Helena Populo1,2, Regina Caldas1,2,3, Jose Manuel Lopes1,2,4,5, Joana Pardal5, Valdemar Maximo1,2,4 & Paula Soares†,2,4
† Istituto per la Ricerca e l’Innovazione nella Salute (Instituto de Investigacao e Inovacao em Saude), Università di Porto, Porto, Portogallo
1 Istituto di Patologia Molecolare e Immunologia dell’Università di Porto (IPATIMUP), Università di Porto, Porto, Portogallo
Tel: +22 557 0700; Fax: +22 557 0799; E-mail: [email protected]
2 Istituto per la Ricerca e l’Innovazione nella Salute (Instituto de Investigacao e Inovac¸aoem Saude), Università di Porto, Porto, Portogallo
3 Facoltà di Medicina, Università di Porto, Porto, Portogallo
4 Dipartimento di Patologia e Oncologia, Facoltà di Medicina, Università di Porto, Porto, Portogallo
5 Servizio di Patologia Anatomica Ospedale Sao Joao, Porto, Portogallo
Pubblicato online: 14 maggio 2015
Abstract
Obiettivo: Ci siamo posti l’obiettivo di verificare se esistono prove per considerare il dicloroacetato (DCA), che inibisce la piruvato deidrogenasi chinasi (PDK) e inverte lo spostamento metabolico delle cellule tumorali dalla glicolisi alla fosforilazione ossidativa, come un farmaco promettente per la terapia dei pazienti affetti da melanoma cutaneo (CM).
Disegno e metodi di ricerca: Abbiamo valutato il profilo di espressione di PDK 1, 2 e 3 in una serie di campioni di melanoma, per verificare se i tumori di melanoma esprimono i bersagli di DCA, se questa espressione correla con l’attivazione di cascate di segnalazione importanti per la melanomagenesi e anche con la prognosi dei pazienti di melanoma. Abbiamo anche stabilito la sensibilità delle linee cellulari di melanoma al trattamento con DCA, valutando le loro alterazioni metaboliche, la proliferazione e la sopravvivenza.
Risultati: Abbiamo osservato che entrambe le isoforme PDK 1 e 2 sono sovraespresse nel CM rispetto ai nevi, espressione associata all’espressione degli effettori della via mTOR e indipendente dallo stato mutazionale di BRAF. Le linee cellulari di melanoma trattate con DCA hanno mostrato un cambiamento nel metabolismo, cioè una diminuzione del consumo di glucosio e della produzione di lattato, una riduzione della proliferazione, un aumento dell’apoptosi e una diminuzione dell’attivazione della via mTOR.
Conclusioni: I nostri risultati suggeriscono che l’espressione di PDK può svolgere un ruolo nello sviluppo del melanoma e che il DCA può essere utile per la terapia del CM, da solo o in combinazione con gli inibitori di mTOR.
Parole chiave: dicloroacetato, melanoma, metabolismo, mTOR, piruvato deidrogenasi chinasi
© 2015 Informa UK, Ltd. ISSN 1472-8222, e-ISSN 1744-7631
INTRODUZIONE
Il melanoma cutaneo (CM) è un tumore maligno molto aggressivo e, nonostante sia il tipo di cancro della pelle meno comune, è responsabile della maggior parte dei decessi legati ai tumori della pelle. Poiché l’incidenza del CM è in aumento, attualmente è il tumore invasivo più probabile da sviluppare prima dei 50 anni nel sesso maschile [1,2]. L’esposizione ai raggi UV è considerata il principale fattore di rischio per la melanomagenesi [2]. Il CM può essere classificato in diversi sottotipi istologici: il più comune è il melanoma a diffusione superficiale (SSM), seguito dal melanoma nodulare (NM), dal melanoma lentigo maligna (LMM) e dal melanoma acrale lentigginoso (ALM). Il SSM e il NM insorgono nella pelle con esposizione solare intermittente, mentre l’LMM si verifica nella pelle cronicamente danneggiata dal sole e l’ALM è limitato alla pelle senza esposizione solare. Questa classificazione istologica non ha valore prognostico [3,4]. La stadiazione del CM tiene conto dello spessore del tumore, dell’ulcerazione, del tasso mitotico, del coinvolgimento dei linfonodi e della presenza di metastasi [5]. Fortunatamente, la maggior parte dei casi di CM viene diagnosticata in fase precoce, con un tasso di sopravvivenza a 5 anni che raggiunge il 98% [1]. Tuttavia, per i pazienti con melanoma metastatico, la sopravvivenza mediana è di soli 8-9 mesi [5].
Il CM è un tumore molto eterogeneo e molte vie di segnalazione cellulare sono deregolate nella melanomagenesi [6,7]. La via MAPK è costitutivamente attivata nella maggior parte dei CM. Mutazioni NRAS sono state segnalate nel 10-20% delle MC, la più comune delle quali è NRASQ61K/R. Le mutazioni BRAF, in particolare la mutazione BRAFV600E, sono state identificate nel 40-60% dei casi. Il nostro gruppo ha osservato un’associazione tra la presenza di mutazioni BRAF e le mutazioni del promotore TERT recentemente descritte nei CM [8]. Mutazioni BRAFV600E e, in misura minore, NRASQ61K/R sono state riportate anche in quasi l’80% dei nevi (lesioni melanocitarie benigne che nel 25% dei casi sono considerate precursori del CM), indicando che l’attivazione della via MAPK può essere necessaria ma non sufficiente per lo sviluppo del melanoma [7,9,10]. Infatti, BRAFV600E è stato correlato alla senescenza indotta dall’oncogene e quindi può portare, in molti nevi, a una condizione di arresto della crescita [11]. L’attivazione della via PI3K-AKTmTOR può superare questo fenotipo di senescenza e favorire la progressione tumorale [12]. Il nostro gruppo ha confermato l’importanza della via PI3K-AKT-mTOR nell’aggressività del CM, associando la sua attivazione alla presenza di mutazioni BRAF e a caratteristiche prognostiche peggiori. Infatti, la sovraespressione degli effettori della via mTOR è stata associata a un tasso mitotico più elevato, a un livello di Clark più alto, a un maggiore spessore del tumore e alla presenza di ulcerazioni cutanee [13].
Prima del 2011, le opzioni terapeutiche per i pazienti con CM avanzato si basavano principalmente sulla chemioterapia convenzionale, che presentava bassi tassi di risposta ed effetti minori sulla sopravvivenza globale (OS) dei pazienti. Da allora, cinque nuovi farmaci sono stati approvati per i pazienti con CM in stadio IV [14]. L’ipilimumab, un anticorpo anti-CTLA4, ha rivelato notevoli miglioramenti nella OS dei pazienti, ma i suoi effetti avversi lo hanno reso non adatto a tutti i pazienti [15]. Vemurafenib e dabrafenib, inibitori selettivi di BRAFV600E, e trametinib, un inibitore di MEK, hanno tutti come bersaglio la via MAPK e sono stati riportati tassi di risposta superiori al 50%, persino migliori di quelli di ipilimumab. Tuttavia, i loro miglioramenti nella OS non superano i 7-8 mesi a causa dello sviluppo di resistenza [16-18]. Recentemente, un altro immuno-modulatore, pembrolizumab, un anticorpo contro il recettore di morte programmata-1 (PD-1), è stato approvato per il trattamento di pazienti con melanoma non resecabile o metastatico e progressione della malattia dopo ipilimumab o, se positivi alla mutazione BRAFV600 , un inibitore BRAF [19]. Sono in corso studi clinici con farmaci che mirano alla via PI3K-AKT-mTOR [4,14], ma sono necessari nuovi approcci terapeutici.
Il metabolismo delle cellule tumorali è diverso da quello delle cellule normali. Nelle cellule normali, a seconda della disponibilità di O2, il glucosio può essere parzialmente metabolizzato a lattato attraverso la glicolisi in condizioni di ipossia, oppure può essere completamente ossidato aCO2 in presenza di O2, attraverso la fosforilazione ossidativa mitocondriale, un processo energetico più efficiente. Le cellule tumorali, invece, metabolizzano la maggior parte del glucosio in lattato, indipendentemente dall’apporto di O2, il cosiddetto effetto Warburg [20,21]. L’acquisizione di questo fenotipo glicolitico nelle cellule tumorali non è del tutto chiara, ma è stato riconosciuto un ruolo essenziale del fattore di trascrizione hypoxia-inducible factor 1a (HIF1-α) [22]. È noto che la proteina HIF1-α si stabilizza in condizioni di ipossia, ma anche le vie oncogeniche, come le vie MAPK e mTOR, sembrano mediare la sua attivazione nel cancro (Figura 1) [22,23]. Una forte evidenza indica che il CM acquisisce questo fenotipo glicolitico, che può essere confermato dalla scansione FDGPET dei pazienti [24-27]. In effetti, HIF1-α sembra essere sovraespresso nella CM, perché la pelle è un ambiente lievemente ipossico e la produzione di melanina stimola indirettamente l’espressione di HIF1-α attraverso la produzione di ROS [28-30]. Tuttavia, la glicolisi citoplasmatica deve essere disaccoppiata dalla fosforilazione ossidativa mitocondriale, in modo che la maggior parte del piruvato possa essere convertito in lattato. Quest’ultimo processo è guidato dalla piruvato deidrogenasi chinasi (PDK), un enzima upregolato da HIF1-α (Figura 1) [31,32].
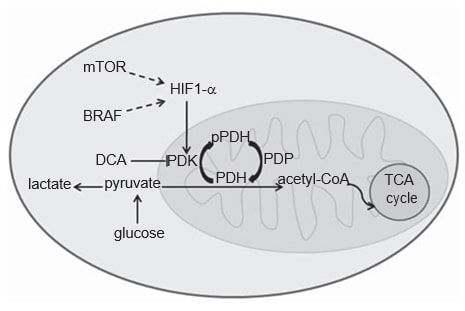
La PDK è un elemento del complesso della piruvato deidrogenasi (PDC) mitocondriale. La PDC comprende l’enzima piruvato deidrogenasi (PDH) e le sue proteine regolatrici: PDK, che fosforilando la PDH agisce da inibitore, e la fosfatasi della piruvato deidrogenasi, che attiva la PDH mediante de-fosforilazione [33]. La PDH defosforilata catalizza la decarbossilazione ossidativa del piruvato in acetil-CoA,CO2 e NADH (H+ ), collegando così la via glicolitica alla via ossidativa del ciclo dell’acido tricarbossilico (Figura 1) [33]. Esistono quattro isoforme di PDK (1, 2, 3, 4) che si differenziano per l’attività intrinseca, la distribuzione tissutale e la sensibilità all’inibitore selettivo, il dicloroacetato (DCA) [33-35]. Sebbene la PDK1 sia l’unica in grado di legarsi a tutti i siti di fosforilazione della PDH, la PDK3 sembra essere l’isoforma più attiva [33]. La PDK2 è la più sensibile all’inibizione da parte del DCA ed è espressa in modo ubiquitario in diversi tessuti umani, mentre le altre sono più specifiche per i tessuti [33,34]. La PDK4 è essenzialmente legata alla flessibilità metabolica fisiologica ed è l’unica isoforma non upregolata da HIF1-α [32,36]. L’espressione delle isoforme PDK è stata valutata in vari tipi di cancro, in particolare nel carcinoma a cellule squamose della testa e del collo (HNSCC) [37], nel carcinoma del colon [38], nel carcinoma a cellule renali [39] e nel carcinoma gastrico [40].
Il DCA è stato utilizzato nel trattamento di condizioni associate all’acidosi lattica nel contesto della disfunzione mitocondriale, come le malattie mitocondriali congenite [35]. Bonnet et al. hanno riferito che il DCA induce l’apoptosi e riduce la crescita cellulare promuovendo l’ossidazione del glucosio, la depolarizzazione della membrana mitocondriale e la produzione di ROS in cellule di cancro al seno e non a piccole cellule [41]. Questi effetti sono stati replicati in diversi modelli di cancro e sembrano essere selettivi nelle cellule tumorali [35,41,42]. Inoltre, il DCA è anche in grado di sopprimere l’angiogenesi attraverso l’inibizione indiretta di HIF1-α [43]. Tutti questi dati, uniti al noto profilo di sicurezza nell’uomo, dimostrano che il DCA è un farmaco promettente per la terapia del cancro. È già stato condotto uno studio clinico per valutare l’effetto del DCA in pazienti affetti da glioblastoma
Il melanoma cutaneo (CM) è un tumore maligno molto aggressivo e, nonostante sia il tipo di tumore della pelle meno comune, è responsabile della maggior parte dei decessi legati al cancro della pelle. Poiché l’incidenza del CM è in aumento, attualmente è il tumore invasivo più probabile da sviluppare prima dei 50 anni nel sesso maschile [1,2]. L’esposizione ai raggi UV è considerata il principale fattore di rischio per la melanomagenesi [2]. Il CM può essere classificato in diversi sottotipi istologici: il più comune è il melanoma a diffusione superficiale (SSM), seguito dal melanoma nodulare (NM), dal melanoma lentigo maligna (LMM) e dal melanoma acrale lentigginoso (ALM). Il SSM e il NM insorgono nella pelle con esposizione solare intermittente, mentre l’LMM si verifica nella pelle cronicamente danneggiata dal sole e l’ALM è limitato alla pelle senza esposizione solare. Questa classificazione istologica non ha valore prognostico [3,4]. La stadiazione del CM tiene conto dello spessore del tumore, dell’ulcerazione, del tasso mitotico, del coinvolgimento dei linfonodi e della presenza di metastasi [5]. Fortunatamente, la maggior parte dei casi di CM viene diagnosticata in fase precoce, con un tasso di sopravvivenza a 5 anni che raggiunge il 98% [1]. Tuttavia, per i pazienti con melanoma metastatico, la sopravvivenza mediana è di soli 8-9 mesi [5]. Il CM è un tumore molto eterogeneo e molte vie di segnalazione cellulare sono deregolate nella melanomagenesi [6,7]. La via MAPK è costitutivamente attivata nella maggior parte dei CM. Mutazioni NRAS sono state segnalate nel 10-20% delle MC, la più comune delle quali è NRASQ61K/R. Le mutazioni BRAF, in particolare la mutazione BRAFV600E, sono state identificate nel 40-60% dei casi. Il nostro gruppo ha osservato un’associazione tra la presenza di mutazioni BRAF e le mutazioni del promotore TERT recentemente descritte nei CM [8]. Mutazioni BRAFV600E e, in misura minore, NRASQ61K/R sono state riportate anche in quasi l’80% dei nevi (lesioni melanocitarie benigne che nel 25% dei casi sono considerate precursori del CM), indicando che l’attivazione della via MAPK può essere necessaria ma non sufficiente per lo sviluppo del melanoma [7,9,10]. Infatti, BRAFV600E è stato correlato alla senescenza indotta dall’oncogene e quindi può portare, in molti nevi, a una condizione di arresto della crescita [11]. L’attivazione della via PI3K-AKTmTOR può superare questo fenotipo di senescenza e favorire la progressione tumorale [12]. Il nostro gruppo ha confermato l’importanza della via PI3K-AKT-mTOR nell’aggressività del CM, associando la sua attivazione alla presenza di mutazioni BRAF e a caratteristiche prognostiche peggiori. Infatti, la sovraespressione degli effettori della via mTOR è stata associata a un tasso mitotico più elevato, a un livello di Clark più alto, a un maggiore spessore del tumore e alla presenza di ulcerazioni cutanee [13].
Prima del 2011, le opzioni terapeutiche per i pazienti con CM avanzato si basavano principalmente sulla chemioterapia convenzionale, che presentava bassi tassi di risposta ed effetti minori sulla sopravvivenza globale (OS) dei pazienti. Da allora, cinque nuovi farmaci sono stati approvati per i pazienti con CM in stadio IV [14]. L’ipilimumab, un anticorpo anti-CTLA4, ha rivelato notevoli miglioramenti nella OS dei pazienti, ma i suoi effetti avversi lo hanno reso non adatto a tutti i pazienti [15]. Vemurafenib e dabrafenib, inibitori selettivi di BRAFV600E, e trametinib, un inibitore di MEK, hanno tutti come bersaglio la via MAPK e sono stati riportati tassi di risposta superiori al 50%, persino migliori di quelli di ipilimumab. Tuttavia, i loro miglioramenti nella OS non superano i 7-8 mesi a causa dello sviluppo di resistenza [16-18]. Recentemente, un altro immuno-modulatore, pembrolizumab, un anticorpo contro il recettore di morte programmata-1 (PD-1), è stato approvato per il trattamento di pazienti con melanoma non resecabile o metastatico e progressione della malattia dopo ipilimumab o, se positivi alla mutazione BRAFV600, un inibitore BRAF [19]. Sono in corso studi clinici con farmaci mirati alla via PI3K-AKT-mTOR [4,14], ma sono necessari nuovi approcci terapeutici. Il metabolismo delle cellule tumorali è diverso da quello delle cellule normali. Nelle cellule normali, a seconda della disponibilità di O2, il glucosio può essere parzialmente metabolizzato a lattato attraverso la glicolisi in condizioni di ipossia, oppure può essere completamente ossidato aCO2 in presenza di O2, attraverso la fosforilazione ossidativa mitocondriale, un processo energetico più efficiente. Le cellule tumorali, invece, metabolizzano la maggior parte del glucosio in lattato, indipendentemente dall’apporto di O2, il cosiddetto effetto Warburg [20,21]. L’acquisizione di questo fenotipo glicolitico nelle cellule tumorali non è del tutto chiara, ma è stato riconosciuto un ruolo essenziale del fattore di trascrizione hypoxia-inducible factor 1a (HIF1-α) [22]. È noto che la proteina HIF1-α si stabilizza in condizioni di ipossia, ma anche le vie oncogeniche, come le vie MAPK e mTOR, sembrano mediare la sua attivazione nel cancro (Figura 1) [22,23]. Una forte evidenza indica che il CM acquisisce questo fenotipo glicolitico, che può essere confermato dalla scansione FDGPET dei pazienti [24-27]. In effetti, HIF1-α sembra essere sovraespresso nella CM, perché la pelle è un ambiente lievemente ipossico e la produzione di melanina stimola indirettamente l’espressione di HIF1-α attraverso la produzione di ROS [28-30]. Tuttavia, la glicolisi citoplasmatica deve essere disaccoppiata dalla fosforilazione ossidativa mitocondriale, in modo che la maggior parte del piruvato possa essere convertito in lattato. Quest’ultimo processo è guidato dalla piruvato deidrogenasi chinasi (PDK), un enzima upregolato da HIF1-α (Figura 1) [31,32]. La PDK è un elemento del complesso della piruvato deidrogenasi (PDC) mitocondriale. La PDC comprende l’enzima piruvato deidrogenasi (PDH) e le sue proteine regolatrici: PDK, che fosforilando la PDH agisce da inibitore, e la fosfatasi della piruvato deidrogenasi, che attiva la PDH mediante de-fosforilazione [33]. La PDH defosforilata catalizza la decarbossilazione ossidativa del piruvato in acetil-CoA,CO2 e NADH (H+ ), collegando così la via glicolitica alla via ossidativa del ciclo dell’acido tricarbossilico (Figura 1) [33]. Esistono quattro isoforme di PDK (1, 2, 3, 4) che si differenziano per l’attività intrinseca, la distribuzione tissutale e la sensibilità all’inibitore selettivo, il dicloroacetato (DCA) [33-35]. Sebbene la PDK1 sia l’unica in grado di legarsi a tutti i siti di fosforilazione della PDH, la PDK3 sembra essere l’isoforma più attiva [33]. La PDK2 è la più sensibile all’inibizione da parte del DCA ed è espressa in modo ubiquitario in diversi tessuti umani, mentre le altre sono più specifiche per i tessuti [33,34]. La PDK4 è essenzialmente legata alla flessibilità metabolica fisiologica ed è l’unica isoforma non upregolata da HIF1-α [32,36]. L’espressione delle isoforme PDK è stata valutata in vari tipi di cancro, in particolare nel carcinoma a cellule squamose della testa e del collo (HNSCC) [37], nel carcinoma del colon [38], nel carcinoma a cellule renali [39] e nel carcinoma gastrico [40]. Il DCA è stato utilizzato nel trattamento di condizioni associate all’acidosi lattica nel contesto della disfunzione mitocondriale, come le malattie mitocondriali congenite [35]. Bonnet et al. hanno riferito che il DCA induce l’apoptosi e riduce la crescita cellulare promuovendo l’ossidazione del glucosio, la depolarizzazione della membrana mitocondriale e la produzione di ROS in cellule di cancro al seno e non a piccole cellule [41]. Questi effetti sono stati replicati in diversi modelli di cancro e sembrano essere selettivi nelle cellule tumorali [35,41,42]. Inoltre, il DCA è anche in grado di sopprimere l’angiogenesi attraverso l’inibizione indiretta di HIF1-α [43]. Tutti questi dati, uniti al noto profilo di sicurezza nell’uomo, dimostrano che il DCA è un farmaco promettente per la terapia del cancro. È già stato condotto uno studio clinico per valutare l’effetto del DCA in pazienti affetti da glioblastoma, che ha riportato un aumento dell’apoptosi e una riduzione dell’angiogenesi, con risultati promettenti in quattro dei cinque pazienti [44].
Nel presente studio è stata valutata l’espressione delle isoforme PDK 1 – 3 nel CM e la sua associazione con l’espressione delle principali cascate di vie di segnalazione per la melanomagenesi e con la prognosi dei pazienti affetti da melanoma. Inoltre, sono stati eseguiti studi in vitro per valutare l’effetto del trattamento con DCA sul metabolismo, la proliferazione e la sopravvivenza di linee cellulari di melanoma con diversi profili genetici.
Materiali e metodi
Selezione dei campioni, parametri clinico-patologici e prognostici
I tessuti fissati in formalina e inclusi in paraffina di 120 casi di CM e di 22 nevi melanocitici (12 nevi composti e 10 nevi di Spitz) sono stati prelevati dal Servizio di Patologia Anatomica dell’Ospedale S. Joa˜o di Porto e dell’Ospedale S. Marcos di Braga. I dati clinico-patologici (Tabella 1) e di follow-up sono stati ottenuti dalle cartelle cliniche dei pazienti e dai registri oncologici dell’Ospedale S. Joa˜o e dell’Ospedale S. Marcos, nonché dal RORENO (Registro Oncologico della Regione Nord). Tutti i casi sono stati rivisti e stadiati secondo la settima edizione dell’AJCC [5]. I dati di follow-up includono le recidive e le metastasi (DFS; n = 108) e il numero di decessi dovuti al melanoma (mortalità malattia-specifica; OS; n = 118). Il tempo medio di follow-up dei pazienti per la DFS è stato di 51 mesi (SE ± 3,59, range 1-195) e per la OS di 55 mesi (SE ± 3,48, range 1 – 207). Questo lavoro è stato approvato dal Comitato etico locale ed è stato conforme alle norme etiche nazionali.
Analisi immunoistochimica
La colorazione delle proteine analizzate è stata eseguita su sezioni di paraffina di 3 µm di aree tumorali rappresentative, montate su vetrini rivestiti di poli-L-lisina. Le sezioni sono state deparaffinate e reidratate, seguite da una procedura di recupero dell’antigene a microonde con tampone citrato di sodio 10 mM a pH 6,0 con 1 mM di EDTA a pH 9,0 (PDK2) o tampone EDTA a pH 9,0 (PDK1 e PDK3). Le sezioni sono state incubate per una notte a 4°C in camera umidificata con gli anticorpi primari PDK1 (policlonale, coniglio, 1:50), PDK2 (policlonale, coniglio, 1:150), PDK3 (monoclonale, topo, 1:500), tutti della Sigma-Aldrich Co. (St. Louis, Missouri, USA). La rilevazione è stata ottenuta con il sistema Envision G/2 System/AP (K5355; Dako, Danimarca). metodo anti-fosfatasi alcalina (APAAP) e il colore è stato sviluppato con cromogeno rosso permanente. I vetrini sono stati controcolorati con ematossilina e quindi montati con un mezzo di montaggio miscibile con acqua. Il metodo APAAP è stato eseguito per evitare l’interferenza della pigmentazione melaninica con l’analisi immunoistochimica. Il tessuto gastrico umano è stato utilizzato come controllo negativo (omissione dell’anticorpo primario) e positivo.
Valutazione immunoistochimica
Tre osservatori (J.M.L., H.P. e R.C.) hanno valutato l’immunoreattività delle cellule tumorali senza conoscere i dati clinici dei casi. L’immunoreattività del tessuto non tumorale adiacente è stata utilizzata come controllo interno. È stato stabilito un punteggio IHC per PDK1 – 3, che risulta dalla moltiplicazione del punteggio di intensità della colorazione (negativo = 0, debole = 1, moderato = 2 e forte = 3) e del punteggio di estensione dell’immunoreattività delle cellule tumorali (0 – 5% = 0, 6 – 25% = 1, 26 – 50% = 2, 51 – 75% = 3, 76 – 100% = 4). I punteggi IHC sono stati poi classificati come negativi/bassi (valore del punteggio £ 4) e moderati/alti (valore del punteggio > 4). Le analisi delle cascate MAPK e PI3KAKT-mTOR in parte della serie sono state effettuate in precedenza [13] e i risultati sono stati utilizzati in questo studio.
Estrazione del DNA e analisi delle mutazioni
L’estrazione del DNA da tumori di dimensioni inferiori a 5 mm è stata eseguita dopo microdissezione con PALM MicroLaser Systems (PALM, Germania) e utilizzando il kit Quiamp DNA micro (Quiagen, Hilden). Nei tumori di dimensioni superiori a 5 mm, l’estrazione del DNA è stata effettuata mediante dissezione manuale di sezioni intere di 10 µm di tessuto incluso in paraffina utilizzando il mini kit Invisorb spin tissue (Invitek, Berlino). I frammenti dell’esone 15 di BRAF e dell’esone 2 di NRAS sono stati amplificati mediante reazione a catena della polimerasi (PCR) utilizzando primer precedentemente descritti [45]. Il DNA genomico (25 – 100 ng) è stato amplificato mediante PCR utilizzando le seguenti condizioni di ciclaggio: 35 s a 94°C, 40 s a 58°C per BRAF e 57°Cper NRAS, e 45 s a 72°Cper 40 cicli.
| Caratteristiche clinico-patologiche | |
| Numero di casi (n) | 120 |
| Età mediana (± SD) | 61.5 ± 17.0 |
| Sesso (n [%]) | |
| Femmina | 68 (56.7) |
| Maschio | 52 (43.3) |
| Esposizione al sole (n [%]) | |
| Assente | 27 (22.7) |
| Intermittente | 72 (60.5) |
| Cronica | 20 (16.8) |
| Sottotipo istologico (n [%]) | |
| LMM | 16 (13.3 |
| ALM | 24 (20.0) |
| NM | 19 (15.8) |
| SSM | 61 (50.8) |
| Spessore mediano (mm) | 3.7 (0 — 70) |
| Ulcerazione epidermica (n [%]) | |
| Assente | 79 (65.8) |
| Presente | 41 (34.2) |
| Tasso mitotico (n [%]) | |
| < 1/mm2 | 45 (37.5) |
| ≥1/mm2 | 75 (62.5 |
| pT (n [%]) | |
| ≤ pT2 | 62 (51.7) |
| >pT2 | 58 (48.3) |
Tutti i prodotti di PCR sono stati purificati e sequenziati direttamente su un sequenziatore automatico ABI Prism 3130 xl (Perkin-Elmer, Foster City, CA) utilizzando il kit di sequenziamento ABI Prism Dye Terminator Cycle (Perkin-Elmer). La reazione di sequenziamento è stata eseguita in direzione forward e nei campioni sospettati di essere portatori di mutazioni è stata eseguita un’amplificazione PCR indipendente, in direzione forward e reverse. Anche in questo caso, l’analisi mutazionale di BRAF e NRAS in parte della serie è stata effettuata in precedenza [13] e i risultati sono stati utilizzati in questo studio.
Linee cellulari e condizioni di coltura
In questo lavoro sono state utilizzate due linee cellulari, la linea cellulare di melanoma cutaneo A375 portatrice di BRAFV600E e la linea cellulare di melanoma cutaneo Mewo portatrice di BRAFwt. Entrambe le linee cellulari sono state analizzate per verificare la presenza di micoplasma.
A375 è stata mantenuta in terreno RPMI (Gibco/BRL – Invitrogen) e Mewo in terreno DMEM (Gibco/BRL – Invitrogen). Tutti i terreni sono stati integrati con il 10% di siero fetale bovino, 100 U/ml di penicillina e 100 µg/ml di streptomicina. Le linee cellulari sono state mantenute in atmosfera umidificata (5%CO2) a 37°C.
Trattamento delle linee cellulari di melanoma con DCA
Il DCAsodico, acquistato da Sigma-Aldrich (St. Louis, MO, EUA), è stato sciolto in dH2Oe aggiunto al terreno di coltura e utilizzato per il trattamento di 24 e 48 ore. Le cellule di melanoma incubate con terreno di coltura integrato con dH2Osono servite come controllo.
Saggio di vitalità cellulare
Gli effetti del DCA sulla crescita delle linee cellulari di melanoma sono stati analizzati mediante il saggio PrestoBlue (PB). Le cellule sono state seminate in piastre da 96 pozzetti a una densità di 5 103 in 200 µl di terreno. Dopo 24 ore, il terreno è stato sostituito con un terreno contenente 5, 20, 40, 60 mM di DCA. Le cellule sono state incubate per 24 e 48 ore, lavate con PBS (pH 7,4) e saggiate per la crescita cellulare con PB secondo le istruzioni del produttore. Durante l’incubazione con le cellule, il reagente PB viene modificato dall’ambiente riducente delle cellule vitali e diventa altamente fluorescente. La fluorescenza è stata misurata con un lettore di micropiastre (Synergy HT Multi-Mode Microplate Reader, BioTek Instruments Inc., Winooski, VT, USA) alle lunghezze d’onda di eccitazione ed emissione di 560 e 590 nm, rispettivamente. L’assorbanza dei pozzetti contenenti terreno di coltura e cellule tumorali è stata utilizzata come controllo e ogni condizione sperimentale è stata valutata in triplo e ripetuta in doppio. Confrontando le misure di fluorescenza/assorbanza dei pozzetti contenenti DCA con quelle dei pozzetti contenenti cellule non trattate, è stato possibile generare profili dose-risposta e determinare i valori di IC50 (la concentrazione che inibisce la sopravvivenza del 50%), utilizzando GraphPadPrism5.0 (GraphPad Software, Inc., La Jolla, CA).
Quantificazione del glucosio e del lattato
Per le misurazioni dei livelli di glucosio e lattato, le cellule di melanoma sono state disposte in piastre a 6 pozzetti a una densità finale di 2 105 cellule/pozzetto e incubate a 37°C per 24 ore. Le cellule sono state poi trattate con 35 mM di DCA. Come controllo, le cellule sono state incubate con il composto veicolo (dH2O). Dopo 24 e 48 ore di trattamento, il terreno di coltura è stato raccolto. I livelli di glucosio presenti nel mezzo condizionato in coltura sono stati quantificati utilizzando il Glucose GOD/PAP Kit (Roche Applied Sciences) e sottratti ai livelli iniziali (0 h). Il lattato è stato quantificato in modo simile, utilizzando il saggio enzimatico colorimetrico LO-POD (Spinreact, Sant Esteve de Bas, Spagna).
Analisi del ciclo cellulare e dell’apoptosi
Per l’analisi del profilo del ciclo cellulare e dell’apoptosi, le cellule di melanoma sono state disposte in piastre a 6 pozzetti a una densità finale di 1 105 cellule/pozzetto e incubate a 37°C per 24 ore. Le cellule sono state poi trattate con DCA a 35 mM per 24 e 48 ore di trattamento. Come controllo, le cellule sono state incubate con il composto veicolo (dH2O). Per l’analisi del ciclo cellulare, le cellule sono state raccolte e fissate per una notte in etanolo al 70% freddo. Successivamente, le cellule sono state risospese in PBS con 0,1 mg/ml di RNasi A e 5 µg/ml di ioduro di propidio, prima dell’analisi. Per le misurazioni dell’apoptosi, le cellule sono state raccolte e i livelli di apoptosi sono stati analizzati mediante citometria a flusso utilizzando l’Annexin-V FITC Apoptosis Kit (Clontech Laboratories, Inc., Saint-Germainen-Laye, Francia) secondo le istruzioni del produttore. L’analisi in citometria a flusso del contenuto di DNA cellulare e dell’esternalizzazione della fosfatidilserina è stata eseguita con un citometro a flusso (BD Accuri C6), tracciando almeno 20.000 eventi per campione. I dati sono stati analizzati con il software FlowJo 7.6.5 (Tree Star, Inc., Ashland, USA).
Analisi Western blot e anticorpi
Le cellule sono state lisate per 15 minuti a 4°C utilizzando il tampone RIPA (1% NP-40 in 150 mM NaCl, 50 mM Tris (pH 7,5), 2 mM EDTA) contenente inibitori di fosfatasi e proteasi. Le proteine sono state quantificate utilizzando un saggio Bradford modificato (Biorad). I campioni di proteine (50 µg) sono stati separati in gel SDS/ PAGE al 10% ed elettroblottati su membrana Hybond ECL (Amersham Biosciences). Abbiamo utilizzato i seguenti anticorpi primari: PDH e pPDH Ser293, da Abcam; PDK1 (Sigma-Aldrich), PDK2 (Sigma-Aldrich), HIF1-α (Transduction Laboratories) e gli effettori della via mTOR pS6 Ser235/236, s6, p-4EBP1 Thr37/46, 4EBP1, tutti da Cell Signaling Technology. Gli anticorpi secondari sono stati coniugati con perossidasi (Santa Cruz Biotechnology) e visualizzati con la soluzione di rilevamento ECL. Le membrane sono state colorate nuovamente con un anticorpo policlonale di capra anti-actina (Santa Cruz Biotechnology) per il controllo del carico proteico. Tutti gli esperimenti e le quantificazioni (utilizzando il software Bio-Rad Quantity One 1-D Analysis (versione 4.6.6)) sono stati eseguiti in triplo.
Analisi statistica
L’analisi statistica è stata eseguita utilizzando STAT VIEW-J 5.0 (SAS Institute, Inc., Cary, NC). La relazione tra il livello medio di espressione (punteggio) dei marcatori immunoistochimici e i parametri clinico-patologici è stata valutata mediante ANOVA. Quando appropriato, sono state eseguite correzioni per confronti multipli utilizzando i test post hoc di Bonferroni e Tamhane. La correlazione tra il punteggio di immunoreattività dei diversi marcatori è stata valutata utilizzando il test esatto di Fisher. I dati degli esperimenti sulle linee cellulari sono stati analizzati con il test t di Student a due code non accoppiate. Il metodo Kaplan-Meier e il test log-rank sono stati utilizzati per valutare i dati di sopravvivenza del melanoma. Sono state eseguite analisi univariate e multivariate per determinare il valore prognostico delle covariate in relazione a OS e DFS utilizzando il modello di regressione di Cox. L’OS e la DFS sono state calcolate dal momento della diagnosi fino alla morte per malattia o metastasi, rispettivamente, o censurate al momento dell’ultimo follow-up o della morte non correlata alla malattia. Un valore p < 0,05 è stato considerato statisticamente significativo.
Risultati
Espressione delle PDK nella CM e nei nevi
Sia nella CM che nelle lesioni nevi, le isoforme PDK 1, 2 e 3 sono state espresse non solo nei melanociti/melanoma, ma anche nei cheratinociti e nelle cellule delle ghiandole sebacee e dei follicoli piliferi. I livelli di espressione di tutte le isoforme PDK analizzate erano positivamente correlati tra loro, suggerendo un’espressione concomitante di queste proteine (Tabella 2).
| Proteine | Livello medio di espressione (± SD) | Correlazione (valore p) | Correlazione (valore p) | Correlazione (valore p) |
| PDK1 | 4.8 ± 3.5 | < 0.01* | < 0.01§ | |
| PDK2 | 4.6 ± 3.6 | < 0.01† | ||
| PDK3 | 7.0 ± 3.2 |
*Tra PDK1 e PDK2; † tra PDK2 e PDK3; § tra PDK1 e PDK3.
Espressione immunoistochimica di PDK1
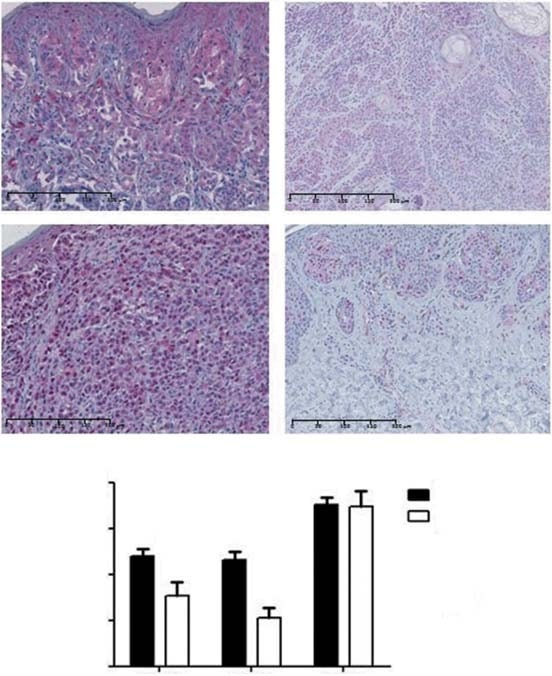
La colorazione citoplasmatica di PDK1 è stata osservata nell’88% dei CM e nel 76% dei nevi (Figura 2A). Il punteggio di colorazione negativo/basso è stato osservato nel 52 e 76% e il punteggio di colorazione moderato/alto è stato osservato nel 48 e 24% dei melanomi e dei nevi, rispettivamente. Pertanto, i melanomi hanno mostrato livelli medi di espressione di PDK1 significativamente più elevati rispetto ai nevi (p = 0,03; Figura 2B). Per quanto riguarda i sottotipi istologici di CM, i LMM hanno mostrato livelli di espressione di PDK1 maggiori rispetto ai NM (p = 0,04) e agli ALM (p = 0,04). Non è stata trovata alcuna associazione tra l’espressione di PDK1 e il tipo di esposizione al sole. Per quanto riguarda i fattori prognostici, PDK1 era maggiormente espressa nei tumori di 1 mm di spessore (p = 0,02). Livelli di espressione di PDK1 più elevati erano associati a stadi tumorali più bassi (p = 0,04), ma non alla sopravvivenza globale e libera da malattia dei pazienti. Abbiamo anche valutato se l’espressione delle isoforme PDK fosse correlata alle vie MAPK e PI3K-AKT-mTOR. Non è stata trovata alcuna associazione tra l’espressione di PDK1 e le mutazioni BRAF/NRAS o con l’attivazione della via MAPK (valutata dall’espressione di pERK). L’espressione di PDK1 è stata correlata positivamente con l’espressione di proteine della cascata PI3K-AKT-mTOR, in particolare mTOR e 4EBP1 (p = 0,02 e p = 0,04, rispettivamente).
Espressione immunoistochimica di PDK2
PDK2 ha mostrato un’espressione sia nucleare che citoplasmatica nell’87% dei CM e nell’83% dei nevi (Figura 2A). Il punteggio di colorazione negativo/basso è stato osservato nel 58 e nell’89% e il punteggio di colorazione moderato/alto è stato osservato nel 42 e nell’11% dei melanomi e dei nevi, rispettivamente. Il livello medio di espressione di PDK2 era più alto nei melanomi rispetto ai nevi (p < 0,01; Figura 2B). Per quanto riguarda i sottotipi istologici di CM, SSM ha mostrato livelli più elevati di espressione di PDK2 rispetto ad ALM (p = 0,02). Non abbiamo trovato alcuna associazione tra il tipo di esposizione al sole e l’espressione di PDK2. Gli stadi tumorali più bassi hanno mostrato livelli significativamente più elevati di PDK2 (p = 0,02), sebbene non sia stata trovata alcuna associazione tra l’espressione di questa isoforma e i principali fattori prognostici del CM, né con la sopravvivenza globale e libera da malattia dei pazienti. L’espressione di PDK2 non è stata associata allo stato mutazionale di BRAF/NRAS o all’attivazione della via MAPK. È stata trovata una correlazione positiva significativa tra l’espressione di PDK2 e l’espressione di AKT della cascata PI3K-AKT-mTOR (p = 0,03).
Espressione immunoistochimica di PDK3
PDK3 ha mostrato un’espressione citoplasmatica in tutti i CM e nevi. Il punteggio di colorazione negativo/basso è stato osservato nel 28 e 22% e il punteggio di colorazione moderato/alto è stato osservato nel 72 e 78% dei CM e dei nevi, rispettivamente. Il livello di espressione dell’isoforma PDK3 era simile tra CM e nevi (Figura 2B). Non sono state riscontrate differenze significative nel confronto del livello medio di espressione di PDK3 tra i sottotipi di CM e il tipo di esposizione al sole. I tumori con spessore £ 1 mm e quelli con stadi inferiori mostravano livelli più elevati di PDK3 (p = 0,01 e p < 0,01, rispettivamente). L’ulcerazione, il tasso mitotico e la sopravvivenza globale e libera da malattia dei pazienti non erano significativamente associati all’espressione di PDK3. Non è stata trovata alcuna associazione tra l’espressione di PDK3 e le mutazioni BRAF/NRAS né l’attivazione della cascata MAPK. Per quanto riguarda la via PI3K-AKT-mTOR, l’espressione di PDK3 è risultata positivamente correlata all’espressione totale di AKT, mTOR e 4EBP1 (rispettivamente p < 0,01, p < 0,01 e p = 0,04). A causa dell’espressione generale di PDK3 in ogni CM e nevi, la successiva analisi in vitro non ha incluso questa isoforma PDK.
Effetto del trattamento con DCA sulla vitalità delle linee cellulari di melanoma
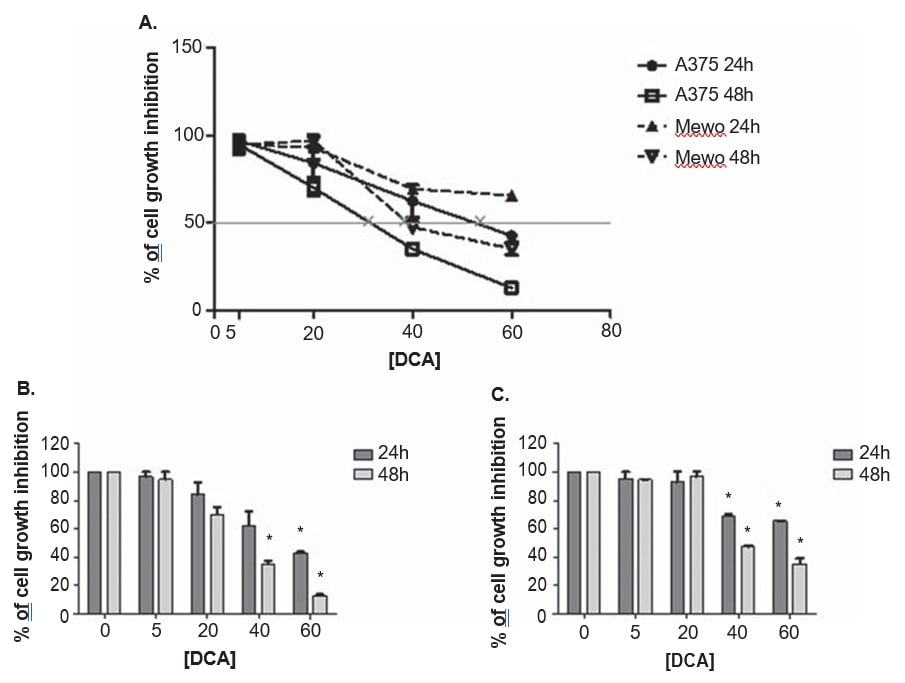
Le linee cellulari di melanoma A375 e Mewo sono state esposte a concentrazioni crescenti di DCA per stabilire l’effetto sulla vitalità cellulare, utilizzando il saggio PrestoBlue. Il DCA ha ridotto la vitalità di entrambe le linee cellulari in modo dose-dipendente dopo 24 e 48 ore di trattamento, anche se la A375 è risultata leggermente più sensibile al trattamento con DCA, con una maggiore inibizione della crescita rispetto alla linea cellulare Mewo (Figura 3). L’effetto del DCA sulla vitalità delle cellule A375 è stato osservato dopo il trattamento con 5 mM e nella linea cellulare Mewo è stato più intenso dopo il trattamento con 20 mM, con valori di inibizione compresi tra il 3 e l’87% in A375 e tra il 5 e il 65% in Mewo. I valori di IC50 sono stati stimati in 33 ± 5,5 mM per A375 e 39,9 ± 2,2 mM per la linea cellulare Mewo, dopo 48 ore di trattamento con DCA (Figura 3).
Effetto del trattamento con DCA sui livelli di consumo di glucosio e produzione di lattato delle linee cellulari di melanoma
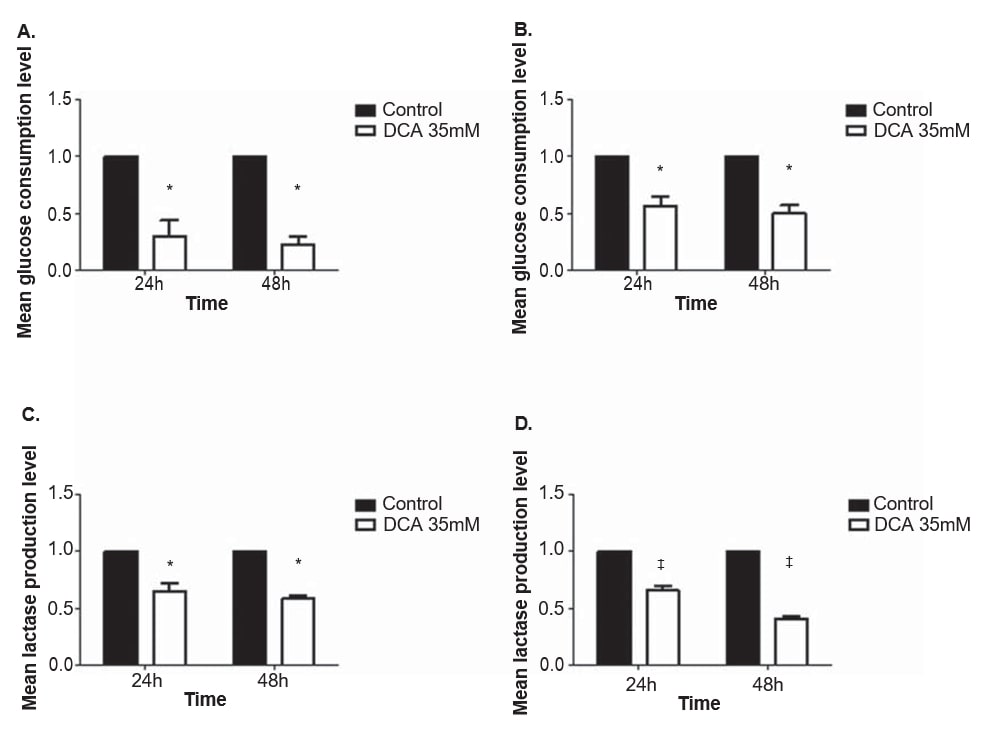
Per valutare l’effetto del DCA sul metabolismo delle linee cellulari di melanoma, sono stati quantificati i livelli di glucosio e lattato nel terreno di coltura dopo 24 e 48 ore di trattamento con 35 mM di DCA, una concentrazione vicina all’IC50 per entrambe le linee cellulari. Il consumo di glucosio e la produzione di lattato sono diminuiti in entrambe le linee cellulari di melanoma dopo il trattamento con DCA (Figura 4). Le linee cellulari A375 e Mewo hanno mostrato una diminuzione significativa del livello di consumo di glucosio sia dopo 24 che dopo 48 ore di trattamento con DCA (p = 0,01 e p < 0,01, rispettivamente). Anche il livello di produzione di lattato è diminuito significativamente dopo 24 e 48 ore nelle linee cellulari A375 (p = 0,01 e p < 0,01, rispettivamente) e Mewo (p < 0,01).
Effetto del trattamento con DCA sugli effettori delle vie PDK e mTOR e sull’espressione di HIF1-α nelle linee cellulari di melanoma
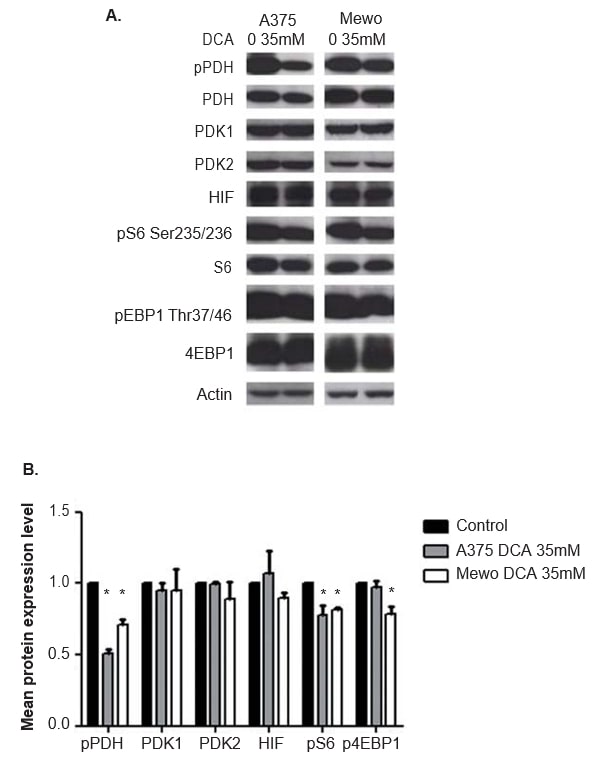
L’efficacia del trattamento con DCA nell’inibire l’attività di PDK è stata valutata analizzando l’espressione dell’effettore a valle fosforilato PDH, mediante western blot. A 24 ore dal trattamento con DCA, è stata osservata una significativa inibizione della fosforilazione di PDH in entrambe le linee cellulari (p < 0,01; Figura 5). L’espressione di PDK1 e PDK2 non è stata alterata dal trattamento con DCA (Figura 5), come previsto. È stata valutata anche l’espressione dei due effettori a valle e indicatori dell’attivazione della via mTOR, S6 e 4EBP1. Sebbene il trattamento con DCA non abbia alterato l’espressione di S6 e 4EBP1, il livello di espressione delle forme fosforilate di entrambe le proteine è diminuito in A375 (p = 0,03 e non significativo, rispettivamente) e in Mewo (p < 0,01 e p = 0,01, rispettivamente; Figura 5). Poiché il DCA può influenzare i livelli di HIF1-α, è stata valutata anche l’espressione di questa proteina e non è stata osservata alcuna variazione dopo il trattamento con DCA (Figura 5). Risultati simili sono stati ottenuti a 48 ore dal trattamento con DCA, che inibisce significativamente la fosforilazione di PDH in entrambe le linee cellulari (p <0,01). Il livello di espressione delle forme fosforilate di S6 e 4EBP1 è diminuito significativamente in A375 (p = 0,01 e p = 0,02, rispettivamente) e in Mewo (p = 0,01 e non significativo, rispettivamente). L’espressione di PDK1, PDK2 e HIF1-α non è stata alterata (dati non mostrati).
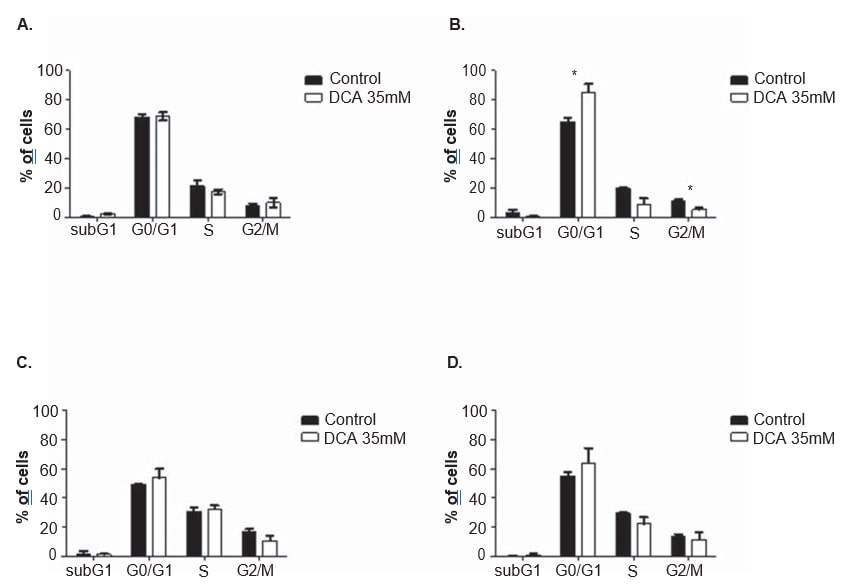
Effetto del trattamento con DCA sul ciclo cellulare e sull’apoptosi delle linee cellulari di melanoma
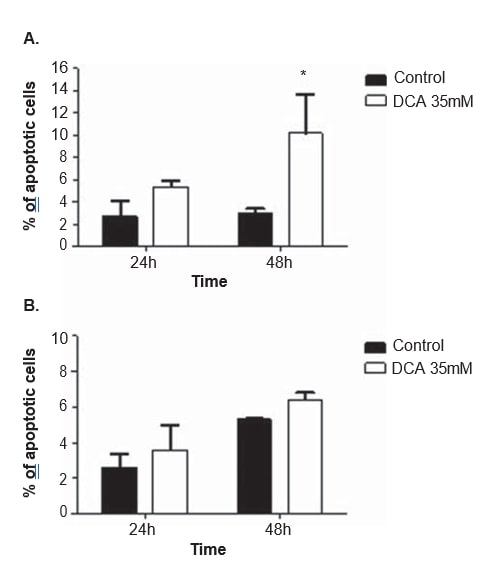
Per chiarire il meccanismo d’azione del DCA, sono state effettuate analisi del ciclo cellulare e misurazioni dell’apoptosi in entrambe le linee cellulari, dopo 24 e 48 ore di trattamento con 35 mM di DCA. Il ciclo cellulare è stato analizzato con lo ioduro di propidio. Dopo 48 ore di trattamento con DCA, la linea cellulare A375 ha mostrato un aumento significativo della percentuale di cellule nella fase G0/G1 del ciclo cellulare (dal 65,0 ± 4,9% osservato nelle cellule non trattate all’85,2 ± 10,2% nelle cellule trattate con DCA; p = 0,04), una diminuzione significativa nella fase G2/M (dall’11.3 ± 1,3% osservato nelle cellule non trattate a 5,2 ± 2,2% nelle cellule trattate con DCA; p = 0,01) e una diminuzione, che non raggiunge la soglia di significatività statistica, nella fase S (da 19,7 ± 1,8% osservato nelle cellule non trattate a 9,1 ± 6,7% nelle cellule trattate con DCA; p = 0,06; Figura 6). La stessa tendenza è stata osservata nella linea cellulare Mewo 48 ore dopo il trattamento con DCA (aumento della percentuale di cellule in G0/G1 e diminuzione in S e G2/M), anche se non ha raggiunto la significatività statistica (Figura 6). L’analisi in citometria a flusso è stata effettuata dopo la colorazione con Annexin V/ioduro di propidio. È stato osservato un aumento significativo del numero di cellule apoptotiche nella linea cellulare A375 dopo 48 ore di trattamento con DCA (dal 3,0 ± 0,5% nelle cellule non trattate al 16,9 ± 4,6% nelle cellule trattate con DCA; p < 0,01; Figura 7). Sono stati osservati aumenti non significativi della percentuale di cellule apoptotiche nella linea cellulare A375 dopo 24 ore e nella linea cellulare Mewo dopo 24 ore e 48 ore di trattamento con DCA.
Discussione
In questo lavoro abbiamo stabilito, per la prima volta, che la CM sovraesprime le proteine PDK1 e PDK2. Le PDK sono proteine chiave, regolate da HIF1-α, che guidano le cellule maligne verso il fenotipo della glicolisi aerobica, un segno distintivo del cancro [20,22,31].
L’effetto Warburg è considerato un fattore di resistenza ai chemioterapici convenzionali [21]. La sovraespressione di PDK riscontrata nel CM ci ha portato a valutare l’effetto del trattamento con DCA nelle linee cellulari di melanoma. Abbiamo osservato che il trattamento con DCA riduce il consumo di glucosio e la produzione di lattato nelle linee cellulari di melanoma, coerentemente con l’effetto del DCA nel passaggio dalla glicolisi aerobica alla fosforilazione ossidativa precedentemente riportato nelle cellule tumorali [41]. Questo spostamento nel metabolismo del glucosio deriva probabilmente dalla diminuzione significativa dell’espressione della forma fosforilata della PDH, che conferma l’efficacia del trattamento con DCA. È importante sottolineare che la lettura corretta dell’effetto del DCA è il livello di PDH fosforilata, poiché inibendo l’attività della PDK e non la sua espressione, il DCA stimola la de-fosforilazione della PDH (Figura 1).
Abbiamo osservato una diminuzione dose-dipendente della vitalità cellulare dopo il trattamento con DCA in entrambe le linee cellulari di melanoma. Inoltre, è stata osservata una riduzione della proliferazione, attraverso l’arresto G0/G1, insieme a un aumento dell’apoptosi in entrambe le linee cellulari, ma solo in modo statisticamente significativo nella linea cellulare A375. Il fatto che la concentrazione di DCA scelta nel presente studio (35 mM) sia leggermente inferiore all’IC50 per la linea cellulare Mewo può spiegare l’effetto meno pronunciato osservato in queste cellule.
La relazione tra DCA e HIF-1a nel cancro non è completamente chiarita, poiché il DCA aumenta il consumo di ossigeno dei tessuti e la produzione di ROS, che dovrebbero portare rispettivamente all’up-regolazione o alla stabilizzazione di HIF-1a [46,47]. Al contrario, alcuni autori hanno riportato che il DCA porta a una diminuzione dell’espressione di HIF1-α [25,48,49]. Nel nostro lavoro non è stata osservata alcuna alterazione del livello di HIF1-α dopo il trattamento con DCA delle linee cellulari di melanoma, in accordo con Shahrzad et al. che hanno riportato che l’espressione di HIF1-α diminuisce solo dopo il trattamento con DCA in condizioni di ipossia [49].
Per superare la senescenza indotta dal mutante BRAF e progredire verso fenotipi più aggressivi, il CM probabilmente attiva altre cascate di segnalazione, come la via mTOR [12]. Questa via contribuisce all’effetto Warburg promuovendo l’attività di HIF1-α, che a sua volta può aumentare l’espressione di PDK [23,31]. In precedenza abbiamo riportato un’attivazione completa di questa via nella CM [13]. In questo studio, abbiamo osservato un’espressione concomitante di PDK 1, 2 e 3 e degli effettori della via mTOR nei CM. L’espressione delle isoforme PDK nella CM è correlata positivamente con l’espressione di mTOR, nonché con gli effettori a monte e a valle di questa via: AKT e 4EBP1. Inoltre, abbiamo osservato una diminuzione significativa di pS6 e p4EBP1 nelle linee cellulari A375 e Mewo dopo il trattamento con DCA.
I nevi condividono alcune alterazioni genetiche con il CM, poiché entrambi presentano un’alta frequenza di mutazioni BRAF [10]. Sebbene a livelli molto più bassi, l’espressione di PDK è stata osservata anche nei nevi. Abbiamo valutato se lo stato mutazionale di BRAF e l’attivazione a valle di ERK potessero essere associati a una maggiore espressione di PDK. Non abbiamo trovato alcuna associazione né nei nevi né nella CM. La linea cellulare A375 presenta la mutazione BRAFV600E, mentre la linea cellulare Mewo è wild type per questo gene; la risposta di entrambe le linee cellulari al trattamento con DCA è stata simile, nonostante l’intensità variabile. In realtà, è già stato riportato che lo stato mutazionale di BRAF non altera la sensibilità al trattamento con DCA [50]. Questi risultati contrastano con quelli ottenuti in precedenza utilizzando inibitori della via mTOR in diverse linee cellulari derivate dal melanoma, dove abbiamo riscontrato una maggiore sensibilità al trattamento con RAD001 nelle linee cellulari CM portatrici della mutazione BRAFV600E [51].
Le isoforme PDK sono state espresse nel citoplasma delle cellule di melanoma, a sostegno della nota funzione mitocondriale di tali isoforme [33]. Per quanto riguarda PDK2, nella maggior parte dei casi di CM è stata osservata un’espressione nucleare. Questa localizzazione nucleare è stata segnalata anche per PDK1 nell’HNSCC [37]. Per quanto ne sappiamo, il ruolo di PDK nel nucleo rimane da chiarire ma, recentemente, è stata avanzata l’ipotesi di una funzione nucleare per PDH, l’effettore a valle di PDK. Sutendra et al. hanno riportato che la PDH è coinvolta nella formazione nucleare di acetil-CoA [52]. In risposta alla stimolazione dei fattori di crescita, la PDH e la sua fosfatasi traslocano dai mitocondri, dove è inibita dalla PDK, al nucleo, ma non è stata identificata una localizzazione nucleare per la PDK [52]. Tuttavia, è dimostrato che alcuni enzimi glicolitici sono impegnati in altre funzioni cellulari oltre alla glicolisi, in particolare nella regolazione trascrizionale [53]. Sono necessari ulteriori studi per chiarire se la PDK svolge anche una funzione nucleare.
Nel carcinoma a cellule renali, è stato riportato che l’espressione di PDK1 diminuisce durante la progressione del tumore [39]. Nel nostro lavoro, invece, PDK è risultata maggiormente espressa negli stadi tumorali più bassi, senza influenzare la sopravvivenza globale e libera da malattia dei pazienti affetti da melanoma. Al contrario, la sovraespressione di PDK è stata identificata come un marcatore di progressione tumorale, prognosi sfavorevole e recidiva, nel HNSCC e nel cancro gastrico [37,40,54]. Sebbene il nostro obiettivo non fosse quello di esplorare il ruolo prognostico delle PDK nel CM, i nostri risultati suggeriscono che le PDK nel CM potrebbero svolgere un ruolo più attivo nello sviluppo del melanoma che nella sua progressione. Siamo consapevoli che la nostra serie è composta principalmente da CM primari e che saranno necessari ulteriori studi nel melanoma avanzato (metastatico) per chiarire questo aspetto.
Il nostro obiettivo in questo studio è stato quello di esplorare i biomarcatori per nuovi approcci terapeutici nei pazienti con CM. I nostri risultati suggeriscono una potenziale utilità del DCA nella gestione dei pazienti con CM, in linea con Abildgaard et al. che hanno riportato una diminuzione dei livelli di ATP e della crescita del melanoma con il trattamento con DCA [50]. Questi autori hanno anche riportato una combinazione sinergica tra DCA e un inibitore di BRAF, vemurafenib [50]. Inoltre, i nostri risultati mostrano l’espressione concomitante di PDK e degli effettori della via mTOR nel CM e la downregulation dell’attività della via mTOR con il trattamento con DCA. Considerando i risultati di Hong et al. che hanno riportato un effetto sinergico della combinazione di DCA con un inibitore di S6K1 (effettore della via mTOR) [55], si può ipotizzare che si possa ottenere un effetto terapeutico sinergico nel CM utilizzando la combinazione di DCA con un inibitore diretto di mTOR.
Conclusioni
Riportiamo per la prima volta la sovraespressione di PDK1 e 2 nel CM rispetto ai nevi. L’espressione delle PDK è risultata associata all’espressione degli effettori della via mTOR e non correlata allo stato mutazionale di BRAF. Il trattamento con DCA porta a uno spostamento del metabolismo, a una riduzione della proliferazione, a un aumento dell’apoptosi e a una riduzione dell’attivazione della via mTOR nelle linee cellulari di melanoma. Considerando tutti questi risultati, concludiamo che l’espressione di PDK può avere un ruolo nello sviluppo del melanoma e che la sua inibizione mediante DCA da solo o in combinazione con inibitori diretti di mTOR può portare benefici ai pazienti affetti da CM.
Ringraziamenti
Siamo grati a tutti i pazienti che hanno partecipato a questo studio e ai medici che hanno fornito informazioni cliniche, patologiche e di follow-up. Ringraziamo la dott.ssa Madalena Pinto, del CEQUIMED, Facoltà di Farmacia, Università di Porto, Portogallo, che ci ha gentilmente fornito la linea cellulare di melanoma cutaneo A375, e il dott. Marc Mareel, del Dipartimento di Radioterapia e Medicina Nucleare, Ospedale Universitario di Gand, Belgio, che ci ha gentilmente fornito la linea cellulare di melanoma cutaneo Mewo. Ringraziamo Gabriela Almeida per gli utili consigli tecnici relativi al saggio PB. Ringraziamo anche il Prof. Manuel Sobrinho Simões per la lettura critica di questo manoscritto. H Pópulo e R Caldas hanno contribuito in egual misura a questo lavoro.
Dichiarazione di interesse
Questo studio è stato sostenuto dalla Fondazione portoghese per la scienza e la tecnologia attraverso una borsa di studio post-doc per HP (rif.: SFRH/BPD/85249/2012). Un ulteriore finanziamento è stato ottenuto dal progetto “Microenvironment, metabolism and cancer”, parzialmente sostenuto dal Programa Operacional Regional do Norte (ON.2 – O Novo Norte) nell’ambito del Quadro de Referência Estratégico Nacional (QREN) e del Fundo Europeu de Desenvolvimento Regional (FEDER). L’IPATIMUP integra l’unità di ricerca i3S, parzialmente sostenuta dalla FCT, la Fondazione portoghese per la scienza e la tecnologia. Questo lavoro è stato finanziato da fondi FEDER attraverso il Programma Operativo per i Fattori di Competitività – COMPETE e da fondi nazionali attraverso la FCT, nell’ambito dei progetti “PEst-C/SAU/LA0003/2013” Gli autori non rivelano potenziali conflitti di interesse. Gli autori non hanno altre affiliazioni o coinvolgimenti finanziari rilevanti con organizzazioni o enti che abbiano un interesse finanziario o un conflitto finanziario con l’argomento o i materiali discussi nel manoscritto, a parte quelli divulgati.
RIFERIMENTI
1 Siegel R, Ma J, Zou Z, et al. Statistiche sul cancro, 2014. CA Cancer J Clin 2014;64(1):9-29
2 Nikolaou V, Stratigos AJ. Tendenze emergenti nell’epidemiologia del melanoma. Br J Dermatol 2014;170(1):11-19
3 Smoller BR. Criteri istologici per la diagnosi di melanoma primario cutaneo maligno. Mod Pathol 2006;19(Suppl 2):S34-40
4 Populo H, Soares P, Lopes JM. Approfondimenti sul melanoma: il bersaglio terapeutico della via mTOR. Expert Opin Ther Targets 2012;16(7):689-705
5 Balch CM, Gershenwald JE, Soong SJ, et al. Versione finale della stadiazione e classificazione del melanoma AJCC 2009. J Clin Oncol 2009;27(36):6199-206
6 Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol 2008;84(2):289-306
7 Bertolotto C. Melanoma: dal melanocita alle alterazioni genetiche e alle opzioni cliniche. Scientifica 2013;2013:22
8 Populo H, Boaventura P, Vinagre J, et al. Mutazioni del promotore TERT nel cancro della pelle: effetti dell’esposizione solare e dell’irradiazione X. J Invest Dermatol 2014;134(8):2251-7
9 Elder DE. Precursori del melanoma e loro mimici: nevi di siti speciali. Modern pathology Inc 2006;19(Suppl 2):S4-20
10 Kumar R, Angelini S, Snellman E, et al. Le mutazioni di BRAF sono eventi somatici comuni
11 nei nevi melanocitici. J Invest Dermatol 2004;122(2):342-8 Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associato all’arresto del ciclo cellulare simile alla senescenza dei naevi umani. Nature 2005;436(7051):720-4
12 Vredeveld LC, Possik PA, Smit MA, et al. L’abrogazione della senescenza indotta da BRAFV600E mediante l’attivazione della via PI3K contribuisce alla melanomagenesi. Genes Dev 2012;26(10):1055-69
13 Populo H, Soares P, Faustino A, et al. L’attivazione della via mTOR nel melanoma cutaneo è associata a caratteristiche di prognosi peggiore. Pigment Cell Melanoma Res 2011;24(1):254-7
14 Olszanski AJ. Ruolo attuale e futuro della terapia mirata e dell’immunoterapia nel melanoma avanzato. J manag Care Spec Pharm 2014;20(4):346-56
15 Hodi FS, O’Day SJ, McDermott DF, et al. Miglioramento della sopravvivenza con ipilimumab in pazienti con melanoma metastatico. N Engl J Med 2010;363(8):711-23
16 Chapman PB, Hauschild A, Robert C, et al. Miglioramento della sopravvivenza con vemurafenib nel melanoma con mutazione BRAF V600E. N Engl J Med 2011;364(26):2507-16
17 Flaherty KT, Robert C, Hersey P, et al. Migliore sopravvivenza con l’inibizione di MEK nel melanoma mutato in BRAF. N Engl J Med 2012;367(2):107-14
18 Ballantyne AD, Garnock-Jones KP. Dabrafenib: prima approvazione globale. Farmaci 2013;73(12):1367-76
19Disponibile da:http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm412861.htm
20 Gillies RJ, Robey I, Gatenby RA. Cause e conseguenze dell’aumento del metabolismo del glucosio nei tumori. J Nucl Med 2008;49(Suppl 2):24S-42S
21 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532
22 Semenza GL. HIF-1 media le risposte metaboliche all’ipossia intratumorale e alle mutazioni oncogeniche. J Clin Invest 2013;123(9):3664-71
23 Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851-64
24 Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 2011;286(49):42626-34
25 Kluza J, Corazao-Rozas P, Touil Y, et al. L’inattivazione dell’asse di segnalazione HIF-1alfa/PDK3 spinge il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res 2012;72(19):5035-47
26 Baudy AR, Dogan T, Flores-Mercado JE, et al. La FDG-PET è un buon biomarcatore della risposta precoce e della resistenza acquisita nei melanomi BRAFV600 mutanti trattati con vemurafenib e l’inibitore MEK GDC-0973. Ricerca EJNMMI 2012;2(1):22
27 Hall A, Meyle KD, Lange MK, et al. La fosforilazione ossidativa disfunzionale rende le cellule di melanoma maligno dipendenti dalla glicolisi guidata dall’oncogene (V600E)BRAF. Oncotarget 2013;4(4):584-99
28 Kumar SM, Yu H, Edwards R, et al. Il BRAF mutante V600E aumenta l’espressione dell’hypoxia inducible factor-1alpha nel melanoma. Cancer Res 2007;67(7):3177-84
29 Kuphal S, Winklmeier A, Warnecke C, et al. Attività costitutiva di HIF-1 nel melanoma maligno. Eur J Cancer 2010;46(6):1159-69
30 Slominski A, Kim TK, Brozyna AA, et al. Il ruolo della melanogenesi nella regolazione del comportamento del melanoma: La melanogenesi porta alla stimolazione dell’espressione di HIF-1alfa e delle vie di comunicazione HIF-dipendenti. Archivi di biochimica e biofisica 2014;563:79-93
31 Kim JW, Tchernyshyov I, Semenza GL, et al. Espressione mediata da HIF-1 della piruvato deidrogenasi chinasi: un interruttore metabolico necessario per l’adattamento cellulare all’ipossia. Cell Metab 2006;3(3):177-85
32 Lu CW, Lin SC, Chen KF, et al. L’induzione della piruvato deidrogenasi chinasi-3 da parte dell’hypoxia-inducible factor-1 promuove lo switch metabolico e la resistenza ai farmaci. J Biol Chem 2008;283(42):28106-14
33 Patel MS, Korotchkina LG. Regolazione del complesso della piruvato deidrogenasi. Biochem Soc Trans 2006;34(Pt 2):217-22
34 Bowker-Kinley MM, Davis WI, Wu P, et al. Evidenza dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J 1998;329(Pt 1):191-6
35 Papandreou I, Goliasova T, Denko NC. Farmaci antitumorali che hanno come bersaglio il metabolismo: il dicloroacetato è il nuovo paradigma? Int J Cancer 2011;128(5):1001-8
36 Zhang S, Hulver MW, McMillan RP, et al. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutrizione e metabolismo 2014;11(1):10
37 Wigfield SM, Winter SC, Giatromanolaki A, et al. PDK-1 regola la produzione di lattato in ipossia ed è associata a una prognosi sfavorevole nel cancro squamoso della testa e del collo. Br J Cancer 2008;98(12):1975-84
38 Lu CW, Lin SC, Chien CW, et al. La sovraespressione della piruvato deidrogenasi chinasi 3 aumenta la resistenza ai farmaci e la recidiva precoce nel cancro del colon. Am J Pathol 2011;179(3):1405-14
39 Baumunk D, Reichelt U, Hildebrandt J, et al. Parametri di espressione dei geni della via metabolica piruvato deidrogenasi chinasi-1 (PDK-1) e DJ-1/PARK7 nel carcinoma a cellule renali (RCC). World J Urol 2013;31(5):1191-6
40 Hur H, Xuan Y, Kim YB, et al. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int J Oncol 2013;42(1):44-54
41 Bonnet S, Archer SL, Allalunis-Turner J, et al. Un asse mitocondriale-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell 2007;11(1):37-51
42 Kankotia S, Stacpoole PW. Dicloroacetato e cancro: Una nuova casa per un farmaco orfano? Biochimica et biophysica acta 2014;1846(2):617-29
43 Sutendra G, Dromparis P, Kinnaird A, et al. L’attivazione mitocondriale mediante l’inibizione di PDKII sopprime la segnalazione di HIF1a e l’angiogenesi nel cancro. Oncogene 2013;32(13):1638-50
44 Michelakis ED, Sutendra G, Dromparis P, et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci Transl Med 2010;2(31):31ra34
45 Castro P, Rebocho AP, Soares RJ, et al. Il riarrangiamento PAX8-PPARgamma è frequentemente rilevato nella variante follicolare del carcinoma papillare della tiroide. J Clin Endocrinol Metab 2006;91(1):213-20
46 Sun W, Zhou S, Chang SS, et al. Mutazioni mitocondriali contribuiscono all’accumulo di HIF1alpha attraverso l’aumento delle specie reattive dell’ossigeno e l’up-regolazione della piruvato deidrogenasi chinasi 2 nel carcinoma a cellule squamose della testa e del collo. Clin Cancer Res 2009;15(2):476-84
47 Cairns RA, Bennewith KL, Graves EE, et al. L’ipossia tumorale aumentata farmacologicamente può essere misurata mediante tomografia a emissione di positroni con 18F-Fluoroazomicina arabinoside e aumenta la risposta tumorale alla citotossina ipossica PR-104. Clin Cancer Res 2009;15(23):7170-4
48 Sun RC, Board PG, Blackburn AC. Il bersaglio del metabolismo con triossido di arsenico e dicloroacetato nelle cellule di cancro al seno. Mol Cancer 2011;10:142
49 Shahrzad S, Lacombe K, Adamcic U, et al. Il dicloroacetato di sodio (DCA) riduce l’apoptosi nell’ipossia del tumore colorettale. Cancer Lett 2010;297(1):75-83
50 Abildgaard C, Dahl C, Basse AL, et al. La modulazione bioenergetica con dicloroacetato riduce la crescita delle cellule di melanoma e potenzia la loro risposta all’inibizione di BRAFV600E. J Transl Med 2014;12:247
51 Populo H, Tavares S, Faustino A, et al. Le mutazioni GNAQ e BRAF mostrano un’attivazione differenziale della via mTOR nelle cellule umane trasformate. Peer J 2013;1:e104
52 Sutendra G, Kinnaird A, Dromparis P, et al. Un complesso nucleare di piruvato deidrogenasi è importante per la generazione di acetil-CoA e l’acetilazione degli istoni. Cell 2014;158(1):84-97
53 Kim JW, Dang CV. I molteplici ruoli degli enzimi glicolitici. Trends Biochem Sci 2005;30(3):142-50
54 Xuan Y, Hur H, Ham IH, et al. Il dicloroacetato attenua la resistenza al 5-fluorouracile indotta dall’ipossia nel cancro gastrico attraverso la regolazione del metabolismo del glucosio. Exp Cell Res 2014;321(2):219-30
55 Hong SE, Shin KS, Lee YH, et al. L’inibizione di S6K1 aumenta la morte cellulare indotta dal dicloroacetato. J Cancer Res Clin Oncol 2014.