Жеральдин Де Претер1, Мари-Алин Невё1, Пьер Данье1, Люси Бриссон2, Валери Л. Пайен2, Паоло Е. Порпорато2, Бенедикт Ф. Джордан1, Пьер Сонво2 и Бернар Галлез1
1 Лувенский институт исследования лекарственных средств (LDRI), Группа исследований биомедицинского магнитного резонанса, Католический университет Лувена (UCL), Брюссель, Бельгия
Институт экспериментальных и клинических исследований (IREC), отделение фармакологии, Католический университет Лувена (UCL), Брюссель, Бельгия
Корреспонденция: Бернар Галлез, e-mail: [email protected]
Получено: 1 июня 2015 г.
Принято: 26 декабря 2020 г.
Опубликовано: 9 декабря 2020 г
Аннотация
Ферментация глюкозы посредством гликолиза даже в присутствии кислорода (эффект Варбурга) является общей особенностью раковых клеток, которые все чаще рассматриваются в качестве привлекательной мишени для клинических разработок. Целью данного исследования был анализ связи между метаболизмом, запасами энергии и скоростью пролиферации в раковых клетках. Мы обнаружили, что клеточная пролиферация, оцениваемая по количественному определению синтеза ДНК, коррелирует с гликолитической эффективностью в шести линиях раковых клеток, а также в изогенных линиях раковых клеток. Для дальнейшего изучения связи между гликолизом и пролиферацией был использован фармакологический ингибитор пентозофосфатного пути (PPP). Мы продемонстрировали, что снижение активности PPP уменьшает пролиферацию раковых клеток, оказывая значительный эффект на раковые клетки с фенотипом Варбурга. Решающая роль ППП в поддержании пролиферации раковых клеток была подтверждена с помощью siRNAs против глюкозо-6-фосфатдегидрогеназы, первого и лимитирующего фермента ППП. Кроме того, мы обнаружили, что дихлорацетат (ДХА), новое клинически испытанное соединение, индуцирует переход гликолитических раковых клеток к более окислительному фенотипу и снижает пролиферацию. Показав, что ДХА снижает активность ППП, мы представили новый механизм, с помощью которого ДХА контролирует пролиферацию раковых клеток.
Ключевые слова: биоэнергетика, гликолиз, дихлорацетат, пентозофосфатный путь, пролиферация
© 2020 авторы. Лицензиат MDPI, Базель, Швейцария. Данная статья является статьей с открытым доступом и распространяется на условиях лицензии Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
ВВЕДЕНИЕ
В последние несколько лет метаболизм вызывает огромный интерес в области изучения рака. Многие исследования были посвящены различным метаболическим профилям различных опухолей [1-3], поскольку метаболическая пластичность вовлечена в прогрессирование рака, лекарственную устойчивость и метастазирование [4-6]. В нормальных клетках гликолиз соединен с окислительным фосфорилированием (OXPHOS) для оптимального синтеза внутриклеточного АТФ из глюкозы [7]. Однако многие раковые клетки претерпевают фундаментальную метаболическую трансформацию, «гликолитический переключатель», в результате которой гликолиз отсоединяется от дыхания и переключается на молочнокислую ферментацию, становясь, таким образом, основным источником производства АТФ в клетке. Переключение на гликолитический метаболизм происходит в основном в условиях гипоксии как механизм спасения для производства энергии. Однако некоторые раковые клетки в дальнейшем принимают особый гликолитический фенотип, впервые описанный Варбургом [8] и получивший название «аэробный гликолиз» [9]. Биологическое обоснование фенотипа Варбурга остается спорным, но недавно было выдвинуто предположение, что пролиферирующие раковые клетки усиливают гликолиз, поскольку он приносит пользу как биоэнергетике, так и биосинтезу [4,10]. Действительно, гликолитический метаболизм потенциально обеспечивает быстрое производство АТФ и дает промежуточные углеродные соединения, которые могут быть направлены в разветвленные биосинтетические пути, что позволяет быстрее наращивать клеточную биомассу. Убедительно, что мутации, происходящие в сигнальных путях, регулирующих как клеточный биосинтез, так и аэробный гликолиз, таких как путь PI3K/Akt/mTOR, являются наиболее распространенным классом мутаций в опухолях человека [11]. Однако экспериментальные данные, связывающие аэробный гликолиз с пролиферацией раковых клеток, отсутствуют, и селективное преимущество, обеспечиваемое этим фенотипом, не совсем ясно. Целью настоящего исследования было выяснить связь между метаболизмом, энергообеспечением и клеточной пролиферацией в различных раковых клетках человека и мыши. Метаболические переключения были индуцированы, чтобы предоставить доказательства того, что биоэнергетика, и в частности гликолиз, непосредственно управляет пролиферацией раковых клеток. В этом ряду мы обнаружили новый терапевтический механизм дихлорацетата (DCA), активатора митохондриального окисления глюкозы, который в настоящее время исследуется в клинических исследованиях [12]. ДХА ингибирует пентозофосфатный путь (ПФП), важнейший биосинтетический путь, разветвляющийся на гликолиз. Мы сообщаем, что PPP перекидывает мост между гликолитическим метаболизмом и пролиферацией раковых клеток.
РЕЗУЛЬТАТЫ
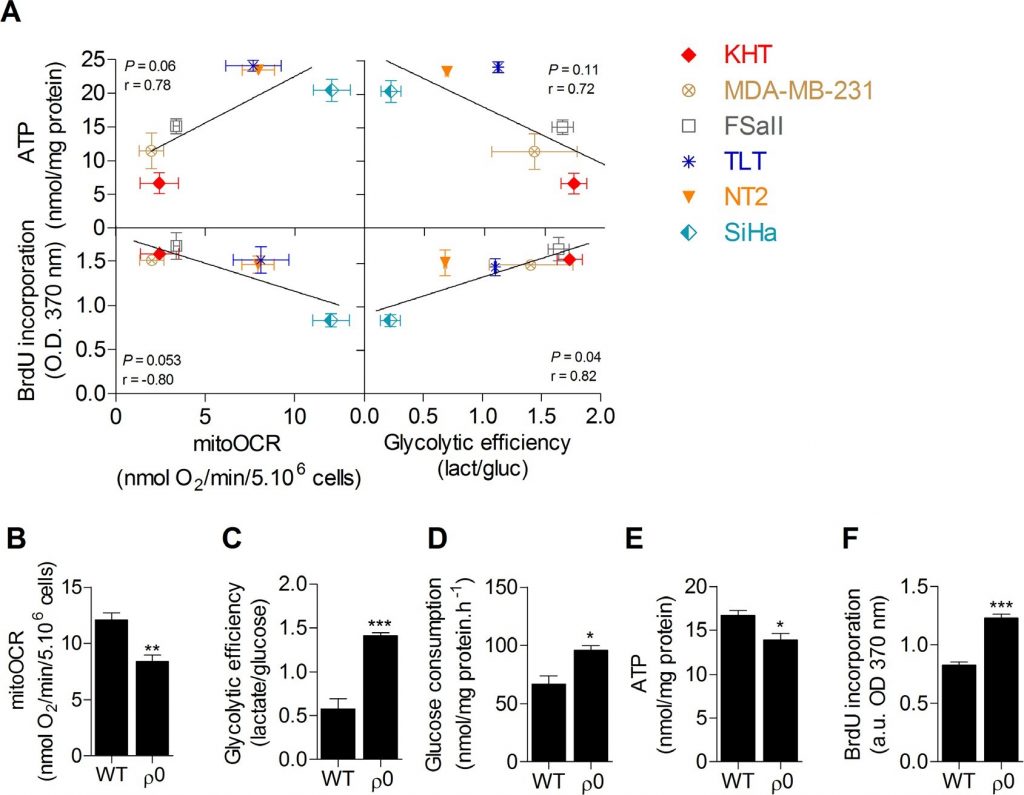
Гликолитическая эффективность положительно связана с пролиферацией, но не с уровнем АТФ в раковых клетках
Сначала был проведен скрининг метаболической активности шести линий раковых клеток для изучения связи между метаболизмом глюкозы, запасами АТФ и способностью к пролиферации. OXPHOS, оцениваемая по скорости потребления кислорода, чувствительной к олигомицину (mitoOCR), и гликолитическая эффективность, измеряемая отношением произведенного лактата к потребленной глюкозе (моль/моль) [13], оценивались отдельно на клетках, инкубированных 24 часа в культуральной среде, содержащей только глюкозу в качестве энергетического топлива. Для изучения влияния метаболического статуса на клеточные запасы энергии также измеряли общий внутриклеточный пул АТФ. Не было обнаружено значительной корреляции между содержанием внутриклеточного АТФ и метаболическими параметрами. Однако мы обнаружили, что пролиферация клеток, оцениваемая по синтезу ДНК, значительно коррелировала с гликолитической эффективностью (Рисунок 1A), где увеличение гликолиза было связано с увеличением пролиферативной способности, и обратно. Значительной корреляции между способностью к пролиферации и митохондриальным дыханием не наблюдалось. Эти данные позволяют предположить, что гликолиз потенциально является ключевым энергетическим путем, поддерживающим пролиферацию раковых клеток. В настоящее время существует гипотеза, что этот путь поддерживает пролиферацию клеток преимущественно за счет поставки предшественников для биосинтетических путей, а не за счет производства АТФ [10,11]. В пользу того, что поступление АТФ не является основным ограничивающим фактором для клеточной пролиферации, свидетельствует отсутствие корреляции между содержанием АТФ и синтезом ДНК в клеточных линиях (Дополнительный рисунок S1). Чтобы подтвердить, что гликолиз способствует пролиферации раковых клеток, и исключить независимые от метаболизма влияния, присущие различным генотипам, способность к пролиферации раковых клеток шейки матки SiHa человека дикого типа (WT) к окислению сравнивали со способностью SiHa с частичным истощением митохондрий (SiHa ρ0) [14,15]. Раковые клетки SiHa ρ0 демонстрировали гликолитический фенотип с ~40% снижением митоОКР (Рисунок 1B), ~2,5-кратным увеличением гликолитической эффективности (Рисунок 1C) и ~50% увеличением потребления глюкозы (Рисунок 1D). Чистое снижение общего пула АТФ на ~20% в клетках SiHa ρ0 подтвердило, что усиленный гликолиз был недостаточен для поддержания запасов АТФ (Рисунок 1E). Проведя количественный анализ синтеза ДНК, мы обнаружили, что усиленный аэробный гликолиз способствует пролиферации клеток, так как гликолитический переход в клетках SiHa ρ0 был связан с увеличением пролиферации клеток на ~45% (Рисунок 1F). Анализ жизнеспособности не выявил различий в количестве жизнеспособных клеток через 24 ч после инкубации клеток в экспериментальной культуральной среде (Дополнительный рисунок S2), вероятно, потому что количественный анализ синтеза ДНК выявляет ранние изменения в скорости пролиферации клеток.
Ингибирование гликолиза с помощью DCA ухудшает пролиферацию раковых клеток
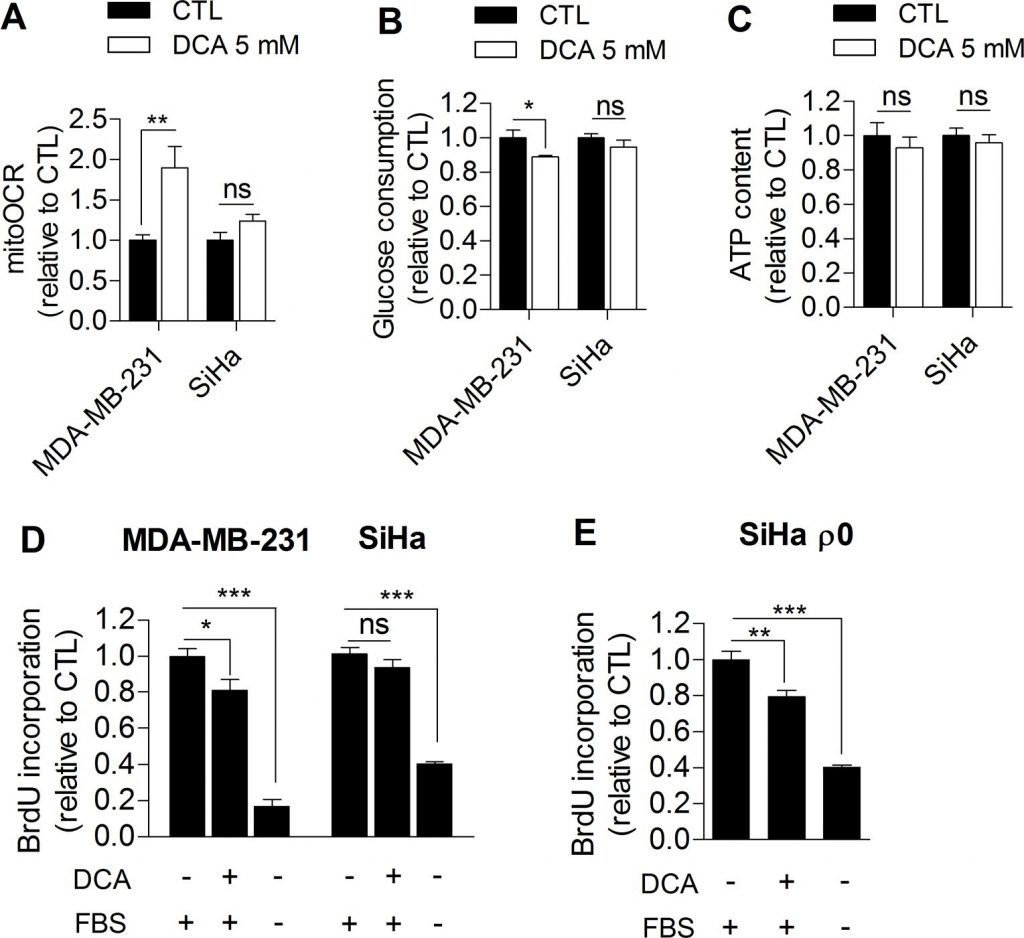
Основываясь на наших наблюдениях, мы далее исследовали, может ли ингибирование гликолиза непосредственно ухудшить пролиферацию раковых клеток. Для этого клетки рака молочной железы человека MDA-MB-231 (фенотип Варбурга, рисунок 1А) и клетки рака шейки матки человека SiHa (окислительный фенотип, рисунок 1А) обрабатывали дихлорацетатом (DCA), ингибитором киназы пируватдегидрогеназы (PDK), который усиливает окислительную активность клеток путем активации пируватдегидрогеназы (PDH), фермента, поддерживающего окисление глюкозы в митохондриях [16]. На сегодняшний день многообещающее терапевтическое действие DCA на раковые клетки во всем мире связывают с нормализацией гиперполяризованного мембранного потенциала митохондрий, характерного для раковых клеток, и с ресенсибилизацией к апоптозу [17]. Здесь мы предположили, что DCA также контролирует пролиферацию опухоли путем ингибирования гликолиза. Для проверки этой гипотезы гликолитические MDA-MB-231 и окислительные SiHa раковые клетки обрабатывали 5 мМ DCA в течение 48 ч и оценивали влияние лечения на метаболизм и пролиферацию. По сравнению с клетками, обработанными транспортным средством, ДКА вызывал переход гликолитических раковых клеток MDA-MB-231 в более окислительный фенотип, о чем свидетельствует увеличение митоОКР (рис. 2A) и снижение потребления глюкозы (рис. 2B). Снижение гликолитической активности, наблюдаемое в этом эксперименте, согласуется с результатами другого недавнего исследования [18] и, вероятно, индуцируется эффектом Пастера [4,19] для поддержания гомеостаза АТФ в клетках (Рисунок 2C). Мы также наблюдали, что ингибирование гликолиза с помощью DCA было связано со снижением скорости пролиферации раковых клеток MDA-MB-231 (Рисунок 2D). В подтверждение того, что ингибирование гликолиза нарушает пролиферацию в этой линии клеток, раковые клетки MDA-MB-231, обработанные 2-дезокси-D-глюкозой, также демонстрировали снижение скорости пролиферации (Дополнительный рисунок S3).
С другой стороны, DCA существенно не изменял метаболическую активность окисленных раковых клеток SiHa (Рисунок 2A-2C) и не оказывал значительного влияния на пролиферацию SiHa (Рисунок 2D). Краткосрочные (1 час) измерения выработки лактата показали, что DCA действительно более эффективен в гликолитической линии раковых клеток, чем в окислительной (Дополнительный рисунок S4). Чтобы выяснить, определяет ли метаболический профиль ответ на DCA, способность к пролиферации гликолитических раковых клеток SiHa ρ0 также была проанализирована после лечения DCA. Мы обнаружили значительное снижение синтеза ДНК в этой линии клеток (Рисунок 2E), эффект, который не наблюдался в SiHa WT. Кроме того, одинаковое количество жизнеспособных раковых клеток MDA-MB-231 и SiHa ρ0 было измерено через 48 ч после обработки транспортным средством или DCA (Дополнительный рисунок S5 A-B), что показывает, что воздействие DCA на метаболические функции и скорость пролиферации не связано с гибелью клеток.
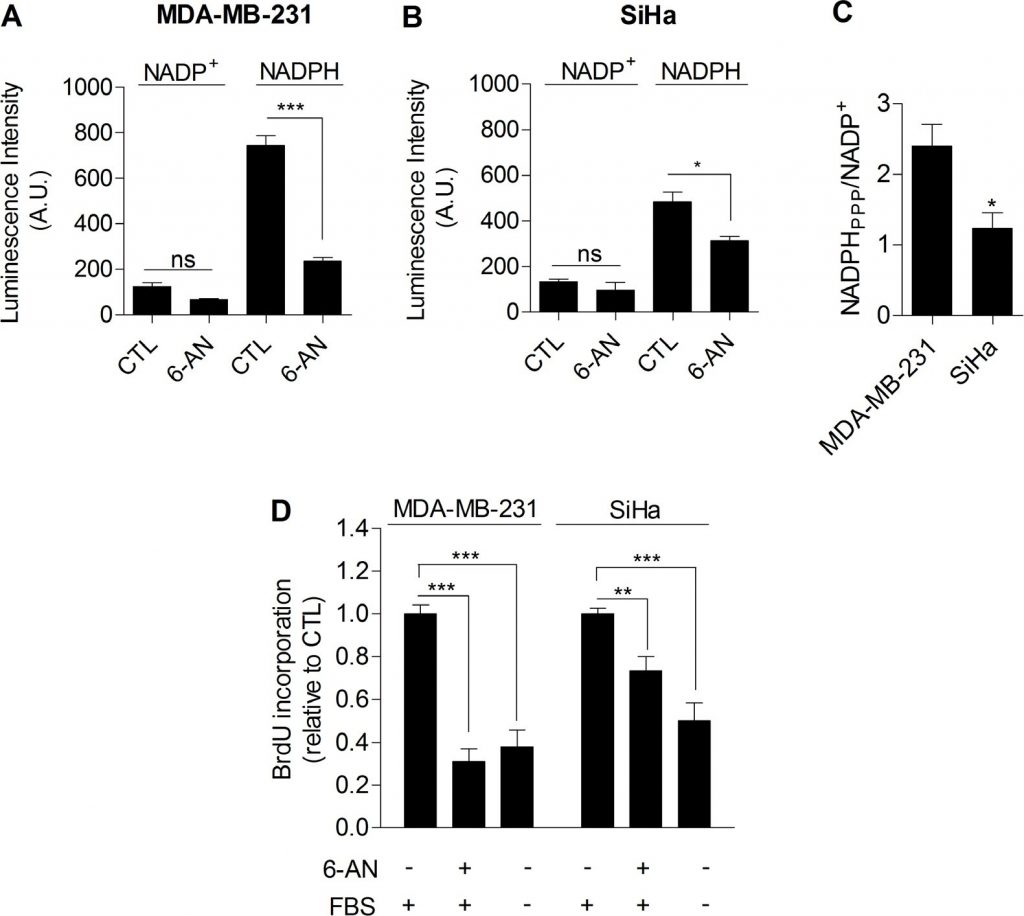
Гликолиз контролирует пролиферацию раковых клеток через пентозофосфатный путь
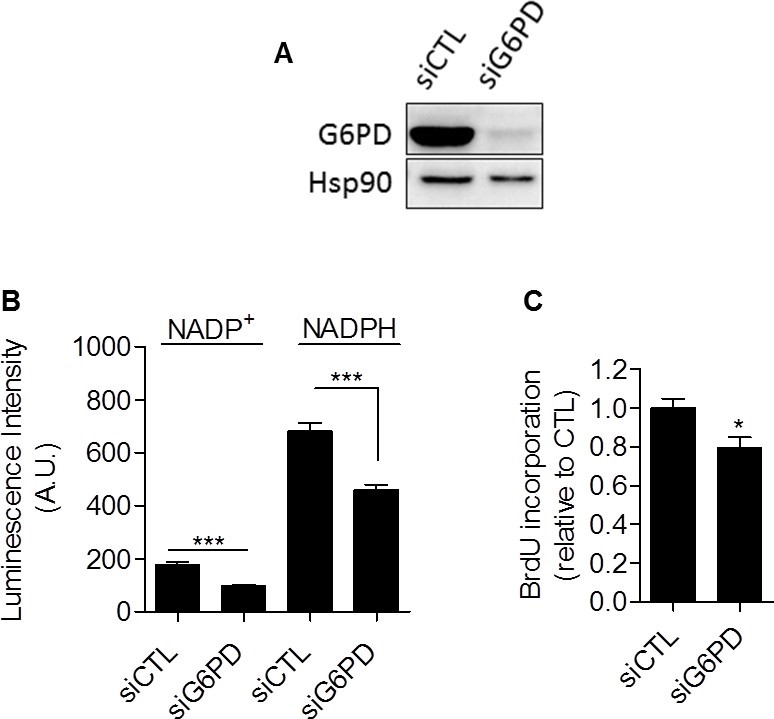
В совокупности наши данные предоставили убедительные экспериментальные доказательства того, что гликолиз как таковой участвует в контроле клеточной пролиферации. Механизм, связывающий гликолиз и пролиферацию, еще предстоит выяснить. Мы предположили, что пентозофосфатный путь (ПФП) может связывать гликолиз с пролиферацией, поскольку ПФП использует промежуточные продукты гликолиза для снабжения клеток нуклеотидами и NADPH, важнейшим восстановителем в анаболических процессах [20]. Для конкретного определения NADPH, образующегося в ППП (NADPHppp), клетки обрабатывали 6-аминоникотинамидом (6-AN), специфическим ингибитором ППП [21,22]. Мы обнаружили, что вклад NADPHppp в общий пул NADPH (NADPHtot) был более преобладающим в раковых клетках MDA-MB-231, чем в раковых клетках SiHa, о чем свидетельствует значительное снижение уровня NADPHtot после обработки 6-AN в гликолитических клетках MDA-MB-231 (Рисунок 3A). Более ограниченный эффект наблюдался в окислительных клетках SiHa (Рисунок 3B). Измерения также показали более высокое соотношение NADPHppp/NADP+ в раковых клетках MDA-MB-231, что свидетельствует о более высоком потоке PPP в этой гликолитической линии клеток по сравнению с SiHa (Рисунок 3C). Чтобы убедиться, что ППП участвует в контроле пролиферации раковых клеток, оценивали синтез ДНК в клетках, обработанных и не обработанных 6-AN. Мы наблюдали, что способность раковых клеток к пролиферации снижалась при ингибировании PPP (Рисунок 3D). Интересно, что более сильный эффект наблюдался в гликолитических MDA-MB-231 (~70 % снижение скорости синтеза ДНК) по сравнению с окислительными раковыми клетками SiHa (~25 % снижение скорости синтеза ДНК) (рис. 3D). Важно отметить, что SiHa ρ0 были более чувствительны к 6-AN, чем SiHa WT, так как наблюдалось снижение синтеза ДНК на ~40 % (Дополнительный рисунок S6). В связи с потенциальными внецелевыми эффектами фармакологических ингибиторов мы дополнили наши исследования 6-AN, используя малые интерферирующие РНК (siRNAs), нацеленные на глюкозо-6-фосфатдегидрогеназу (G6PD), первый и лимитирующий фермент ППП [23]. В трансфицированных клетках MDA-MB-231 мы подтвердили глушение G6PD с помощью иммуноблотинга (Рисунок 4A) и продемонстрировали, что, как и 6-AN, ингибирование G6PD снижает активность ППП (Рисунок 4B) и синтез ДНК (Рисунок 4C). Эти результаты указывают на преобладающий вклад ППП в поддержание пролиферации раковых клеток варбургского фенотипа.
DCA ингибирует пентозофосфатный путь
Наконец, мы исследовали, может ли DCA ингибировать ППП, что объясняет связь между ингибированием гликолиза и снижением скорости пролиферации раковых клеток, обработанных DCA. Как показано на рисунке 5, мы наблюдали, что DCA (5 мМ, 48 ч) значительно снижал уровень NADPHtot в раковых клетках MDA-MB-231 (рисунок 5A). Более того, хотя при совместном воздействии DCA и 6-AN на клетки дополнительного снижения NADPHtot не происходило (Рисунок 5A), мы показали, что DCA специфически снижает уровень NADPHPPP. Аналогичные результаты были получены при молекулярном ингибировании G6PD (Рисунок 5B). Эти результаты демонстрируют участие PPP в воздействии DCA на гликолитические раковые клетки.
ДИСКУССИЯ
Данное исследование подчеркивает преобладающую роль пентозофосфатного пути в биологии рака. Эффект Варбурга (ферментация глюкозы до лактата в присутствии физиологической концентрации O2 ), наблюдаемый в многочисленных раковых клетках, был предложен для удовлетворения энергетических (АТФ) и биосинтетических потребностей [4,10]. В данном исследовании мы показали, что усиленный аэробный гликолиз был недостаточен для поддержания уровня АТФ в раковых клетках. Следовательно, низкий уровень внутриклеточного АТФ был связан с усилением гликолиза. Более того, гликолитический переключатель, индуцированный в раковых клетках с дефицитом митохондрий, не мог обеспечить адекватное снабжение АТФ по сравнению с клетками дикого типа. Существует предположение, что с биоэнергетической точки зрения гликолитический метаболизм менее эффективен, чем окислительный, но гликолиз может быть выгоден для быстрого производства АТФ для удовлетворения кратковременных энергетических потребностей [24]. Наши результаты также подтверждают, что раковые клетки с Варбургским фенотипом развивают ряд адаптаций для поддержания низкого уровня АТФ, чтобы избежать аллостерического ингибирования лимитирующих скорость гликолитических ферментов (эффект Пастера) и поддерживать повышенную скорость гликолитического потока [10]. С другой стороны, мы показали, что гликолиз способствует пролиферации раковых клеток, что подтверждает клинически опробованные стратегии, ингибирующие эффект Варбурга [12,25]. Положительная связь между гликолитической эффективностью (моль лактата, произведенного на моль потребленной глюкозы) и скоростью пролиферации раковых клеток показала, что быстро делящиеся клетки ферментируют большое количество глюкозы в лактат. Недавние исследования показали, что производство лактата позволяет раковым клеткам эффективно восстанавливать NAD+ с помощью фермента лактатдегидрогеназы [26]. Поддерживая редокс-баланс NAD+/NADH, раковые клетки обеспечивают более быстрый поток глюкозы через гликолиз, а также более быстрое включение метаболитов глюкозы в биосинтетические пути, тем самым обеспечивая преимущества для пролиферации [10].
Чтобы проверить, способствует ли усиленный аэробный гликолиз пролиферации раковых клеток путем подпитки анаболических процессов, было оценено участие пентозофосфатного пути (PPP). Действительно, известно, что ППП использует глюкозо-6-фосфат (продукт гликолитического фермента гексокиназы) для снабжения клеток нуклеотидами и NADPH, важнейшим восстановителем в анаболических реакциях [20]. В нашем исследовании мы показали, что поток ППП, оцениваемый по соотношению NADPHppp/NADP+, был повышен в клетках варбургского фенотипа по сравнению с раковыми клетками окислительного типа. Для подтверждения этой разницы в потоке ППП можно было бы также провести сравнение активности ферментов ППП. Тем не менее, наши результаты подтверждают, что активация гликолиза сопровождается увеличением активности ППП для биосинтеза в быстро делящихся клетках [20]. Убедительно, что ингибирование ППП снижало пролиферацию раковых клеток, причем наибольший эффект наблюдался в раковых клетках варбургского фенотипа. Различная чувствительность гликолитических и окислительных раковых клеток к ингибированию ППП, вероятно, связана с различной зависимостью от этого пути синтеза макромолекул, таких как липиды и нуклеотиды. Кроме того, раковые клетки с повышенной митохондриальной активностью могут компенсировать уровень NADPH, используя ферменты, связанные с циклом ТСА (яблочные ферменты и изоцитратдегидрогеназы), для пополнения пула NADPH [20]. В совокупности эти результаты показывают, что раковые клетки, особенно агрессивные раковые клетки варбургского фенотипа, полагаются на пентозофосфатный путь для оптимальной пролиферации.
Чтобы оценить терапевтическую значимость этих результатов, было исследовано действие DCA. DCA уже продемонстрировал интересные противораковые свойства in vitro и in vivo
[17], и в настоящее время проходит клинические испытания I-II фазы [12]. На сегодняшний день DCA объясняет свои терапевтические свойства ресенсибилизацией раковых клеток к апоптозу [17]. Однако, как недавно отметили Стоквин и другие [27], DCA относительно неактивен в отношении жизнеспособности клеток и вызывает апоптоз только при высоких концентрациях. Стоквин и др. также сообщили, что DCA был преимущественно активен в клетках с дефектами митохондрий. Поэтому мы предположили, что при более низкой дозе DCA также снижает пролиферацию раковых клеток за счет уменьшения гликолиза. Мы обнаружили, что 5 мМ DCA был более эффективен в раковых клетках с фенотипом Варбурга, снижая пролиферацию клеток за счет уменьшения гликолиза. Различная чувствительность клеточных линий с разным метаболическим фенотипом может быть результатом способности ДКА достигать PDK в матриксе митохондрий. Действительно, как и пируват, ДКА ионизирован и не может проходить через мембраны. Любопытно, что имеется лишь несколько сообщений о транспорте ДКА в цитозоль клеток млекопитающих [28,29]. Хотя это еще не исследовано, уровень экспрессии митохондриальных транспортеров пирувата (MPC) также может определять эффективность DCA. Кроме того, поскольку ДКА имеет различный Kiдля каждого из четырех изоферментов PDK [30], различная чувствительность раковых клеток может быть результатом дифференциальной экспрессии PDK.
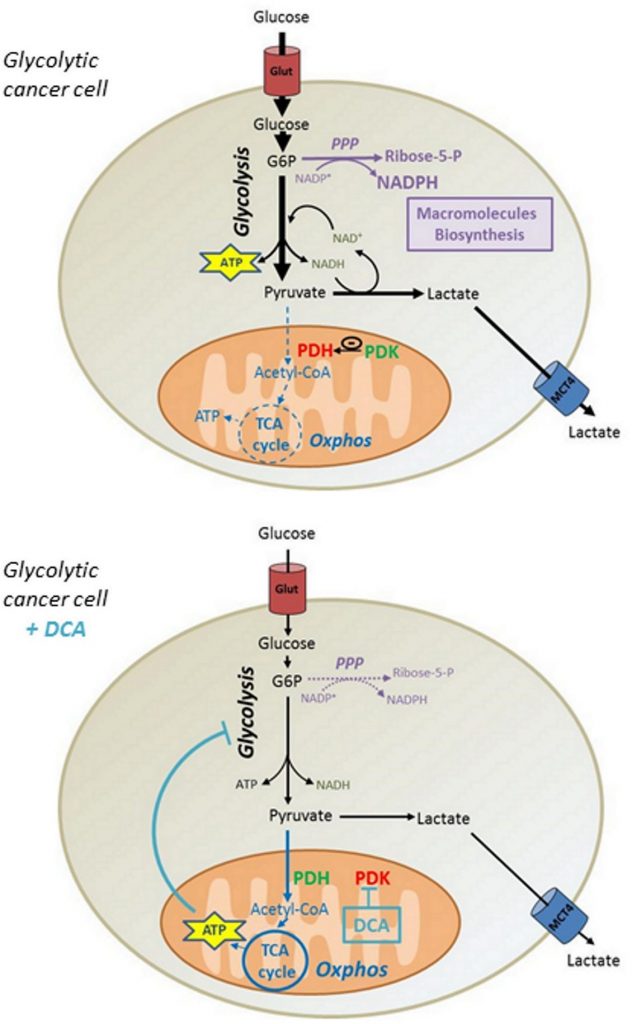
Хотя в некоторых отчетах показано, что DCA в сопоставимых концентрациях способствует цитотоксичности других клеточных линий [31,32], мы показали, что DCA не влияет на жизнеспособность раковых клеток MDA-MB-231 и SiHa. Основываясь на наших данных, мы предполагаем, что реактивация производства АТФ в митохондриях, индуцированная DCA, снижает гликолитический поток раковых клеток, тем самым уменьшая включение глюкозы. Следовательно, снижение синтеза гликолитических промежуточных продуктов снижает активность биосинтетических путей, таких как ППП, что ставит под угрозу пролиферацию раковых клеток варбургского фенотипа (рис. 6). Тем не менее, можно предположить, что в антипролиферативном действии DCA могут участвовать и другие механизмы, поскольку другие исследования показали остановку клеточного цикла при сопоставимых концентрациях DCA [33, 34].
В целом, наше исследование показало, что PPP связывает гликолиз и пролиферацию в агрессивных раковых клетках, и требует учета метаболизма рака для выбора адекватных терапевтических противораковых стратегий. В частности, присоединяясь к выводам других авторов [27], мы предполагаем, что клиническая разработка DCA может выиграть от отбора пациентов с высокогликолитическими опухолями.
МАТЕРИАЛЫ И МЕТОДЫ
Культура клеток и реактивы
SiHa плоскоклеточная карцинома шейки матки человека (ATCC), MDA-MB-231 рак молочной железы человека (ATCC), TLT (трансплантируемая опухоль печени) гепатокарцинома мыши [35], FSaII фибросаркома мыши [36], KHT саркома мыши [37], NT2 опухоль молочной железы мыши [38] выращивались в соответствии с рекомендациями поставщика или согласно описанию. SiHa с частичным истощением митохондрий (ρ0) получали путем хронического воздействия низких концентраций бромистого этидия, как описано ранее [14,39]. Все культуры содержались при 37°C в атмосфере 5%CO2. За 24 часа до начала экспериментов клетки инкубировали в уникальной экспериментальной среде (DMEM без глутамина [Invitrogen], содержащей 4,5 г/л глюкозы, дополненной 10% термоинактивированным FBS и 1% пенициллином-стрептомицином). Когда SiHa ρ0 сравнивали с SiHa дикого типа, использовали DMEM без глютамина (Invitrogen), содержащую 4,5 г/л глюкозы, дополненную 1% пирувата, 10% термоинактивированного FBS, 1% пенициллина-стрептомицина и 50 нг/мл уридина. Если не указано иное, эксперименты проводились на конфлюентных клетках. Олигомицин — ингибитор АТФ-синтазы. Дихлорацетат (DCA) — ингибитор киназы пируватдегидрогеназы (PDK). 6-аминоникотинамид (6-AN) является ингибитором NADP+-зависимых ферментов пентозофосфатного пути, глюкозо-6-фосфатдегидрогеназы (G6PD) и 6-фосфоглюконатдегидрогеназы (6-PGD). Все химические вещества были приобретены у компании Sigma. Олигомицин был растворен в ДМСО. DCA и 6-AN были непосредственно растворены в культуральной среде.
трансфекция siRNA
ON-TARGETplus SMARTpool siRNA против человеческого G6PD и ON-TARGETplus Non-targeting siRNA были получены от Dharmacon. Конечная концентрация siRNA составляла 25 нМ. Все siRNA были трансфецированы с помощью RNAi/MAX в соответствии с инструкциями производителя (Invitrogen).
Вестерн-блоттинг
Цельные лизаты клеток собирали и подвергали иммуноблот-анализу, как описано ранее [40]. Первичными антителами были человеческие моноклональные антитела против G6PD или Hsp90 (Sigma).
Потребление кислорода митохондриями
Скорость потребления кислорода (СПК) интактных целых клеток измеряли с помощью ЭПР-спектрометра Bruker EMX, работающего на частоте 9,5 ГГц, как описано ранее [41]. Адгезивные клетки собирали в свежей экспериментальной среде (107 клеток/мл). 100 мкл клеточной суспензии смешивали со 100 мкл 20% декстрана, чтобы избежать агломерации, и клетки запечатывали в стеклянную капиллярную трубку в присутствии 0,2 мМ нитроксидного зонда, действующего как датчик кислорода (15N4-оксо-2,2,6,6-тетраметилпиперидин-d16-15N-1-оксил, CDN isotopes, Pointe-Claire, Quebec, Canada). Во время получения спектров клетки поддерживали при 37°C. Ширину линии ЭПР измеряли каждую минуту и наносили на калибровочную кривую для получения концентрации кислорода. OCR определяли по абсолютному значению наклона снижения концентрации кислорода в закрытой капиллярной трубке с течением времени [42]. Чтобы получить OCR от окислительного фосфорилирования, OCR в присутствии обработки 1 мкМ олигомицином вычитали из общего OCR клеток.
Потребление глюкозы и продукция лактата
Потребление глюкозы и продукцию лактата измеряли в супернатантах культивируемых клеток. Концентрацию метаболитов определяли количественно в депротеинизированных образцах с помощью специфических ферментативных анализов на анализаторе CMA600 (CMA Microdialysis AB, Solna, Швеция). Потребление глюкозы и производство лактата нормировали на содержание белка с помощью анализатора Pierce BCA Protein assay (Thermo Scientific). Для эффективного выявления различий в концентрации метаболитов между супернатантами SiHa WT и SiHa ρ0 использовали среду с низким содержанием глюкозы (1 г/л).
Количественное определение внутриклеточного АТФ
Общий внутриклеточный АТФ измеряли с помощью набора ATP Determination Kit (Invitrogen) в соответствии с протоколом производителя. Клетки дважды промывали PBS и лизировали в буфере, рекомендованном производителем (10 мМ Трис, 1 мМ ЭДТА, 100 мМ NaCl, 0,01% Тритон Х-100). Лизаты клеток добавляли в реакционную смесь, содержащую люциферазу и люциферин, для измерения биолюминесценции с помощью планшетного ридера (SpectraMax M2e, Molecular Devices). Стандартная кривая была построена с использованием известных концентраций АТФ в тех же условиях. Часть клеточных лизатов систематически использовалась для количественного определения белка (Pierce BCA Protein assay, Thermo Scientific) для нормализации уровня АТФ к содержанию белка.
Пролиферация
Пролиферацию клеток определяли с помощью 5-бром-2′-дезоксиуридина (BrdU)-ELISA набора (Roche) в соответствии с инструкциями поставщика. Субконфлюэнтные клетки инкубировали в присутствии BrdU (аналог нуклеотида) в течение 4 часов. При сравнении пролиферации SiHa WT и SiHa ρ0 клетки инкубировали в течение 6 часов в присутствии BrdU. Количество BrdU, включенного в клетки, оценивали колориметрически с помощью планшетного ридера (SpectraMax M2e, Molecular Devices), что позволяло количественно оценить синтез ДНК в реплицирующихся клетках.
Измерения NADPH и NADP+
Уровни NADP+ и NADPH определяли по отдельности из 6000 собранных клеток с помощью набора для определения (NADP/NADPH-Glo Assay, Promega) в соответствии с инструкциями производителя.
Статистика
Все результаты выражены как среднее значение ± стандартная ошибка среднего (SEM). Статистический анализ проводили с помощью программы GraphPad Prism 5. Статистически значимым считалось значение P<0,05.
БЛАГОДАРНОСТИ
Данное исследование было поддержано грантами Национального фонда научных исследований (F.R.S.-FNRS, PDR T.0107.13), Фонда Жозефа Мейсина, Концертной программы научных исследований ARC 14/19-058 и стартовым грантом Европейского исследовательского совета (ERC № 243188 TUMETABO для П. Сонво). BFJ и PS являются научными сотрудниками, LB и PEP — постдокторскими исследователями, PD — постдокторским стипендиатом Télévie. VLP — научный сотрудник F.R.S.-FNRS. ВВП — научный сотрудник программы Télévie PhD.
КОНФЛИКТЫ ИНТЕРЕСОВ
Авторы заявили об отсутствии конфликта интересов.
ССЫЛКИ
1 Морено-Санчес Р, Марин-Эрнандес А, Сааведра Е, Пардо Ж.П., Ральф С.Дж. и Родригес-Энрикес С. Кто контролирует поступление АТФ в раковые клетки? Уроки биохимии для понимания энергетического метаболизма рака. Int J Biochem Cell Biol. 2014; 50:10-23.2 Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL and Smith JW. Сравнительное профилирование метаболических потоков клеточных линий меланомы: за пределами эффекта Варбурга. J Biol Chem. 2011; 286:42626-42634.
3 Jose C, Bellance N и Rossignol R. Выбор между гликолизом и окислительным фосфорилированием: дилемма опухоли? Biochim Biophys Acta. 2011; 1807:552-561.
4 Gatenby RA and Gillies RJ. Почему раковые опухоли имеют высокий аэробный гликолиз? Nat Rev Cancer. 2004; 4:891-899.
5 Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O and Dewhirst MW. Нацеливание на лактат-заправленное дыхание избирательно убивает гипоксические опухолевые клетки у мышей. J Clin Invest. 2008; 118:3930-3942.
6 Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B and Sonveaux P. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014; 8:754-766.
7 Rolfe DF and Brown GC. Утилизация энергии клетками и молекулярное происхождение стандартной скорости метаболизма у млекопитающих. Physiol Rev. 1997; 77:731-758.
8 Warburg O. Uber den Stoffwechsel der Carcinomzelle. Klin Wochenschr. 1925;4:534-536..
9 Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P and Zhong H. ‘The metabolism of tumours’: 70 years later. Novartis Found Symp. 2001; 240:251-260; обсуждение 260-254.
10 Lunt SY and Vander Heiden MG. Аэробный гликолиз: удовлетворение метаболических потребностей клеточной пролиферации. Annu Rev Cell Dev Biol. 2011; 27:441-464.
11 DeBerardinis RJ, Lum JJ, Hatzivassiliou G and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7:11-20.
12 Tennant DA, Duran RV и Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010; 10:267-277.
13 Zancan P, Sola-Penna M, Furtado CM and Da Silva D. Дифференциальная экспрессия изоформ фосфофруктокиназы-1 коррелирует с гликолитической эффективностью клеток рака молочной железы. Молекулярная генетика и метаболизм. 2010; 100:372-378.
14 De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O и Sonveaux P. Лактат активирует HIF-1 в окислительных, но не в опухолевых клетках человека варбургского фенотипа. PLoS One. 2012; 7:e46571.
15 Bol V, Bol A, Bouzin C, Labar D, Lee JA, Janssens G, Porporato PE, Sonveaux P, Feron O and Gregoire V. Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation. Acta Oncol. 2014:1-9.
16 Stacpoole PW and Felts JM. Влияние диизопропиламмония дихлорацетата на метаболизм глюкозы. Proc West Pharmacol Soc. 1969; 12:111-113.
17 Kankotia S and Stacpoole PW. Дихлорацетат и рак: Новый дом для сиротского препарата? Biochim Biophys Acta. 2014; 1846:617-629.
18 Abildgaard C, Dahl C, Basse AL, Ma T and Guldberg P. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition. J Transl Med. 2014; 12:247.
19 Neveu MA, Bol V, Bol A, Bouzin C, Gregoire V, Feron O, Jordan BF and Gallez B. Увеличение оксигенации опухоли при карбогенном дыхании вызывает снижение поглощения [F]-фтор-дезокси-глюкозы. Radiother Oncol. 2015; 116:400-3.
20 Jiang P, Du W и Wu M. Регуляция пентозофосфатного пути при раке. Protein Cell. 2014; 5:592-602.
21 Koutcher JA, Alfieri AA, Matei C, Meyer KL, Street JC and Martin DS. Влияние 6-аминоникотинамида на пентозофосфатный путь: исследования 31P ЯМР и задержки роста опухоли. Magn Reson Med. 1996; 36:887-892.
22 Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, Sharpe MA, Xin L and Frigo DE. Регуляция пентозофосфатного пути механизмом, опосредованным андрогенным рецептором и МТОР, и его роль в росте клеток рака простаты. Онкогенез. 2014; 3:e103.
23 Krebs HA and Eggleston LV. Регуляция пентозофосфатного цикла в печени крысы. Adv Enzyme Regul. 1974; 12:421-434.
24 Epstein T, Xu L, Gillies RJ and Gatenby RA. Разделение метаболического предложения и спроса: аэробный гликолиз как нормальный физиологический ответ на колебания энергетических потребностей в мембране. Cancer Metab. 2014; 2:7.
25 Chen X, Qian Y and Wu S. The Warburg effect: Эволюционирующие интерпретации устоявшейся концепции. Free Radic Biol Med. 2015; 79:253-263.
26 Porporato PE, Dhup S, Dadhich RK, Copetti T and Sonveaux P. Противораковые мишени в гликолитическом метаболизме опухолей: комплексный обзор. Front Pharmacol. 2011; 2:49.
27 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG and Newton DL. Дихлорацетат натрия избирательно нацелен на клетки с дефектами митохондриального ЭТЦ. Int J Cancer. 2010; 127:2510-2519.
28 Babu E, Ramachandran S, CoothanKandaswamy V, Elangovan S, Prasad PD, Ganapathy V и Thangaraju M. Роль SLC5A8, транспортера плазматической мембраны и опухолевого супрессора, в противоопухолевой активности дихлорацетата. Oncogene. 2011; 30:4026-4037.
29 Jackson VN and Halestrap AP. Кинетика, субстрат и ингибиторная специфичность монокарбоксилатного (лактатного) транспортера клеток печени крысы, определенные с помощью флуоресцентного внутриклеточного индикатора pH, 2′,7′-бис(карбоксиэтил)-5(6)-карбоксифлуоресцеина. J Biol Chem. 1996; 271:861-868.
30 Bowker-Kinley MM, Davis WI, Wu P, Harris RA and Popov KM. Доказательства существования тканеспецифической регуляции комплекса пируватдегидрогеназы млекопитающих. Biochem J. 1998; 329:191-196.
31 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B and Michelakis ED. Ось митохондрий-K+ каналов подавляется при раке, и ее нормализация способствует апоптозу и подавляет рост рака. Cancer Cell. 2007; 11:37-51.
32 Pajuelo-Reguera D, Alan L, Olejar T и Jezek P. Дихлорацетат стимулирует изменения в морфологии митохондриальной сети через частичную митофагию в клетках нейробластомы человека SH-SY5Y. Int J Oncol. 2015; 46:2409-2418.
33 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS and Michelakis ED. Активация митохондрий путем ингибирования PDKII подавляет сигнализацию HIF1a и ангиогенез при раке. Oncogene. 2013; 32:1638-1650.
34 Hong SE, Shin KS, Lee YH, Seo SK, Yun SM, Choe TB, Kim HA, Kim EK, Noh WC, Kim JI, Hwang CS, Lee JK, Hwang SG, Jin HO and Park IC. Ингибирование S6K1 усиливает вызванную дихлорацетатом гибель клеток. J Cancer Res Clin Oncol. 2015; 141:1171-1179.
35 Taper HS, Woolley GW, Teller MN and Lardis MP. Новая трансплантируемая опухоль печени мыши спонтанного происхождения. Cancer Res. 1966; 26:143-148.
36 Volpe JP, Hunter N, Basic I и Milas L. Метастатические свойства мышиных сарком и карцином. I. Положительная корреляция с колонизацией легких и отсутствие корреляции с опухолевым дублем. Clin Exp Metastasis. 1985; 3:281-294.
37 Rockwell S and Kallman RF. Кинетика роста и клеточной популяции одиночных и множественных сарком KHT. Cell Tissue Kinet. 1972; 5:449-457.
38 Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH and Jaffee EM. HER-2/neu является мишенью отторжения опухоли у толерантных HER-2/neu трансгенных мышей. Cancer Res. 2000; 60:3569-3576.
39 King MP and Attardi G. Человеческие клетки, лишенные мтДНК: репопуляция экзогенными митохондриями путем комплементации. Science. 1989; 246:500-503.
40 Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA и Michel T. Нацеливание эндотелиальной синтазы оксида азота на кавеолы. Специфические взаимодействия с изоформами кавеолина в сердечных миоцитах и эндотелиальных клетках. J Biol Chem. 1996; 271:22810-22814.
41 Diepart C, Verrax J, Calderon PB, Feron O, Jordan BF и Gallez B. Сравнение методов измерения потребления кислорода в опухолевых клетках in vitro. Anal Biochem. 2010; 396:250-256.
42 Jordan BF, Gregoire V, Demeure RJ, Sonveaux P, Feron O, O’Hara J, Vanhulle VP, Delzenne N and Gallez B. Insulin increases the sensitivity of tumors to irradiation: involvement of an increase in tumor oxygenation mediated by nitric oxide-dependent decrease of the tumor cells oxygen consumption. Cancer Res. 2002; 62:3555-3561.
Связанный контент: