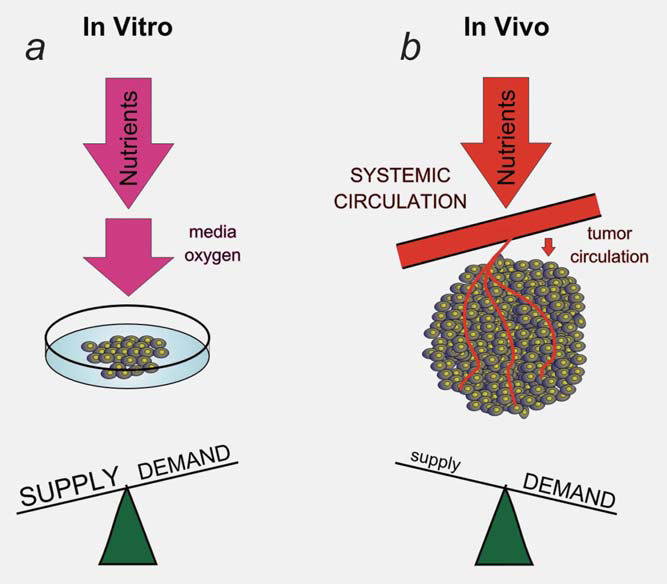
È stato proposto il concetto che le alterazioni metaboliche sono una risposta a richieste uniche all’interno del tumore,3 anche se è difficile quantificare tali richieste. Esiste un’interazione tra i cambiamenti oncogeni nella cellula tumorale e gli aspetti unici del microambiente tumorale che hanno un impatto sul metabolismo cellulare e viceversa (Fig. 1). È quindi difficile stabilire le esatte richieste metaboliche all’interno del tumore studiando le cellule del tumore coltivato ex vivo. Le condizioni ambientali utilizzate per far crescere le cellule in coltura sono molto diverse da quelle in vivo. Il terreno di coltura Dulbecco modificato Eagles ad alto contenuto di glucosio e un’atmosfera con il 21% di ossigeno sono molto diversi dalle condizioni ipossiche e/o ipoglicemiche riscontrate nel tumore.4,5 La concentrazione di glucosio di 25 mM è circa cinque volte quella dei normali livelli ematici e la tensione di ossigeno è almeno quattro volte superiore a quella riscontrata in vivo. Il fatto che le cellule siano immerse in questi substrati metabolici altera in modo significativo i loro programmi metabolici intrinseci.4,6 L’elevata concentrazione di glucosio favorisce la glicolisi (effetto Crabtree7 ), mentre l’elevata ossigenazione produce un aumento dei sottoprodotti dell’ossigeno e riduce la durata della vita cellulare.8 Il metabolismo del glucosio illustra l’interazione di questi tre fattori nel tumore. La trasformazione oncogenica spinge la proliferazione delle cellule tumorali più della capacità vascolare, generando ipossia. L’ipossia all’interno del microambiente tumorale aumenta il metabolismo glicolitico, in gran parte attraverso l’attivazione del fattore di trascrizione hypoxiainducible factor 1 (HIF1).9 L’aumento della glicolisi porta a una maggiore produzione di lattato, che contribuisce a un pH extracellulare acido e a ulteriori cambiamenti nell’espressione genica.10 Sia l’ipossia che l’acidosi possono contribuire a un aumento dei livelli di mutazioni somatiche che possono guidare ulteriormente la progressione del tumore.11,12 È chiaramente difficile riprodurre queste complesse interazioni nelle cellule coltivate in vitro.
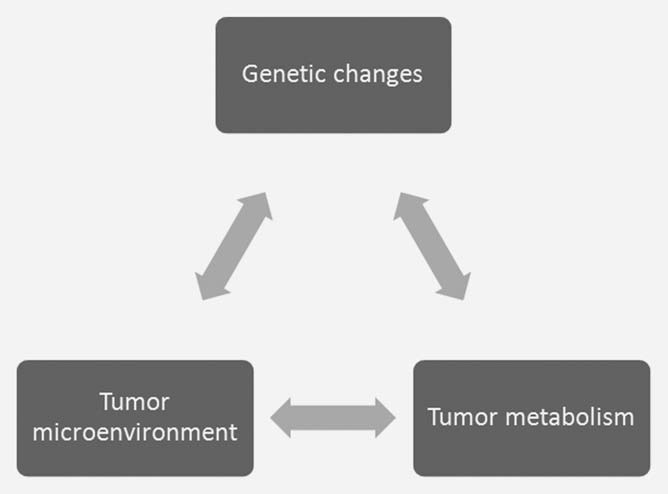
Parte dell’interazione tra il microambiente e il metabolismo delle cellule tumorali è generata da una risposta adattativa ai cambiamenti dinamici della domanda e dell’offerta di metaboliti da parte delle cellule. Il semplice fatto che si possano misurare regioni di ipossia e acidosi all’interno dei tumori indica che i vasi tumorali non mantengono un ambiente costante per la crescita delle cellule tumorali.13 La vascolarizzazione tumorale rappresenta un collo di bottiglia nell’apporto di sostanze nutritive e nella rimozione di prodotti di scarto dal tumore.4 L’apporto inadeguato da parte dei vasi tumorali avvia una risposta adattativa da parte delle cellule tumorali, volta a diminuire la richiesta di metaboliti limitati. Questo processo dinamico è difficile da modellare in vitro (Fig. 2). Ad esempio, bassi livelli di ossigeno all’interno del tumore inducono il fattore di trascrizione HIF1 e il suo programma metabolico.9 Parte del programma metabolico avviato da HIF1 consiste nel ridurre la richiesta di ossigeno diminuendo la funzione mitocondriale. Parte di questa risposta è mediata dall’induzione HIF1-dipendente di PDK1 e PDK3 all’interno delle cellule tumorali e dalla riduzione dell’ossidazione del piruvato all’interno dei mitocondri.14-17 Questa risposta adattativa è responsabile dell’avvicinamento della domanda di ossigeno all’offerta limitata.
Il dicloroacetato (DCA) è in grado di interferire con questo adattamento all’ipossia tumorale inibendo la funzione delle PDK (Fig. 3). Il blocco della risposta adattativa all’ipossia si osserva più chiaramente quando il tumore nel suo complesso è in deficit di ossigeno e la vascolarizzazione tumorale non può rispondere a questa maggiore richiesta.18 Anche quando le cellule tumorali sono poste in ipossia, c’è abbastanza ossigeno nell’ambiente (1-2%) per mantenere una condizione di ossigeno stabile, anche se bassa, all’interno della cellula anche con l’aggiunta di DCA. Il tasso di diffusione all’interno delle cellule è più veloce del tasso di consumo, quindi il livello intracellulare di ossigeno non dipende dal tasso di consumo (a meno che non si utilizzi un numero molto elevato di cellule in piatti di vetro che impediscono la diffusione dell’ossigeno attraverso la plastica14). In questo articolo, presentiamo un’analisi dei dati pubblicati relativi al DCA come esempio di farmaco progettato per influenzare il metabolismo tumorale, a sostegno della nostra ipotesi che questa classe di farmaci abbia un effetto molto diverso sulle cellule tumorali che crescono in vivo con il collo di bottiglia metabolico indotto dalla vascolarizzazione, rispetto al loro effetto sulle cellule cresciute in vitro.
Il complesso della piruvato deidrogenasi
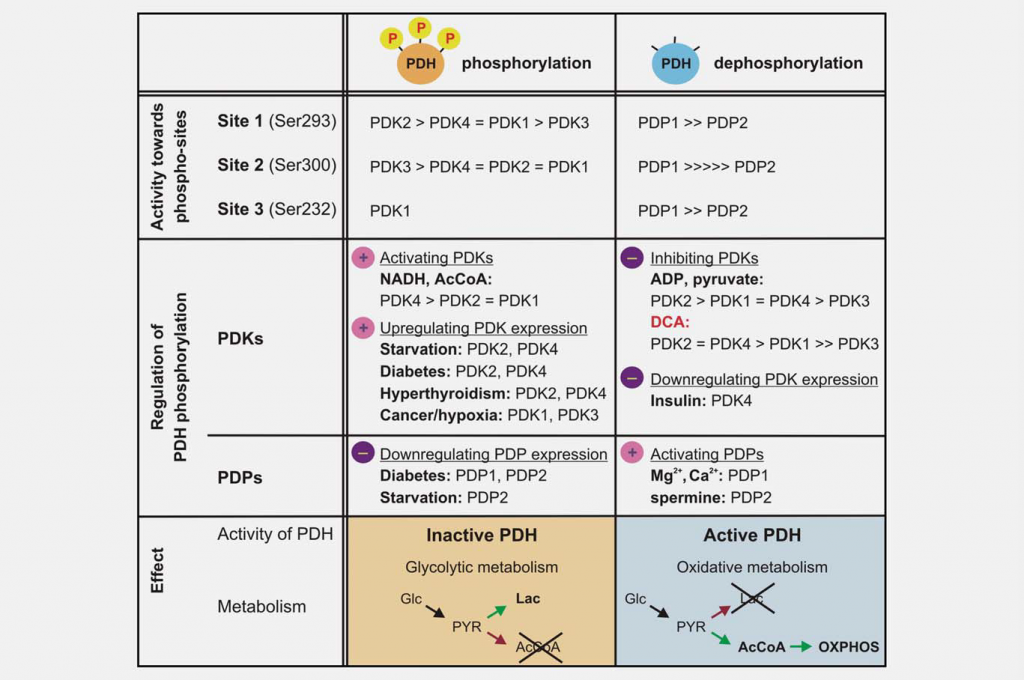
L’inibizione della funzione mitocondriale è importante quanto l’aumento della glicolisi per produrre l’effetto Warburg. Uno dei principali regolatori della funzione mitocondriale è il complesso della piruvato deidrogenasi (PDC), che catalizza la decarbossilazione irreversibile del piruvato in acetil-CoA, CO2 e NADH. Il controllo della PDC controlla l’ingresso dei carboni derivati dai carboidrati nei mitocondri. Questa reazione svolge un ruolo centrale nella regolazione delle vie di produzione energetica mitocondriale (ciclo degli acidi tricarbossilici (TCA) e fosforilazione ossidativa (OXPHOS)) e nella generazione di intermedi biosintetici, come il citrato. La PDC è costituita da tre componenti catalitici, la piruvato deidrogenasi (E1), la diidrolipoamide transacetilasi (E2) e la diidrolipoamide deidrogenasi (E3), organizzati in grandi complessi multimerici insieme alla subunità strutturale E3 binding protein (E3BP). Il nucleo di base della piruvato deidrogenasi E1 è un eterotetramero di due subunità alfa e due beta (α2β2) e catalizza la prima fase della decarbossilazione del piruvato. L’attività del complesso è ampiamente regolata dalla fosforilazione reversibile di tre residui di serina di E1α. Le chinasi della piruvato deidrogenasi (PDK1-4) inattivano la PDC e le fosfatasi della piruvato deidrogenasi (PDP1-2) la attivano/riattivano (Fig. 3).
Le diverse isoforme PDK variano nelle loro proprietà regolatorie, nella distribuzione tissutale e nella modulazione da parte dei segnali metabolici a monte. Ne consegue un controllo dinamico e organo-specifico della funzione mitocondriale e della produzione di energia. I saggi enzimatici hanno dimostrato che le PDK differiscono nella loro specificità verso i tre siti bersaglio E1α e nei parametri cinetici della fosforilazione. La PDK1 è l’unica isoforma in grado di fosforilare tutti e tre i siti, mentre le PDK2-4 fosforilano i siti 1 e 2 con velocità diverse in vitro. 19,20 La fosforilazione di anche uno solo dei sei siti E1a nell’eterotetramero è sufficiente a inattivare la PDC e si ritiene che al massimo tre dei possibili sei siti del tetramero E1 possano essere fosforilati in qualsiasi momento.21,22 Anche i cambiamenti spazio-temporali nei livelli e nell’attività delle fosfatasi PDC (PDP1-2) sono regolatori. La fosforilazione in vitro sembra essere casuale; entrambi gli isozimi sono in grado di fosforilare i tre siti E1 di substrati mutati ricombinanti.23
Dicloroacetato: Un inibitore PDK
Il DCA è stato identificato come attivatore della piruvato deidrogenasi grazie alla sua capacità di stimolare l’attività enzimatica della PDC in un modello di cuore di ratto perfuso.24 È ora noto che questo piruvato mimetico agisce inibendo l’azione delle PDK. È stata ottenuta la struttura cristallina della PDK2 in complesso con il DCA, che mostra che il DCA occupa il sito di legame del piruvato nel dominio regolatore (R) N-terminale.25 I quattro isoenzimi variano notevolmente nella loro sensibilità all’inibizione da parte del DCA. La PDK2 è la più sensibile, la PDK3 la più resistente, mentre la PDK1 e la PDK4 sono relativamente sensibili.26,27
Trattamento dell’acidosi lattica con DCA
La sicurezza e l’efficacia del DCA sono state valutate in casi di acidosi lattica congenita e acquisita. Il trattamento con DCA riduce efficacemente i livelli di lattato in circolo stimolando l’ossidazione del piruvato; tuttavia, non è ancora noto se il DCA possa migliorare la prognosi dei pazienti affetti da queste sindromi.28,29 È stato proposto che i bambini piccoli con deficit di PDH possano trarre il massimo beneficio dal trattamento cronico con DCA.30 L’effetto avverso più significativo della somministrazione di DCA a lungo termine è una neuropatia periferica reversibile.31,32 La gravità della tossicità sembra dipendere dall’età, con i pazienti adulti più suscettibili dei bambini.31,33 Le ragioni di questa discrepanza non sono del tutto chiare, ma sono probabilmente legate alla diversa farmacocinetica e al diverso metabolismo del DCA nei due gruppi di età.34 Il DCA è stato utilizzato anche in studi clinici per le malattie cardiache, tra cui l’insufficienza cardiaca congestizia e la cardiopatia ischemica, mostrando risultati positivi e migliorando le prestazioni del miocardio.35,36
DCA come potenziale terapia antitumorale
Negli ultimi anni, il DCA ha attirato l’attenzione come mezzo potenzialmente semplice ed economico per colpire i tumori glicolitici, producendo al contempo effetti collaterali limitati negli organi sani ossidativi. L’interesse per questo farmaco da parte della comunità scientifica, dei pazienti oncologici e dei media si è acceso nel 2007, dopo che un gruppo dell’Università di Alberta ha riferito che il DCA era unicamente tossico per le linee cellulari tumorali umane e inibiva la crescita di xenotrapianti di tumore polmonare A549 nei ratti.37 Da allora, i rapporti emergenti sull’efficacia del DCA in vitro e in vivo rivelano alcune caratteristiche interessanti e anche sconcertanti, che distinguono il caso del DCA dalla maggior parte dei farmaci sviluppati come agenti antitumorali (Tabella 1). Il numero di tipi di tumore e di strategie sperimentali testate finora è troppo limitato per poter trarre conclusioni generalizzate sull’efficacia dei DCA contro tutti i tipi di tumore. Con questa avvertenza, un confronto qualitativo della letteratura suggerisce che il DCA mostra un maggiore effetto antitumorale in vivo rispetto all’effetto antitumorale delle cellule in vitro.
| Tipo di tumore | Riferimenti | Effetto sulla sopravvivenza e sulla crescita |
| Studi in vitro | ||
| Polmone, Glioblastoma, Seno | 37 | Apoptosi in vitro, inibizione della crescita di xenotrapianti |
| Prostata | 38 | Inibizione della crescita in vitro, moderata radiosensibilizzazione |
| Endometrio | 39 | Inibizione della crescita |
| Cervicale | 40 | Vantaggio di crescita in condizioni di ipossia in vitro |
| Testa e collo | 41 | Inibizione della crescita in vitro solo in cellule con mutND2 sovraespresso |
| Pediatrico | 42 | Apoptosi ad alte concentrazioni in vitro, qualche influenza sulla risposta alla chemioterapia |
| Colorettale | 43 | Apoptosi a concentrazioni molto elevate |
| Modelli preclinici | ||
| Colorettale | 44 | Scarso effetto sulla crescita, aumento dell’ipossia da parte della HRE-luciferasi, sensibilizzazione alle citotossine ipossiche |
| Colorettale | 18 | Scarso effetto sulla crescita, aumento dell’ipossia mediante 18F-FAZA PET, sensibilizzazione alla citotossina ipossica |
| Modelli preclinici | 17 | Inibizione della crescita dello xenotrapianto |
| Colorettale | 45 | Inibizione della crescita dello xenotrapianto |
| Colorettale | 46 | Protegge dall’anossia in vitro, promuove la crescita di xenotrapianti di SW480 |
| Colorettale, Seno, PML, Prostata | 47 | Attivo solo contro le cellule con catena di trasporto degli elettroni difettosa |
| Seno | 48 | Inibizione della crescita di xenotrapianti e delle metastasi |
| Dati su pazienti umani | ||
| Glioblastoma | 49 | Malattia clinicamente stabile in vivo, diminuzione di HIF1, aumento di p53 ex vivo |
Studi in vitro. È stato riportato che il DCA ha effetti citotossici in vitro, 37-40 con alcune risposte a concentrazioni clinicamente rilevanti (0,5-1 mM), mentre altre richiedono livelli sovra-farmacologici (10-100 mM) e altri gruppi ancora non hanno riscontrato alcuna tossicità diretta in vitro. 18,41,42,47 Una condizione identificata che sensibilizza le cellule al DCA è rappresentata da mutazioni che perturbano la funzione respiratoria mitocondriale,41,47 suggerendo che l’utilizzo forzato dell’OXPHOS difettosa sia tossico. Dato che anche la privazione di ossigeno riduce la funzione mitocondriale, sembrava ragionevole ipotizzare che le cellule ipossiche sarebbero state più sensibili al DCA. Tuttavia, questa ipotesi non è stata supportata, almeno nel numero limitato di linee cellulari testate finora. Un’ipossia moderata in vitro non ha influenzato il profilo del ciclo cellulare delle cellule del colon-retto trattate con DCA43 o la vitalità riproduttiva delle cellule del cancro del pancreas trattate con DCA (nostre osservazioni non pubblicate). È interessante notare che un altro studio ha riscontrato che una grave ipossia (anossia?) può essere protettiva contro l’apoptosi indotta dal DCA nelle cellule di cancro del colon-retto.46 La ragione di queste discrepanze non è chiara.
Nel complesso, la maggior parte dei dati supporta l’idea che concentrazioni clinicamente rilevanti di DCA (inferiori a 1 mM) non siano direttamente citotossiche in vitro. La ragione di questa apparente resistenza cellulare non è l’inattivazione del DCA in condizioni di coltura tissutale o l’incapacità di inattivare le PDK, poiché è stato dimostrato che il DCA aumenta transitoriamente l’attività mitocondriale e fa collassare il potenziale di membrana mitocondriale.18,37,50 Pertanto, la base del limitato effetto antitumorale del DCA in coltura risiede probabilmente nella complessa fisiologia cellulare e nell’enorme eccesso di metaboliti presenti nei mezzi di coltura.
Modelli preclinici. I rapporti sull’attività del DCA contro i tumori modello coltivati nei roditori sono incoraggianti, sebbene vi siano alcuni casi di linee tumorali che non rispondono al trattamento e persino un esempio di crescita tumorale accelerata in risposta al DCA.46 Il primo rapporto sull’attività antitumorale del DCA è stato quello di Bonnet et al. 37 Gli autori hanno riferito che gli xenotrapianti di adenocarcinoma polmonare A549 coltivati in ratti nudi hanno mostrato un significativo ritardo nella crescita tumorale dopo il trattamento con il DCA, con alcuni gruppi sperimentali che hanno addirittura mostrato una regressione del tumore. Questi effetti erano associati a un aumento dell’apoptosi e a una riduzione della proliferazione. Utilizzando le stesse cellule A549, Stockwin et al. 47 hanno recentemente confermato che il DCA è in grado di sopprimere la crescita nei tumori modello, pur avendo riscontrato una scarsa tossicità in vitro.
Il nostro gruppo ha riportato che il trattamento quotidiano con DCA di topi con xenotrapianti pancreatici SU86.86 ha causato un significativo ritardo nella crescita del tumore e un aumento della frazione ipossica dei tumori. A sostegno di questo modello, il DCA ha aumentato l’entità dell’ipossia tumorale negli xenotrapianti del colon-retto RKO, come valutato dai reporter della luciferasi HRE (hypoxia response element)44 o dalla tomografia a emissione di positroni con 18F-fluoroazomicinaarabinoside18 Questo modello RKO ha mostrato una riduzione molto modesta della crescita dei tumori trattati con DCA, ma i cambiamenti acuti nell’ossigenazione dopo il DCA li hanno sensibilizzati al trattamento con citotossine attivate dall’ipossia, come la tirapazamina44 o il PR-104.18 Ulteriori lavori su modelli di cancro del colon-retto, svolti da altri gruppi, hanno rivelato una significativa eterogeneità nella risposta al DCA. Alcune linee cellulari hanno ridotto il tasso di crescita del tumore,45 mentre altre non hanno risposto o sono addirittura cresciute più rapidamente.46 Inoltre, in un modello di ratto di adenocarcinoma mammario, un programma intensivo di trattamento con DCA è stato in grado di ridurre il numero di metastasi polmonari macroscopiche.48 Questi modelli preclinici supportano il concetto che il DCA ha la capacità di modulare il metabolismo tumorale in vivo, determinando effetti antitumorali maggiori o minori a seconda del modello testato.
Dati di sperimentazione clinica. I primi dati sulla valutazione del DCA per il trattamento del cancro umano sono stati riportati recentemente.49 In questo studio, il DCA è stato utilizzato in combinazione con la chirurgia, la temozolomide e le radiazioni per il trattamento di cinque pazienti con glioblastoma multiforme. Sebbene gli autori riportino risultati clinici promettenti in quattro dei cinque pazienti, l’enfasi della relazione è stata posta sull’analisi ex vivo delle cellule tumorali prima e dopo il trattamento con DCA. Gli autori riportano cambiamenti nel potenziale di membrana mitocondriale, un aumento della quantità di radicali dell’ossigeno generati dai mitocondri e un aumento dell’apoptosi delle cellule tumorali. Gli studi meccanicistici hanno rilevato livelli alterati di segnalazione HIF1, attivazione di p53 e diminuzione dell’angiogenesi. Questi dati suggeriscono che il DCA ha molti meccanismi d’azione a valle della sua inibizione delle PDK. Ovviamente, per poter generalizzare questi risultati entusiasmanti, è necessario trattare un numero maggiore di pazienti, preferibilmente provenienti da siti diversi.
Regolazione organica del metabolismo
Anche i fattori umorali di crescita e nutrizionali potrebbero influenzare la risposta del tumore alla riprogrammazione metabolica. La regolazione dell’attività della PDC tumorale in vivo non è stata ancora studiata sistematicamente, ma le conoscenze acquisite sul metabolismo dei tessuti normali e sui campi di ricerca dell’endocrinologia e dell’obesità forniscono alcune ipotesi interessanti. L’inedia aumenta l’espressione delle PDK e diminuisce l’attività delle PDH negli organi periferici come strategia per mantenere una fornitura stabile di carboidrati al cervello e ad altri tessuti neuronali. Ad esempio, la fame attiva trascrizionalmente PDK4 e PDK2 nel fegato, nel rene e in altri tessuti.51,52 Anche i glucocorticoidi, l’ormone tiroideo T3 e gli acidi grassi liberi aumentano i livelli di PDK4.53 La rialimentazione e/o l’aumento dei livelli di insulina diminuiscono la trascrizione di PDK4 e riattivano la PDC. Allo stesso modo, è stato dimostrato che l’inedia e il diabete riducono i livelli di mRNA e di proteina di PDP2, effetti che vengono invertiti dalla rialimentazione o dal trattamento con insulina.54
Pertanto, i numerosi segnali in ingresso che influenzano l’attività della PDH in vivo potrebbero influenzare l’efficacia del DCA e di altri farmaci a bersaglio metabolico. La somministrazione del farmaco durante il digiuno potrebbe avere un effetto molto diverso rispetto allo stato di alimentazione. Le risposte alle condizioni umorali sono ovviamente diverse per i vari tessuti e tipi di tumore e quindi difficili da imitare in vitro. È quindi possibile valutare l’effetto antitumorale di molecole come il DCA in vitro? Molti anni di studi su un gran numero di linee cellulari tumorali coltivate in vitro dimostrano che esse mantengono le caratteristiche anomale della glicolisi aerobica in vitro e forniscono quindi un valido strumento per lo studio del metabolismo del cancro.55 Tuttavia, esistono anche limitazioni intrinseche in questi sistemi, come dimostrano i dati incoerenti presenti in letteratura quando si confrontano gli effetti del DCA in coltura cellulare con i risultati preclinici e clinici.
L’analisi precedente presenta il modello che un farmaco antitumorale mirato al metabolismo e poco tossico in vitro possa avere un potenziale significativo in vivo. Abbiamo evidenziato i substrati metabolici limitati nel tumore e la regolazione sistemica del metabolismo da parte di fattori umorali che potrebbero aumentare l’efficacia del farmaco in vivo. È possibile che sia vero anche il contrario, cioè che un farmaco con una buona attività in vitro possa avere scarso successo in vivo. Gli effetti sulla tossicità dei tessuti normali o sulla cooperazione metabolica tra tipi di cellule potrebbero limitare l’efficacia di un farmaco in vivo. L’inibitore metabolico 2-deossiglucosio, ampiamente testato, ha una ragionevole attività antitumorale in vitro, ma non può essere utilizzato nei pazienti a causa dei suoi effetti negativi sui tessuti normali che dipendono dal consumo di glucosio. La tossicità neurologica che limita la dose si verifica a un livello di farmaco ben al di sotto di quello necessario per gli effetti antitumorali nei tumori dei roditori.56,57 In alternativa, è possibile che la cooperazione metabolica tra tipi di cellule o tra cellule normali e tumorali possa aggirare il blocco metabolico indotto dal farmaco. Per esempio, mentre il lattato è spesso considerato un prodotto di scarto del metabolismo, in alcune cellule può essere utilizzato come combustibile per alimentare la funzione mitocondriale.58,59
Per quanto riguarda la combinazione del DCA con le terapie esistenti, i dati preclinici finora non mostrano un modello evidente di interazione che consenta una selezione semplice e razionale dei regimi terapeutici. I modelli animali continueranno a essere il miglior mezzo di sperimentazione per determinare empiricamente le combinazioni più promettenti. Abbiamo dimostrato che, grazie alla sua capacità di aumentare il consumo di ossigeno, il DCA aumenta l’ipossia tumorale e sensibilizza i tumori del pancreas e del colon xenotrapiantati alle citotossineipossiche18,44, per cui è interessante immaginare un piano di trattamento che includa queste due classi di farmaci, entrambe progettate per sfruttare l’esclusivo microambiente ipossico del tumore.
Non è stata riportata l’interazione del DCA con altri modulatori metabolici. Un potenziale bersaglio per la terapia combinata è la lattato deidrogenasi A (LDHA). È stato dimostrato che l’inibizione genetica o farmacologica della LDHA aumenta la funzione mitocondriale e inibisce la formazione e la progressione dei tumori modello.60,61 In questo schema di combinazione, l’inibitore della LDHA bloccherebbe la conversione del piruvato in lattato e il DCA dirotterebbe il piruvato accumulato verso l’ossidazione mitocondriale. Se l’effetto antitumorale del DCA deriva dall’aumento della funzione mitocondriale, è possibile che la combinazione degli inibitori di PDK e LDHA forzi un tasso di ossidazione mitocondriale ancora maggiore e comprometta la crescita tumorale in modo più efficiente.
Anche i punti nodali di vie chiave per la sopravvivenza, come la via PI3KAkt-mTOR, sono oggetto di intensi sforzi per lo sviluppo di farmaci.62 Parte delle proprietà di promozione della crescita di questa via deriva dalla sua capacità di regolare il metabolismo e la produzione di energia attraverso meccanismi diretti o indiretti. Ad esempio, l’attivazione oncogenica di PI3K-Akt stimola l’assorbimento del glucosio e la glicolisi aerobica,63,64 mentre l’attivazione di Akt e mTORC1 aumenta la traduzione dell’mRNA di Hif-1a in condizioni di ipossia.65 Promettenti inibitori di Akt e inibitori di mTOR di nuova generazione sono in fase di sperimentazione clinica66,67 e rappresentano validi candidati per la terapia di combinazione con DCA per modulare il metabolismo aerobico e ipossico.
Conclusioni
In conclusione, le recenti conoscenze sul peculiare metabolismo dei tumori solidi hanno identificato diverse nuove vie farmacologiche che possono essere utilizzate preferenzialmente nelle cellule tumorali rispetto alle cellule normali. L’analisi dei candidati farmaci antitumorali progettati per colpire queste vie metaboliche richiederà un’attenta progettazione sperimentale, sia in vitro che in vivo. L’analisi dei rapporti pubblicati sullo studio del DCA mostra una gamma confusa, e talvolta contraddittoria, di effetti in vitro e in vivo. Studi genetici in tumori modello offrono prove convincenti che questa via è un buon candidato per il targeting terapeutico.68 Sarebbe molto utile nell’analisi della potenziale utilità del DCA se ci fosse una firma molecolare in grado di predire la senescenza al farmaco, sia nei tumori modello che eventualmente nei pazienti. Forse un’attenta analisi del presunto bersaglio del DCA, la fosforilazione della subunità E1α della piruvato deidrogenasi, potrebbe offrire questa firma.
RIFERIMENTI
1 1. Warburg O, Wind F, Negelein E. Il metabolismo dei tumori nell’organismo. J Gen Physiol 1927;8:519-30.
2 Pan JG, Mak TW. Il targeting metabolico come strategia antitumorale: l’alba di una nuova era? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Mattone dopo mattone: metabolismo e crescita delle cellule tumorali. Curr Opin Genet Dev 2008;18:54-61.
4 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004;14:198-206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas: a novel guideline for clinical oncologists? J Cancer Res Clin Oncol 2003;129:321-6.
6 Gstraunthaler G, Seppi T, Pfaller W. Impatto delle condizioni di coltura, dei volumi dei terreni di coltura e del contenuto di glucosio sulle proprietà metaboliche delle colture di cellule epiteliali renali. Le cellule renali in coltura tissutale sono ipossiche? Cell Physiol Biochem 1999;9:150-72.
7 Bloch-Frankenthal L, Ram D. La relazione tra l’effetto Crabtree e il metabolismo ossidativo del glucosio e degli intermedi carboidrati nelle cellule tumorali di ascite. Cancer Res 1959;19:835-42.
8ParrinelloS, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. La sensibilità all’ossigeno limita fortemente la durata della vita replicativa dei fibroblasti murini. Nat Cell Biol 2003;5:741-7.
9 Denko NC. Ipossia, HIF1 e metabolismo del glucosio nel tumore solido. Nat Rev Cancer 2008;8:705-13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. Analisi genomica dell’acidosi lattica e della risposta all’acidosi nei tumori umani. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Down-regulation indotta dall’ipossia dell’espressione di BRCA1 da parte di E2F. Cancer Res 2005;65: 11597-604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Adattamenti cellulari all’ipossia e all’acidosi durante l’evoluzione somatica del cancro al seno. Br J Cancer 2007;97:646-53.
13 Dewhirst MW, Cao Y, Moeller B. L’ipossia ciclica e i radicali liberi regolano l’angiogenesi e la risposta alla radioterapia. Nat Rev Cancer 2008;8:425-37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. L’HIF-1 media l’adattamento all’ipossia attraverso un’attiva downregolazione del consumo di ossigeno mitocondriale. Cell Metab 2006;3:187-97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. L’induzione della piruvato deidrogenasi chinasi-3 da parte dell’hypoxia-inducible factor-1 promuove lo switch metabolico e la resistenza ai farmaci. J Biol Chem 2008;283:28106-14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Espressione mediata da HIF-1 della piruvato deidrogenasi chinasi: un interruttore metabolico necessario per l’adattamento cellulare all’ipossia. Cell Metab 2006;3:177-85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Il consumo di ossigeno può regolare la crescita dei tumori, una nuova prospettiva sull’effetto Warburg. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. L’ipossia tumorale aumentata farmacologicamente può essere misurata mediante tomografia a emissione di positroni con 18F-fluoroazomicina arabinoside e aumenta la risposta tumorale alla citotossina ipossica PR-104. Clin Cancer Res 2009;15: 7170-4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regolazione dell’attività della piruvato deidrogenasi attraverso la fosforilazione in più siti. Biochem J 2001;358:69-77.
20 Korotchkina LG, Patel MS. Specificità del sito di quattro isoenzimi della piruvato deidrogenasi chinasi verso i tre siti di fosforilazione della piruvato deidrogenasi umana. J Biol Chem 2001;276: 37223-9.
21 Sugden PH, Randle PJ. Regolazione della piruvato deidrogenasi del cuore di maiale mediante fosforilazione. Studi sulla subunità e sulle stoicheiometrie di fosforilazione. Biochem J 1978;173:659-68.
22 Korotchkina LG, Patel MS. Studi di mutagenesi dei siti di fosforilazione della piruvato deidrogenasi umana ricombinante. Regolazione sito-specifica. J Biol Chem 1995;270:14297-304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Caratterizzazione degli isoenzimi della piruvato deidrogenasi fosfatasi: implicazioni per la regolazione dell’attività della piruvato deidrogenasi. Biochim Biophys Acta 2003;1652:126-35.
24 Whitehouse S, Randle PJ. Attivazione della piruvato deidrogenasi nel cuore di ratto perfuso da parte del dicloroacetato (breve comunicazione). Biochem J 1973;134: 651-3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Ruoli regolatori del dominio N-terminale basati su strutture cristalline della piruvato deidrogenasi chinasi 2 umana contenenti ligandi fisiologici e sintetici. Biochimica 2006;45:402-15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Prove dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J 1998;329(Pt 1):191-6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marcate differenze tra due isoforme della piruvato deidrogenasi chinasi umana. J Biol Chem 2000; 275:15773-81.
28 Stacpoole PW, Greene YJ. Dicloroacetato. Diabetes Care 1992;15:785-91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Trattamento dell’acidosi lattica con dicloroacetato. N Engl J Med 1983;309:390-6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Ruolo del dicloroacetato nel trattamento delle malattie genetiche mitocondriali. Adv Drug Deliv Rev 2008;60:1478-87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Il dicloroacetato causa neuropatia tossica nel MELAS: uno studio clinico randomizzato e controllato. Neurology 2006;66: 324-30.
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Farmacocinetica, metabolismo e tossicologia del dicloroacetato. Drug Metab Rev 1998;30: 499-539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Valutazione del trattamento a lungo termine di bambini con acidosi lattica congenita con dicloroacetato. Pediatrics 2008;121:e1223-8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Cinetica e metabolismo del dicloroacetato in funzione dell’età: possibile rilevanza per la tossicità. J Pharmacol Exp Ther 2008;324:1163-71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Effetti metabolici ed emodinamici del dicloroacetato nel miocardio nella malattia coronarica. Am J Cardiol 1988;61:65-70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Miglioramento della funzione emodinamica e dell’efficienza meccanica nell’insufficienza cardiaca congestizia con il dicloroacetato di sodio. J Am Coll Cardiol 1994;23:1617-24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2 in vitro. Prostate 2008;68:1223-31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol 2008;109: 394-402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. Effetti in vitro del dicloroacetato e della CO2 sulle cellule HeLa ipossiche. Anticancer Res 2009;29:4579-88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Le mutazioni mitocondriali contribuiscono all’accumulo di HIF1alfa attraverso l’aumento delle specie reattive dell’ossigeno e l’up-regolazione della piruvato deidrogenasi chinasi 2 nel carcinoma a cellule squamose della testa e del collo. Clin Cancer Res 2009;15:476-84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. La terapia metabolicamente mirata con dicloroacetato sconfigge la citotossicità dei farmaci antitumorali standard. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Il dicloroacetato induce apoptosi e arresto del ciclo cellulare nelle cellule di cancro del colon-retto. Br J Cancer 2010;102:1746-52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Il targeting metabolico dell’ipossia e dell’HIF1 nei tumori solidi può migliorare la chemioterapia citotossica. Proc Natl Acad Sci USA 2007;104:9445-50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. La selezione di cellule tumorali con mitocondri repressi innesca la progressione del cancro del colon. Carcinogenesi 2010;31:567-76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Il dicloroacetato di sodio (DCA) riduce l’apoptosi nell’ipossia del tumore colorettale. Cancer Lett 2010;287:75-83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Il dicloroacetato di sodio (DCA) colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer 2010;127:2510-19.
48SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. L’inversione del fenotipo glicolitico mediante dicloroacetato inibisce la crescita delle cellule metastatiche di cancro al seno in vitro e in vivo. Breast Cancer Res Treat 2010;120:253-60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci Transl Med 2010;2: 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoraggio della fosforilazione del complesso piruvato deidrogenasi. Anal Biochem 2009;389:157-64.
51 Holness MJ, Sugden MC. Attività della piruvato deidrogenasi durante la transizione tra alimentazione e fame e durante la rialimentazione dopo una fame acuta o prolungata. Biochem J 1989;258:529-33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regolazione dell’espressione della piruvato deidrogenasi chinasi da parte di ligandi del recettore perossisome proliferator-activated receptoralpha, glucocorticoidi e insulina. Diabetes 2002;51:276-83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regolazione della piruvato deidrogenasi chinasi 4 (PDK4) da parte dell’ormone tiroideo: ruolo del coattivatore del recettore gamma del perossisoma proliferatore attivato (PGC-1 alfa). J Biol Chem 2010;285:2375-85.
54 Huang B, Wu P, Popov KM, Harris RA. La fame e il diabete riducono la quantità di piruvato deidrogenasi fosfatasi nel cuore e nel rene di ratto. Diabetes 2003;52: 1371-6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. L’analisi metabolica multiparametrica rivela uno stretto legame tra l’attenuazione della funzione bioenergetica mitocondriale e la maggiore dipendenza dalla glicolisi nelle cellule tumorali umane. Am J Physiol Cell Physiol 2007;292:C125-36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507-14.
57 Maher JC, Krishan A, Lampidis TJ. Maggiore inibizione del ciclo cellulare e citotossicità indotta dal 2-deossi-D-glucosio nelle cellule tumorali trattate in condizioni ipossiche rispetto a quelle aerobiche. Cancer Chemother Pharmacol 2004;53:116-22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. La respirazione mirata alimentata dal lattato uccide selettivamente le cellule tumorali ipossiche nei topi. J Clin Invest 2008;118:3930-42.
59KoukourakisMI, Giatromanolaki A, Harris AL, Sivridis E. Confronto delle vie metaboliche tra cellule tumorali e cellule stromali nei carcinomi colorettali: un ruolo di sopravvivenza metabolica per lo stroma associato al tumore. Cancer Res 2006;66:632-7.
60 Fantin VR, St-Pierre J, Leder P. L’attenuazione dell’espressione di LDHA scopre un legame tra glicolisi, fisiologia mitocondriale e mantenimento del tumore. Cancer Cell 2006;9:425-34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. L’inibizione della lattato deidrogenasi A induce stress ossidativo e inibisce la progressione tumorale. Proc Natl Acad Sci USA 2010;107:2037-42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235-56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimola la glicolisi aerobica nelle cellule tumorali. Cancer Res 2004; 64:3892-9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. La dipendenza dal glucosio delle cellule trasformate in Akt può essere invertita dall’attivazione farmacologica della beta-ossidazione degli acidi grassi. Oncogene 2005; 24:4165-73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. La segnalazione di HER2 (neu) aumenta il tasso di sintesi dell’hypoxia-inducible factor 1alpha (HIF-1alpha): un nuovo meccanismo per l’espressione del fattore di crescita endoteliale vascolare mediata da HIF-1. Mol Cell Biol 2001;21:3995-4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Studio di fase II dell’inibitore orale del target mammifero della rapamicina everolimus nella macroglobulinemia di Waldenstrom recidivata o refrattaria. J Clin Oncol 2010;28:1408-14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Prendere di mira la rete di segnalazione mTOR per la terapia del cancro. J Clin Oncol 2009;27: 2278-87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. L’attività del complesso della piruvato deidrogenasi controlla il fenotipo metabolico e maligno delle cellule tumorali. J Biol Chem 2008;283:22700-8.
Contenuti correlati: