Géraldine De Preter1, Marie-Aline Neveu1, Pierre Danhier1, Lucie Brisson2, Valéry L. Payen2, Paolo E. Porporato2, Bénédicte F. Jordan1, Pierre Sonveaux2 und Bernard Gallez1
1 Louvain Drug Research Institute (LDRI), Biomedical Magnetic Resonance Research Group, Université Catholique de Louvain (UCL), Brüssel, Belgien2
Institut de Recherche Expérimentale et Clinique (IREC), Pole of Pharmacology, Université Catholique de Louvain (UCL), Brüssel, Belgien
Korrespondenz: Bernard Gallez, E-Mail: [email protected]
Eingereicht am: 1. Juni 2015Akzeptiert
: 26. Dezember 2020Veröffentlicht
: 9. Dezember 2020
Zusammenfassung
Die Glukosegärung durch Glykolyse auch in Gegenwart von Sauerstoff (Warburg-Effekt) ist ein häufiges Merkmal von Krebszellen, die zunehmend als verlockendes Ziel für die klinische Entwicklung angesehen werden. Ziel dieser Studie war es, den Zusammenhang zwischen Stoffwechsel, Energiespeichern und Proliferationsraten in Krebszellen zu analysieren. Wir fanden heraus, dass die Zellproliferation, bewertet durch die Quantifizierung der DNA-Synthese, in sechs Krebszelllinien sowie in isogenen Krebszelllinien mit der glykolytischen Effizienz korreliert ist. Zur weiteren Untersuchung des Zusammenhangs zwischen Glykolyse und Proliferation wurde ein pharmakologischer Inhibitor des Pentosephosphatwegs (PPP) eingesetzt. Wir konnten zeigen, dass eine Verringerung der PPP-Aktivität die Proliferation von Krebszellen verringert, wobei die Wirkung bei Krebszellen des Warburg-Phänotyps besonders ausgeprägt ist. Die entscheidende Rolle des PPP bei der Aufrechterhaltung der Proliferation von Krebszellen wurde durch den Einsatz von siRNAs gegen Glukose-6-Phosphat-Dehydrogenase, das erste und ratenlimitierende Enzym des PPP, bestätigt. Darüber hinaus stellten wir fest, dass Dichloracetat (DCA), eine neue klinisch getestete Verbindung, eine Umstellung der glykolytischen Krebszellen auf einen stärker oxidativen Phänotyp und eine verringerte Proliferation bewirkte. Durch den Nachweis, dass DCA die Aktivität der PPP verringert, haben wir einen neuen Mechanismus gefunden, durch den DCA die Proliferation von Krebszellen kontrolliert.
Stichworte: Bioenergetik, Glykolyse, Dichloracetat, Pentosephosphatweg, Proliferation
© 2020 by the authors. Lizenznehmer MDPI, Basel, Schweiz. Dieser Artikel ist ein Open-Access-Artikel, der unter den Bedingungen der Creative Commons Attribution (CC BY) Lizenz verbreitet wird (http://creativecommons.org/licenses/by/4.0/).
EINLEITUNG
In den letzten Jahren hat der Stoffwechsel auf dem Gebiet der Krebsforschung ein enormes Interesse geweckt. Viele Studien konzentrierten sich auf die unterschiedlichen Stoffwechselprofile verschiedener Tumoren [1-3], da die metabolische Plastizität an der Krebsentwicklung, der Arzneimittelresistenz und der Metastasierung beteiligt ist [4-6]. In normalen Zellen ist die Glykolyse an die oxidative Phosphorylierung (OXPHOS) gekoppelt, um intrazelluläres ATP optimal aus Glukose zu synthetisieren [7]. Viele Krebszellen durchlaufen jedoch eine grundlegende metabolische Umwandlung, den „glykolytischen Switch“, bei dem die Glykolyse von der Atmung abgekoppelt und auf Milchsäuregärung umgestellt wird, wodurch sie zur primären Quelle der ATP-Produktion der Zelle wird. Die Umstellung auf einen glykolytischen Stoffwechsel erfolgt in erster Linie unter Hypoxie als Rettungsmechanismus für die Energieproduktion. Einige Krebszellen nehmen jedoch darüber hinaus einen besonderen glykolytischen Phänotyp an, der erstmals von Warburg [8] beschrieben und als „aerobe Glykolyse“ bezeichnet wurde [9]. Die biologische Grundlage des Warburg-Phänotyps ist nach wie vor umstritten, doch wurde kürzlich vorgeschlagen, dass proliferierende Krebszellen die Glykolyse verstärken, weil sie sowohl der Bioenergetik als auch der Biosynthese zugute kommt [4,10]. Ein glykolytischer Stoffwechsel ermöglicht in der Tat eine schnelle ATP-Produktion und liefert Kohlenstoff-Zwischenprodukte, die in verzweigte Biosynthesewege geleitet werden können, was eine schnellere Expansion der zellulären Biomasse ermöglicht. Es ist überzeugend, dass Mutationen in Signalwegen, die sowohl die zelluläre Biosynthese als auch die aerobe Glykolyse regulieren, wie z. B. der PI3K/Akt/mTOR-Weg, die häufigste Mutationsklasse in menschlichen Tumoren sind [11]. Es gibt jedoch keine experimentellen Belege für einen Zusammenhang zwischen aerober Glykolyse und der Vermehrung von Krebszellen, und der selektive Vorteil dieses Phänotyps ist nicht ganz klar. Ziel der vorliegenden Studie war es, die Kopplung zwischen Stoffwechsel, Energieversorgung und Zellproliferation in verschiedenen menschlichen und murinen Krebszellen aufzuklären. Es wurden Stoffwechselumschaltungen herbeigeführt, um nachzuweisen, dass die Bioenergetik, insbesondere die Glykolyse, die Proliferation von Krebszellen direkt steuert. In diesem Zusammenhang entdeckten wir einen neuen therapeutischen Mechanismus von Dichloracetat (DCA), einem Aktivator der mitochondrialen Oxidation von Glukose, der derzeit in klinischen Studien untersucht wird [12]. DCA hemmt den Pentosephosphatweg (PPP), einen zentralen, mit der Glykolyse verzweigten Biosyntheseweg. Wir berichten, dass der PPP die Lücke zwischen einem glykolytischen Stoffwechsel und der Proliferation von Krebszellen überbrückt.
ERGEBNISSE
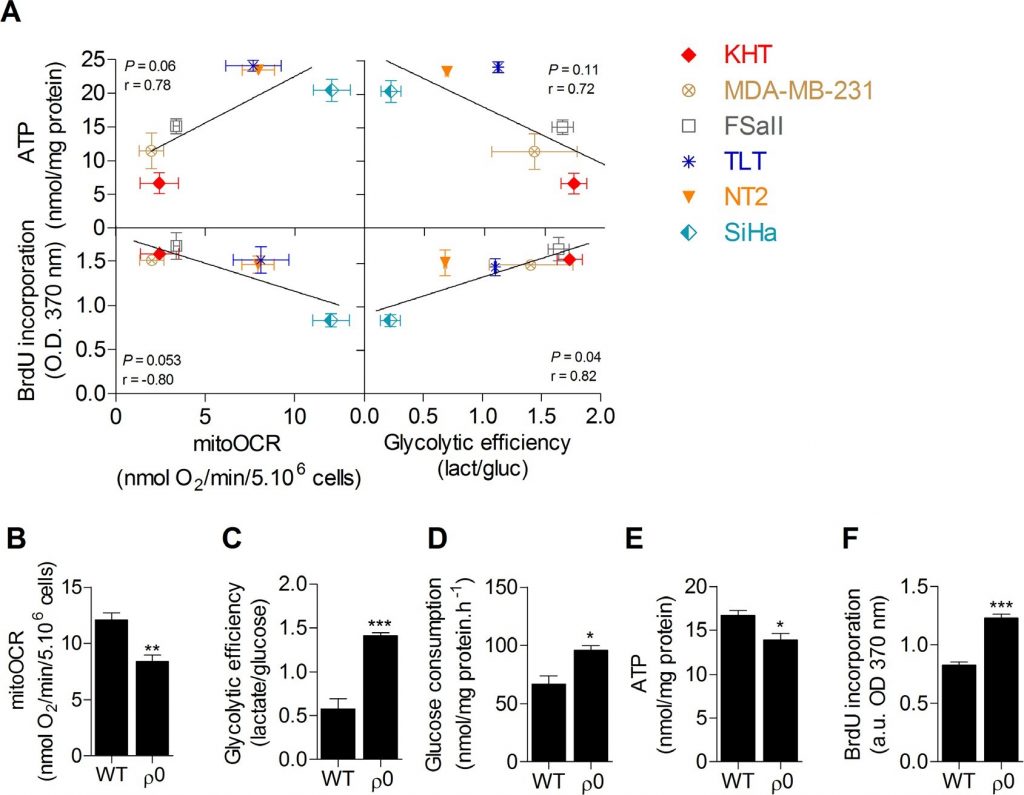
Dieglykolytische Effizienz steht in einem positiven Zusammenhang mit der Proliferation, nicht aber mit dem ATP-Gehalt von Krebszellen
Zunächst wurdeeinScreening der Stoffwechselaktivitäten von sechs Krebszelllinien durchgeführt, um den Zusammenhang zwischen Glukosestoffwechsel, ATP-Speicher und Proliferationsfähigkeit zu untersuchen. Die OXPHOS, die anhand der Oligomycin-sensitiven Sauerstoffverbrauchsrate (mitoOCR) gemessen wird, und die glykolytische Effizienz, die anhand des Verhältnisses von produziertem Laktat zu verbrauchter Glukose (mol/mol) [13] gemessen wird, wurden getrennt an Zellen untersucht, die 24 Stunden in einem Kulturmedium bebrütet wurden, das nur Glukose als Energieträger enthält. Um den Einfluss des Stoffwechselstatus auf die zellulären Energiespeicher zu untersuchen, wurde auch der gesamte intrazelluläre ATP-Pool gemessen. Es wurde keine signifikante Korrelation zwischen dem intrazellulären ATP-Gehalt und den Stoffwechselparametern festgestellt. Wir fanden jedoch heraus, dass die Zellproliferation, die anhand der DNA-Synthese bewertet wurde, signifikant mit der glykolytischen Effizienz korreliert war (Abbildung 1A), wobei eine erhöhte Glykolyse mit einer erhöhten Proliferationskapazität verbunden war, und umgekehrt. Zwischen der Proliferationskapazität und der mitochondrialen Atmung wurde keine signifikante Korrelation festgestellt. Diese Daten deuten darauf hin, dass die Glykolyse möglicherweise der wichtigste Energiestoffwechselweg ist, der die Proliferation von Krebszellen unterstützt. Gegenwärtig wird angenommen, dass dieser Weg die Zellproliferation vor allem durch die Bereitstellung von Vorläufersubstanzen für biosynthetische Wege und nicht durch die ATP-Produktion unterstützt [10,11]. Als Beleg dafür, dass die ATP-Versorgung nicht der wichtigste limitierende Faktor für die Zellproliferation ist, wurde in den Zelllinien keine Korrelation zwischen ATP-Gehalt und DNA-Synthese gefunden (ergänzende Abbildung S1). Um zu bestätigen, dass die Glykolyse die Proliferation von Krebszellen fördert, und um stoffwechselunabhängige Einflüsse aufgrund unterschiedlicher Genotypen auszuschließen, wurde die Proliferationskapazität menschlicher oxidativer SiHa-Gebärmutterhalskrebszellen vom Wildtyp (WT) mit der von SiHa-Zellen mit partieller Mitochondriendepletion (SiHa ρ0) verglichen [14,15]. SiHa ρ0-Krebszellen wiesen einen glykolytischen Phänotyp mit einer ~40%igen Abnahme der MitoOCR (Abbildung 1B), einem ~2,5-fachen Anstieg der glykolytischen Effizienz (Abbildung 1C) und einem ~50%igen Anstieg des Glukoseverbrauchs (Abbildung 1D) auf. Die Nettoabnahme des Gesamt-ATP-Pools in SiHa-ρ0-Zellen um 20 % bestätigte, dass die erhöhte Glykolyse nicht ausreichte, um die ATP-Speicher zu erhalten (Abbildung 1E). Durch die Quantifizierung der DNA-Synthese konnten wir feststellen, dass die verstärkte aerobe Glykolyse die Zellproliferation fördert, da die Umstellung der Glykolyse in SiHa-ρ0-Zellen mit einem Anstieg der Zellproliferation um etwa 45 % verbunden war (Abbildung 1F). Viabilitätsuntersuchungen ergaben keinen Unterschied in der Anzahl der lebensfähigen Zellen 24 Stunden nach der Inkubation der Zellen im experimentellen Kulturmedium (ergänzende Abbildung S2), wahrscheinlich weil die Quantifizierung der DNA-Synthese frühe Veränderungen in der Proliferationsrate der Zellen erkennt.
Glykolysehemmung durch DCA beeinträchtigt die Proliferation von Krebszellen
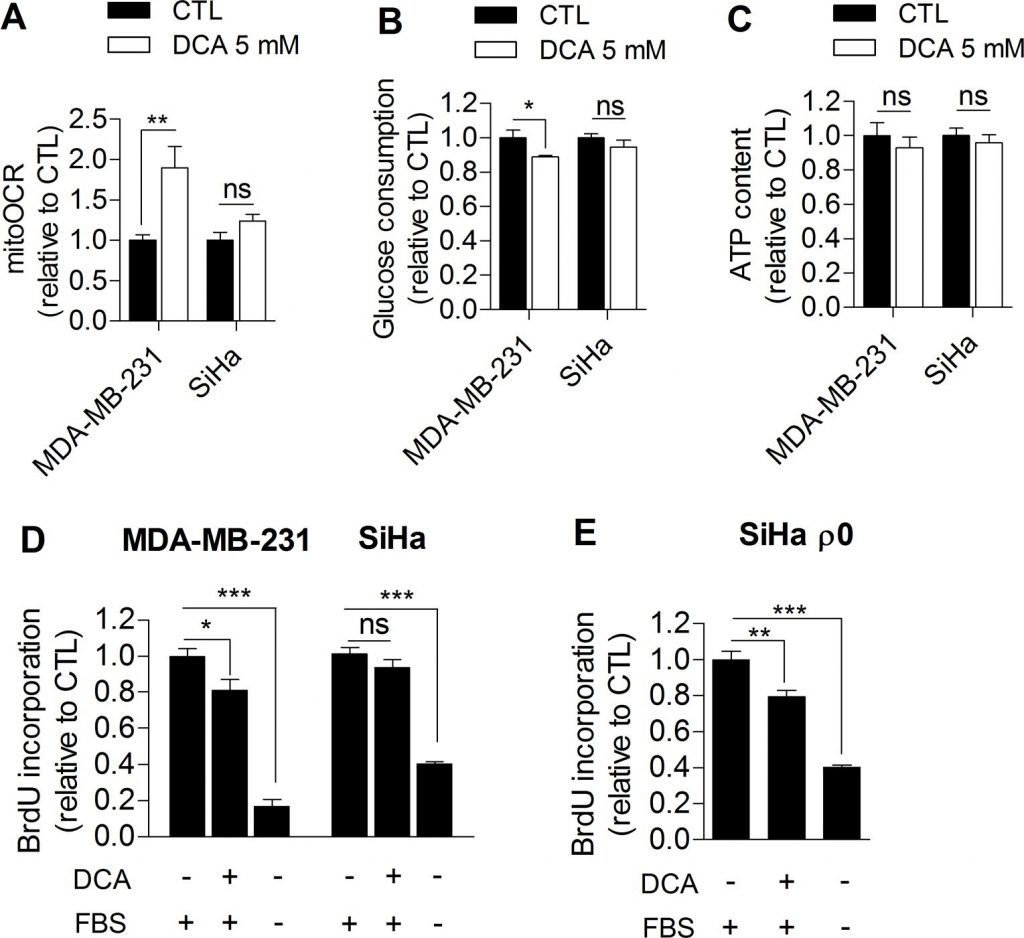
Auf der Grundlage unserer Beobachtungen haben wir weiter untersucht, ob die Glykolysehemmung die Proliferation von Krebszellen direkt beeinträchtigen könnte. Zu diesem Zweck wurden menschliche Brustkrebszellen MDA-MB-231 (Warburg-Phänotyp, Abbildung 1A) und menschliche Gebärmutterhalskrebszellen SiHa (oxidativer Phänotyp, Abbildung 1A) mit Dichloracetat (DCA) behandelt, einem Pyruvat-Dehydrogenase-Kinase (PDK)-Inhibitor, der die oxidative Aktivität der Zellen durch Aktivierung der Pyruvat-Dehydrogenase (PDH), des Gatekeeping-Enzyms der Glukoseoxidation in den Mitochondrien, steigert [16]. Bislang wird die vielversprechende therapeutische Wirkung von DCA auf Krebszellen allgemein auf eine Normalisierung des für Krebszellen charakteristischen hyperpolarisierten mitochondrialen Membranpotenzials und auf eine Re-Sensibilisierung für Apoptose zurückgeführt [17]. Wir haben hier postuliert, dass DCA auch die Tumorproliferation durch Hemmung der Glykolyse kontrolliert. Um diese Hypothese zu testen, wurden glykolytische MDA-MB-231- und oxidative SiHa-Krebszellen 48 Stunden lang mit 5 mM DCA behandelt, und die Auswirkungen der Behandlung auf den Stoffwechsel und die Proliferation wurden bewertet. Im Vergleich zu den mit dem Vehikel behandelten Zellen führte DCA bei den glykolytischen MDA-MB-231-Krebszellen zu einer Umstellung auf einen stärker oxidativen Phänotyp, was durch einen Anstieg des MitoOCR (Abbildung 2A) und einen Rückgang des Glukoseverbrauchs (Abbildung 2B) belegt wird. Die in diesem Experiment beobachtete Abnahme der glykolytischen Aktivität stimmt mit einer anderen kürzlich durchgeführten Studie [18] überein und wird wahrscheinlich durch den Pasteur-Effekt [4,19] induziert, um die ATP-Homöostase in den Zellen aufrechtzuerhalten (Abbildung 2C). Wir beobachteten auch, dass die Hemmung der Glykolyse durch DCA mit einer verringerten Proliferationsrate der MDA-MB-231-Krebszellen verbunden war (Abbildung 2D). Als Beleg dafür, dass die Hemmung der Glykolyse die Proliferation in dieser Zelllinie beeinträchtigt, wiesen mit 2-Deoxy-D-Glucose behandelte MDA-MB-231-Krebszellen ebenfalls eine verringerte Proliferationsrate auf (ergänzende Abbildung S3).
Andererseits veränderte DCA die Stoffwechselaktivitäten der oxidativen SiHa-Krebszellen nicht signifikant (Abbildung 2A-2C) und hatte keine signifikanten Auswirkungen auf die SiHa-Proliferation (Abbildung 2D). Kurzfristige (1 Stunde) Messungen der Laktatproduktion zeigten, dass DCA in der glykolytischen Krebszelllinie tatsächlich wirksamer war als in der oxidativen (ergänzende Abbildung S4). Um zu untersuchen, ob das Stoffwechselprofil die Reaktion auf DCA bestimmt, wurde auch die Proliferationskapazität der glykolytischen SiHa ρ0-Krebszellen nach DCA-Behandlung analysiert. Wir stellten eine signifikante Abnahme der DNA-Synthese in dieser Zelllinie fest (Abbildung 2E), ein Effekt, der bei SiHa WT nicht beobachtet wurde. Darüber hinaus wurde die gleiche Anzahl lebensfähiger MDA-MB-231- und SiHa ρ0-Krebszellen 48 Stunden nach der Behandlung mit Vehikel oder DCA gemessen (ergänzende Abbildung S5 A-B), was zeigt, dass die Auswirkungen von DCA auf Stoffwechselfunktionen und Proliferationsraten nicht auf Zellsterblichkeit zurückzuführen sind.
Die Glykolyse steuert die Proliferation von Krebszellen über den Pentosephosphatweg
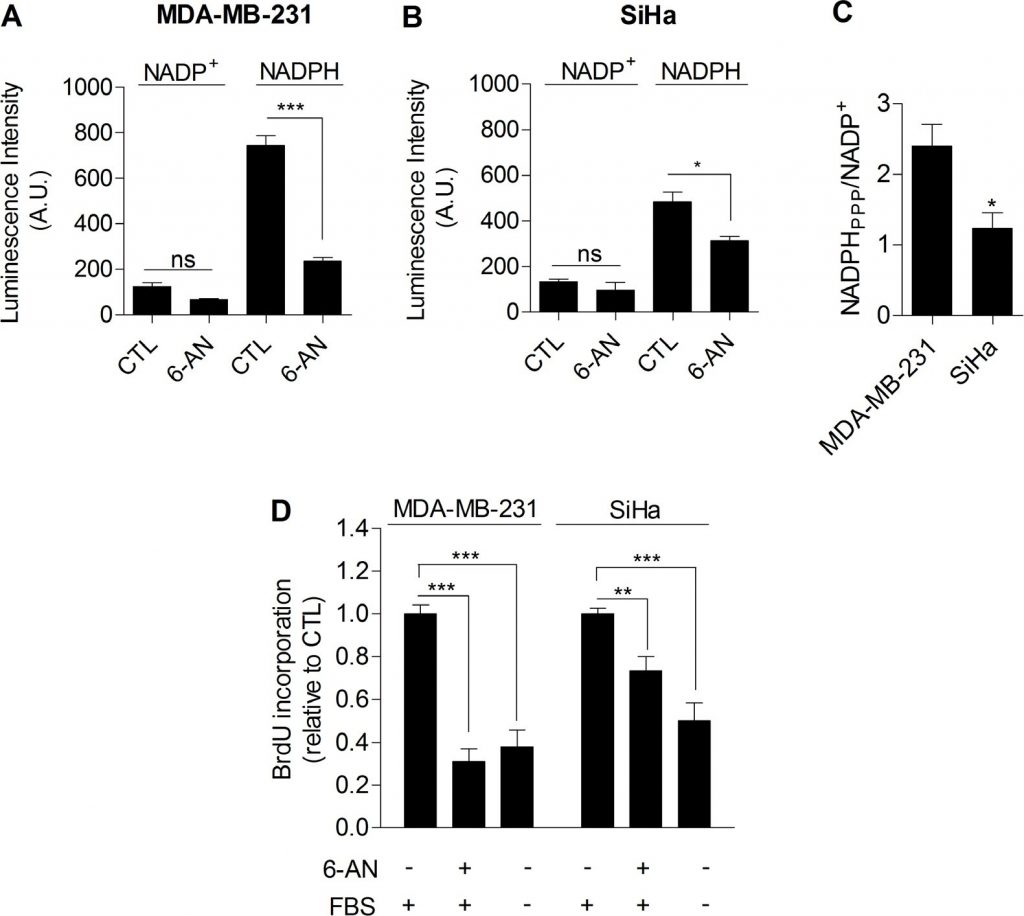
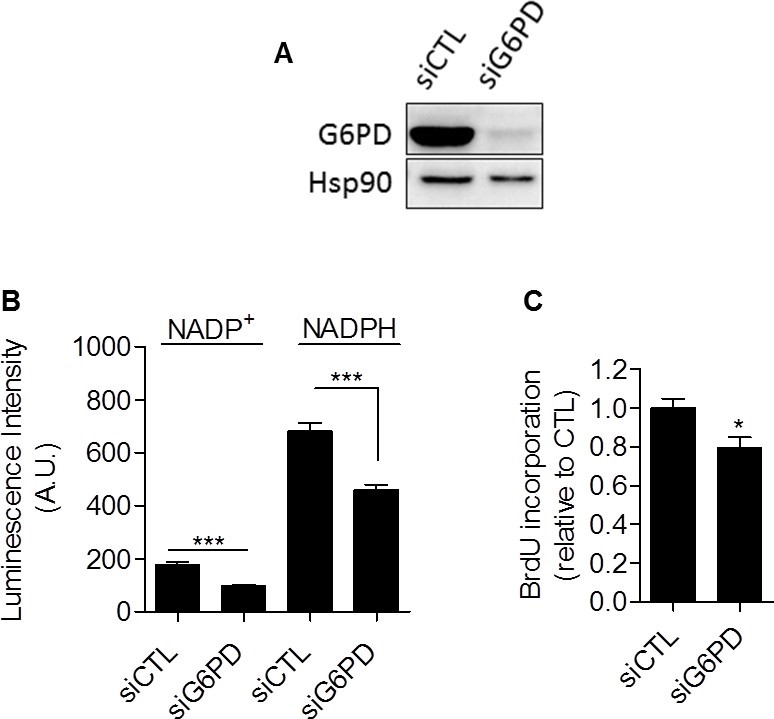
Zusammengenommen lieferten unsere Daten umfassende experimentelle Beweise dafür, dass die Glykolyse per se an der Kontrolle der Zellproliferation beteiligt ist. Der Mechanismus, der die Glykolyse und die Proliferation miteinander verbindet, musste jedoch noch geklärt werden. Wir postulierten, dass der Pentosephosphatweg (PPP) die Glykolyse mit der Proliferation verbinden könnte, da der PPP glykolytische Zwischenprodukte nutzt, um Zellen mit Nukleotiden und NADPH zu versorgen, einem entscheidenden Reduktionsmittel bei anabolen Prozessen [20]. Um das von der PPP produzierte NADPH (NADPHppp) spezifisch zu bestimmen, wurden die Zellen mit 6-Aminonicotinamid (6-AN) behandelt, einem spezifischen Inhibitor der PPP [21,22]. Wir stellten fest, dass der Beitrag von NADPHppp zum gesamten NADPH-Pool (NADPHtot) in den MDA-MB-231-Krebszellen stärker ausgeprägt war als in den SiHa-Krebszellen, was durch einen starken Rückgang des NADPHtot-Spiegels nach der Behandlung mit 6-AN in den glykolytischen MDA-MB-231-Zellen belegt wird (Abbildung 3A). In den oxidativen SiHa-Zellen wurden geringere Auswirkungen festgestellt (Abbildung 3B). Die Messungen ergaben auch ein höheres NADPHppp/NADP+-Verhältnis in MDA-MB-231-Krebszellen, was auf einen höheren PPP-Fluss in dieser glykolytischen Zelllinie im Vergleich zu SiHa hinweist (Abbildung 3C). Um zu überprüfen, ob die PPP an der Kontrolle der Proliferation von Krebszellen beteiligt ist, wurde die DNA-Synthese in 6-AN-behandelten und unbehandelten Zellen untersucht. Wir stellten fest, dass die Proliferationsfähigkeit der Krebszellen beeinträchtigt war, wenn die PPP gehemmt wurde (Abbildung 3D). Interessanterweise zeigte sich eine stärkere Wirkung bei glykolytischen MDA-MB-231-Krebszellen (~70 % Rückgang der DNA-Syntheserate) im Vergleich zu oxidativen SiHa-Krebszellen (~25 % Rückgang der DNA-Syntheserate) (Abbildung 3D). Wichtig ist, dass SiHa ρ0 empfindlicher als SiHa WT auf 6-AN reagierten, da ein Rückgang der DNA-Syntheserate um 40 % festgestellt wurde (ergänzende Abbildung S6). Aufgrund der potenziellen Off-Target-Effekte pharmakologischer Inhibitoren ergänzten wir unsere 6-AN-Studien mit kleinen interferierenden RNAs (siRNAs), die auf Glukose-6-Phosphat-Dehydrogenase (G6PD), das erste und ratenlimitierende Enzym der PPP, abzielen [23]. In transfizierten MDA-MB-231-Zellen bestätigten wir das Silencing von G6PD durch Immunoblotting (Abbildung 4A) und zeigten, dass die Hemmung von G6PD wie 6-AN die PPP-Aktivität (Abbildung 4B) und die DNA-Synthese (Abbildung 4C) verringerte. Diese Ergebnisse weisen auf den überwiegenden Beitrag der PPP zur Aufrechterhaltung der Proliferation von Krebszellen des Warburg-Phänotyps hin.
DCA hemmt den Pentosephosphatweg
Schließlich untersuchten wir, ob DCA den PPP hemmen könnte, was den Zusammenhang zwischen der Glykolysehemmung und der verringerten Proliferationsrate von DCA-behandelten Krebszellen erklären würde. Wie in Abbildung 5 dargestellt, beobachteten wir, dass DCA (5 mM, 48 h) den NADPHtot-Spiegel in MDA-MB-231-Krebszellen signifikant senkte (Abbildung 5A). Während keine zusätzliche Verringerung von NADPHtot erreicht wurde, wenn die Zellen DCA und 6-AN zusammen ausgesetzt wurden (Abbildung 5A), zeigten wir, dass DCA spezifisch NADPHPPP verringerte. Ähnliche Ergebnisse wurden bei der molekularen Hemmung von G6PD gefunden (Abbildung 5B). Diese Ergebnisse zeigen, dass die PPP bei den Auswirkungen von DCA auf glykolytische Krebszellen eine Rolle spielt.
DISKUSSION
Diese Studie unterstreicht die vorherrschende Rolle des Pentosephosphatweges in der Krebsbiologie. Der Warburg-Effekt (die Fermentation von Glukose zu Laktat in Anwesenheit einer physiologischenO2-Konzentration ), der in zahlreichen Krebszellen beobachtet wurde, soll sowohl energetische (ATP) als auch biosynthetische Anforderungen erfüllen [4,10]. In dieser Studie haben wir gezeigt, dass eine verstärkte aerobe Glykolyse nicht ausreicht, um den ATP-Spiegel in Krebszellen aufrechtzuerhalten. Ein niedriger intrazellulärer ATP-Spiegel war also mit einer erhöhten Glykolyse verbunden. Darüber hinaus war die in Krebszellen mit Mitochondrienmangel induzierte glykolytische Umschaltung im Vergleich zu Wildtypzellen nicht in der Lage, eine angemessene ATP-Versorgung sicherzustellen. Es wird vermutet, dass aus bioenergetischer Sicht ein glykolytischer Stoffwechsel weniger effizient ist als ein oxidativer, dass aber die Glykolyse vorteilhaft sein kann, wenn es darum geht, schnell ATP zu produzieren, um kurzfristigen Energiebedarf zu decken [24]. Unsere Ergebnisse bestätigen auch, dass Krebszellen des Warburg-Phänotyps mehrere Anpassungen entwickeln, um niedrige ATP-Spiegel aufrechtzuerhalten, um die allosterische Hemmung der ratenlimitierenden glykolytischen Enzyme (den Pasteur-Effekt) zu vermeiden und erhöhte glykolytische Flussraten aufrechtzuerhalten [10]. Andererseits haben wir gezeigt, dass die Glykolyse die Proliferation von Krebszellen fördert, was die klinisch getesteten Strategien zur Hemmung des Warburg-Effekts unterstützt [12,25]. Die positive Beziehung zwischen der glykolytischen Effizienz (Mol Laktat pro Mol verbrauchter Glukose) und der Proliferationsrate von Krebszellen unterstreicht, dass sich schnell teilende Zellen große Mengen an Glukose in Laktat umwandeln. Jüngste Studien haben vorgeschlagen, dass die Laktatproduktion den Krebszellen eine effiziente Regeneration von NAD+ durch das Enzym Laktatdehydrogenase ermöglicht [26]. Durch die Aufrechterhaltung des NAD+/NADH-Redox-Gleichgewichts ermöglichen Krebszellen einen schnelleren Glukosefluss durch die Glykolyse sowie einen schnelleren Einbau von Glukosemetaboliten in Biosynthesewege, was Vorteile für die Proliferation mit sich bringt [10].
Um zu prüfen, ob eine verstärkte aerobe Glykolyse die Proliferation von Krebszellen fördert, indem sie anabole Prozesse in Gang setzt, wurde die Beteiligung des Pentosephosphatwegs (PPP) untersucht. Es ist bekannt, dass der PPP Glukose-6-Phophat (das Produkt des glykolytischen Enzyms Hexokinase) verwendet, um die Zellen mit Nukleotiden und NADPH zu versorgen, einem entscheidenden Reduktionsmittel bei anabolen Reaktionen [20]. In unserer Studie konnten wir zeigen, dass der PPP-Fluss, der anhand des NADPHppp/NADP+-Verhältnisses gemessen wird, in Krebszellen vom Warburg-Phänotyp im Vergleich zu oxidativen Zellen erhöht war. Um diesen Unterschied im PPP-Fluss zu bestätigen, hätte auch ein Vergleich der Aktivität der PPP-Enzyme durchgeführt werden können. Nichtsdestotrotz unterstützen unsere Ergebnisse, dass die Aktivierung der Glykolyse mit einer Erhöhung der PPP-Aktivität für die Biosynthese in sich schnell teilenden Zellen einhergeht [20]. Überzeugenderweise konnten wir beobachten, dass die Hemmung der PPP die Proliferation von Krebszellen verringerte, wobei die Wirkung bei Krebszellen des Warburg-Phänotyps am größten war. Die unterschiedliche Empfindlichkeit von glykolytischen und oxidativen Krebszellen gegenüber einer PPP-Hemmung ist wahrscheinlich darauf zurückzuführen, dass sie für die Synthese von Makromolekülen wie Lipiden und Nukleotiden unterschiedlich stark auf diesen Weg angewiesen sind. Darüber hinaus können Krebszellen mit erhöhter mitochondrialer Aktivität den NADPH-Spiegel ausgleichen, indem sie TCA-Zyklus-assoziierte Enzyme (Apfelsäureenzyme und Isocitrat-Dehydrogenasen) einsetzen, um den NADPH-Pool wieder aufzufüllen [20]. Zusammengenommen zeigen diese Ergebnisse, dass Krebszellen, insbesondere aggressive Krebszellen vom Warburg-Phänotyp, für eine optimale Vermehrung auf den Pentosephosphatweg angewiesen sind.
Um die therapeutische Relevanz dieser Ergebnisse zu bewerten, wurden die Auswirkungen von DCA untersucht. DCA hat in vitro und in vivo bereits interessante krebshemmende Eigenschaften gezeigt
[17] und wird derzeit in klinischen Studien der Phase I-II getestet [12]. Bislang verdankt DCA seine therapeutischen Eigenschaften der Re-Sensibilisierung von Krebszellen für die Apoptose [17]. Wie Stockwin et al. [27] kürzlich feststellten, ist DCA jedoch relativ unwirksam für die Lebensfähigkeit der Zellen und löst nur bei hohen Konzentrationen Apoptose aus. Stockwin et al. berichteten außerdem, dass DCA bevorzugt in Zellen mit mitochondrialem Defekt aktiv ist. Daher postulierten wir, dass DCA in niedrigeren Dosen auch die Proliferation von Krebszellen verringert, indem es die Glykolyse reduziert. Wir fanden heraus, dass 5 mM DCA in Krebszellen des Warburg-Phänotyps wirksamer war und die Zellproliferation durch Verringerung der Glykolyse reduzierte. Die unterschiedliche Empfindlichkeit der Zelllinien mit unterschiedlichem Stoffwechselphänotyp könnte auf die Fähigkeit von DCA zurückzuführen sein, PDK in der Matrix der Mitochondrien zu erreichen. Wie Pyruvat ist DCA nämlich ionisiert und kann nicht durch die Membranen gelangen. Seltsamerweise gibt es nur wenige Berichte über den Transport von DCA im Zytosol von Säugetierzellen [28,29]. Obwohl dies noch nicht untersucht wurde, könnte das Ausmaß der Expression mitochondrialer Pyruvat-Transporter (MPCs) auch die Wirksamkeit von DCA bestimmen. Da DCA für jedes der vier PDK-Isoenzyme einen anderen Ki-Werthat [30], könnte die unterschiedliche Empfindlichkeit der Krebszellen auf die unterschiedliche Expression der PDKs zurückzuführen sein.
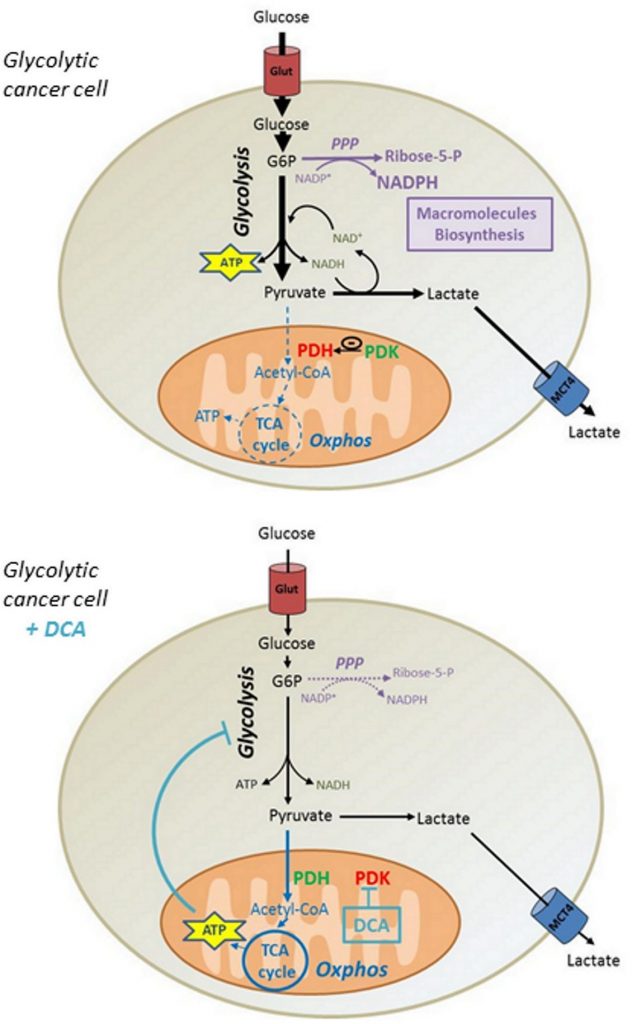
Obwohl einige Berichte zeigen, dass DCA in vergleichbaren Konzentrationen die Zytotoxizität bei anderen Zelllinien fördert [31,32], haben wir gezeigt, dass DCA keinen Einfluss auf die Lebensfähigkeit von MDA-MB-231- und SiHa-Krebszellen hat. Auf der Grundlage unserer Daten schlagen wir vor, dass die durch DCA induzierte Reaktivierung der ATP-Produktion in den Mitochondrien den glykolytischen Fluss der Krebszellen senkt und dadurch den Einbau von Glukose verringert. Der Rückgang der Synthese von glykolytischen Zwischenprodukten reduziert folglich die Aktivität von Biosynthesewegen wie dem PPP und beeinträchtigt die Proliferation von Krebszellen des Warburg-Phänotyps (Abbildung 6). Es ist jedoch denkbar, dass auch andere Mechanismen an der proliferationshemmenden Wirkung von DCA beteiligt sind, da andere Studien bei vergleichbaren DCA-Konzentrationen einen Zellzyklus-Stillstand gezeigt haben [33, 34].
Insgesamt hat unsere Studie gezeigt, dass das PPP eine Brücke zwischen Glykolyse und Proliferation in aggressiven Krebszellen schlägt, und plädiert dafür, den Krebsstoffwechsel bei der Wahl geeigneter therapeutischer Krebsbekämpfungsstrategien zu berücksichtigen. Wir schließen uns insbesondere den Schlussfolgerungen anderer Autoren [27] an und schlagen vor, dass die klinische Entwicklung von DCA von der Auswahl von Patienten mit stark glykolytischen Tumoren profitieren könnte.
MATERIALIEN UND METHODEN
Zellkultur und Reagenzien
SiHa, menschliches Plattenepithelkarzinom des Gebärmutterhalses (ATCC), MDA-MB-231, menschlicher Brustkrebs (ATCC), TLT (transplantierbarer Lebertumor), Hepatokarzinom der Maus [35], FSaII, Fibrosarkom der Maus [36], KHT, Sarkom der Maus [37] und NT2, Brusttumor der Maus [38 ] wurden nach den Empfehlungen des Herstellers oder wie beschrieben gezüchtet. SiHa mit partieller mitochondrialer Depletion (ρ0) wurden durch chronische Exposition gegenüber niedrigen Konzentrationen von Ethidiumbromid gewonnen, wie zuvor beschrieben [14,39]. Alle Kulturen wurden bei 37°C in einer Atmosphäre von 5%CO2 gehalten. Die Zellen wurden 24 Stunden vor den Experimenten in einem speziellen Versuchsmedium (DMEM ohne Glutamin [Invitrogen], mit 4,5 g/L Glukose, ergänzt mit 10 % hitzeinaktiviertem FBS und 1 % Penicillin-Streptomycin) inkubiert. Für den Vergleich von SiHa ρ0 mit SiHa Wildtyp wurde DMEM ohne Glutamin (Invitrogen) verwendet, das 4,5 g/L Glukose, ergänzt mit 1 % Pyruvat, 10 % hitzeinaktiviertem FBS, 1 % Penicillin-Streptomycin und 50 ng/ml Uridin enthält. Sofern nicht anders angegeben, wurden die Experimente an konfluenten Zellen durchgeführt. Oligomycin ist ein ATP-Synthase-Inhibitor. Dichloracetat (DCA) ist ein Pyruvat-Dehydrogenase-Kinase (PDK)-Inhibitor. 6-Aminonicotinamid (6-AN) ist ein Inhibitor der NADP+-abhängigen Enzyme des Pentosephosphatweges, der Glucose-6-Phosphat-Dehydrogenase (G6PD) und der 6-Phosphogluconat-Dehydrogenase (6-PGD). Alle Chemikalien wurden von Sigma bezogen. Oligomycin wurde in DMSO aufgelöst. DCA und 6-AN wurden direkt in den Kulturmedien aufgelöst.
siRNA-Transfektion
ON-TARGETplus SMARTpool siRNA gegen humanes G6PD und ON-TARGETplus Non-targeting siRNA stammen von Dharmacon. Die endgültige siRNA-Konzentration betrug 25 nM. Alle siRNAs wurden mit RNAi/MAX gemäß den Anweisungen des Herstellers (Invitrogen) transfiziert.
Western Blotting
Ganze Zelllysate wurden gesammelt und einer Immunoblot-Analyse unterzogen, wie zuvor beschrieben [40]. Primäre Antikörper waren humane Monoklonale gegen G6PD oder Hsp90 (Sigma).
Mitochondrialer Sauerstoffverbrauch
Die Sauerstoffverbrauchsrate (OCR) intakter ganzer Zellen wurde mit einem Bruker EMX EPR-Spektrometer bei 9,5 GHz gemessen, wie zuvor beschrieben [41]. Adhärente Zellen wurden in frischem Versuchsmedium (107 Zellen/ml) geerntet. 100 µl der Zellsuspension wurden mit 100 µl 20%igem Dextran gemischt, um eine Agglomeration zu vermeiden, und die Zellen wurden in einem Glaskapillarröhrchen in Gegenwart von 0,2 mM einer Nitroxid-Sonde versiegelt, die als Sauerstoffsensor dient (15N4-oxo-2,2,6,6-tetramethylpiperidin-d16-15N-1-oxyl, CDN isotopes, Pointe-Claire, Quebec, Kanada). Die Zellen wurden während der Erfassung der Spektren bei 37°C gehalten. Die EPR-Linienbreite wurde jede Minute gemessen und in eine Kalibrierungskurve eingetragen, um die Sauerstoffkonzentration zu erhalten. Die OCR wurde durch den absoluten Wert der Steigung der Abnahme der Sauerstoffkonzentration im geschlossenen Kapillarrohr über die Zeit bestimmt [42]. Um die OCR aus der oxidativen Phosphorylierung zu erhalten, wurde die OCR in Gegenwart einer 1 µM Oligomycin-Behandlung von der Gesamt-OCR der Zellen subtrahiert.
Glukoseverbrauch und Laktatproduktion
Der Glukoseverbrauch und die Laktatproduktion wurden aus den Überständen der kultivierten Zellen gemessen. Die Metaboliten-Konzentrationen wurden an deproteinisierten Proben mit Hilfe spezifischer enzymatischer Assays auf einem CMA600-Analysegerät (CMA Microdialysis AB, Solna, Schweden) quantifiziert. Der Glukoseverbrauch und die Laktatproduktion wurden mit Hilfe des Pierce BCA-Protein-Assays (Thermo Scientific) auf den Proteingehalt normiert. Um Unterschiede in den Metaboliten-Konzentrationen zwischen SiHa WT- und SiHa ρ0-Überständen effizient nachzuweisen, wurde ein Medium mit niedriger Glukose (1 g/L) verwendet.
Intrazelluläre ATP-Quantifizierung
Das gesamte intrazelluläre ATP wurde mit dem ATP-Bestimmungskit (Invitrogen) nach dem Protokoll des Herstellers gemessen. Die Zellen wurden zweimal mit PBS gewaschen und in dem vom Hersteller empfohlenen Puffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0,01% Triton X-100) lysiert. Die Zelllysate wurden einer Reaktionsmischung zugesetzt, die Luziferase und Luziferin enthielt, um die Biolumineszenz mit einem Plattenlesegerät (SpectraMax M2e, Molecular Devices) zu messen. Eine Standardkurve wurde mit bekannten ATP-Konzentrationen unter denselben Bedingungen erstellt. Eine Fraktion der Zelllysate wurde systematisch für die Proteinquantifizierung (Pierce BCA Protein Assay, Thermo Scientific) verwendet, um den ATP-Gehalt auf den Proteingehalt zu normalisieren.
Proliferation
Die Zellproliferation wurde mit einem 5-Brom-2′-Deoxyuridin (BrdU)-ELISA-Kit (Roche) gemäß den Anweisungen des Herstellers untersucht. Subkonfluente Zellen wurden in Gegenwart von BrdU (einem Nukleotidanalogon) 4 Stunden lang inkubiert. Beim Vergleich der Proliferation von SiHa WT und SiHa ρ0 wurden die Zellen 6 Stunden lang in Gegenwart von BrdU inkubiert. Die Menge des in die Zellen aufgenommenen BrdU wurde durch kolorimetrische Messungen mit einem Plattenlesegerät (SpectraMax M2e, Molecular Devices) bestimmt, das die Quantifizierung der DNA-Synthese in sich replizierenden Zellen ermöglichte.
NADPH- und NADP+-Messungen
Die NADP+-und NADPH-Konzentrationen wurden mit einem Detektionskit (NADP/NADPH-Glo Assay, Promega) gemäß den Anweisungen des Herstellers an 6 000 geernteten Zellen einzeln bestimmt.
Statistik
Alle Ergebnisse sind als Mittelwerte ± Standardfehler des Mittelwerts (SEM) angegeben. Die statistischen Analysen wurden mit der Software GraphPad Prism 5 durchgeführt. P<0,05 wurde als statistisch signifikant angesehen.
DANKSAGUNGEN
Diese Studie wurde durch Zuschüsse des Fonds National de la Recherche Scientifique (F.R.S.-FNRS, PDR T.0107.13), des Fonds Joseph Maisin, der Action de Recherches Concertées ARC 14/19-058 und eines Starting Grant des Europäischen Forschungsrats (ERC Nr. 243188 TUMETABO an P. Sonveaux) unterstützt. BFJ und PS sind Research Associates, LB und PEP sind Postdoktoranden, PD ist Postdoctoral Télévie Fellow. VLP ist ein PhD-Stipendiat des F.R.S.-FNRS. GDP ist ein Télévie-Doktorandenstipendiat.
INTERESSENKONFLIKTE
Die Autoren haben erklärt, dass keine Interessenkonflikte bestehen.
REFERENZEN
1 Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ und Rodriguez-Enriquez S. Who controls the ATP supply in cancer cells? Biochemische Lektionen zum Verständnis des Energiestoffwechsels von Krebs. Int J Biochem Cell Biol. 2014; 50:10-23.2 Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL und Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011; 286:42626-42634.
3 Jose C, Bellance N und Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta. 2011; 1807:552-561.
4 Gatenby RA und Gillies RJ. Warum haben Krebse eine hohe aerobe Glykolyse? Nat Rev Cancer. 2004; 4:891-899.
5 Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O und Dewhirst MW. Die gezielte Laktatatmung tötet selektiv hypoxische Tumorzellen in Mäusen ab. J Clin Invest. 2008; 118:3930-3942.
6 Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B und Sonveaux P. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014; 8:754-766.
7 Rolfe DF und Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997; 77:731-758.
8 Warburg O. Über den Stoffwechsel der Carcinomzelle. Klin Wochenschr. 1925;4:534-536..
9 Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P und Zhong H. „Der Stoffwechsel von Tumoren“: 70 Jahre später. Novartis Found Symp. 2001; 240:251-260; Diskussion 260-254.
10 Lunt SY und Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011; 27:441-464.
11 DeBerardinis RJ, Lum JJ, Hatzivassiliou G und Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7:11-20.
12 Tennant DA, Duran RV und Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010; 10:267-277.
13 Zancan P, Sola-Penna M, Furtado CM und Da Silva D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Molekulare Genetik und Metabolismus. 2010; 100:372-378.
14 De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O und Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012; 7:e46571.
15 Bol V, Bol A, Bouzin C, Labar D, Lee JA, Janssens G, Porporato PE, Sonveaux P, Feron O und Gregoire V. Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation. Acta Oncol. 2014:1-9.
16 Stacpoole PW and Felts JM. Auswirkungen von Diisopropylammoniumdichloracetat auf den Glukosestoffwechsel. Proc West Pharmacol Soc. 1969; 12:111-113.
17 Kankotia S und Stacpoole PW. Dichloracetat und Krebs: Neue Heimat für ein verwaistes Medikament? Biochim Biophys Acta. 2014; 1846:617-629.
18 Abildgaard C, Dahl C, Basse AL, Ma T and Guldberg P. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition. J Transl Med. 2014; 12:247.
19 Neveu MA, Bol V, Bol A, Bouzin C, Gregoire V, Feron O, Jordan BF und Gallez B. The increase in tumor oxygenation under carbogen breathing induces a decrease in the uptake of [F]-fluoro-deoxy-glucose. Radiother Oncol. 2015; 116:400-3.
20 Jiang P, Du W und Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014; 5:592-602.
21 Koutcher JA, Alfieri AA, Matei C, Meyer KL, Street JC und Martin DS. Effect of 6-aminonicotinamide on the pentose phosphate pathway: 31P NMR and tumor growth delay studies. Magn Reson Med. 1996; 36:887-892.
22 Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, Sharpe MA, Xin L und Frigo DE. Regulierung des Pentosephosphatwegs durch einen Androgenrezeptor-MTOR-vermittelten Mechanismus und seine Rolle beim Wachstum von Prostatakrebszellen. Oncogenesis. 2014; 3:e103.
23 Krebs HA und Eggleston LV. The regulation of the pentose phosphate cycle in rat liver. Adv Enzyme Regul. 1974; 12:421-434.
24 Epstein T, Xu L, Gillies RJ und Gatenby RA. Trennung von Stoffwechselangebot und -nachfrage: aerobe Glykolyse als normale physiologische Reaktion auf schwankende energetische Anforderungen in der Membran. Cancer Metab. 2014; 2:7.
25 Chen X, Qian Y und Wu S. The Warburg effect: Evolving Interpretations of an established concept. Free Radic Biol Med. 2015; 79:253-263.
26 Porporato PE, Dhup S, Dadhich RK, Copetti T und Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol. 2011; 2:49.
27 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG und Newton DL. Natriumdichloracetat wirkt selektiv auf Zellen mit Defekten im mitochondrialen ETC. Int J Cancer. 2010; 127:2510-2519.
28 Babu E, Ramachandran S, CoothanKandaswamy V, Elangovan S, Prasad PD, Ganapathy V und Thangaraju M. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene. 2011; 30:4026-4037.
29 Jackson VN und Halestrap AP. Kinetik, Substrat und Inhibitorspezifität des Monocarboxylat-(Laktat-)Transporters von Rattenleberzellen, bestimmt mit dem fluoreszierenden intrazellulären pH-Indikator 2′,7′-Bis(carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996; 271:861-868.
30 Bowker-Kinley MM, Davis WI, Wu P, Harris RA und Popov KM. Beweise für die Existenz einer gewebespezifischen Regulierung des Pyruvat-Dehydrogenase-Komplexes bei Säugetieren. Biochem J. 1998; 329:191-196.
31 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B und Michelakis ED. Eine Mitochondrien-K+-Kanal-Achse ist bei Krebs unterdrückt und ihre Normalisierung fördert die Apoptose und hemmt das Krebswachstum. Krebszelle. 2007; 11:37-51.
32 Pajuelo-Reguera D, Alan L, Olejar T und Jezek P. Dichloracetat stimuliert Veränderungen in der Morphologie des mitochondrialen Netzwerks durch partielle Mitophagie in menschlichen SH-SY5Y Neuroblastomzellen. Int J Oncol. 2015; 46:2409-2418.
33 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS und Michelakis ED. Mitochondriale Aktivierung durch Hemmung von PDKII unterdrückt HIF1a-Signalisierung und Angiogenese bei Krebs. Oncogene. 2013; 32:1638-1650.
34 Hong SE, Shin KS, Lee YH, Seo SK, Yun SM, Choe TB, Kim HA, Kim EK, Noh WC, Kim JI, Hwang CS, Lee JK, Hwang SG, Jin HO und Park IC. Die Hemmung von S6K1 verstärkt den Dichloracetat-induzierten Zelltod. J Cancer Res Clin Oncol. 2015; 141:1171-1179.
35 Taper HS, Woolley GW, Teller MN und Lardis MP. Ein neuer transplantierbarer Mauslebertumor spontanen Ursprungs. Cancer Res. 1966; 26:143-148.
36 Volpe JP, Hunter N, Basic I und Milas L. Metastatic properties of murine sarcomas and carcinomas. I. Positive Korrelation mit der Lungenbesiedlung und fehlende Korrelation mit der s.c. Tumoraufnahme. Clin Exp Metastasis. 1985; 3:281-294.
37 Rockwell S und Kallman RF. Wachstum und Zellpopulationskinetik von einzelnen und multiplen KHT-Sarkomen. Cell Tissue Kinet. 1972; 5:449-457.
38 Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH und Jaffee EM. HER-2/neu ist ein Ziel der Tumorabwehr in tolerierten HER-2/neu-transgenen Mäusen. Cancer Res. 2000; 60:3569-3576.
39 King MP und Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989; 246:500-503.
40 Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA und Michel T. Endothelial nitric oxide synthase targeting to caveolae. Spezifische Interaktionen mit Caveolin-Isoformen in kardialen Myozyten und Endothelzellen. J Biol Chem. 1996; 271:22810-22814.
41 Diepart C, Verrax J, Calderon PB, Feron O, Jordan BF und Gallez B. Comparison of methods for measuring oxygen consumption in tumor cells in vitro. Anal Biochem. 2010; 396:250-256.
42 Jordan BF, Gregoire V, Demeure RJ, Sonveaux P, Feron O, O’Hara J, Vanhulle VP, Delzenne N und Gallez B. Insulin erhöht die Empfindlichkeit von Tumoren gegenüber Bestrahlung: Beteiligung eines Anstiegs der Tumoroxygenierung, vermittelt durch eine Stickoxid-abhängige Abnahme des Sauerstoffverbrauchs der Tumorzellen. Cancer Res. 2002; 62:3555-3561.
Ähnliche Inhalte: