Géraldine De Preter1, Marie-Aline Neveu1, Pierre Danhier1, Lucie Brisson2, Valéry L. Payen2, Paolo E. Porporato2, Bénédicte F. Jordan1, Pierre Sonveaux2 et Bernard Gallez1
1 Louvain Drug Research Institute (LDRI), Biomedical Magnetic Resonance Research Group, Université Catholique de Louvain (UCL), Bruxelles, Belgique
2 Institut de Recherche Expérimentale et Clinique (IREC), Pôle de Pharmacologie, Université Catholique de Louvain (UCL), Bruxelles, Belgique
Correspondance : Bernard Gallez, courriel : [email protected]
Reçu : 1er juin 2015
Accepté : 26 décembre 2020
Publié : 9 décembre 2020
Résumé
La fermentation du glucose par glycolyse même en présence d’oxygène (effet Warburg) est une caractéristique commune des cellules cancéreuses de plus en plus considérées comme une cible séduisante en développement clinique. Cette étude visait à analyser le lien entre le métabolisme, les réserves d’énergie et les taux de prolifération dans les cellules cancéreuses. Nous avons constaté que la prolifération cellulaire, évaluée par la quantification de la synthèse d’ADN, est corrélée à l’efficacité glycolytique dans six lignées cellulaires cancéreuses ainsi que dans des lignées cellulaires cancéreuses isogéniques. Pour approfondir le lien entre la glycolyse et la prolifération, nous avons utilisé un inhibiteur pharmacologique de la voie du pentose phosphate (PPP). Nous avons démontré que la réduction de l’activité de la PPP diminue la prolifération des cellules cancéreuses, avec un effet profond dans les cellules cancéreuses de phénotype Warburg. Le rôle crucial de la PPP dans le maintien de la prolifération des cellules cancéreuses a été confirmé par l’utilisation de siRNA contre la glucose-6-phosphate déshydrogénase, la première enzyme de la PPP et celle qui en limite la vitesse. En outre, nous avons constaté que le dichloroacétate (DCA), un nouveau composé testé en clinique, induit un changement des cellules cancéreuses glycolytiques vers un phénotype plus oxydatif et diminue la prolifération. En démontrant que le DCA diminue l’activité de la PPP, nous fournissons un nouveau mécanisme par lequel le DCA contrôle la prolifération des cellules cancéreuses.
Mots clés : bioénergétique, glycolyse, dichloroacétate, voie du pentose phosphate, prolifération
2020 par les auteurs. Licencié MDPI, Bâle, Suisse. Cet article est un article en accès libre distribué selon les termes et conditions de la licence Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
INTRODUCTION
Ces dernières années, le métabolisme a suscité un intérêt considérable dans le domaine de la recherche sur le cancer. De nombreuses études se sont concentrées sur les différents profils métaboliques de différentes tumeurs [1-3] car la plasticité métabolique est impliquée dans la progression du cancer, la résistance aux médicaments et les métastases [4-6]. Dans les cellules normales, la glycolyse est couplée à la phosphorylation oxydative (OXPHOS) pour synthétiser de manière optimale l’ATP intracellulaire à partir du glucose [7]. Cependant, de nombreuses cellules cancéreuses subissent une transformation métabolique fondamentale, le « switch glycolytique », par lequel la glycolyse est découplée de la respiration et recouplée à la fermentation lactique, devenant ainsi la principale source de production d’ATP cellulaire. Le passage à un métabolisme glycolytique se produit principalement en cas d’hypoxie, comme mécanisme de secours pour la production d’énergie. Cependant, certaines cellules cancéreuses adoptent un phénotype glycolytique particulier, décrit pour la première fois par Warburg [8] et baptisé « glycolyse aérobie » [9]. La justification biologique du phénotype de Warburg reste controversée, mais il a été récemment proposé que les cellules cancéreuses en prolifération augmentent la glycolyse car elle est bénéfique à la fois pour la bioénergétique et la biosynthèse [4,10]. En effet, un métabolisme glycolytique permet potentiellement une production rapide d’ATP et fournit des intermédiaires carbonés qui peuvent être dirigés vers des voies de biosynthèse ramifiées, permettant une expansion plus rapide de la biomasse cellulaire. De manière convaincante, les mutations survenant dans les voies de signalisation régulant à la fois la biosynthèse cellulaire et la glycolyse aérobie, telles que la voie PI3K/Akt/mTOR, constituent la classe de mutations la plus répandue dans les tumeurs humaines [11]. Cependant, les preuves expérimentales reliant la glycolyse aérobie à la prolifération des cellules cancéreuses font défaut, et l’avantage sélectif procuré par ce phénotype n’est pas entièrement clair. L’objectif de la présente étude était d’élucider le couplage entre le métabolisme, l’approvisionnement en énergie et la prolifération cellulaire dans diverses cellules cancéreuses humaines et murines. Des commutations métaboliques ont été induites afin de fournir la preuve que la bioénergétique, et plus particulièrement la glycolyse, dirige directement la prolifération des cellules cancéreuses. Dans cette optique, nous avons découvert un nouveau mécanisme thérapeutique du dichloroacétate (DCA), un activateur de l’oxydation mitochondriale du glucose actuellement étudié dans des études cliniques [12]. Le DCA inhibe la voie du pentose phosphate (PPP), une voie de biosynthèse pivotante ramifiée à la glycolyse. Nous rapportons que la PPP fait le lien entre un métabolisme glycolytique et la prolifération des cellules cancéreuses.
RÉSULTATS
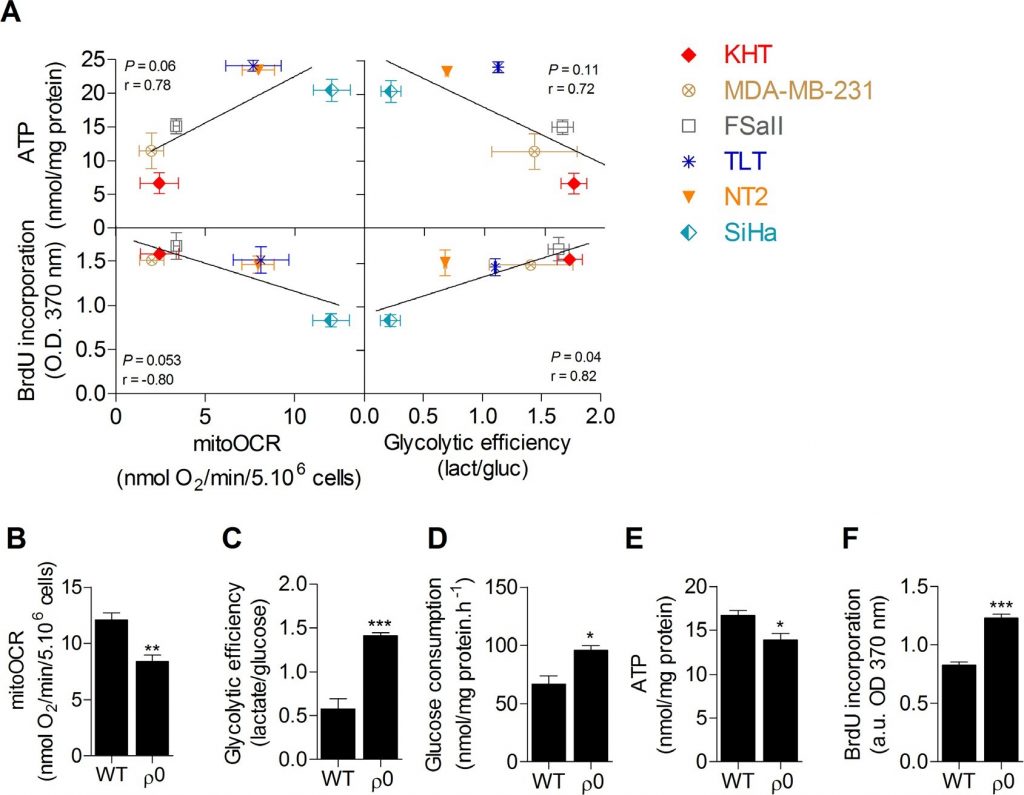
L‘efficacité glycolytique est positivement liée à la prolifération mais pas aux niveaux d’ATP dans les cellules cancéreuses
Un screening des activités métaboliques de six lignées cellulaires cancéreuses a d’abord été réalisé afin d’étudier le lien entre le métabolisme du glucose, les réserves d’ATP et la capacité de prolifération. L’OXPHOS, évaluée par le taux de consommation d’oxygène sensible à l’oligomycine (mitoOCR), et l’efficacité glycolytique, mesurée par le rapport entre le lactate produit et le glucose consommé (mol/mol) [13], ont été évaluées séparément sur des cellules incubées pendant 24 heures dans un milieu de culture contenant uniquement du glucose comme carburant énergétique. Pour étudier l’influence du statut métabolique sur les réserves d’énergie cellulaire, le pool total d’ATP intracellulaire a également été mesuré. Aucune corrélation significative n’a été trouvée entre le contenu en ATP intracellulaire et les paramètres métaboliques. Cependant, nous avons constaté que la prolifération cellulaire évaluée par la synthèse d’ADN était significativement corrélée à l’efficacité glycolytique (Figure 1A), où une glycolyse accrue était associée à une capacité de prolifération accrue, et inversement. Aucune corrélation significative n’a été observée entre la capacité de prolifération et la respiration mitochondriale. Ces données suggèrent que la glycolyse est potentiellement la voie énergétique clé soutenant la prolifération des cellules cancéreuses. L’hypothèse actuelle est que cette voie soutient la prolifération cellulaire principalement en fournissant des précurseurs pour les voies de biosynthèse, plutôt que par la production d’ATP [10,11]. L’absence de corrélation entre la teneur en ATP et la synthèse d’ADN dans les lignées cellulaires (figure supplémentaire S1) confirme que l’approvisionnement en ATP n’est pas le principal facteur limitant de la prolifération cellulaire. Pour confirmer que la glycolyse favorise la prolifération des cellules cancéreuses et exclure les influences indépendantes du métabolisme inhérentes aux différents génotypes, la capacité de prolifération des cellules cancéreuses cervicales SiHa oxydatives de type sauvage (WT) humain a été comparée à celle des SiHa présentant une déplétion mitochondriale partielle (SiHa ρ0) [14,15]. Les cellules cancéreuses SiHa ρ0 ont présenté un phénotype glycolytique avec une diminution de ~40% de la mitoOCR (Figure 1B), une augmentation de ~2,5 fois de l’efficacité glycolytique (Figure 1C) et une augmentation de ~50% de la consommation de glucose (Figure 1D). La diminution nette de ~20% du pool total d’ATP dans les cellules SiHa ρ0 a confirmé que l’augmentation de la glycolyse n’était pas suffisante pour maintenir les réserves d’ATP (Figure 1E). En effectuant une quantification de la synthèse de l’ADN, nous avons constaté que l’augmentation de la glycolyse aérobie favorise la prolifération cellulaire, car le changement glycolytique dans les cellules SiHa ρ0 était associé à une augmentation d’environ 45 % de la prolifération cellulaire (Figure 1F). Les tests de viabilité n’ont révélé aucune différence dans le nombre de cellules viables 24 heures après l’incubation des cellules dans le milieu de culture expérimental (figure supplémentaire S2), probablement parce que la quantification de la synthèse d’ADN détecte les changements précoces dans le taux de prolifération des cellules.
L’inhibition de la glycolyse par le DCA entrave la prolifération des cellules cancéreuses
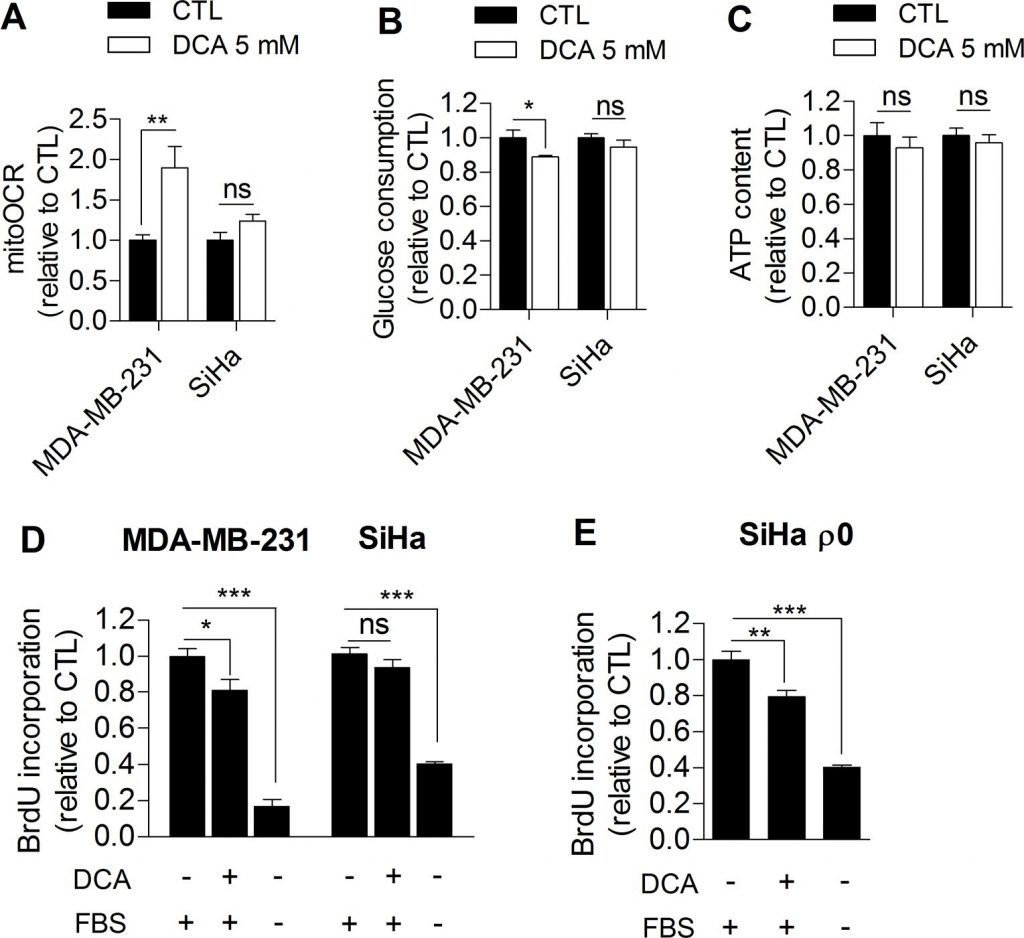
Sur la base de nos observations, nous avons cherché à savoir si l’inhibition de la glycolyse pouvait entraver directement la prolifération des cellules cancéreuses. À cette fin, nous avons traité des cellules humaines de cancer du sein MDA-MB-231 (phénotype de Warburg, figure 1A) et des cellules humaines de cancer du col de l’utérus SiHa (phénotype oxydatif, figure 1A) avec du dichloroacétate (DCA), un inhibiteur de la pyruvate déshydrogénase kinase (PDK) qui augmente l’activité oxydative des cellules en activant la pyruvate déshydrogénase (PDH), l’enzyme de maintien de l’oxydation du glucose dans les mitochondries [16]. À ce jour, l’effet thérapeutique prometteur du DCA sur les cellules cancéreuses est globalement attribué à une normalisation du potentiel de membrane mitochondrial hyperpolarisé qui caractérise les cellules cancéreuses et à une re-sensibilisation à l’apoptose [17]. Ici, nous avons postulé que le DCA contrôle également la prolifération tumorale en inhibant la glycolyse. Pour tester cette hypothèse, des cellules cancéreuses glycolytiques MDA-MB-231 et oxydatives SiHa ont été traitées avec 5 mM de DCA pendant 48 h, et les effets du traitement sur le métabolisme et la prolifération ont été évalués. Par rapport aux cellules traitées par un véhicule, le DCA a induit un passage des cellules cancéreuses glycolytiques MDA-MB-231 à un phénotype plus oxydatif, comme en témoignent une augmentation de la mitoOCR (figure 2A) et une diminution de la consommation de glucose (figure 2B). La diminution de l’activité glycolytique observée dans cette expérience est cohérente avec une autre étude récente [18] et est probablement induite par l’effet Pasteur [4,19] pour maintenir l’homéostasie de l’ATP dans les cellules (Figure 2C). Nous avons également observé que l’inhibition de la glycolyse par le DCA était associée à une diminution du taux de prolifération des cellules cancéreuses MDA-MB-231 (figure 2D). Pour confirmer que l’inhibition de la glycolyse nuit à la prolifération de cette lignée cellulaire, les cellules cancéreuses MDA-MB-231 traitées au 2-Deoxy-D-glucose ont également présenté un taux de prolifération réduit (figure supplémentaire S3).
En revanche, le DCA n’a pas modifié de manière significative les activités métaboliques des cellules cancéreuses SiHa oxydées (figures 2A-2C) et n’a eu aucun effet significatif sur la prolifération des SiHa (figure 2D). Les mesures de la production de lactate à court terme (1 heure) ont montré que le DCA était effectivement plus efficace sur la lignée de cellules cancéreuses glycolytiques que sur la lignée oxydative (figure supplémentaire S4). Afin de déterminer si le profil métabolique détermine la réponse au DCA, la capacité de prolifération des cellules cancéreuses glycolytiques SiHa ρ0 a également été analysée après le traitement au DCA. Nous avons constaté une diminution significative de la synthèse d’ADN dans cette lignée cellulaire (figure 2E), un effet qui n’a pas été observé dans les SiHa WT. En outre, le même nombre de cellules cancéreuses viables MDA-MB-231 et SiHa ρ0 a été mesuré 48 h après le traitement avec le véhicule ou le DCA (figure supplémentaire S5 A-B), ce qui montre que les effets du DCA sur les fonctions métaboliques et les taux de prolifération ne sont pas dus à la mortalité cellulaire.
La glycolyse contrôle la prolifération des cellules cancéreuses par la voie du pentose phosphate
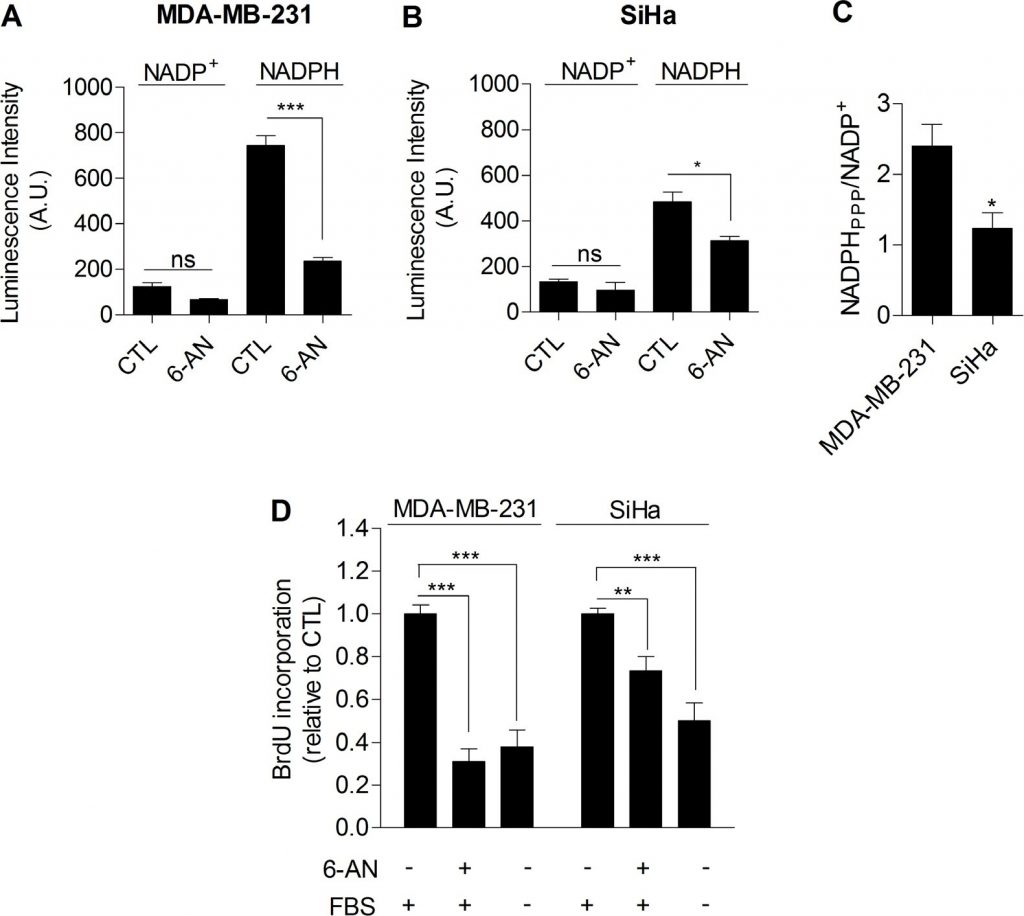
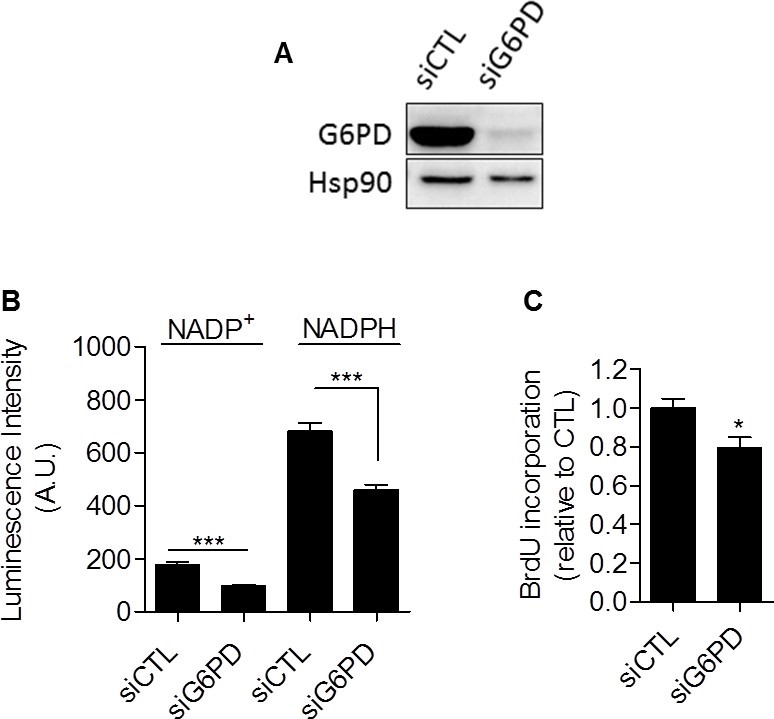
Dans l’ensemble, nos données ont fourni des preuves expérimentales compilées que la glycolyse en soi est impliquée dans le contrôle de la prolifération cellulaire. Le mécanisme reliant la glycolyse et la prolifération restait encore à établir. Nous avons postulé que la voie du pentose phosphate (PPP) pouvait relier la glycolyse à la prolifération, car la PPP utilise les intermédiaires glycolytiques pour fournir aux cellules des nucléotides et du NADPH, un réducteur crucial dans les processus anaboliques [20]. Pour déterminer spécifiquement le NADPH produit par la PPP (NADPHppp), les cellules ont été traitées avec du 6-aminonicotinamide (6-AN), un inhibiteur spécifique de la PPP [21,22]. Nous avons constaté que la contribution de la NADPHppp au pool total de NADPH (NADPHtot) était plus prédominante dans les cellules cancéreuses MDA-MB-231 que dans les cellules cancéreuses SiHa, comme le montre une diminution importante du niveau de NADPHtot après le traitement au 6-AN dans les cellules glycolytiques MDA-MB-231 (Figure 3A). Des effets plus limités ont été observés dans les cellules oxydatives SiHa (figure 3B). Les mesures ont également révélé un rapport NADPHppp/NADP+ plus élevé dans les cellules cancéreuses MDA-MB-231, mettant en évidence un flux de PPP plus important dans cette lignée cellulaire glycolytique par rapport à SiHa (Figure 3C). Pour vérifier que la PPP est impliquée dans le contrôle de la prolifération des cellules cancéreuses, la synthèse de l’ADN dans les cellules traitées et non traitées par le 6-AN a été évaluée. Nous avons observé que la capacité de prolifération des cellules cancéreuses était altérée lorsque le PPP était inhibé (Figure 3D). De manière intéressante, un effet plus fort a été mis en évidence dans les cellules cancéreuses glycolytiques MDA-MB-231 (~70 % de diminution du taux de synthèse d’ADN) par rapport aux cellules cancéreuses oxydatives SiHa (~25 % de diminution du taux de synthèse d’ADN) (Figure 3D). Il est important de noter que les SiHa ρ0 étaient plus sensibles que les SiHa WT au 6-AN, car une diminution de ~40 % de la synthèse d’ADN a été observée (figure supplémentaire S6). En raison des effets hors cible potentiels des inhibiteurs pharmacologiques, nous avons complété nos études sur le 6-AN en utilisant des petits ARN interférents (ARNi) ciblant la glucose-6-phosphate déshydrogénase (G6PD), la première enzyme du PPP et celle qui en limite la vitesse [23]. Dans les cellules transfectées MDA-MB-231, nous avons confirmé l’inhibition de la G6PD par immunoblotting (figure 4A) et démontré que, comme le 6-AN, l’inhibition de la G6PD diminuait l’activité de la PPP (figure 4B) et la synthèse de l’ADN (figure 4C). Ces résultats soulignent la contribution prédominante de la PPP au maintien de la prolifération des cellules cancéreuses de phénotype Warburg.
LeDCA inhibe la voie du pentose phosphate
Enfin, nous avons cherché à savoir si le DCA pouvait inhiber la PPP, ce qui expliquerait le lien entre l’inhibition de la glycolyse et la diminution du taux de prolifération des cellules cancéreuses traitées au DCA. Comme le montre la figure 5, nous avons observé que le DCA (5 mM, 48 h) diminuait considérablement le niveau de NADPHtot dans les cellules cancéreuses MDA-MB-231 (figure 5A). En outre, alors qu’aucune diminution supplémentaire du NADPHtot n’a été obtenue lorsque les cellules ont été exposées à la fois au DCA et au 6-AN (figure 5A), nous avons montré que le DCA diminuait spécifiquement le NADPHPPP. Des résultats similaires ont été obtenus en utilisant l’inhibition moléculaire de la G6PD (figure 5B). Ces résultats démontrent l’implication de la PPP dans les effets du DCA sur les cellules cancéreuses glycolytiques.
DISCUSSION
Cette étude souligne le rôle prédominant de la voie des pentoses phosphates dans la biologie du cancer. L’effet Warburg (la fermentation du glucose en lactate en présence d’une concentration physiologique d’O2 ) observé dans de nombreuses cellules cancéreuses a été proposé pour répondre aux demandes énergétiques (ATP) et biosynthétiques [4,10]. Dans cette étude, nous avons montré que l’augmentation de la glycolyse aérobie n’était pas suffisante pour maintenir les niveaux d’ATP dans les cellules cancéreuses. Ainsi, un faible niveau d’ATP intracellulaire était associé à une augmentation de la glycolyse. De plus, le changement glycolytique induit dans les cellules cancéreuses déficientes en mitochondries n’était pas en mesure de fournir un approvisionnement adéquat en ATP par rapport aux cellules de type sauvage. Il est actuellement suggéré que, d’un point de vue bioénergétique, un métabolisme glycolytique est moins efficace qu’un métabolisme oxydatif, mais que la glycolyse peut être avantageuse pour produire rapidement de l’ATP afin de répondre à des demandes énergétiques à court terme [24]. Nos résultats corroborent également le fait que les cellules cancéreuses de phénotype Warburg développent plusieurs adaptations pour maintenir de faibles niveaux d’ATP afin d’éviter l’inhibition allostérique des enzymes glycolytiques limitant la vitesse de production (effet Pasteur) et de conserver des taux de flux glycolytiques élevés [10]. D’autre part, nous avons montré que la glycolyse favorise la prolifération des cellules cancéreuses, soutenant les stratégies testées cliniquement qui inhibent l’effet Warburg [12,25]. La relation positive entre l’efficacité glycolytique (moles de lactate produites par mole de glucose consommée) et le taux de prolifération des cellules cancéreuses a mis en évidence que les cellules qui se divisent rapidement fermentent de grandes quantités de glucose en lactate. Des études récentes ont proposé que la production de lactate permette aux cellules cancéreuses de régénérer efficacement le NAD+ par l’enzyme lactate déshydrogénase [26]. En maintenant l’équilibre redox NAD+/NADH, les cellules cancéreuses permettent un flux de glucose plus rapide à travers la glycolyse, ainsi qu’une incorporation plus rapide des métabolites du glucose dans les voies de biosynthèse, conférant ainsi des avantages pour la prolifération [10].
Pour vérifier si l’augmentation de la glycolyse aérobie favorise la prolifération des cellules cancéreuses en alimentant les processus anaboliques, l’implication de la voie des pentoses phosphates (PPP) a été évaluée. En effet, il est connu que la PPP utilise le glucose-6-phophate (le produit de l’enzyme glycolytique hexokinase) pour fournir aux cellules des nucléotides et du NADPH, un réducteur crucial dans les réactions anaboliques [20]. Dans notre étude, nous avons montré que le flux de PPP, évalué par le rapport NADPHppp/NADP+, était accru dans les cellules cancéreuses de phénotype Warburg par rapport à celles de phénotype oxydatif. Une comparaison de l’activité des enzymes PPP aurait également pu être effectuée pour confirmer cette différence de flux PPP. Néanmoins, nos résultats confirment que l’activation de la glycolyse s’accompagne d’une augmentation de l’activité PPP pour la biosynthèse dans les cellules en division rapide [20]. De manière convaincante, nous avons observé que l’inhibition de la PPP diminuait la prolifération des cellules cancéreuses, avec un effet majeur dans les cellules cancéreuses de phénotype Warburg. La sensibilité différente des cellules cancéreuses glycolytiques et oxydatives à l’inhibition de la PPP est probablement due à la dépendance variable de cette voie pour la synthèse des macromolécules, comme les lipides et les nucléotides. En outre, les cellules cancéreuses présentant une activité mitochondriale élevée peuvent compenser les niveaux de NADPH en utilisant des enzymes associées au cycle TCA (enzymes maliques et isocitrate déshydrogénases) pour reconstituer le pool de NADPH [20]. L’ensemble de ces résultats démontre que les cellules cancéreuses, en particulier les cellules cancéreuses agressives de phénotype Warburg, dépendent de la voie du pentose phosphate pour une prolifération optimale.
Pour évaluer la pertinence thérapeutique de ces résultats, les effets du DCA ont été étudiés. Le DCA a déjà montré des propriétés anticancéreuses intéressantes in vitro et in vivo
[17], et est actuellement testé dans des essais cliniques de phase I-II [12]. À ce jour, le DCA doit ses propriétés thérapeutiques à la re-sensibilisation des cellules cancéreuses à l’apoptose [17]. Cependant, comme l’ont récemment souligné Stockwin et al [27], le DCA est relativement inactif sur la viabilité des cellules et n’induit l’apoptose qu’à des concentrations élevées. Stockwin et al. ont en outre signalé que le DCA était préférentiellement actif dans les cellules présentant un défaut mitochondrial. Par conséquent, nous avons postulé que, à faible dose, le DCA diminue également la prolifération des cellules cancéreuses en réduisant la glycolyse. Nous avons constaté que le DCA 5 mM était plus efficace dans les cellules cancéreuses de phénotype Warburg, réduisant la prolifération cellulaire en diminuant la glycolyse. Les différentes sensibilités entre les lignées cellulaires ayant un phénotype métabolique différent peuvent résulter de la capacité du DCA à atteindre le PDK dans la matrice des mitochondries. En effet, comme le pyruvate, le DCA est ionisé et ne peut pas traverser les membranes. Curieusement, il n’y a eu que peu de rapports sur le transport du DCA dans le cytosol des cellules de mammifères [28,29]. Bien qu’il n’ait pas encore été étudié, le niveau d’expression des transporteurs de pyruvate mitochondriaux (MPC) pourrait également définir l’efficacité du DCA. En outre, comme le DCA a un Kidifférent pour chacune des quatre isoenzymes PDK [30], la sensibilité variable des cellules cancéreuses peut résulter de l’expression différentielle des PDK.
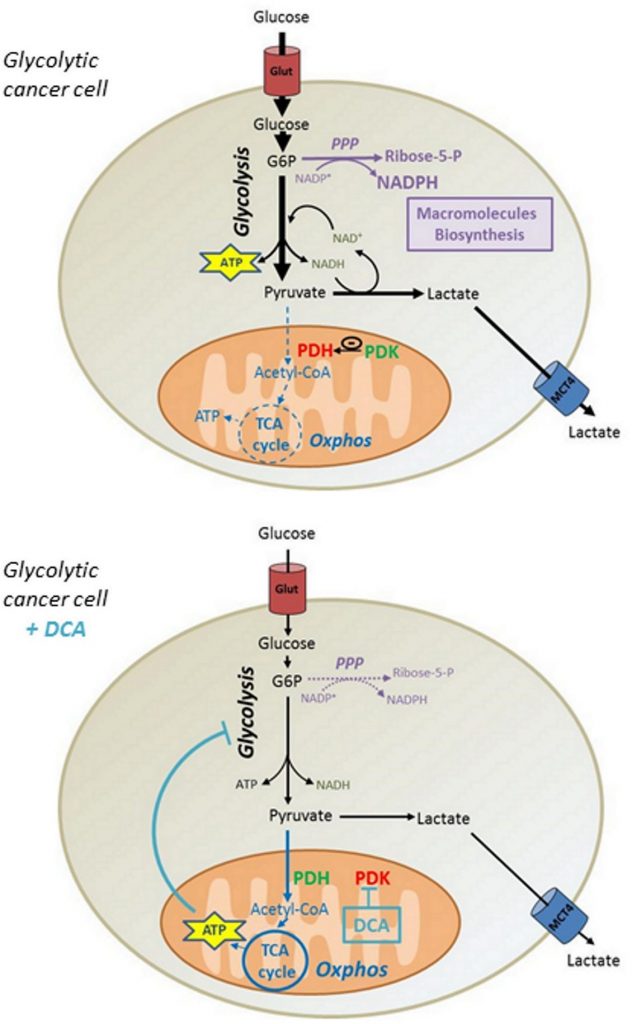
Bien que certains rapports aient montré que le DCA à des concentrations comparables favorise la cytotoxicité dans d’autres lignées cellulaires [31,32], nous avons montré que le DCA n’avait aucune incidence sur la viabilité des cellules cancéreuses MDA-MB-231 et SiHa. Sur la base de nos données, nous proposons que la réactivation de la production d’ATP dans les mitochondries induite par le DCA diminue le flux glycolytique des cellules cancéreuses, réduisant ainsi l’incorporation de glucose. Par conséquent, la diminution de la synthèse des intermédiaires glycolytiques réduit l’activité des voies de biosynthèse telles que la PPP, compromettant ainsi la prolifération des cellules cancéreuses de phénotype Warburg (Figure 6). Néanmoins, il est concevable que d’autres mécanismes puissent participer aux effets anti-prolifératifs du DCA, car d’autres études ont montré un arrêt du cycle cellulaire à des concentrations comparables de DCA [33, 34].
Dans l’ensemble, notre étude a montré que le PPP relie la glycolyse et la prolifération dans les cellules cancéreuses agressives, et plaide pour la prise en compte du métabolisme du cancer pour le choix de stratégies thérapeutiques anticancéreuses adéquates. En particulier, rejoignant les conclusions d’autres chercheurs [27], nous suggérons que le développement clinique du DCA pourrait bénéficier de la sélection de patients présentant des tumeurs hautement glycolytiques.
MATÉRIEL ET MÉTHODES
Culture cellulaire et réactifs
Le carcinome épidermoïde du col de l’utérus humain SiHa (ATCC), le cancer du sein humain MDA-MB-231 (ATCC), l’hépatocarcinome de souris TLT (transplantable liver tumor) [35], le fibrosarcome de souris FSaII [36], le sarcome de souris KHT [37], la tumeur mammaire de souris NT2 [38] ont été cultivés selon les recommandations du fournisseur ou comme décrit. Les SiHa présentant une déplétion mitochondriale partielle (ρ0) ont été obtenus par exposition chronique à de faibles concentrations de bromure d’éthidium, comme décrit précédemment [14,39]. Toutes les cultures ont été maintenues à 37°C dans une atmosphère deCO2 à 5%. Les cellules ont été incubées dans un milieu expérimental unique 24 heures avant les expériences (DMEM sans glutamine [Invitrogen], contenant 4,5 g/L de glucose complété par 10 % de FBS inactivé par la chaleur et 1 % de pénicilline-streptomycine). Lorsque les SiHa ρ0 ont été comparés aux SiHa de type sauvage, on a utilisé du DMEM sans glutamine (Invitrogen), contenant 4,5 g/L de glucose complété par 1 % de pyruvate, 10 % de FBS inactivé à la chaleur, 1 % de pénicilline-streptomycine et 50 ng/ml d’uridine. Sauf indication contraire, les expériences ont été réalisées sur des cellules confluentes. L’oligomycine est un inhibiteur de l’ATP synthase. Le dichloroacétate (DCA) est un inhibiteur de la pyruvate déshydrogénase kinase (PDK). le 6-aminonicotinamide (6-AN) est un inhibiteur des enzymes NADP+-dépendantes de la voie des pentoses phosphates, la glucose-6-phosphate déshydrogénase (G6PD) et la 6-phosphogluconate déshydrogénase (6-PGD). Tous les produits chimiques ont été achetés chez Sigma. L’oligomycine a été dissoute dans du DMSO. Le DCA et le 6-AN ont été directement dissous dans les milieux de culture.
transfection du siRNA
Le siRNA SMARTpool ON-TARGETplus contre la G6PD humaine et le siRNA non ciblé ON-TARGETplus proviennent de Dharmacon. La concentration finale du siRNA était de 25 nM. Tous les siRNA ont été transfectés en utilisant RNAi/MAX selon les instructions du fabricant (Invitrogen).
Western blotting
Des lysats de cellules entières ont été collectés et soumis à une analyse immunoblot comme décrit précédemment [40]. Les anticorps primaires étaient des monoclonaux humains contre G6PD ou Hsp90 (Sigma).
Consommation d’oxygène par les mitochondries
Le taux de consommation d’oxygène (OCR) des cellules entières intactes a été mesuré à l’aide d’un spectromètre RPE Bruker EMX fonctionnant à 9,5 GHz, comme décrit précédemment [41]. Les cellules adhérentes ont été récoltées dans un milieu expérimental frais (107 cellules/ml). 100 µl de la suspension cellulaire ont été mélangés avec 100 µl de dextran à 20% pour éviter l’agglomération et les cellules ont été scellées dans un tube capillaire en verre en présence de 0,2 mM d’une sonde nitroxyde agissant comme un capteur d’oxygène (15N4-oxo-2,2,6,6-tétraméthylpipéridine-d16-15N-1-oxyl, CDN isotopes, Pointe-Claire, Québec, Canada). Les cellules ont été maintenues à 37°C pendant l’acquisition des spectres. La largeur de la ligne RPE a été mesurée toutes les minutes et reportée sur une courbe d’étalonnage pour obtenir la concentration en oxygène. L’OCR a été déterminée par la valeur absolue de la pente de la diminution de la concentration d’oxygène dans le tube capillaire fermé en fonction du temps [42]. Pour obtenir la RCO provenant de la phosphorylation oxydative, la RCO en présence d’un traitement à l’oligomycine 1 µM a été soustraite de la RCO totale des cellules.
Consommation de glucose et production de lactate
La consommation de glucose et la production de lactate ont été mesurées à partir des surnageants des cellules cultivées. Les concentrations de métabolites ont été quantifiées sur des échantillons déprotéinés à l’aide de tests enzymatiques spécifiques sur un analyseur CMA600 (CMA Microdialysis AB, Solna, Suède). La consommation de glucose et la production de lactate ont été normalisées par rapport à la teneur en protéines à l’aide du test Pierce BCA Protein (Thermo Scientific). Pour détecter efficacement les différences de concentrations de métabolites entre les surnageants de SiHa WT et de SiHa ρ0, un milieu pauvre en glucose (1 g/L) a été utilisé.
Quantification de l’ATP intracellulaire
L’ATP intracellulaire total a été mesuré par le kit de détermination de l’ATP (Invitrogen) selon le protocole du fabricant. Les cellules ont été lavées deux fois avec du PBS et lysées dans le tampon recommandé par le fabricant (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0,01% Triton X-100). Les lysats cellulaires ont été ajoutés à un mélange réactionnel contenant de la luciférase et de la luciférine pour les mesures de bioluminescence à l’aide d’un lecteur de plaques (SpectraMax M2e, Molecular Devices). Une courbe standard a été générée avec des concentrations connues d’ATP dans les mêmes conditions. Une fraction des lysats cellulaires a été systématiquement utilisée pour la quantification des protéines (Pierce BCA Protein assay, Thermo Scientific) afin de normaliser le niveau d’ATP par rapport au contenu en protéines.
Prolifération
La prolifération cellulaire a été testée à l’aide d’un kit à base de 5-bromo-2′-déoxyuridine (BrdU)-ELISA (Roche) en suivant les instructions du fournisseur. Des cellules subconfluentes ont été incubées en présence de BrdU (un analogue de nucléotide) pendant 4 heures. Lorsque la prolifération de SiHa WT et SiHa ρ0 a été comparée, les cellules ont été incubées pendant 6 heures en présence de BrdU. La quantité de BrdU incorporée dans les cellules a été évaluée par des mesures colorimétriques à l’aide d’un lecteur de plaque (SpectraMax M2e, Molecular Devices), ce qui a permis de quantifier la synthèse d’ADN dans les cellules en réplication.
Mesures du NADPH et du NADP+
Les niveaux de NADP+ et de NADPH ont été détectés individuellement à partir de 6 000 cellules récoltées à l’aide d’un kit de détection (NADP/NADPH-Glo Assay, Promega) selon les instructions du fabricant.
Statistiques
Tous les résultats sont exprimés en tant que moyennes ± erreur standard de la moyenne (SEM). Les analyses statistiques ont été réalisées à l’aide du logiciel GraphPad Prism 5. P<0,05 a été considéré comme statistiquement significatif.
REMERCIEMENTS
Cette étude a été soutenue par des subventions du Fonds National de la Recherche Scientifique (F.R.S.-FNRS, PDR T.0107.13), du Fonds Joseph Maisin, de l’Action de Recherches Concertées ARC 14/19-058, et par une Starting Grant du Conseil Européen de la Recherche (ERC No. 243188 TUMETABO à P. Sonveaux). BFJ et PS sont des chercheurs associés, LB et PEP sont des chercheurs postdoctoraux, PD est un boursier Télévie postdoctoral. VLP est titulaire d’une bourse de doctorat du F.R.S.-FNRS. GDP est un boursier Télévie.
CONFLITS D’INTÉRÊTS
Les auteurs ont déclaré qu’il n’y a pas de conflit d’intérêts.
RÉFÉRENCES
1 Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ et Rodriguez-Enriquez S. Who controls the ATP supply in cancer cells ? Des leçons de biochimie pour comprendre le métabolisme énergétique du cancer. Int J Biochem Cell Biol. 2014 ; 50:10-23.2 Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL et Smith JW. Profilage comparatif des flux métaboliques des lignées cellulaires de mélanome : au-delà de l’effet Warburg. J Biol Chem. 2011 ; 286:42626-42634.
3 Jose C, Bellance N et Rossignol R. Choosing between glycolysis and oxidative phosphorylation : a tumor’s dilemma ? Biochim Biophys Acta. 2011 ; 1807:552-561.
4 Gatenby RA et Gillies RJ. Pourquoi les cancers ont-ils une glycolyse aérobie élevée ? Nat Rev Cancer. 2004 ; 4:891-899.
5 Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O et Dewhirst MW. Le ciblage de la respiration alimentée par le lactate tue sélectivement les cellules tumorales hypoxiques chez les souris. J Clin Invest. 2008 ; 118:3930-3942.
6 Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B et Sonveaux P. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014 ; 8:754-766.
7 Rolfe DF et Brown GC. Utilisation de l’énergie cellulaire et origine moléculaire du taux métabolique standard chez les mammifères. Physiol Rev. 1997 ; 77:731-758.
8 Warburg O. Uber den Stoffwechsel der Carcinomzelle. Klin Wochenschr. 1925;4:534-536..
9 Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P et Zhong H. ‘The metabolism of tumours’ : 70 years later. Novartis Found Symp. 2001 ; 240:251-260 ; discussion 260-254.
10 Lunt SY et Vander Heiden MG. Glycolyse aérobie : répondre aux exigences métaboliques de la prolifération cellulaire. Annu Rev Cell Dev Biol. 2011 ; 27:441-464.
11 DeBerardinis RJ, Lum JJ, Hatzivassiliou G et Thompson CB. The biology of cancer : metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008 ; 7:11-20.
12 Tennant DA, Duran RV et Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010 ; 10:267-277.
13 Zancan P, Sola-Penna M, Furtado CM et Da Silva D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Génétique moléculaire et métabolisme. 2010 ; 100:372-378.
14 De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O et Sonveaux P. Le lactate active HIF-1 dans les cellules tumorales humaines de phénotype oxydatif mais pas dans celles de phénotype Warburg. PLoS One. 2012 ; 7:e46571.
15 Bol V, Bol A, Bouzin C, Labar D, Lee JA, Janssens G, Porporato PE, Sonveaux P, Feron O et Gregoire V. Reprogrammer le métabolisme tumoral en ciblant les mitochondries améliore la réponse tumorale à l’irradiation. Acta Oncol. 2014:1-9.
16 Stacpoole PW et Felts JM. Effets du dichloroacétate de diisopropylammonium sur le métabolisme du glucose. Proc West Pharmacol Soc. 1969 ; 12:111-113.
17 Kankotia S et Stacpoole PW. Dichloroacetate et cancer : New home for an orphan drug ? Biochim Biophys Acta. 2014 ; 1846:617-629.
18 Abildgaard C, Dahl C, Basse AL, Ma T et Guldberg P. La modulation bioénergétique avec le dichloroacétate réduit la croissance des cellules de mélanome et potentialise leur réponse à l’inhibition de BRAFV600E. J Transl Med. 2014 ; 12:247.
19 Neveu MA, Bol V, Bol A, Bouzin C, Gregoire V, Feron O, Jordan BF et Gallez B. L’augmentation de l’oxygénation de la tumeur sous respiration carbogène induit une diminution de la captation du [F]-fluoro-deoxy-glucose. Radiother Oncol. 2015 ; 116:400-3.
20 Jiang P, Du W et Wu M. Régulation de la voie du pentose phosphate dans le cancer. Protein Cell. 2014 ; 5:592-602.
21 Koutcher JA, Alfieri AA, Matei C, Meyer KL, Street JC et Martin DS. Effet de la 6-aminonicotinamide sur la voie du pentose phosphate : études de la RMN 31P et du retard de croissance tumorale. Magn Reson Med. 1996 ; 36:887-892.
22 Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, Sharpe MA, Xin L et Frigo DE. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenèse. 2014 ; 3:e103.
23 Krebs HA et Eggleston LV. La régulation du cycle des pentoses phosphates dans le foie de rat. Adv Enzyme Regul. 1974 ; 12:421-434.
24 Epstein T, Xu L, Gillies RJ et Gatenby RA. Separation of metabolic supply and demand : aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014 ; 2:7.
25 Chen X, Qian Y et Wu S. The Warburg effect : Interprétations évolutives d’un concept établi. Free Radic Biol Med. 2015 ; 79:253-263.
26 Porporato PE, Dhup S, Dadhich RK, Copetti T et Sonveaux P. Cibles anticancéreuses dans le métabolisme glycolytique des tumeurs : une revue complète. Front Pharmacol. 2011 ; 2:49.
27 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG et Newton DL. Le dichloroacétate de sodium cible sélectivement les cellules présentant des défauts dans l’ETC mitochondrial. Int J Cancer. 2010 ; 127:2510-2519.
28 Babu E, Ramachandran S, Coothan-Kandaswamy V, Elangovan S, Prasad PD, Ganapathy V et Thangaraju M. Role of SLC5A8, a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate. Oncogene. 2011 ; 30:4026-4037.
29 Jackson VN et Halestrap AP. La cinétique, le substrat et la spécificité des inhibiteurs du transporteur de monocarboxylate (lactate) des cellules hépatiques de rat déterminés en utilisant l’indicateur de pH intracellulaire fluorescent, 2′,7′-bis(carboxyéthyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996 ; 271:861-868.
30 Bowker-Kinley MM, Davis WI, Wu P, Harris RA et Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998 ; 329:191-196.
31 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B et Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007 ; 11:37-51.
32 Pajuelo-Reguera D, Alan L, Olejar T et Jezek P. Le dichloroacétate stimule les changements dans la morphologie du réseau mitochondrial via une mitophagie partielle dans les cellules de neuroblastome humain SH-SY5Y. Int J Oncol. 2015 ; 46:2409-2418.
33 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS et Michelakis ED. L’activation mitochondriale par l’inhibition de PDKII supprime la signalisation de HIF1a et l’angiogenèse dans le cancer. Oncogene. 2013 ; 32:1638-1650.
34 Hong SE, Shin KS, Lee YH, Seo SK, Yun SM, Choe TB, Kim HA, Kim EK, Noh WC, Kim JI, Hwang CS, Lee JK, Hwang SG, Jin HO et Park IC. L’inhibition de S6K1 améliore la mort cellulaire induite par le dichloroacétate. J Cancer Res Clin Oncol. 2015 ; 141:1171-1179.
35 Taper HS, Woolley GW, Teller MN et Lardis MP. Une nouvelle tumeur du foie de souris transplantable d’origine spontanée. Cancer Res. 1966 ; 26:143-148.
36 Volpe JP, Hunter N, Basic I et Milas L. Metastatic properties of murine sarcomas and carcinomas. I. Positive correlation with lung colonization and lack of correlation with s.c. tumor take. Clin Exp Metastasis. 1985 ; 3:281-294.
37 Rockwell S et Kallman RF. Cinétique de croissance et de population cellulaire des sarcomes KHT simples et multiples. Cell Tissue Kinet. 1972 ; 5:449-457.
38 Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH et Jaffee EM. HER-2/neu est une cible de rejet des tumeurs chez les souris transgéniques HER-2/neu tolérées. Cancer Res. 2000 ; 60:3569-3576.
39 King MP et Attardi G. Human cells lacking mtDNA : repopulation with exogenous mitochondria by complementation. Science. 1989 ; 246:500-503.
40 Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA et Michel T. Endothelial nitric oxide synthase targeting to caveolae. Interactions spécifiques avec les isoformes de cavéolines dans les myocytes cardiaques et les cellules endothéliales. J Biol Chem. 1996 ; 271:22810-22814.
41 Diepart C, Verrax J, Calderon PB, Feron O, Jordan BF et Gallez B. Comparison of methods for measuring oxygen consumption in tumor cells in vitro. Anal Biochem. 2010 ; 396:250-256.
42 Jordan BF, Gregoire V, Demeure RJ, Sonveaux P, Feron O, O’Hara J, Vanhulle VP, Delzenne N and Gallez B. L’insuline augmente la sensibilité des tumeurs à l’irradiation : implication d’une augmentation de l’oxygénation de la tumeur médiée par une diminution de la consommation d’oxygène des cellules tumorales dépendante de l’oxyde nitrique. Cancer Res. 2002 ; 62:3555-3561.
Contenu connexe :