Géraldine De Preter1, Marie-Aline Neveu1, Pierre Danhier1, Lucie Brisson2, Valéry L. Payen2, Paolo E. Porporato2, Bénédicte F. Jordan1, Pierre Sonveaux2 e Bernard Gallez1
1 Istituto di Ricerca sui Farmaci di Louvain (LDRI), Gruppo di Ricerca sulla Risonanza Magnetica Biomedica, Université Catholique de Louvain (UCL), Bruxelles, Belgio
2 Institut de Recherche Expérimentale et Clinique (IREC), Polo di Farmacologia, Université Catholique de Louvain (UCL), Bruxelles, Belgio
Corrispondenza: Bernard Gallez, e-mail: [email protected]
Ricevuto: 1 giugno 2015
Accettato: 26 dicembre 2020
Pubblicato: 9 dicembre 2020
Abstract
La fermentazione del glucosio attraverso la glicolisi anche in presenza di ossigeno (effetto Warburg) è una caratteristica comune delle cellule tumorali, sempre più considerate un bersaglio allettante nello sviluppo clinico. Questo studio si proponeva di analizzare il legame tra metabolismo, riserve energetiche e tassi di proliferazione nelle cellule tumorali. Abbiamo scoperto che la proliferazione cellulare, valutata mediante quantificazione della sintesi del DNA, è correlata all’efficienza glicolitica in sei linee cellulari tumorali e in linee cellulari tumorali isogeniche. Per approfondire il legame tra glicolisi e proliferazione, è stato utilizzato un inibitore farmacologico della via del pentoso fosfato (PPP). Abbiamo dimostrato che la riduzione dell’attività della PPP riduce la proliferazione delle cellule tumorali, con un effetto profondo nelle cellule tumorali a fenotipo Warburg. Il ruolo cruciale della PPP nel sostenere la proliferazione delle cellule tumorali è stato confermato utilizzando siRNA contro la glucosio-6-fosfato deidrogenasi, il primo enzima e quello che limita la velocità della PPP. Inoltre, abbiamo scoperto che il dicloroacetato (DCA), un nuovo composto testato clinicamente, ha indotto un passaggio delle cellule tumorali glicolitiche a un fenotipo più ossidativo e ha ridotto la proliferazione. Dimostrando che il DCA riduce l’attività della PPP, forniamo un nuovo meccanismo attraverso il quale il DCA controlla la proliferazione delle cellule tumorali.
Parole chiave: bioenergetica, glicolisi, dicloroacetato, via del pentoso fosfato, proliferazione
© 2020 dagli autori. Licenziatario MDPI, Basilea, Svizzera. Questo articolo è un articolo ad accesso libero distribuito secondo i termini e le condizioni della licenza Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
INTRODUZIONE
Negli ultimi anni il metabolismo ha suscitato un enorme interesse nel campo della ricerca sul cancro. Molti studi si sono concentrati sui vari profili metabolici di diversi tumori [1-3] perché la plasticità metabolica è coinvolta nella progressione del cancro, nella resistenza ai farmaci e nelle metastasi [4-6]. Nelle cellule normali, la glicolisi è accoppiata alla fosforilazione ossidativa (OXPHOS) per sintetizzare in modo ottimale l’ATP intracellulare dal glucosio [7]. Tuttavia, molte cellule tumorali subiscono una trasformazione metabolica fondamentale, lo “switch glicolitico”, attraverso il quale la glicolisi viene disaccoppiata dalla respirazione e reimpostata sulla fermentazione lattica, diventando così la fonte primaria di produzione di ATP cellulare. Il passaggio a un metabolismo glicolitico avviene principalmente in condizioni di ipossia come meccanismo di salvataggio per la produzione di energia. Tuttavia, alcune cellule tumorali adottano un particolare fenotipo glicolitico, descritto per la prima volta da Warburg [8] e denominato “glicolisi aerobica” [9]. Il razionale biologico alla base del fenotipo di Warburg rimane controverso, ma recentemente è stato proposto che le cellule tumorali in proliferazione aumentino la glicolisi perché ne beneficiano sia la bioenergetica che la biosintesi [4,10]. Infatti, un metabolismo glicolitico consente potenzialmente una rapida produzione di ATP e fornisce intermedi di carbonio che possono essere indirizzati a vie biosintetiche ramificate, consentendo una più rapida espansione della biomassa cellulare. È convincente che le mutazioni che si verificano nelle vie di segnalazione che regolano sia la biosintesi cellulare sia la glicolisi aerobica, come la via PI3K/Akt/mTOR, siano la classe di mutazioni più prevalente nei tumori umani [11]. Tuttavia, mancano prove sperimentali che colleghino la glicolisi aerobica alla proliferazione delle cellule tumorali e il vantaggio selettivo fornito da questo fenotipo non è del tutto chiaro. Lo scopo del presente studio è stato quello di chiarire l’accoppiamento tra metabolismo, approvvigionamento energetico e proliferazione cellulare in diverse cellule tumorali umane e murine. Sono stati indotti interruttori metabolici per dimostrare che la bioenergetica, e in particolare la glicolisi, guida direttamente la proliferazione delle cellule tumorali. In questa linea, abbiamo scoperto un nuovo meccanismo terapeutico del dicloroacetato (DCA), un attivatore dell’ossidazione mitocondriale del glucosio attualmente studiato in studi clinici [12]. Il DCA ha inibito la via del pentoso fosfato (PPP), una via biosintetica cardine ramificata alla glicolisi. Abbiamo riferito che la PPP colma il divario tra il metabolismo glicolitico e la proliferazione delle cellule tumorali.
RISULTATI
L’efficienza glicolitica è positivamente legata alla proliferazione ma non ai livelli di ATP nelle cellule tumorali
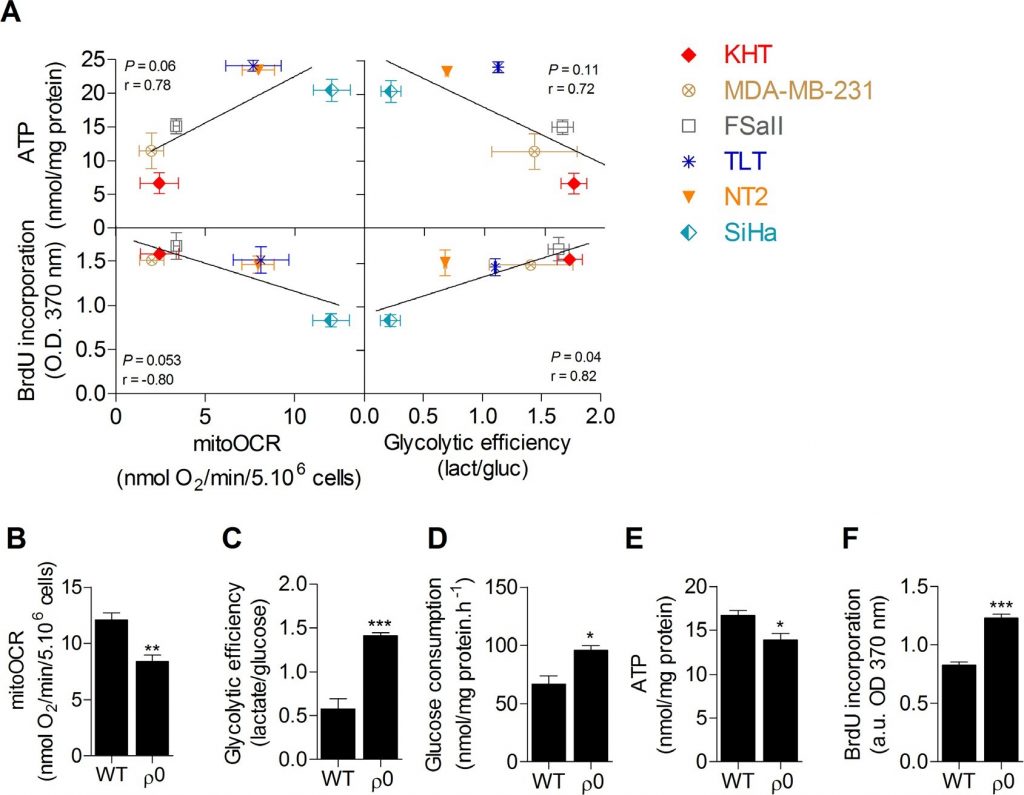
È stato dapprima eseguitounoscreening delle attività metaboliche di sei linee cellulari tumorali per indagare il legame tra metabolismo del glucosio, riserve di ATP e capacità di proliferazione. L’OXPHOS, valutata mediante il tasso di consumo di ossigeno sensibile all’oligomicina (mitoOCR), e l’efficienza glicolitica, misurata mediante il rapporto tra lattato prodotto e glucosio consumato (mol/mol) [13], sono state valutate separatamente su cellule incubate 24 ore in un terreno di coltura contenente solo glucosio come combustibile energetico. Per indagare l’influenza dello stato metabolico sulle riserve energetiche cellulari, è stato misurato anche il pool totale di ATP intracellulare. Non è stata trovata alcuna correlazione significativa tra il contenuto intracellulare di ATP e i parametri metabolici. Tuttavia, abbiamo riscontrato che la proliferazione cellulare, valutata attraverso la sintesi di DNA, era significativamente correlata all’efficienza glicolitica (Figura 1A), dove un aumento della glicolisi era associato a un aumento della capacità di proliferazione, e inversamente. Non è stata osservata alcuna correlazione significativa tra la capacità di proliferazione e la respirazione mitocondriale. Questi dati suggeriscono che la glicolisi è potenzialmente la via energetica chiave che supporta la proliferazione delle cellule tumorali. Attualmente si ipotizza che questa via sostenga la proliferazione cellulare prevalentemente fornendo precursori per le vie biosintetiche, piuttosto che attraverso la produzione di ATP [10,11]. A sostegno del fatto che l’apporto di ATP non è il principale fattore limitante per la proliferazione cellulare, nelle linee cellulari non è stata trovata alcuna correlazione tra il contenuto di ATP e la sintesi di DNA (Figura S1 supplementare). Per confermare che la glicolisi promuove la proliferazione delle cellule tumorali ed escludere le influenze indipendenti dal metabolismo inerenti ai diversi genotipi, la capacità di proliferazione delle cellule di cancro cervicale SiHa ossidative umane wild-type (WT) è stata confrontata con quella delle SiHa con parziale deplezione mitocondriale (SiHa ρ0) [14,15]. Le cellule tumorali SiHa ρ0 hanno mostrato un fenotipo glicolitico con una diminuzione di circa il 40% del mitoOCR (Figura 1B), un aumento di circa 2,5 volte dell’efficienza glicolitica (Figura 1C) e un aumento di circa il 50% del consumo di glucosio (Figura 1D). La diminuzione netta del 20% del pool totale di ATP nelle cellule SiHa ρ0 ha confermato che l’aumento della glicolisi non è sufficiente a mantenere le riserve di ATP (Figura 1E). Eseguendo la quantificazione della sintesi del DNA, abbiamo scoperto che l’aumento della glicolisi aerobica promuove la proliferazione cellulare, in quanto il passaggio alla glicolisi nelle cellule SiHa ρ0 è stato associato a un aumento del 45% circa della proliferazione cellulare (Figura 1F). I test di vitalità non hanno rivelato differenze nel numero di cellule vitali 24 ore dopo l’incubazione delle cellule nel terreno di coltura sperimentale (Figura supplementare S2), probabilmente perché la quantificazione della sintesi del DNA rileva cambiamenti precoci nel tasso di proliferazione delle cellule.
L’inibizione della glicolisi da parte del DCA compromette la proliferazione delle cellule tumorali
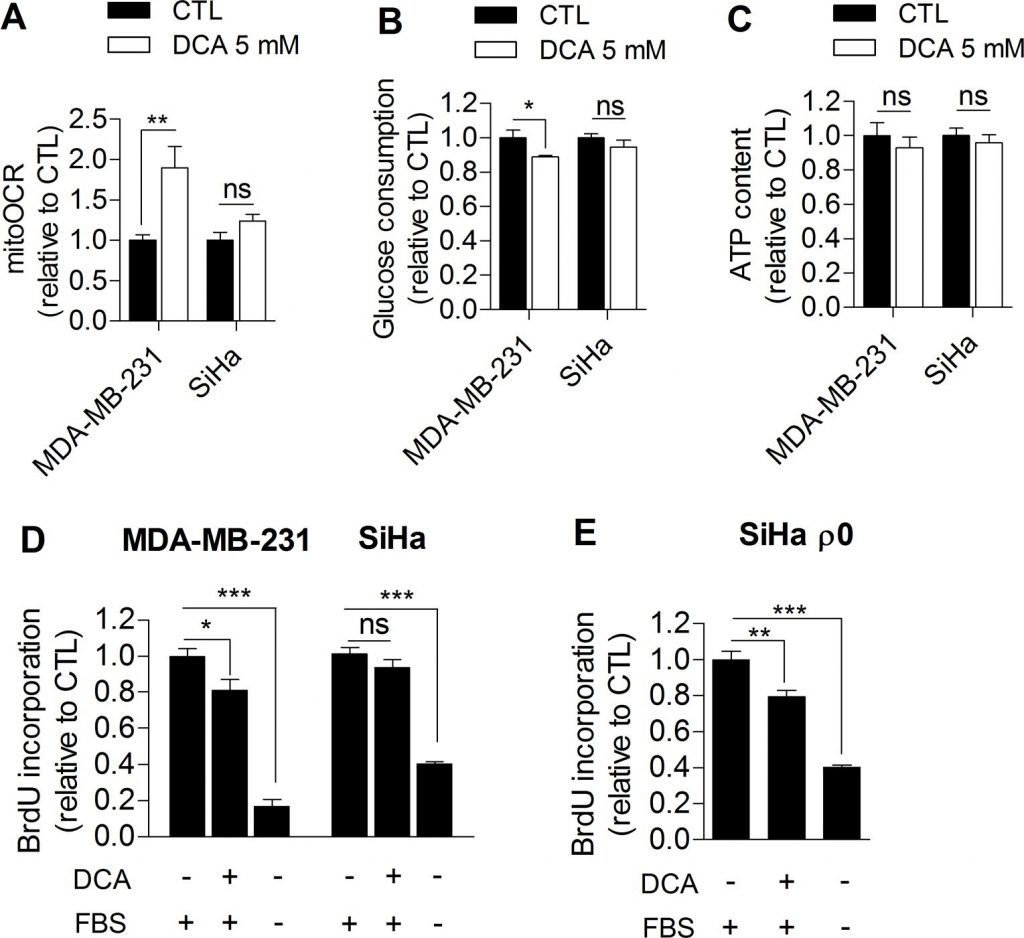
Sulla base delle nostre osservazioni, abbiamo ulteriormente indagato se l’inibizione della glicolisi potesse compromettere direttamente la proliferazione delle cellule tumorali. A tal fine, le cellule di carcinoma mammario umano MDA-MB-231 (fenotipo Warburg, Figura 1A) e le cellule di carcinoma cervicale umano SiHa (fenotipo ossidativo, Figura 1A) sono state trattate con dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi (PDK) che aumenta l’attività ossidativa delle cellule attivando la piruvato deidrogenasi (PDH), l’enzima di mantenimento dell’ossidazione del glucosio nei mitocondri [16]. Ad oggi, il promettente effetto terapeutico del DCA sulle cellule tumorali è globalmente attribuito alla normalizzazione del potenziale di membrana mitocondriale iperpolarizzato che caratterizza le cellule tumorali e alla risensibilizzazione all’apoptosi [17]. In questa sede, abbiamo ipotizzato che il DCA controlli anche la proliferazione tumorale inibendo la glicolisi. Per verificare questa ipotesi, cellule tumorali MDA-MB-231 glicolitiche e SiHa ossidative sono state trattate con 5 mM di DCA per 48 ore e sono stati valutati gli effetti del trattamento sul metabolismo e sulla proliferazione. Rispetto alle cellule trattate con il veicolo, il DCA ha indotto un passaggio delle cellule tumorali MDA-MB-231 glicolitiche a un fenotipo più ossidativo, come evidenziato da un aumento del mitoOCR (Figura 2A) e da una diminuzione del consumo di glucosio (Figura 2B). La diminuzione dell’attività glicolitica osservata in questo esperimento è coerente con un altro studio recente [18] ed è probabilmente indotta dall’effetto Pasteur [4,19] per mantenere l’omeostasi dell’ATP nelle cellule (Figura 2C). Abbiamo anche osservato che l’inibizione della glicolisi da parte del DCA era associata a una diminuzione del tasso di proliferazione delle cellule tumorali MDA-MB-231 (Figura 2D). A sostegno del fatto che l’inibizione della glicolisi compromette la proliferazione in questa linea cellulare, anche le cellule tumorali MDA-MB-231 trattate con 2-deossi-D-glucosio hanno mostrato una riduzione del tasso di proliferazione (Figura supplementare S3).
D’altra parte, il DCA non ha alterato significativamente le attività metaboliche delle cellule tumorali SiHa ossidative (Figura 2A-2C) e non ha avuto effetti significativi sulla proliferazione delle SiHa (Figura 2D). Le misurazioni della produzione di lattato a breve termine (1 ora) hanno dimostrato che il DCA è stato effettivamente più efficace nella linea di cellule tumorali glicolitiche rispetto a quella ossidativa (Figura S4 supplementare). Per verificare se il profilo metabolico determina la risposta al DCA, è stata analizzata anche la capacità di proliferazione delle cellule glicolitiche del cancro SiHa ρ0 dopo il trattamento con DCA. Abbiamo riscontrato una significativa diminuzione della sintesi di DNA in questa linea cellulare (Figura 2E), un effetto che non è stato osservato nelle SiHa WT. Inoltre, lo stesso numero di cellule tumorali vitali MDA-MB-231 e SiHa ρ0 è stato misurato 48 ore dopo il trattamento con veicolo o DCA (Figura supplementare S5 A-B), dimostrando che gli effetti del DCA sulle funzioni metaboliche e sui tassi di proliferazione non sono dovuti alla mortalità delle cellule.
Laglicolisi controlla la proliferazione delle cellule tumorali attraverso la via del pentoso fosfato
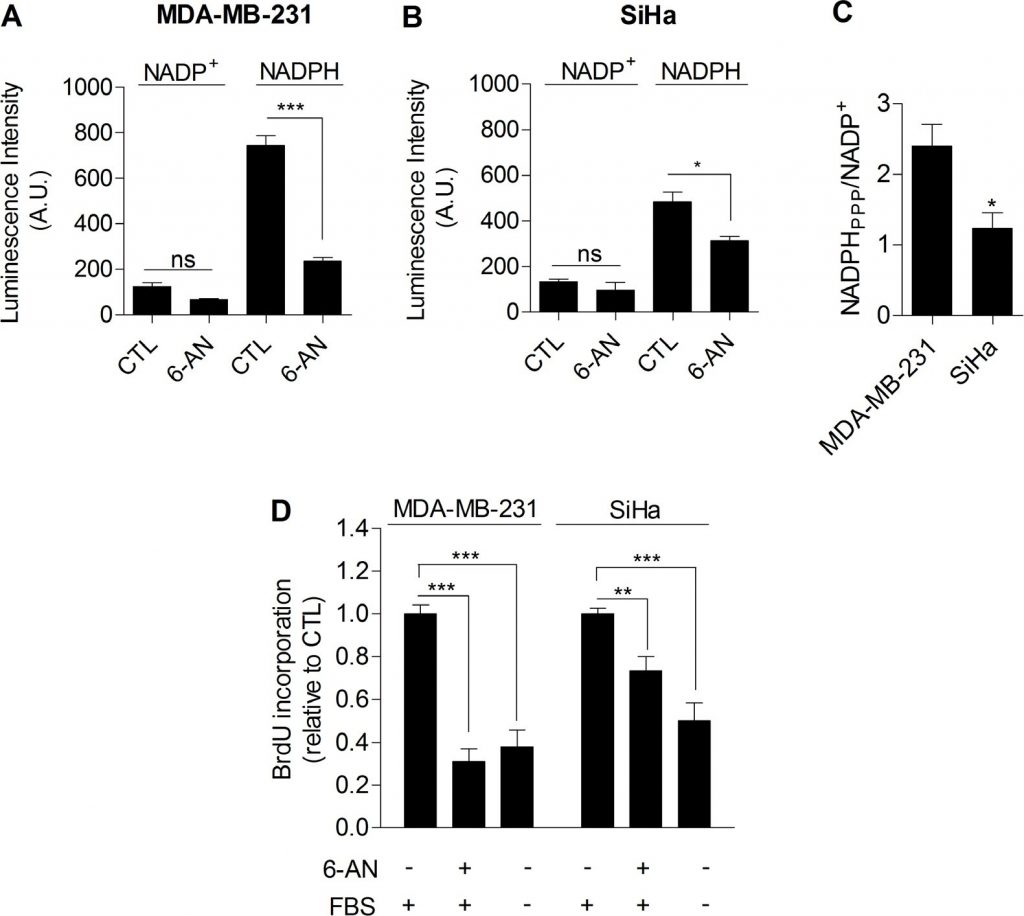
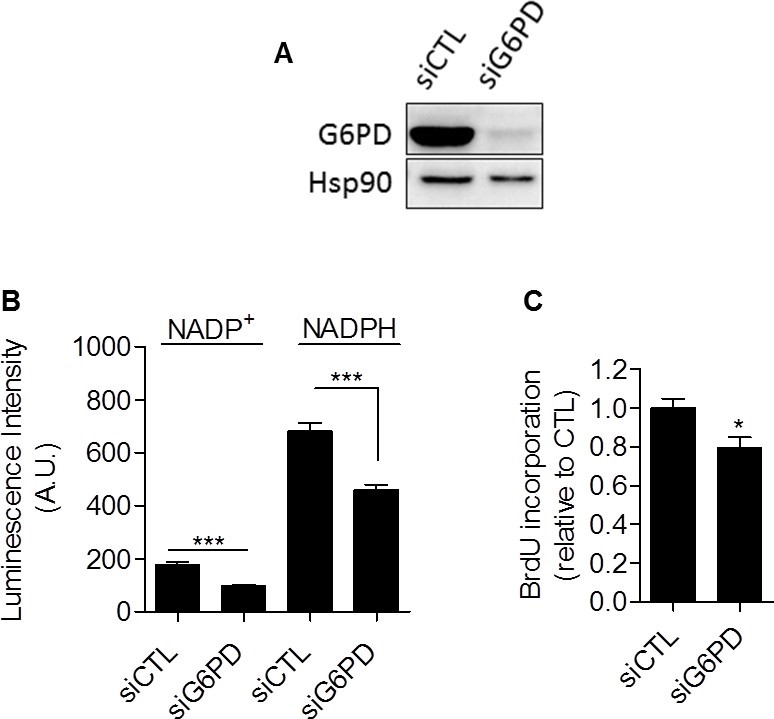
Nel complesso, i nostri dati hanno fornito una serie di prove sperimentali che dimostrano che la glicolisi di per sé è coinvolta nel controllo della proliferazione cellulare. Il meccanismo che collega glicolisi e proliferazione deve ancora essere stabilito. Abbiamo ipotizzato che la via del pentoso fosfato (PPP) potesse collegare la glicolisi alla proliferazione, poiché la PPP utilizza gli intermedi glicolitici per fornire alle cellule nucleotidi e NADPH, un riduttore cruciale nei processi anabolici [20]. Per determinare in modo specifico il NADPH prodotto dalla PPP (NADPHppp), le cellule sono state trattate con 6-aminonicotinamide (6-AN), un inibitore specifico della PPP [21,22]. Abbiamo riscontrato che il contributo di NADPHppp al pool totale di NADPH (NADPHtot) era più predominante nelle cellule tumorali MDA-MB-231 rispetto alle cellule tumorali SiHa, come evidenziato da un’importante diminuzione del livello di NADPHtot in seguito al trattamento con 6-AN nelle cellule glicolitiche MDA-MB-231 (Figura 3A). Effetti più limitati sono stati osservati nelle cellule ossidative SiHa (Figura 3B). Le misurazioni hanno anche rivelato un rapporto NADPHppp/NADP+ più elevato nelle cellule tumorali MDA-MB-231, evidenziando un flusso di PPP più elevato in questa linea cellulare glicolitica rispetto alle SiHa (Figura 3C). Per verificare che la PPP sia coinvolta nel controllo della proliferazione delle cellule tumorali, è stata valutata la sintesi di DNA in cellule trattate e non trattate con 6-AN. Abbiamo osservato che la capacità di proliferazione delle cellule tumorali era ridotta quando la PPP veniva inibita (Figura 3D). È interessante notare che l’effetto è stato più forte nelle cellule glicolitiche MDA-MB-231 (riduzione del 70% del tasso di sintesi del DNA) rispetto alle cellule ossidative SiHa (riduzione del 25% del tasso di sintesi del DNA) (Figura 3D). È importante notare che le SiHa ρ0 erano più sensibili delle SiHa WT al 6-AN, poiché è stata riscontrata una diminuzione del 40% della sintesi di DNA (Figura S6 supplementare). A causa dei potenziali effetti fuori bersaglio degli inibitori farmacologici, abbiamo integrato i nostri studi con 6-AN utilizzando piccoli RNA interferenti (siRNA) che hanno come bersaglio la glucosio-6-fosfato deidrogenasi (G6PD), il primo enzima e l’enzima limitante della PPP [23]. Nelle cellule MDA-MB-231 trasfettate, abbiamo confermato il silenziamento della G6PD mediante immunoblotting (Figura 4A) e abbiamo dimostrato che, come il 6-AN, l’inibizione della G6PD ha ridotto l’attività della PPP (Figura 4B) e la sintesi di DNA (Figura 4C). Questi risultati evidenziano il contributo predominante della PPP nel sostenere la proliferazione delle cellule tumorali a fenotipo Warburg.
IlDCA inibisce la via del pentoso fosfato
Infine, abbiamo indagato se il DCA potesse inibire la PPP, il che spiegherebbe il legame tra l’inibizione della glicolisi e la diminuzione del tasso di proliferazione delle cellule tumorali trattate con DCA. Come mostrato nella Figura 5, abbiamo osservato che il DCA (5 mM, 48 ore) ha ridotto significativamente il livello di NADPHtot nelle cellule tumorali MDA-MB-231 (Figura 5A). Inoltre, mentre non è stata ottenuta un’ulteriore diminuzione del NADPHtot quando le cellule sono state esposte a DCA e 6-AN insieme (Figura 5A), abbiamo dimostrato che il DCA ha ridotto specificamente il NADPHPPP. Risultati simili sono stati ottenuti utilizzando l’inibizione molecolare della G6PD (Figura 5B). Questi risultati dimostrano l’implicazione della PPP negli effetti del DCA sulle cellule tumorali glicolitiche.
DISCUSSIONE
Questo studio sottolinea il ruolo predominante della via del pentoso fosfato nella biologia del cancro. L’effetto Warburg (fermentazione del glucosio in lattato in presenza di una concentrazione fisiologica di O2 ), osservato in numerose cellule tumorali, è stato proposto per soddisfare le esigenze energetiche (ATP) e biosintetiche [4,10]. In questo studio abbiamo dimostrato che il potenziamento della glicolisi aerobica non è sufficiente a mantenere i livelli di ATP nelle cellule tumorali. Pertanto, un basso livello di ATP intracellulare era associato a un aumento della glicolisi. Inoltre, lo switch glicolitico indotto nelle cellule tumorali con deficit di mitocondri non è stato in grado di fornire un adeguato apporto di ATP rispetto alle cellule wild-type. È stato suggerito che, dal punto di vista bioenergetico, un metabolismo glicolitico è meno efficiente di uno ossidativo, ma che la glicolisi può essere vantaggiosa per produrre rapidamente ATP per soddisfare richieste energetiche a breve termine [24]. I nostri risultati confermano anche che le cellule tumorali a fenotipo Warburg sviluppano diversi adattamenti per mantenere bassi i livelli di ATP, al fine di evitare l’inibizione allosterica degli enzimi glicolitici limitanti il tasso (effetto Pasteur) e di mantenere elevati tassi di flusso glicolitico [10]. D’altra parte, abbiamo dimostrato che la glicolisi promuove la proliferazione delle cellule tumorali, a sostegno delle strategie clinicamente testate che inibiscono l’effetto Warburg [12,25]. La relazione positiva tra l’efficienza glicolitica (moli di lattato prodotte per mole di glucosio consumato) e il tasso di proliferazione delle cellule tumorali ha evidenziato che le cellule in rapida divisione fermentano grandi quantità di glucosio in lattato. Studi recenti hanno proposto che la produzione di lattato consenta alle cellule tumorali di rigenerare in modo efficiente il NAD+ grazie all’enzima lattato deidrogenasi [26]. Mantenendo l’equilibrio redox NAD+/NADH, le cellule tumorali consentono un flusso più rapido di glucosio attraverso la glicolisi e una più rapida incorporazione dei metaboliti del glucosio nelle vie biosintetiche, conferendo così vantaggi alla proliferazione [10].
Per verificare se l’aumento della glicolisi aerobica promuova la proliferazione delle cellule tumorali alimentando i processi anabolici, è stato valutato il coinvolgimento della via del pentoso fosfato (PPP). È noto, infatti, che la PPP utilizza il glucosio-6-fosfato (prodotto dell’enzima glicolitico esochinasi) per rifornire le cellule di nucleotidi e NADPH, un riduttore cruciale nelle reazioni anaboliche [20]. Nel nostro studio abbiamo dimostrato che il flusso di PPP, valutato dal rapporto NADPHppp/NADP+, è aumentato nelle cellule tumorali a fenotipo Warburg rispetto a quelle ossidative. Per confermare questa differenza nel flusso di PPP, si sarebbe potuto analizzare anche il confronto dell’attività degli enzimi PPP. Tuttavia, i nostri risultati confermano che l’attivazione della glicolisi è accompagnata da un aumento dell’attività della PPP per la biosintesi nelle cellule in rapida divisione [20]. In modo convincente, abbiamo osservato che l’inibizione della PPP riduce la proliferazione delle cellule tumorali, con un effetto maggiore nelle cellule tumorali a fenotipo Warburg. La diversa sensibilità delle cellule tumorali glicolitiche e ossidative all’inibizione della PPP è probabilmente dovuta alla diversa dipendenza da questa via per la sintesi di macromolecole, come lipidi e nucleotidi. Inoltre, le cellule tumorali con un’elevata attività mitocondriale possono compensare i livelli di NADPH utilizzando enzimi associati al ciclo TCA (enzimi malici e isocitrato deidrogenasi) per ricostituire il pool di NADPH [20]. Nel complesso, questi risultati dimostrano che le cellule tumorali, soprattutto quelle aggressive a fenotipo Warburg, si affidano alla via del pentoso fosfato per una proliferazione ottimale.
Per valutare la rilevanza terapeutica di questi risultati, sono stati studiati gli effetti del DCA. Il DCA ha già dimostrato interessanti proprietà antitumorali in vitro e in vivo
[17] ed è attualmente testato in studi clinici di fase I-II [12]. Ad oggi, il DCA deve le sue proprietà terapeutiche alla risensibilizzazione delle cellule tumorali all’apoptosi [17]. Tuttavia, come recentemente sottolineato da Stockwin et al. [27], il DCA è relativamente inattivo sulla vitalità cellulare e induce apoptosi solo ad alte concentrazioni. Stockwin et al. hanno inoltre riportato che il DCA era preferenzialmente attivo nelle cellule con un difetto mitocondriale. Pertanto, abbiamo ipotizzato che, a dosi inferiori, il DCA riduca anche la proliferazione delle cellule tumorali riducendo la glicolisi. Abbiamo scoperto che il DCA 5 mM era più efficace nelle cellule tumorali con fenotipo Warburg, riducendo la proliferazione cellulare attraverso la diminuzione della glicolisi. La diversa sensibilità tra linee cellulari con diverso fenotipo metabolico può derivare dalla capacità del DCA di raggiungere la PDK nella matrice dei mitocondri. Infatti, come il piruvato, il DCA è ionizzato e non può passare attraverso le membrane. Curiosamente, ci sono stati solo pochi rapporti sul trasporto del DCA nel citosol delle cellule di mammifero [28,29]. Sebbene non sia stato ancora studiato, anche il livello di espressione dei trasportatori mitocondriali del piruvato (MPC) potrebbe definire l’efficacia del DCA. Inoltre, poiché il DCA ha un Kidiverso per ciascuno dei quattro isoenzimi PDK [30], la sensibilità variabile delle cellule tumorali potrebbe derivare dall’espressione differenziale delle PDK.
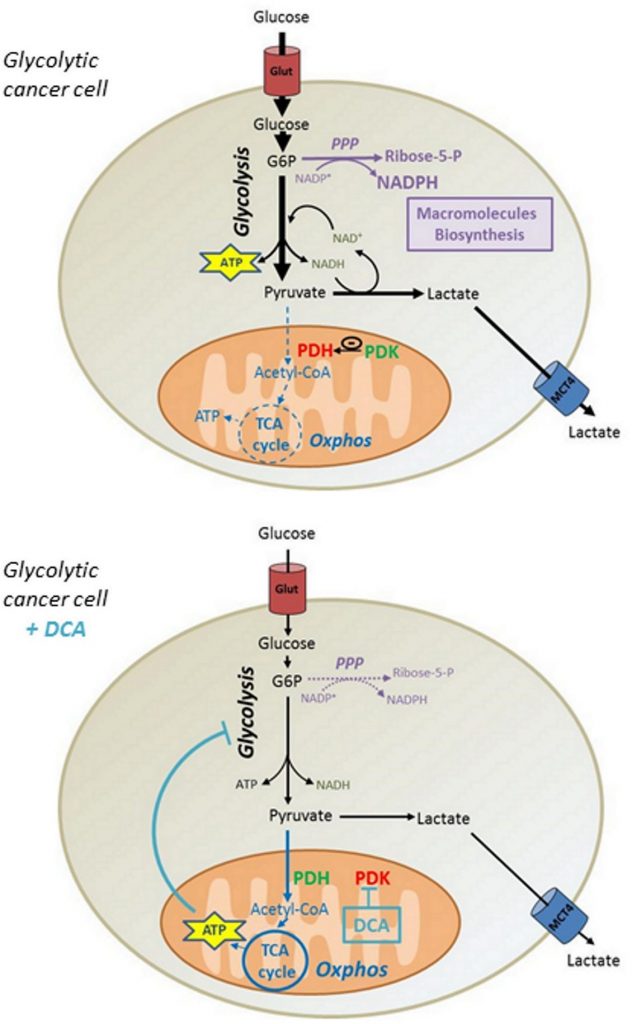
Sebbene alcuni rapporti abbiano mostrato che il DCA in concentrazioni comparabili promuove la citotossicità in altre linee cellulari [31,32], abbiamo dimostrato che il DCA non ha avuto alcuna incidenza sulla vitalità delle cellule tumorali MDA-MB-231 e SiHa. Sulla base dei nostri dati, proponiamo che la riattivazione della produzione di ATP nei mitocondri indotta dal DCA abbassi il flusso glicolitico delle cellule tumorali, riducendo così l’incorporazione di glucosio. Di conseguenza, la diminuzione della sintesi di intermedi glicolitici riduce l’attività di vie biosintetiche come la PPP, compromettendo la proliferazione delle cellule tumorali a fenotipo Warburg (Figura 6). Tuttavia, è ipotizzabile che altri meccanismi possano partecipare agli effetti antiproliferativi del DCA, dato che altri studi hanno dimostrato un arresto del ciclo cellulare a concentrazioni comparabili di DCA [33, 34].
Nel complesso, il nostro studio ha dimostrato che la PPP collega glicolisi e proliferazione nelle cellule tumorali aggressive e invita a considerare il metabolismo del cancro per la scelta di adeguate strategie terapeutiche antitumorali. In particolare, unendoci alle conclusioni di altri [27], suggeriamo che lo sviluppo clinico del DCA potrebbe trarre beneficio dalla selezione di pazienti con tumori altamente glicolitici.
MATERIALI E METODI
Coltura cellulare e reagenti
Il carcinoma squamoso della cervice umana SiHa (ATCC), il carcinoma mammario umano MDA-MB-231 (ATCC), l’epatocarcinoma di topo TLT (transplantable liver tumor) [35], il fibrosarcoma di topo FSaII [36], il sarcoma di topo KHT [37], il tumore mammario di topo NT2 [38] sono stati coltivati secondo le raccomandazioni del fornitore o come descritto. I SiHa con parziale deplezione mitocondriale (ρ0) sono stati ottenuti mediante esposizione cronica a basse concentrazioni di bromuro di etidio come descritto in precedenza [14,39]. Tutte le colture sono state mantenute a 37°C in atmosfera diCO2 al 5%. Le cellule sono state incubate in un unico terreno sperimentale 24 ore prima degli esperimenti (DMEM senza glutammina [Invitrogen], contenente 4,5 g/L di glucosio integrato con il 10% di FBS inattivato al calore e l’1% di penicillina-streptomicina). Quando i SiHa ρ0 sono stati confrontati con i SiHa wild-type, è stato utilizzato DMEM senza glutammina (Invitrogen), contenente 4,5 g/L di glucosio integrato con 1% di piruvato, 10% di FBS inattivato al calore, 1% di penicillina-streptomicina e 50 ng/ml di uridina. Se non diversamente specificato, gli esperimenti sono stati eseguiti su cellule confluenti. L’oligomicina è un inibitore dell’ATP sintasi. Il dicloroacetato (DCA) è un inibitore della piruvato deidrogenasi chinasi (PDK). la 6-aminonicotinamide (6-AN) è un inibitore degli enzimi NADP+ dipendenti della via del pentoso fosfato, la glucosio-6-fosfato deidrogenasi (G6PD) e la 6-fosfogluconato deidrogenasi (6-PGD). Tutti i prodotti chimici sono stati acquistati da Sigma. L’oligomicina è stata disciolta in DMSO. DCA e 6-AN sono stati disciolti direttamente nei terreni di coltura.
trasfezione di siRNA
ON-TARGETplus SMARTpool siRNA contro la G6PD umana e ON-TARGETplus Non-targeting siRNA sono stati prodotti da Dharmacon. La concentrazione finale di siRNA era di 25 nM. Tutti i siRNA sono stati trasfettati utilizzando RNAi/MAX secondo le istruzioni del produttore (Invitrogen).
Western blotting
I lisati di cellule intere sono stati raccolti e sottoposti ad analisi immunoblot come precedentemente descritto [40]. Gli anticorpi primari erano monoclonali umani contro G6PD o Hsp90 (Sigma).
Consumo di ossigeno mitocondriale
Il tasso di consumo di ossigeno (OCR) di cellule intere intatte è stato misurato utilizzando uno spettrometro Bruker EMX EPR operante a 9,5 GHz come precedentemente descritto [41]. Le cellule aderenti sono state raccolte in terreno sperimentale fresco (107 cellule/ml). 100 µl della sospensione cellulare sono stati mescolati con 100 µl di destrano al 20% per evitare agglomerazioni e le cellule sono state sigillate in un tubo capillare di vetro in presenza di 0,2 mM di una sonda nitrossido che funge da sensore di ossigeno (15N4-oxo-2,2,6,6-tetrametilpiperidina-d16-15N-1-oxil, CDN isotopes, Pointe-Claire, Quebec, Canada). Le cellule sono state mantenute a 37°C durante l’acquisizione degli spettri. La larghezza di linea EPR è stata misurata ogni minuto e riportata su una curva di calibrazione per ottenere la concentrazione di ossigeno. L’OCR è stato determinato dal valore assoluto della pendenza della diminuzione della concentrazione di ossigeno nel tubo capillare chiuso nel tempo [42]. Per ottenere l’OCR da fosforilazione ossidativa, l’OCR in presenza di trattamento con oligomicina 1 µM è stato sottratto dall’OCR totale delle cellule.
Consumo di glucosio e produzione di lattato
Il consumo di glucosio e la produzione di lattato sono stati misurati dai surnatanti delle cellule coltivate. Le concentrazioni di metaboliti sono state quantificate su campioni deproteinizzati utilizzando specifici saggi enzimatici su un analizzatore CMA600 (CMA Microdialysis AB, Solna, Svezia). Il consumo di glucosio e la produzione di lattato sono stati normalizzati rispetto al contenuto proteico utilizzando il saggio Pierce BCA Protein (Thermo Scientific). Per rilevare efficacemente le differenze nelle concentrazioni di metaboliti tra i surnatanti di SiHa WT e SiHa ρ0, è stato utilizzato un terreno a basso contenuto di glucosio (1 g/L).
Quantificazione dell’ATP intracellulare
L’ATP intracellulare totale è stato misurato con il kit di determinazione dell’ATP (Invitrogen) secondo il protocollo del produttore. Le cellule sono state lavate due volte con PBS e lisate nel tampone raccomandato dal produttore (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0,01% Triton X-100). I lisati cellulari sono stati aggiunti a una miscela di reazione contenente luciferasi e luciferina per la misurazione della bioluminescenza mediante un lettore di piastre (SpectraMax M2e, Molecular Devices). Una curva standard è stata generata con concentrazioni note di ATP nelle stesse condizioni. Una frazione dei lisati cellulari è stata sistematicamente utilizzata per la quantificazione delle proteine (Pierce BCA Protein assay, Thermo Scientific) per normalizzare il livello di ATP al contenuto proteico.
Proliferazione
La proliferazione delle cellule è stata analizzata con un kit 5-bromo-2′-deossiuridina (BrdU)-ELISA (Roche) seguendo le istruzioni del fornitore. Le cellule subconfluenti sono state incubate in presenza di BrdU (un analogo del nucleotide) per 4 ore. Quando è stata confrontata la proliferazione di SiHa WT e SiHa ρ0, le cellule sono state incubate per 6 ore in presenza di BrdU. La quantità di BrdU incorporata nelle cellule è stata valutata mediante misurazioni colorimetriche con un lettore di piastre (SpectraMax M2e, Molecular Devices), che ha permesso di quantificare la sintesi di DNA nelle cellule in replicazione.
Misurazioni di NADPH e NADP+
I livelli di NADP+ e NADPH sono stati rilevati singolarmente da 6.000 cellule raccolte utilizzando un kit di rilevamento (NADP/NADPH-Glo Assay, Promega) secondo le istruzioni del produttore.
Statistiche
Tutti i risultati sono espressi come media ± errore standard della media (SEM). Le analisi statistiche sono state eseguite utilizzando il software GraphPad Prism 5. P<0,05 è stato considerato statisticamente significativo.
RICONOSCIMENTI
Questo studio è stato sostenuto da sovvenzioni del Fonds National de la Recherche Scientifique (F.R.S.-FNRS, PDR T.0107.13), del Fonds Joseph Maisin, dell’Action de Recherches Concertées ARC 14/19-058 e da una sovvenzione di avviamento del Consiglio europeo della ricerca (ERC No. 243188 TUMETABO a P. Sonveaux). BFJ e PS sono associati alla ricerca, LB e PEP sono ricercatori post-dottorato, PD è borsista post-dottorato di Télévie. VLP è un borsista di dottorato del F.R.S.-FNRS. GDP è un borsista Télévie.
CONFLITTI DI INTERESSE
Gli autori hanno dichiarato che non esistono conflitti di interesse.
RIFERIMENTI
1 Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ e Rodriguez-Enriquez S. Chi controlla la fornitura di ATP nelle cellule tumorali? Lezioni di biochimica per comprendere il metabolismo energetico del cancro. Int J Biochem Cell Biol. 2014; 50:10-23.2 Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL e Smith JW. Profilazione comparativa del flusso metabolico di linee cellulari di melanoma: oltre l’effetto Warburg. J Biol Chem. 2011; 286:42626-42634.
3 Jose C, Bellance N e Rossignol R. Scegliere tra glicolisi e fosforilazione ossidativa: un dilemma del tumore? Biochim Biophys Acta. 2011; 1807:552-561.
4 Gatenby RA e Gillies RJ. Perché i tumori hanno un’elevata glicolisi aerobica? Nat Rev Cancer. 2004; 4:891-899.
5 Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O e Dewhirst MW. Il bersaglio della respirazione alimentata dal lattato uccide selettivamente le cellule tumorali ipossiche nei topi. J Clin Invest. 2008; 118:3930-3942.
6 Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B e Sonveaux P. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014; 8:754-766.
7 Rolfe DF e Brown GC. Utilizzo dell’energia cellulare e origine molecolare del tasso metabolico standard nei mammiferi. Physiol Rev. 1997; 77:731-758.
8 Warburg O. Uber den Stoffwechsel der Carcinomzelle. Klin Wochenschr. 1925;4:534-536..
9 Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P e Zhong H. “Il metabolismo dei tumori”: 70 anni dopo. Novartis Found Symp. 2001; 240:251-260; discussione 260-254.
10 Lunt SY e Vander Heiden MG. Glicolisi aerobica: soddisfare i requisiti metabolici della proliferazione cellulare. Annu Rev Cell Dev Biol. 2011; 27:441-464.
11 DeBerardinis RJ, Lum JJ, Hatzivassiliou G e Thompson CB. La biologia del cancro: la riprogrammazione metabolica alimenta la crescita e la proliferazione cellulare. Cell Metab. 2008; 7:11-20.
12 Tennant DA, Duran RV e Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010; 10:267-277.
13 Zancan P, Sola-Penna M, Furtado CM e Da Silva D. L’espressione differenziale delle isoforme di fosfofruttochinasi-1 è correlata all’efficienza glicolitica delle cellule di cancro al seno. Genetica molecolare e metabolismo. 2010; 100:372-378.
14 De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O e Sonveaux P. Il lattato attiva HIF-1 nelle cellule tumorali umane a ossidazione ma non a fenotipo Warburg. PLoS One. 2012; 7:e46571.
15 Bol V, Bol A, Bouzin C, Labar D, Lee JA, Janssens G, Porporato PE, Sonveaux P, Feron O e Gregoire V. La riprogrammazione del metabolismo tumorale attraverso il bersaglio dei mitocondri migliora la risposta del tumore all’irradiazione. Acta Oncol. 2014:1-9.
16 Stacpoole PW e Felts JM. Effetti del dicloroacetato di diisopropilammonio sul metabolismo del glucosio. Proc West Pharmacol Soc. 1969; 12:111-113.
17 Kankotia S e Stacpoole PW. Dicloroacetato e cancro: Una nuova casa per un farmaco orfano? Biochim Biophys Acta. 2014; 1846:617-629.
18 Abildgaard C, Dahl C, Basse AL, Ma T e Guldberg P. La modulazione bioenergetica con dicloroacetato riduce la crescita delle cellule di melanoma e potenzia la loro risposta all’inibizione di BRAFV600E. J Transl Med. 2014; 12:247.
19 Neveu MA, Bol V, Bol A, Bouzin C, Gregoire V, Feron O, Jordan BF e Gallez B. L’aumento dell’ossigenazione del tumore sotto respirazione di carbogeno induce una diminuzione dell’assorbimento di [F]-fluoro-deossiglucosio. Radiother Oncol. 2015; 116:400-3.
20 Jiang P, Du W e Wu M. Regolazione della via del pentoso fosfato nel cancro. Protein Cell. 2014; 5:592-602.
21 Koutcher JA, Alfieri AA, Matei C, Meyer KL, Street JC e Martin DS. Effetto della 6-aminonicotinamide sulla via del pentoso fosfato: studi di 31P NMR e di ritardo della crescita tumorale. Magn Reson Med. 1996; 36:887-892.
22 Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, Sharpe MA, Xin L e Frigo DE. Regolazione della via del pentoso fosfato attraverso un meccanismo mediato dal recettore degli androgeni e dalla MTOR e suo ruolo nella crescita delle cellule del cancro alla prostata. Oncogenesi. 2014; 3:e103.
23 Krebs HA e Eggleston LV. La regolazione del ciclo del pentoso fosfato nel fegato di ratto. Adv Enzyme Regul. 1974; 12:421-434.
24 Epstein T, Xu L, Gillies RJ e Gatenby RA. Separazione della domanda e dell’offerta metabolica: la glicolisi aerobica come normale risposta fisiologica a richieste energetiche fluttuanti nella membrana. Cancer Metab. 2014; 2:7.
25 Chen X, Qian Y e Wu S. L’effetto Warburg: Evoluzione delle interpretazioni di un concetto consolidato. Free Radic Biol Med. 2015; 79:253-263.
26 Porporato PE, Dhup S, Dadhich RK, Copetti T e Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol. 2011; 2:49.
27 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG e Newton DL. Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer. 2010; 127:2510-2519.
28 Babu E, Ramachandran S, CoothanKandaswamy V, Elangovan S, Prasad PD, Ganapathy V e Thangaraju M. Ruolo di SLC5A8, un trasportatore della membrana plasmatica e un soppressore tumorale, nell’attività antitumorale del dicloroacetato. Oncogene. 2011; 30:4026-4037.
29 Jackson VN e Halestrap AP. Cinetica, substrato e specificità degli inibitori del trasportatore di monocarbossilato (lattato) delle cellule epatiche di ratto, determinati utilizzando l’indicatore fluorescente del pH intracellulare, 2′,7′-bis(carbossietil)-5(6)-carbossifluoresceina. J Biol Chem. 1996; 271:861-868.
30 Bowker-Kinley MM, Davis WI, Wu P, Harris RA e Popov KM. Prove dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J. 1998; 329:191-196.
31 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B e Michelakis ED. L’asse mitocondrio-canale K+ è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del tumore. Cancer Cell. 2007; 11:37-51.
32 Pajuelo-Reguera D, Alan L, Olejar T e Jezek P. Il dicloroacetato stimola cambiamenti nella morfologia della rete mitocondriale attraverso una parziale mitofagia in cellule di neuroblastoma umano SH-SY5Y. Int J Oncol. 2015; 46:2409-2418.
33 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS e Michelakis ED. L’attivazione mitocondriale tramite l’inibizione di PDKII sopprime la segnalazione di HIF1a e l’angiogenesi nel cancro. Oncogene. 2013; 32:1638-1650.
34 Hong SE, Shin KS, Lee YH, Seo SK, Yun SM, Choe TB, Kim HA, Kim EK, Noh WC, Kim JI, Hwang CS, Lee JK, Hwang SG, Jin HO e Park IC. L’inibizione di S6K1 aumenta la morte cellulare indotta dal dicloroacetato. J Cancer Res Clin Oncol. 2015; 141:1171-1179.
35 Taper HS, Woolley GW, Teller MN e Lardis MP. Un nuovo tumore epatico di topo trapiantabile di origine spontanea. Cancer Res. 1966; 26:143-148.
36 Volpe JP, Hunter N, Basic I e Milas L. Proprietà metastatiche di sarcomi e carcinomi murini. I. Correlazione positiva con la colonizzazione polmonare e mancanza di correlazione con l’assunzione di tumori in s.c.. Clin Exp Metastasis. 1985; 3:281-294.
37 Rockwell S e Kallman RF. Cinetica di crescita e popolazione cellulare di sarcomi KHT singoli e multipli. Cell Tissue Kinet. 1972; 5:449-457.
38 Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH e Jaffee EM. HER-2/neu è un bersaglio del rigetto del tumore in topi transgenici HER-2/neu tollerati. Cancer Res. 2000; 60:3569-3576.
39 King MP e Attardi G. Cellule umane prive di mtDNA: ripopolamento con mitocondri esogeni mediante complementazione. Science. 1989; 246:500-503.
40 Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA e Michel T. Il targeting dell’ossido nitrico sintasi endoteliale alle caveole. Interazioni specifiche con le isoforme di caveolina nei miociti cardiaci e nelle cellule endoteliali. J Biol Chem. 1996; 271:22810-22814.
41 Diepart C, Verrax J, Calderon PB, Feron O, Jordan BF e Gallez B. Confronto tra metodi di misurazione del consumo di ossigeno in cellule tumorali in vitro. Anal Biochem. 2010; 396:250-256.
42 Jordan BF, Gregoire V, Demeure RJ, Sonveaux P, Feron O, O’Hara J, Vanhulle VP, Delzenne N e Gallez B. L’insulina aumenta la sensibilità dei tumori all’irradiazione: coinvolgimento di un aumento dell’ossigenazione tumorale mediato da una diminuzione del consumo di ossigeno delle cellule tumorali dipendente dall’ossido nitrico. Cancer Res. 2002; 62:3555-3561.
Contenuti correlati: