Ioanna Papandreou, Tereza Goliasova, et Nicholas C. Denko
Département de radio-oncologie, Division de la biologie des rayonnements et du cancer, École de médecine de l’Université de Stanford, Stanford, CA
Mots-clés : métabolisme tumoral, pyruvate déshydrogénase, effet Warburg, inhibiteurs métaboliques
Abréviations : DCA : dichloroacétate ; HIF1 : hypoxia-inducible factor 1 ; LDH : lactate déshydrogénase ; PDC : complexe pyruvate déshydrogénase ; PDH : pyruvate déshydrogénase ; PDK : pyruvate déshydrogénase kinase ; PDP : pyruvate déshydrogénase phosphatase
Correspondance avec : Nicholas C. Denko, Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University School of Medicine, Stanford, CA 94305, USA
Tél. : 650-724-5066, Fax : 650-723-7382,
E-mail : [email protected]
Reçu : 22 Jul 2010
Accepté : Accepté le 30 septembre 2010
En ligne : 18 octobre 2010
DOI : 10.1002/ijc.25728
Au cours des 20 dernières années, le nombre d’articles contenant le terme » métabolisme tumoral » est passé de 3 à 28 par an, et le nombre de fois où ces articles ont été cités est passé de 23 à 929 par an (statistiques ISI, Thompson Reuters). Le regain d’intérêt pour la compréhension des mécanismes et des conséquences de l’altération du métabolisme tumoral a clairement captivé l’imagination de la communauté scientifique. L’idée que les tumeurs ont un métabolisme modifié a été reconnue pour la première fois par le biochimiste Otto Warburg, lauréat du prix Nobel, lorsqu’il a décrit le métabolisme du glucose.1 Plus récemment, le concept selon lequel les tumeurs ont un métabolisme différent s’est étendu à d’autres caractéristiques, comme la glutaminolyse, l’oxydation des acides gras et la biogénèse des lipides. Il est clair qu’une demande métabolique différente entraîne ces changements dans les cellules qui se divisent continuellement, par rapport aux cellules différenciées en phase terminale. La découverte de ces altérations a soulevé la possibilité de les cibler thérapeutiquement en raison de leur importance unique pour les cellules cancéreuses.2
Le concept selon lequel les changements métaboliques sont une réponse à des demandes uniques au sein de la tumeur a été proposé,3 même s’il est difficile de quantifier ces demandes. Il existe une interaction entre les changements oncogènes dans la cellule tumorale et les aspects uniques du microenvironnement tumoral qui ont un impact sur le métabolisme cellulaire et vice versa (Fig. 1). Il est donc difficile d’établir les exigences métaboliques exactes au sein de la tumeur en étudiant les cellules de la tumeur cultivée ex vivo. Les conditions environnementales utilisées pour cultiver les cellules en culture sont très différentes des conditions environnementales in vivo. Le milieu Dulbecco’s modified Eagles à haute teneur en glucose et une atmosphère de 21% d’oxygène sont très différents des conditions hypoxiques et/ou hypoglycémiques trouvées dans la tumeur.4,5 La concentration de glucose de 25 mM est environ cinq fois celle du sang normal, et la tension d’oxygène est au moins quatre fois supérieure à celle trouvée in vivo. Le fait que les cellules baignent dans ces substrats métaboliques modifie de manière significative leurs programmes métaboliques inhérents.4,6 Des concentrations élevées de glucose favorisent la glycolyse (effet Crabtree7 ) tandis qu’une oxygénation élevée produit une augmentation des sous-produits de l’oxygène et réduit la durée de vie cellulaire.8 Le métabolisme du glucose illustre l’interaction de ces trois facteurs dans la tumeur. La transformation oncogène entraîne la prolifération des cellules tumorales plus que la capacité vasculaire, ce qui génère une hypoxie. L’hypoxie dans le microenvironnement tumoral renforce le métabolisme glycolytique, en grande partie par l’activation du facteur de transcription HIF1 (hypoxiainducible factor 1).9 L’augmentation de la glycolyse entraîne une production accrue de lactate, qui contribue à un pH extracellulaire acide et à d’autres modifications de l’expression génétique.10 L’hypoxie et l’acidose peuvent toutes deux contribuer à l’augmentation des niveaux de mutation somatique qui peuvent favoriser la progression de la tumeur.11,12 Il est clairement difficile de reproduire ces interactions complexes dans des cellules cultivées in vitro.
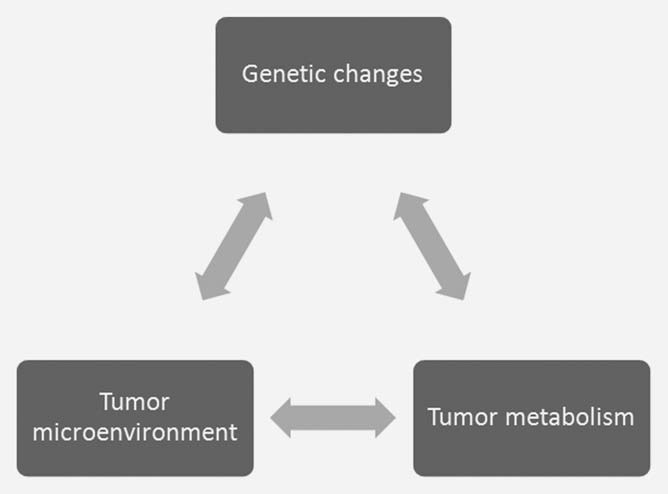
Une partie de l’interaction entre le microenvironnement et le métabolisme des cellules tumorales est générée par une réponse adaptative aux changements dynamiques de l’offre et de la demande de métabolites par les cellules. Le simple fait que nous puissions mesurer des régions d’hypoxie et d’acidose dans les tumeurs indique que les vaisseaux tumoraux ne maintiennent pas un environnement constant pour la croissance des cellules tumorales.13 Le système vasculaire tumoral représente un goulot d’étranglement dans l’apport de nutriments et l’élimination des déchets de la tumeur.4 L’apport inadéquat des vaisseaux tumoraux initie une réponse adaptative des cellules tumorales conçue pour diminuer la demande en métabolites limités. Ce processus dynamique est difficile à modéliser in vitro (Fig. 2). Par exemple, les faibles niveaux d’oxygène dans la tumeur induisent le facteur de transcription HIF1 et son programme métabolique.9 Une partie du programme métabolique initié par HIF1 consiste à réduire la demande en oxygène en diminuant la fonction mitochondriale. Une partie de cette réponse est médiée par l’induction dépendante de HIF1 de PDK1 et PDK3 dans les cellules tumorales et une réduction de l’oxydation du pyruvate dans les mitochondries.14-17 Cette réponse adaptative est responsable du rapprochement de la demande en oxygène de l’offre limitée.
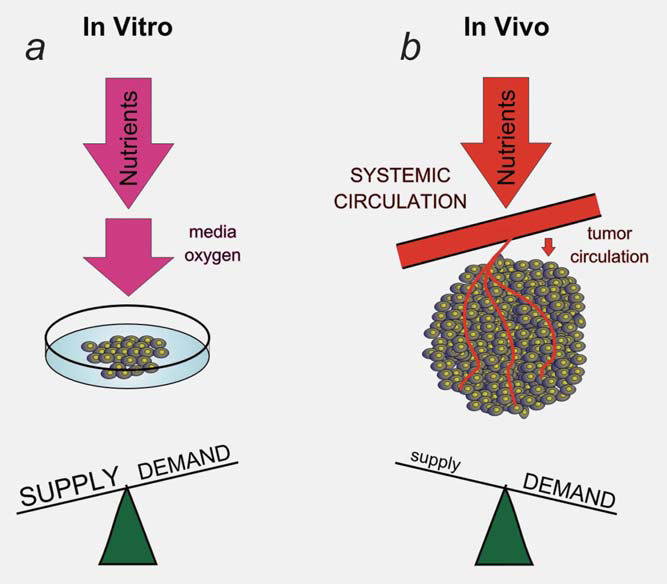
Le dichloroacétate (DCA) est capable d’interférer avec cette adaptation à l’hypoxie tumorale en inhibant la fonction des PDK (Fig. 3). Le blocage d’une réponse adaptative à l’hypoxie est le plus clairement observé lorsque la tumeur dans son ensemble est en déficit d’oxygène et que le système vasculaire de la tumeur ne peut pas répondre à cette demande accrue.18 Même lorsque les cellules tumorales sont placées en hypoxie, il y a suffisamment d’oxygène dans l’environnement (1-2%) pour maintenir une condition stable, bien que faible en oxygène, à l’intérieur de la cellule, même avec l’ajout de DCA. Le taux de diffusion dans les cellules est plus rapide que le taux de consommation, de sorte que le niveau intracellulaire d’oxygène ne dépend pas du taux de consommation (sauf si un nombre très élevé de cellules est utilisé dans des plats en verre qui empêchent la diffusion de l’oxygène à travers le plastique14). Dans cet article, nous présentons une analyse des données publiées concernant le DCA en tant qu’exemple de médicament conçu pour avoir un impact sur le métabolisme des tumeurs, qui soutient notre hypothèse selon laquelle cette classe de médicaments a un effet très différent sur les cellules tumorales se développant in vivo avec le goulot d’étranglement métabolique produit par le système vasculaire, par rapport à leur effet sur les cellules cultivées in vitro.
Le complexe pyruvate déshydrogénase
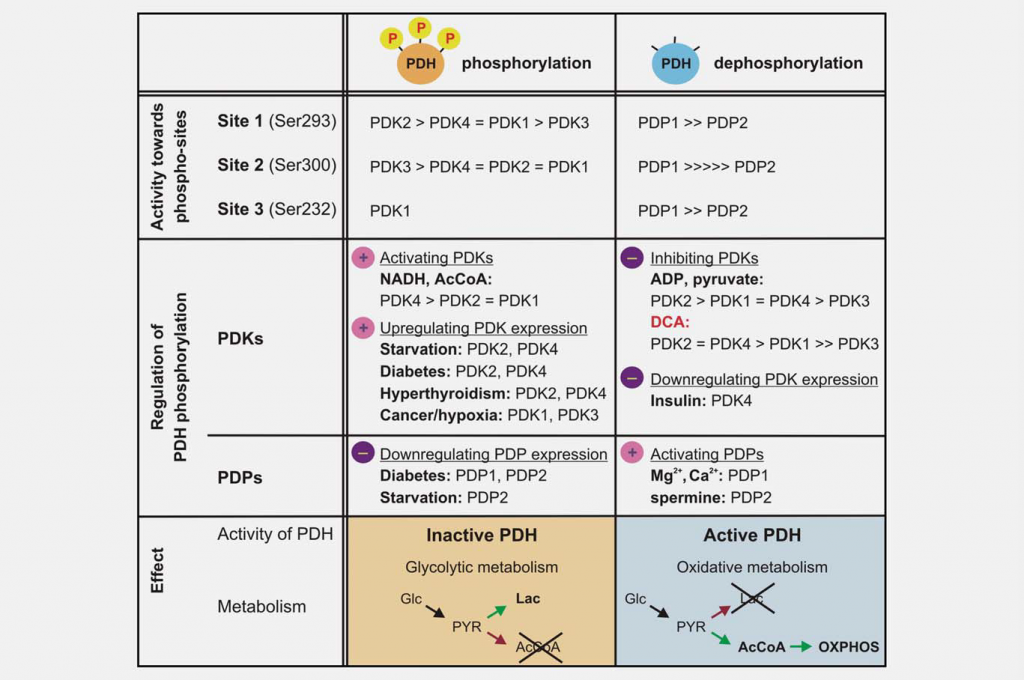
L’inhibition de la fonction mitochondriale est aussi importante que l’augmentation de la glycolyse pour produire l’effet Warburg. L’un des principaux régulateurs de la fonction mitochondriale est le complexe pyruvate déshydrogénase (PDC), qui catalyse la décarboxylation irréversible du pyruvate en acétyl-CoA, CO2 et NADH. Le contrôle de la PDC permet de contrôler l’entrée des carbones dérivés des hydrates de carbone dans la mitochondrie. Cette réaction joue un rôle central dans la régulation des deux voies mitochondriales de production d’énergie (cycle de l’acide tricarboxylique (TCA) et phosphorylation oxydative (OXPHOS)) et de la génération d’intermédiaires de biosynthèse, tels que le citrate. La PDC est constituée de trois composants catalytiques, la pyruvate déshydrogénase (E1), la dihydrolipoamide transacétylase (E2) et la dihydrolipoamide déshydrogénase (E3), qui sont organisés en grands complexes multimériques avec la sous-unité structurelle E3 binding protein (E3BP). Le noyau de base du composant pyruvate déshydrogénase E1 est un hétérotétramère de deux sous-unités alpha et deux sous-unités bêta (α2β2), et il catalyse la première étape de la décarboxylation du pyruvate. L’activité du complexe est largement régulée par la phosphorylation réversible de trois résidus sérine de E1α. Les pyruvate déshydrogénase kinases (PDK1-4) inactivent la PDC et les pyruvate déshydrogénase phosphatases (PDP1-2) l’activent/réactivent (Fig. 3).
Les différentes isoformes de PDK varient dans leurs propriétés régulatrices, leur distribution tissulaire et leur modulation par des signaux métaboliques en amont. Il en résulte un contrôle dynamique et spécifique à chaque organe de la fonction mitochondriale et de la production d’énergie. Des essais enzymatiques ont montré que les PDK diffèrent dans leur spécificité envers les trois sites cibles E1α et les paramètres cinétiques de la phosphorylation. La PDK1 est la seule isoforme capable de phosphoryler les trois sites, tandis que les PDK2-4 phosphorylent les sites 1 et 2 à des vitesses différentes in vitro. 19,20 La phosphorylation d’un seul des six sites E1a dans l’hétérotétramère est suffisante pour inactiver la PDC, et l’on pense qu’au maximum trois des six sites possibles du tétramère E1 peuvent être phosphorylés à un moment donné.21,22 Les changements spatio-temporels dans les niveaux et l’activité des phosphatases PDC (PDP1-2) sont également régulateurs. La déphosphorylation in vitro semble être aléatoire ; les deux isozymes sont capables de déphosphoryler les trois sites E1 de substrats mutésrecombinants23
Dichloroacétate : Un inhibiteur de la PDK
Le DCA a été identifié comme un activateur de la pyruvate déshydrogénase par sa capacité à stimuler l’activité enzymatique de la PDC dans un modèle de cœur de rat perfusé.24 On sait maintenant que ce pyruvate mimétique agit en inhibant l’action des PDK. On sait maintenant que ce pyruvate mimétique agit en inhibant l’action des PDK. La structure cristalline de PDK2 en complexe avec le DCA a été obtenue, et elle montre que le DCA occupe le site de liaison du pyruvate dans le domaine régulateur (R) N-terminal.25 Les quatre isozymes varient considérablement dans leur sensibilité à l’inhibition par le DCA. La PDK2 est la plus sensible, la PDK3 la plus résistante, tandis que la PDK1 et la PDK4 sont relativement sensibles.26,27
Traitement de l’acidose lactique par le DCA
La sécurité et l’efficacité du DCA ont été évaluées dans des cas d’acidose lactique congénitale et acquise. Le traitement par DCA réduit efficacement les niveaux de lactate dans la circulation en stimulant l’oxydation du pyruvate ; cependant, on ne sait toujours pas si le DCA peut améliorer le pronostic des patients atteints de ces syndromes.28,29 Il a été proposé que les jeunes enfants présentant un déficit en PDH soient ceux qui bénéficient le plus d’un traitement chronique par DCA.30 L’effet indésirable le plus important de l’administration de DCA à long terme est une neuropathie périphérique réversible.31,32 La gravité de la toxicité semble dépendre de l’âge, les patients adultes étant plus sensibles que les enfants.31,33 Les raisons de cette différence ne sont pas entièrement claires, mais elles sont probablement liées à la différence de pharmacocinétique et de métabolisme du DCA dans les deux groupes d’âge.34 Le DCA a également été utilisé dans des essais cliniques pour les maladies cardiaques, y compris l’insuffisance cardiaque congestive et la cardiopathie ischémique, montrant des résultats positifs et améliorant la performance du myocarde.35,36
Le DCA comme thérapie potentielle du cancer
Ces dernières années, le DCA a attiré l’attention comme moyen potentiellement simple et économique de cibler les tumeurs glycolytiques tout en produisant des effets secondaires limités dans les organes sains oxydatifs. L’intérêt de la communauté scientifique, des patients atteints de cancer et des médias pour ce médicament a été éveillé en 2007, après qu’un groupe de l’université d’Alberta a signalé que le DCA était uniquement toxique pour les lignées cellulaires cancéreuses humaines et qu’il inhibait la croissance des xénogreffes de tumeurs pulmonaires A549 chez le rat.37 Depuis lors, les nouveaux rapports sur l’efficacité du DCA in vitro et in vivo révèlent certaines caractéristiques intéressantes, mais aussi déroutantes, qui distinguent le cas du DCA de la majorité des médicaments développés comme agents anticancéreux (tableau 1). Le nombre de types de cancers différents et de stratégies expérimentales testées à ce jour est trop limité pour permettre de tirer des conclusions généralisées sur l’efficacité du DCA contre tous les types de tumeurs. Avec cette mise en garde, une comparaison qualitative de la littérature suggère que le DCA montre plus d’effet anticancéreux in vivo que d’effet sur les cellules cancéreuses in vitro.
| Type de cancer | Références | Effet sur la survie et la croissance |
| Études in vitro | ||
| Poumon, Glioblastome, Sein | 37 | Apoptose in vitro, inhibition de la croissance des xénogreffes |
| Prostate | 38 | Inhibition de la croissance in vitro, radiosensibilisation modérée |
| Endomètre | 39 | Inhibition de la croissance |
| Col de l’utérus | 40 | Avantage de croissance sous hypoxie in vitro |
| Tête et cou | 41 | Inhibition de la croissance in vitro uniquement dans les cellules surexprimant mutND2 |
| Pédiatrie | 42 | Apoptose à des concentrations élevées in vitro, une certaine influence sur la réponse à la chimiothérapie |
| Colorectal | 43 | Apoptose à des concentrations très élevées |
| Modèles précliniques | ||
| Colorectal | 44 | Peu d’effet sur la croissance, augmentation de l’hypoxie par HRE-luciférase, sensibilisation aux cytotoxines hypoxiques |
| Colorectal | 18 | Peu d’effet sur la croissance, hypoxie accrue par la TEP au 18F-FAZA, sensibilisation aux cytotoxines hypoxiques |
| Modèles précliniques | 17 | Inhibition de la croissance des xénogreffes |
| Colorectal | 45 | Inhibition de la croissance des xénogreffes |
| Colorectal | 46 | Protège de l’anoxie in vitro, favorise la croissance des xénogreffes de SW480 |
| Colorectal, Sein, PML, Prostate | 47 | Actif uniquement contre les cellules dont la chaîne de transport d’électrons est défectueuse |
| Sein | 48 | Inhibition de la croissance des xénogreffes et des métastases |
| Données sur les patients humains | ||
| Glioblastome | 49 | Maladie cliniquement stable in vivo, diminution de HIF1, augmentation de p53 ex vivo |
Études in vitro. On a signalé que le DCA avait des effets cytotoxiques in vitro, 37-40 avec certaines réponses à des concentrations cliniquement pertinentes (0,5-1 mM), tandis que d’autres nécessitent des niveaux suprapharmacologiques (10-100 mM) et que d’autres groupes encore n’ont trouvé aucune toxicité directe in vitro. 18,41,42,47 On a identifié une condition qui sensibilise les cellules au DCA : les mutations qui perturbent la fonction respiratoire mitochondriale,41,47 ce qui laisse penser que l’utilisation forcée d’une OXPHOS défectueuse est toxique. Comme la privation d’oxygène régule également à la baisse la fonction mitochondriale, il semblait raisonnable d’émettre l’hypothèse que les cellules hypoxiques seraient plus sensibles au DCA. Cependant, cette hypothèse n’a pas été confirmée, du moins dans le nombre limité de lignées cellulaires testées à ce jour. Une hypoxie modérée in vitro n’a pas influencé le profil du cycle cellulaire des cellules colorectales traitées au DCA43 ni la viabilité de reproduction des cellules cancéreuses pancréatiques traitées au DCA (nos observations non publiées). Il est intéressant de noter qu’une autre étude a montré que l’hypoxie sévère (anoxie ?) peut être protectrice contre l’apoptose induite par le DCA dans les cellules cancéreuses colorectales.46 La raison de ces divergences n’est pas claire.
Dans l’ensemble, la majorité des données appuient l’idée que les concentrations cliniquement pertinentes de DCA (moins de 1 mM) ne sont pas directement cytotoxiques in vitro. La raison de cette résistance cellulaire apparente n’est pas une inactivation du DCA dans des conditions de culture de tissus ou une incapacité à inactiver les PDK, car il a été démontré que le DCA augmente transitoirement l’activité mitochondriale et fait chuter le potentiel de la membrane mitochondriale.18,37,50 Par conséquent, la base de l’effet anticancéreux limité du DCA en culture réside probablement dans la physiologie cellulaire complexe et l’énorme excès de métabolites présents dans les milieux de culture.
Modèles précliniques. Les rapports sur l’activité du DCA contre des tumeurs modèles cultivées chez les rongeurs sont encourageants, bien qu’il y ait certains cas de lignées tumorales qui ne répondent pas au traitement, et même un exemple de croissance tumorale accélérée en réponse au DCA.46 Le premier rapport sur l’activité antitumorale du DCA est celui de Bonnet et al. 37 Les auteurs ont rapporté que des xénogreffes d’adénocarcinome pulmonaire A549 cultivées chez des rats nus présentaient un retard significatif de la croissance tumorale après traitement par le DCA, certains groupes expérimentaux présentant même une régression tumorale. Ces effets étaient associés à une augmentation de l’apoptose et à une réduction de la prolifération. En utilisant les mêmes cellules A549, Stockwin et al. 47 ont récemment confirmé que le DCA avait un effet suppresseur de croissance sur des tumeurs modèles, même s’ils ont constaté une faible toxicité in vitro.
Notre groupe a signalé que le traitement quotidien au DCA de souris porteuses de xénogreffes pancréatiques SU86.86 entraînait un retard significatif de la croissance tumorale, ainsi qu’une augmentation de la fraction hypoxique des tumeurs. Nous avons émis l’hypothèse que l’augmentation de la consommation d’oxygène par les mitochondries entraînait une plus grande hypoxie qui inhibait la croissance de la tumeur.17 À l’appui de ce modèle, le DCA a augmenté l’étendue de l’hypoxie tumorale dans les xénogreffes colorectales RKO, évaluée par des rapporteurs de luciférase pilotés par l’élément de réponse à l’hypoxie (HRE)44 ou par la tomographie par émission de positons au 18F-fluoroazomycinearabinoside.18 Ce modèle RKO a montré une réduction très modeste de la croissance des tumeurs traitées au DCA, mais les changements aigus de l’oxygénation après le DCA les ont sensibilisés au traitement par des cytotoxines activées par l’hypoxie telles que la Tirapazamine44 ou le PR-104.18 D’autres travaux sur des modèles de cancer colorectal, réalisés par d’autres groupes, ont révélé une hétérogénéité significative de la réponse au DCA. Certaines lignées cellulaires ont réduit leur taux de croissance tumorale,45 tandis que d’autres n’ont pas réagi ou ont même connu une croissance plus rapide.46 De plus, dans un modèle de rat d’adénocarcinome mammaire, un traitement intensif au DCA a permis de réduire le nombre de métastases pulmonaires macroscopiques.48 Ces modèles précliniques soutiennent le concept selon lequel le DCA a la capacité de moduler le métabolisme tumoral in vivo, ce qui entraîne des effets antitumoraux plus ou moins importants selon le modèle testé.
Données d’essais cliniques. Les premières données sur l’évaluation du DCA pour le traitement du cancer chez l’homme ont été rapportées récemment.49 Dans cette étude, le DCA a été utilisé en combinaison avec la chirurgie, le temozolomide et la radiation pour le traitement de cinq patients atteints de glioblastome multiforme. Si les auteurs font état de résultats cliniques prometteurs chez quatre des cinq patients, l’accent est mis sur l’analyse ex vivo des cellules tumorales avant et après le traitement au DCA. Ils signalent des modifications du potentiel de la membrane mitochondriale, une augmentation de la quantité de radicaux oxygénés générés par la mitochondrie et une augmentation de l’apoptose des cellules tumorales. Les études mécanistes ont révélé une modification des niveaux de signalisation HIF1, une activation de p53 et une diminution de l’angiogenèse. Ces données suggèrent que le DCA possède de nombreux mécanismes d’action en aval de son inhibition des PDK. Il est évident qu’un plus grand nombre de patients, provenant de préférence de plusieurs sites différents, doivent être traités pour pouvoir généraliser ces résultats passionnants.
Régulation du métabolisme par l’organisme
Des facteurs humoraux de croissance et de nutrition pourraient également influencer la réponse des tumeurs à la reprogrammation métabolique. La régulation de l’activité des PDC tumoraux in vivo n’a pas encore été étudiée de manière systématique, mais les connaissances que nous avons acquises sur le métabolisme des tissus normaux et dans les domaines de l’endocrinologie et de la recherche sur l’obésité fournissent quelques hypothèses intéressantes. La privation de nourriture augmente l’expression des PDK et diminue l’activité des PDH dans les organes périphériques, ce qui constitue une stratégie pour maintenir un approvisionnement stable en hydrates de carbone dans le cerveau et les autres tissus neuronaux. Par exemple, la privation de nourriture active la transcription de PDK4 et PDK2 dans le foie, les reins et d’autres tissus.51,52 Les glucocorticoïdes, l’hormone thyroïdienne T3 et les acides gras libres augmentent également les niveaux de PDK4.53 La réalimentation et/ou l’augmentation des niveaux d’insuline diminuent la transcription de PDK4 et réactivent la PDH. De même, il a été démontré que la famine et le diabète diminuent les niveaux d’ARNm et de protéine de la PDP2, effets qui sont inversés par la réalimentation ou le traitement à l’insuline.54
Par conséquent, les nombreux signaux d’entrée qui ont un impact sur l’activité de la PDH in vivo pourraient influencer l’efficacité du DCA et d’autres médicaments à visée métabolique. L’administration du médicament pendant le jeûne pourrait avoir un effet très différent par rapport à l’état d’alimentation. Les réponses aux conditions humorales sont évidemment différentes selon les tissus et les types de tumeurs et sont donc difficiles à imiter in vitro. Alors, est-il possible d’évaluer l’effet antitumoral de molécules comme le DCA in vitro ? De nombreuses années d’étude d’un grand nombre de lignées cellulaires tumorales cultivées montrent qu’elles conservent les caractéristiques anormales de la glycolyse aérobie in vitro et constituent donc un outil précieux pour l’étude du métabolisme du cancer.55 Cependant, ces systèmes présentent aussi des limites inhérentes, comme le montrent les données incohérentes de la littérature lorsqu’on compare l’effet du DCA en culture cellulaire aux résultats précliniques et cliniques.
L’analyse qui précède présente le modèle selon lequel un médicament anticancéreux qui cible le métabolisme et présente une faible toxicité in vitro peut avoir un potentiel important in vivo. Nous avons souligné le nombre limité de substrats métaboliques dans la tumeur et la régulation systémique du métabolisme par des facteurs humoraux qui pourraient augmenter l’efficacité du médicament in vivo. Il est possible que l’inverse soit également vrai, à savoir qu’un médicament ayant une bonne activité in vitro puisse avoir peu de succès in vivo. Les effets sur la toxicité des tissus normaux ou la coopération métabolique entre les types de cellules pourraient limiter l’efficacité d’un médicament in vivo. Le 2-désoxyglucose, un inhibiteur métabolique largement testé, présente une activité anticancéreuse raisonnable in vitro mais ne peut être utilisé chez les patients en raison de ses effets négatifs sur les tissus normaux qui dépendent de la consommation de glucose. La toxicité neurologique limitant la dose se produit à un niveau de médicament bien inférieur à celui nécessaire pour obtenir des effets anticancéreux sur les tumeurs des rongeurs.56,57 Il est également possible que la coopération métabolique entre les types de cellules ou entre les cellules normales et tumorales permette de contourner le blocage métabolique induit par le médicament. Par exemple, alors que le lactate est souvent considéré comme un déchet métabolique, il peut être utilisé dans certaines cellules comme un carburant pour alimenter la fonction mitochondriale.58,59
En ce qui concerne la combinaison du DCA avec les thérapies existantes, les données précliniques ne montrent pas jusqu’à présent un schéma d’interaction évident qui permettrait une sélection facile et rationnelle des régimes thérapeutiques. Les modèles animaux resteront le meilleur moyen d’effectuer des tests pour déterminer empiriquement les combinaisons les plus prometteuses. Nous avons montré qu’en raison de sa capacité à augmenter la consommation d’oxygène, le DCA augmentait l’hypoxie tumorale et sensibilisait les tumeurs du pancréas et du côlon xénogreffées aux cytotoxineshypoxiques18,44; il est donc intriguant d’envisager un plan de traitement incluant ces deux classes de médicaments qui sont toutes deux conçues pour exploiter le microenvironnement hypoxique unique de la tumeur.
L’interaction du DCA avec d’autres modulateurs métaboliques n’a pas été rapportée. Une cible potentielle pour une thérapie combinée est la lactate déshydrogénase A (LDHA). Il a été démontré que l’inhibition génétique ou pharmacologique de la LDHA augmente la fonction mitochondriale et inhibe la formation et la progression de tumeurs modèles.60,61 Dans ce schéma d’association, l’inhibiteur de la LDHA bloquerait la conversion du pyruvate en lactate et le DCA détournerait le pyruvate accumulé vers l’oxydation mitochondriale. Si l’effet antitumoral du DCA provient d’une augmentation de la fonction mitochondriale, il est possible que l’association des inhibiteurs de la PDK et de la LDHA force un taux d’oxydation mitochondriale encore plus élevé et entrave plus efficacement la croissance tumorale.
Les points nodaux des principales voies de survie, comme la voie PI3KAkt-mTOR, font également l’objet d’intenses efforts de développement de médicaments.62 Une partie des propriétés de promotion de la croissance de cette voie provient de sa capacité à réguler le métabolisme et la production d’énergie par des mécanismes directs ou indirects. Par exemple, l’activation oncogène de la PI3K-Akt stimule l’absorption du glucose et la glycolyse aérobie,63,64 tandis que l’activation d’Akt et de mTORC1 augmente la traduction de l’ARNm de l’Hif-1a sous hypoxie.65 Des inhibiteurs d’Akt prometteurs et des inhibiteurs de mTOR de nouvelle génération sont testés dans des essais cliniques66,67 et représentent des candidats viables pour une thérapie combinée avec le DCA pour moduler le métabolisme aérobie et hypoxique.
Conclusions
En conclusion, les connaissances récentes sur le métabolisme unique des tumeurs solides ont permis d’identifier plusieurs nouvelles voies médicamenteuses qui peuvent être utilisées de manière préférentielle dans les cellules tumorales par rapport aux cellules normales. L’analyse des médicaments anticancéreux candidats conçus pour cibler ces voies métaboliques nécessitera une conception expérimentale minutieuse, à la fois in vitro et in vivo. L’analyse des rapports publiés sur le DCA montre une gamme d’effets in vitro et in vivo déroutants et parfois contradictoires. Des études génétiques sur des tumeurs modèles montrent de façon convaincante que cette voie est un bon candidat pour le ciblage thérapeutique.68 Il serait très utile pour l’analyse de l’utilité potentielle du DCA de disposer d’une signature moléculaire permettant de prédire la sensibilité au médicament, à la fois dans les tumeurs modèles et éventuellement chez les patients. Une analyse attentive de la cible présumée du DCA, la phosphorylation de la sous-unité E1α de la pyruvate déshydrogénase, pourrait peut-être offrir cette signature.
RÉFÉRENCES
1 1. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927;8:519-30.
2 Pan JG, Mak TW. Le ciblage métabolique comme stratégie anticancéreuse : l’aube d’une nouvelle ère ? Sci STKE 2007;2007:pe14.
3 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brique par brique : métabolisme et croissance des cellules tumorales. Curr Opin Genet Dev 2008;18:54-61.
4 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004;14:198-206.
5 Walenta S, Chau TV, Schroeder T, Lehr HA, Kunz-Schughart LA, Fuerst A, Mueller-Klieser W. Metabolic classification of human rectal adenocarcinomas : a novel guideline for clinical oncologists ? J Cancer Res Clin Oncol 2003;129:321-6.
6 Gstraunthaler G, Seppi T, Pfaller W. Impact of culture conditions, culture media volumes, and glucose content on metabolic properties of renal epithelial cell cultures. Les cellules rénales en culture tissulaire sont-elles hypoxiques ? Cell Physiol Biochem 1999;9:150-72.
7 Bloch-Frankenthal L, Ram D. The relationship between the Crabtree effect and the oxidative metabolism of glucose and carbohydrate intermediates in ascites tumor cells. Cancer Res 1959;19:835-42.
8ParrinelloS, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. La sensibilité à l’oxygène limite sévèrement la durée de vie réplicative des fibroblastes murins. Nat Cell Biol 2003;5:741-7.
9 Denko NC. Hypoxie, HIF1 et métabolisme du glucose dans la tumeur solide. Nat Rev Cancer 2008;8:705-13.
10 Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. L’analyse génomique de l’acidose lactique et de la réponse à l’acidose dans les cancers humains. PLoS Genet 2008;4:e1000293.
11 Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. Hypoxiainduced down-regulation of BRCA1 expression by E2Fs. Cancer Res 2005;65 : 11597-604.
12 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Adaptations cellulaires à l’hypoxie et à l’acidose pendant l’évolution somatique du cancer du sein. Br J Cancer 2007;97:646-53.
13 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008;8:425-37.
14 Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187-97.
15 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008;283:28106-14.
16 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177-85.
17 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. La consommation d’oxygène peut réguler la croissance des tumeurs, une nouvelle perspective sur l’effet Warburg. PLoS One 2009;4:e7033.
18 Cairns RA, Bennewith KL, Graves EE, Giaccia AJ, Chang DT, Denko NC. L’hypoxie tumorale pharmacologiquement accrue peut être mesurée par tomographie par émission de positons au 18F-fluoroazomycine arabinoside et améliore la réponse tumorale à la cytotoxine hypoxique PR-104. Clin Cancer Res 2009;15 : 7170-4.
19 Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358:69-77.
20 Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 2001;276 : 37223-9.
21 Sugden PH, Randle PJ. Regulation of pig heart pyruvate dehydrogenase by phosphorylation. Studies on the subunit and phosphorylation stoicheiometries. Biochem J 1978;173:659-68.
22 Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem 1995;270:14297-304.
23 Karpova T, Danchuk S, Kolobova E, Popov KM. Characterization of the isozymes of pyruvate dehydrogenase phosphatase : implications for the regulation of pyruvate dehydrogenase activity. Biochim Biophys Acta 2003;1652:126-35.
24 Whitehouse S, Randle PJ. Activation de la pyruvate déshydrogénase dans le cœur de rat perfusé par le dichloroacétate (Communication brève). Biochem J 1973;134 : 651-3.
25 Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006;45:402-15.
26 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6.
27 Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem 2000 ; 275:15773-81.
28 Stacpoole PW, Greene YJ. Dichloroacetate. Diabetes Care 1992;15:785-91.
29 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N Engl J Med 1983;309:390-6.
30 Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 2008;60:1478-87.
31 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS : a randomized, controlled clinical trial. Neurology 2006;66 : 324-30.
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacocinétique, métabolisme et toxicologie du dichloroacétate. Drug Metab Rev 1998;30 : 499-539.
33 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Évaluation du traitement à long terme des enfants atteints d’acidose lactique congénitale par le dichloroacétate. Pediatrics 2008;121:e1223-8.
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate : possible relevance to toxicity. J Pharmacol Exp Ther 2008;324:1163-71.
35 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Effets métaboliques et hémodynamiques myocardiques du dichloroacétate dans la maladie coronarienne. Am J Cardiol 1988;61:65-70.
36 Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994;23:1617-24.
37 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
38 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 2008;68:1223-31.
39 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Le dichloroacétate induit l’apoptose dans les cellules cancéreuses de l’endomètre. Gynecol Oncol 2008;109 : 394-402.
40 Anderson KM, Jajeh J, Guinan P, Rubenstein M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 2009;29:4579-88.
41 Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Les mutations mitochondriales contribuent à l’accumulation de HIF1alpha par l’augmentation des espèces réactives de l’oxygène et la régulation de la pyruvate déhydrogénase kinase 2 dans le carcinome épidermoïde de la tête et du cou. Clin Cancer Res 2009;15:476-84.
42 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. La thérapie métabolique ciblée par le dichloroacétate déjoue la cytotoxicité des médicaments anticancéreux standard. Cancer Chemother Pharmacol 2010.
43 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Le dichloroacétate induit l’apoptose et l’arrêt du cycle cellulaire dans les cellules cancéreuses colorectales. Br J Cancer 2010;102:1746-52.
44 Cairns RA, Papandreou I, Sutphin PD, Denko NC. Le ciblage métabolique de l’hypoxie et de HIF1 dans les tumeurs solides peut améliorer la chimiothérapie cytotoxique. Proc Natl Acad Sci USA 2007;104:9445-50.
45 Sanchez-Arago M, Chamorro M, Cuezva JM. La sélection de cellules cancéreuses avec des mitochondries réprimées déclenche la progression du cancer du côlon. Carcinogenesis 2010;31:567-76.
46 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;287:75-83.
47 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Le dichloroacétate de sodium (DCA) cible sélectivement les cellules présentant des défauts dans l’ETC mitochondrial. Int J Cancer 2010;127:2510-19.
48SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. L’inversion du phénotype glycolytique par le dichloroacétate inhibe la croissance des cellules métastatiques du cancer du sein in vitro et in vivo. Breast Cancer Res Treat 2010;120:253-60.
49 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2 : 31ra34.
50 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Suivi de la phosphorylation du complexe pyruvate déshydrogénase. Anal Biochem 2009;389:157-64.
51 Holness MJ, Sugden MC. Activités de la pyruvate déshydrogénase pendant la transition entre l’alimentation et la famine et lors de la réalimentation après une famine aiguë ou prolongée. Biochem J 1989;258:529-33.
52 Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptoralpha ligands, glucocorticoids, and insulin. Diabetes 2002;51:276-83.
53 Attia RR, Connnaughton S, Boone LR, Wang F, Elam MB, Ness GC, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone : role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J Biol Chem 2010;285:2375-85.
54 Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003;52 : 1371-6.
55 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. L’analyse métabolique multiparamétrique révèle un lien étroit entre la fonction bioénergétique mitochondriale atténuée et la dépendance accrue à la glycolyse dans les cellules tumorales humaines. Am J Physiol Cell Physiol 2007;292:C125-36.
56 Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507-14.
57 Maher JC, Krishan A, Lampidis TJ. Plus grande inhibition du cycle cellulaire et cytotoxicité induite par le 2-déoxy-D-glucose dans les cellules tumorales traitées dans des conditions hypoxiques vs aérobies. Cancer Chemother Pharmacol 2004;53:116-22.
58 Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930-42.
59KoukourakisMI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas : a metabolic survival role for tumor-associated stroma. Cancer Res 2006;66:632-7.
60 Fantin VR, St-Pierre J, Leder P. Attenuation of LDHA expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006;9:425-34.
61 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. L’inhibition de la lactate déshydrogénase A induit un stress oxydatif et inhibe la progression tumorale. Proc Natl Acad Sci USA 2010;107:2037-42.
62 Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway : molecular targets for anti-cancer drug development. Curr Cancer Drug Targets 2004;4:235-56.
63 Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimule la glycolyse aérobie dans les cellules cancéreuses. Cancer Res 2004 ; 64:3892-9.
64 Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. La dépendance au glucose des cellules transformées par Akt peut être inversée par l’activation pharmacologique de la bêta-oxydation des acides gras. Oncogene 2005 ; 24:4165-73.
65 Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis : novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 2001;21:3995-4004.
66 Ghobrial IM, Gertz M, Laplant B, Camoriano J, Hayman S, Lacy M, Chuma S, Harris B, Leduc R, Rourke M, Ansell SM, Deangelo D, et al. Essai de phase II de l’évérolimus, un inhibiteur oral de la cible mammalienne de la rapamycine, dans la macroglobulinémie de Waldenstrom récidivante ou réfractaire. J Clin Oncol 2010;28:1408-14.
67 Meric-Bernstam F, Gonzalez-Angulo AM. Cibler le réseau de signalisation mTOR pour le traitement du cancer. J Clin Oncol 2009;27 : 2278-87.
68 McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem 2008;283:22700-8.
Contenu connexe :