Helena Populo1,2, Regina Caldas1,2,3, Jose Manuel Lopes1,2,4,5, Joana Pardal5, Valdemar Maximo1,2,4 & Paula Soares†,2,4
† Institut de recherche et d’innovation en santé (Instituto de Investigacao e Inovacao em Saude), Université de Porto, Porto, Portugal
1 Institut de pathologie moléculaire et d’immunologie de l’Université de Porto (IPATIMUP), Université de Porto, Porto, Portugal
Tél. : +22 557 0700 ; Fax : +22 557 0799 ; E-mail : [email protected]
2 Institut de recherche et d’innovation en santé (Instituto de Investigacao e Inovac¸aoem Saude), Université de Porto, Porto, Portugal
3 Faculté de médecine, Université de Porto, Porto, Portugal
4 Département de pathologie et d’oncologie, Faculté de médecine, Université de Porto, Porto, Portugal
5 Service de pathologie anatomique Hôpital Sao Joao, Porto, Portugal
Publié en ligne : 14 mai 2015
Résumé
Objectif : Nous avons voulu vérifier s’il existe des preuves pour considérer le dichloroacétate (DCA), qui inhibe la pyruvate déshydrogénase kinase (PDK) et inverse le changement métabolique des cellules cancéreuses de la glycolyse à la phosphorylation oxydative, comme un médicament prometteur pour la thérapie des patients atteints de mélanome cutané (MC).
Conception et méthodes de recherche : Nous avons évalué le profil d’expression des PDK 1, 2 et 3 dans une série d’échantillons de mélanome, afin de vérifier si les tumeurs de mélanome expriment les cibles du DCA, si cette expression est corrélée avec l’activation de cascades de signalisation importantes pour la mélanomagenèse et également avec le pronostic des patients atteints de mélanome. Nous avons également établi la sensibilité des lignées cellulaires de mélanome au traitement par DCA, en évaluant leurs altérations métaboliques, leur prolifération et leur survie.
Résultats : Nous avons observé que les isoformes PDK 1 et 2 sont surexprimées dans les CM par rapport aux naevus, cette expression étant associée à l’expression des effecteurs de la voie mTOR et indépendante du statut mutationnel BRAF. Les lignées cellulaires de mélanome traitées avec du DCA ont montré un changement dans le métabolisme, c’est-à-dire une diminution de la consommation de glucose et de la production de lactate, une régulation à la baisse de la prolifération, une augmentation de l’apoptose et une diminution de l’activation de la voie mTOR.
Conclusion : Nos résultats suggèrent que l’expression de la PDK peut jouer un rôle dans le développement du mélanome et que le DCA peut être utile pour la thérapie du CM, seul ou en combinaison avec des inhibiteurs de mTOR.
Mots clés : dichloroacétate, mélanome, métabolisme, mTOR, pyruvate déshydrogénase kinase
© 2015 Informa UK, Ltd. ISSN 1472-8222, e-ISSN 1744-7631
INTRODUCTION
Le mélanome cutané (MC) est une tumeur maligne très agressive et, bien qu’il soit le type de cancer de la peau le moins fréquent, il est responsable de la majorité des décès liés au cancer de la peau. L’incidence du mélanome cutané étant en augmentation, il est actuellement le cancer invasif le plus susceptible de se développer avant l’âge de 50 ans chez les hommes [1,2]. L’exposition aux rayons UV est considérée comme le principal facteur de risque de la mélanomagenèse [2]. La MC peut être classée en différents sous-types histologiques, le plus courant étant le mélanome superficiel étalé (MSE), suivi du mélanome nodulaire (MN), du mélanome lentigo maligna (LMM) et du mélanome acral lentigineux (MAL). Le SSM et le NM apparaissent sur une peau exposée au soleil de façon intermittente, tandis que le LMM apparaît sur une peau endommagée par le soleil de façon chronique et que l’ALM se limite à une peau non exposée au soleil. Cette classification histologique n’a aucune valeur pronostique [3,4]. La stadification du CM prend en compte l’épaisseur de la tumeur, l’ulcération, le taux de mitose, l’atteinte ganglionnaire et la présence de métastases [5]. Heureusement, la plupart des cas de CM sont diagnostiqués à un stade précoce, avec un taux de survie à 5 ans atteignant 98 % [1]. Néanmoins, pour les patients atteints de mélanome métastatique, la survie médiane n’est que de 8 à 9 mois [5].
Le MC est une tumeur très hétérogène et de nombreuses voies de signalisation cellulaire sont dérégulées dans la mélanomagenèse [6,7]. La voie MAPK est activée de manière constitutive dans la majorité des CM. Des mutations NRAS ont été signalées dans 10 à 20 % des CM, NRASQ61K/R étant la plus fréquente. Les mutations BRAF, et en particulier la mutation BRAFV600E, ont été identifiées dans 40 à 60% des cas. Notre groupe a observé une association entre la présence de mutations BRAF et les mutations du promoteur TERT récemment décrites dans le CM [8]. Les mutations BRAFV600E et, dans une moindre mesure, NRASQ61K/R ont également été signalées dans près de 80 % des naevus (lésions mélanocytaires bénignes considérées comme précurseurs du CM dans 25 % des cas), ce qui indique que l’activation de la voie MAPK peut être nécessaire mais pas suffisante pour le développement du mélanome [7,9,10]. En fait, BRAFV600E a été associé à la sénescence induite par l’oncogène et peut donc conduire, dans de nombreux naevus, à un arrêt de la croissance [11]. L’activation de la voie PI3K-AKTmTOR peut surmonter ce phénotype de sénescence et stimuler la progression tumorale [12]. Notre groupe a confirmé l’importance de la voie PI3K-AKT-mTOR dans l’agressivité des MC en associant son activation à la présence de mutations BRAF et à des caractéristiques de plus mauvais pronostic. En effet, la surexpression des effecteurs de la voie mTOR était associée à un taux de mitose plus élevé, un niveau de Clark plus important, une épaisseur tumorale accrue et la présence d’ulcérations cutanées [13].
Avant 2011, les options de traitement des patients atteints de CM avancé reposaient principalement sur la chimiothérapie conventionnelle, qui présentait de faibles taux de réponse et des effets mineurs sur la survie globale (OS) des patients. Depuis, cinq nouveaux médicaments ont été approuvés pour les patients atteints de CM de stade IV [14]. L’ipilimumab, un anticorps anti-CTLA4, a révélé des améliorations marquées de la SG des patients, mais ses effets indésirables font qu’il ne convient pas à tous les patients [15]. Le vémurafénib et le dabrafénib, des inhibiteurs sélectifs de BRAFV600E, et le trametinib, un inhibiteur de MEK, ciblent tous la voie MAPK et ont été signalés comme permettant d’obtenir des taux de réponse supérieurs à 50 %, voire meilleurs que ceux de l’ipilimumab. Cependant, leurs améliorations de la SG ne dépassent pas 7 à 8 mois en raison du développement de la résistance [16-18]. Récemment, un autre modulateur immunitaire, le pembrolizumab, un anticorps dirigé contre le récepteur-1 de la mort programmée (PD-1), a été approuvé pour le traitement des patients atteints de mélanome non résécable ou métastatique et dont la maladie a progressé après l’ipilimumab ou, si la mutation BRAFV600 est positive, après un inhibiteur BRAF [19]. Des essais cliniques avec des médicaments ciblant la voie PI3K-AKT-mTOR sont en cours [4,14], mais de nouvelles approches thérapeutiques sont nécessaires.
Le métabolisme des cellules cancéreuses diffère de celui des cellules normales. Dans les cellules normales, en fonction de la disponibilité de l’oxygène, le glucose peut être partiellement métabolisé en lactate par la glycolyse dans des conditions hypoxiques, ou être entièrement oxydé enCO2 en présence d’oxygène, par la phosphorylation oxydative mitochondriale, un processus énergétique plus efficace. Les cellules cancéreuses, en revanche, métabolisent la plupart du glucose en lactate, quel que soit leur apport en O2, ce que l’on appelle l’effet Warburg [20,21]. L’acquisition de ce phénotype glycolytique dans les cellules cancéreuses n’est pas entièrement comprise, mais un rôle essentiel a été reconnu pour le facteur de transcription hypoxia-inducible factor 1a (HIF1-α) [22]. La protéine HIF1-α est connue pour être stabilisée sous hypoxie, mais les voies oncogènes, telles que les voies MAPK et mTOR, semblent également médier son activation dans le cancer (Figure 1) [22,23]. Des preuves solides indiquent que le CM acquiert ce phénotype glycolytique, ce qui peut être confirmé par la scintigraphie FDGPET des patients [24-27]. En fait, HIF1-α semble être surexprimé dans le CM, car la peau est un environnement hypoxique doux et la production de mélanine stimule indirectement l’expression de HIF1-α par la production de ROS [28-30]. Pourtant, la glycolyse cytoplasmique doit être découplée de la phosphorylation oxydative mitochondriale, afin que la majeure partie du pyruvate puisse être convertie en lactate. Ce dernier processus est piloté par la pyruvate déshydrogénase kinase (PDK), une enzyme régulée par HIF1-α (Figure 1) [31,32].
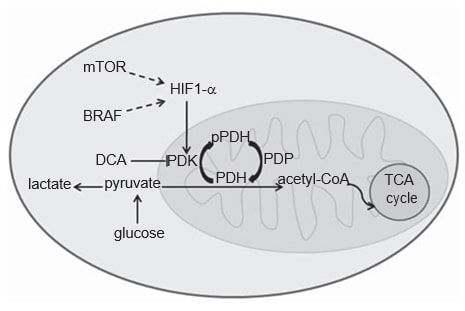
La PDK est un élément du complexe pyruvate déshydrogénase (PDC) mitochondrial. Le PDC comprend l’enzyme pyruvate déshydrogénase (PDH) et ses protéines régulatrices : PDK qui, en phosphorylant la PDH, agit comme un inhibiteur, et la phosphatase de la pyruvate déshydrogénase qui active la PDH par déphosphorylation [33]. La PDH déphosphorylée catalyse la décarboxylation oxydative du pyruvate en acétyl-CoA,CO2 et NADH (H+), reliant ainsi la voie glycolytique à la voie oxydative du cycle de l’acide tricarboxylique (Figure 1) [33]. Il existe quatre isoformes de PDK (1, 2, 3, 4) qui diffèrent par leur activité intrinsèque, leur distribution tissulaire et leur sensibilité à leur inhibiteur sélectif, le dichloroacétate (DCA) [33-35]. Bien que la PDK1 soit la seule capable de se lier à tous les sites de phosphorylation de la PDH, la PDK3 semble être l’isoforme la plus active [33]. La PDK2 est la plus sensible à l’inhibition par le DCA et est exprimée de manière ubiquitaire dans différents tissus humains, tandis que les autres sont plus spécifiques aux tissus [33,34]. PDK4 est essentiellement liée à la flexibilité métabolique physiologique et c’est la seule isoforme qui n’est pas régulée à la hausse par HIF1-α [32,36]. L’expression des isoformes de PDK a été évaluée dans différents types de cancer, à savoir le carcinome épidermoïde de la tête et du cou (HNSCC) [37], le cancer du côlon [38], le carcinome des cellules rénales [39] et le cancer gastrique [40].
Le DCA a été utilisé dans le traitement d’affections associées à l’acidose lactique dans le cadre d’un dysfonctionnement mitochondrial, comme les maladies mitochondriales congénitales [35]. Dans des cellules de cancer du poumon non à petites cellules, de glioblastome et du sein, Bonnet et al. ont signalé que le DCA induit l’apoptose et diminue la croissance cellulaire en favorisant l’oxydation du glucose, la dépolarisation de la membrane mitochondriale et la production de ROS [41]. Ces effets ont été reproduits dans différents modèles de cancer et semblent être sélectifs dans les cellules cancéreuses [35,41,42]. En outre, le DCA est également capable de supprimer l’angiogenèse par une inhibition indirecte de HIF1-α [43]. Toutes ces données, associées au profil de sécurité bien connu chez l’homme, prouvent que le DCA est un médicament prometteur pour le traitement du cancer. Un essai clinique visant à évaluer l’effet du DCA sur des patients atteints de glioblastome a déjà été réalisé
Le mélanome cutané (MC) est une tumeur maligne très agressive et, bien qu’il s’agisse du type de cancer de la peau le moins fréquent, il est responsable de la majorité des décès liés au cancer de la peau. L’incidence du mélanome cutané étant en augmentation, il est actuellement le cancer invasif le plus susceptible de se développer avant l’âge de 50 ans chez les hommes [1,2]. L’exposition aux rayons UV est considérée comme le principal facteur de risque de la mélanomagenèse [2]. La MC peut être classée en différents sous-types histologiques, le plus courant étant le mélanome superficiel étalé (MSE), suivi du mélanome nodulaire (MN), du mélanome lentigo maligna (LMM) et du mélanome acral lentigineux (MAL). Le SSM et le NM apparaissent sur une peau exposée au soleil de façon intermittente, tandis que le LMM apparaît sur une peau endommagée par le soleil de façon chronique et que l’ALM se limite à une peau non exposée au soleil. Cette classification histologique n’a aucune valeur pronostique [3,4]. La stadification du CM prend en compte l’épaisseur de la tumeur, l’ulcération, le taux de mitose, l’atteinte ganglionnaire et la présence de métastases [5]. Heureusement, la plupart des cas de CM sont diagnostiqués à un stade précoce, avec un taux de survie à 5 ans atteignant 98 % [1]. Néanmoins, pour les patients atteints de mélanome métastatique, la survie médiane n’est que de 8 à 9 mois [5]. Le MC est une tumeur très hétérogène et de nombreuses voies de signalisation cellulaire sont dérégulées dans la mélanomagenèse [6,7]. La voie MAPK est activée de manière constitutive dans la majorité des CM. Des mutations NRAS ont été signalées dans 10 à 20 % des CM, NRASQ61K/R étant la plus fréquente. Les mutations BRAF, et en particulier la mutation BRAFV600E, ont été identifiées dans 40 à 60% des cas. Notre groupe a observé une association entre la présence de mutations BRAF et les mutations du promoteur TERT récemment décrites dans le CM [8]. Les mutations BRAFV600E et, dans une moindre mesure, NRASQ61K/R ont également été signalées dans près de 80 % des naevus (lésions mélanocytaires bénignes considérées comme précurseurs du CM dans 25 % des cas), ce qui indique que l’activation de la voie MAPK peut être nécessaire mais pas suffisante pour le développement du mélanome [7,9,10]. En fait, BRAFV600E a été associé à la sénescence induite par l’oncogène et peut donc conduire, dans de nombreux naevus, à un arrêt de la croissance [11]. L’activation de la voie PI3K-AKTmTOR peut surmonter ce phénotype de sénescence et stimuler la progression tumorale [12]. Notre groupe a confirmé l’importance de la voie PI3K-AKT-mTOR dans l’agressivité des MC en associant son activation à la présence de mutations BRAF et à des caractéristiques de plus mauvais pronostic. En effet, la surexpression des effecteurs de la voie mTOR était associée à un taux de mitose plus élevé, un niveau de Clark plus important, une épaisseur tumorale accrue et la présence d’ulcérations cutanées [13].
Avant 2011, les options de traitement des patients atteints de CM avancé reposaient principalement sur la chimiothérapie conventionnelle, qui présentait de faibles taux de réponse et des effets mineurs sur la survie globale (OS) des patients. Depuis, cinq nouveaux médicaments ont été approuvés pour les patients atteints de CM de stade IV [14]. L’ipilimumab, un anticorps anti-CTLA4, a révélé des améliorations marquées de la SG des patients, mais ses effets indésirables font qu’il ne convient pas à tous les patients [15]. Le vémurafénib et le dabrafénib, des inhibiteurs sélectifs de BRAFV600E, et le trametinib, un inhibiteur de MEK, ciblent tous la voie MAPK et ont été signalés comme permettant d’obtenir des taux de réponse supérieurs à 50 %, voire meilleurs que ceux de l’ipilimumab. Cependant, leurs améliorations de la SG ne dépassent pas 7 à 8 mois en raison du développement de la résistance [16-18]. Récemment, un autre modulateur immunitaire, le pembrolizumab, un anticorps dirigé contre le récepteur-1 de la mort programmée (PD-1), a été approuvé pour le traitement des patients atteints de mélanome non résécable ou métastatique et dont la maladie a progressé après l’ipilimumab ou, si la mutation BRAFV600 est positive, après un inhibiteur BRAF [19]. Des essais cliniques avec des médicaments ciblant la voie PI3K-AKT-mTOR sont en cours [4,14], mais de nouvelles approches thérapeutiques sont nécessaires. Le métabolisme des cellules cancéreuses diffère de celui des cellules normales. Dans les cellules normales, en fonction de la disponibilité de l’oxygène, le glucose peut être partiellement métabolisé en lactate par la glycolyse dans des conditions hypoxiques, ou être entièrement oxydé enCO2 en présence d’oxygène, par la phosphorylation oxydative mitochondriale, un processus énergétique plus efficace. Les cellules cancéreuses, en revanche, métabolisent la plupart du glucose en lactate, quel que soit leur apport en O2, ce que l’on appelle l’effet Warburg [20,21]. L’acquisition de ce phénotype glycolytique dans les cellules cancéreuses n’est pas entièrement comprise, mais un rôle essentiel a été reconnu pour le facteur de transcription hypoxia-inducible factor 1a (HIF1-α) [22]. La protéine HIF1-α est connue pour être stabilisée sous hypoxie, mais les voies oncogènes, telles que les voies MAPK et mTOR, semblent également médier son activation dans le cancer (Figure 1) [22,23]. Des preuves solides indiquent que le CM acquiert ce phénotype glycolytique, ce qui peut être confirmé par la scintigraphie FDGPET des patients [24-27]. En fait, HIF1-α semble être surexprimé dans le CM, car la peau est un environnement hypoxique doux et la production de mélanine stimule indirectement l’expression de HIF1-α par la production de ROS [28-30]. Pourtant, la glycolyse cytoplasmique doit être découplée de la phosphorylation oxydative mitochondriale, afin que la majeure partie du pyruvate puisse être convertie en lactate. Ce dernier processus est piloté par la pyruvate déshydrogénase kinase (PDK), une enzyme régulée par HIF1-α (Figure 1) [31,32]. La PDK est un élément du complexe pyruvate déshydrogénase (PDC) mitochondrial. Le PDC comprend l’enzyme pyruvate déshydrogénase (PDH) et ses protéines régulatrices : PDK qui, en phosphorylant la PDH, agit comme un inhibiteur, et la phosphatase de la pyruvate déshydrogénase qui active la PDH par déphosphorylation [33]. La PDH déphosphorylée catalyse la décarboxylation oxydative du pyruvate en acétyl-CoA,CO2 et NADH (H+), reliant ainsi la voie glycolytique à la voie oxydative du cycle de l’acide tricarboxylique (Figure 1) [33]. Il existe quatre isoformes de PDK (1, 2, 3, 4) qui diffèrent par leur activité intrinsèque, leur distribution tissulaire et leur sensibilité à leur inhibiteur sélectif, le dichloroacétate (DCA) [33-35]. Bien que la PDK1 soit la seule capable de se lier à tous les sites de phosphorylation de la PDH, la PDK3 semble être l’isoforme la plus active [33]. La PDK2 est la plus sensible à l’inhibition par le DCA et est exprimée de manière ubiquitaire dans différents tissus humains, tandis que les autres sont plus spécifiques aux tissus [33,34]. PDK4 est essentiellement liée à la flexibilité métabolique physiologique et c’est la seule isoforme qui n’est pas régulée à la hausse par HIF1-α [32,36]. L’expression des isoformes PDK a été évaluée dans différents types de cancer, à savoir le carcinome épidermoïde de la tête et du cou (HNSCC) [37], le cancer du côlon [38], le carcinome des cellules rénales [39] et le cancer gastrique [40]. Le DCA a été utilisé dans le traitement d’affections associées à l’acidose lactique dans le cadre d’un dysfonctionnement mitochondrial, comme les maladies mitochondriales congénitales [35]. Dans des cellules de cancer du poumon non à petites cellules, de glioblastome et du sein, Bonnet et al. ont signalé que le DCA induit l’apoptose et diminue la croissance cellulaire en favorisant l’oxydation du glucose, la dépolarisation de la membrane mitochondriale et la production de ROS [41]. Ces effets ont été reproduits dans différents modèles de cancer et semblent être sélectifs dans les cellules cancéreuses [35,41,42]. En outre, le DCA est également capable de supprimer l’angiogenèse par une inhibition indirecte de HIF1-α [43]. Toutes ces données, associées au profil de sécurité bien connu chez l’homme, prouvent que le DCA est un médicament prometteur pour le traitement du cancer. Un essai clinique visant à évaluer l’effet du DCA chez des patients atteints de glioblastome a déjà été réalisé, rapportant une augmentation de l’apoptose et une diminution de l’angiogenèse, avec des résultats prometteurs chez quatre des cinq patients [44].
Dans la présente étude, l’expression des isoformes PDK 1 à 3 dans le CM a été évaluée et son association avec l’expression des principales cascades de voies de signalisation pour la mélanomagénèse et avec le pronostic des patients atteints de mélanome a été évaluée. De plus, des études in vitro ont été réalisées pour évaluer l’effet du traitement au DCA sur le métabolisme, la prolifération et la survie des lignées cellulaires de mélanome avec différents profils génétiques.
Matériel et méthodes
Sélection des échantillons, paramètres cliniques-pathologiques et pronostiques
Des tissus fixés au formol et inclus en paraffine de 120 cas de CM et de 22 naevus mélanocytaires (12 naevus composés et 10 naevus de Spitz) ont été prélevés dans le service de pathologie anatomique de l’hôpital S. Joa˜o, Porto, et de l’hôpital S. Marcos, Braga. Les données clinico-pathologiques (tableau 1) et de suivi ont été obtenues à partir des dossiers des patients et des registres d’oncologie de l’hôpital S. Joa˜o et de l’hôpital S. Marcos, et de RORENO (registre d’oncologie de la région Nord). Tous les cas ont été révisés et stadifiés selon la 7ème édition de l’AJCC [5]. Les données de suivi comprennent les récidives et les métastases (DFS ; n = 108) et le nombre de décès dus au mélanome (mortalité spécifique à la maladie ; OS ; n = 118). La durée moyenne de suivi des patients était de 51 mois (SE ± 3,59, intervalle 1-195) pour la DFS et de 55 mois (SE ± 3,48, intervalle 1 – 207) pour la OS. Ce travail a été approuvé par le comité d’éthique local et était conforme aux règles nationales d’éthique.
Analyse immunohistochimique
La coloration des protéines analysées a été réalisée sur des coupes de paraffine de 3 µm de zones tumorales représentatives, montées sur des lames recouvertes de poly-L-lysine. Les sections ont été déparaffinées et réhydratées, suivies d’une procédure de récupération de l’antigène par micro-ondes avec un tampon citrate de sodium 10 mM pH 6,0 avec 1 mM EDTA pH 9,0 (PDK2), ou un tampon EDTA pH 9,0 (PDK1 et PDK3). Les sections ont été incubées pendant une nuit à 4°C dans une chambre humidifiée avec les anticorps primaires PDK1 (polyclonal, lapin, 1:50), PDK2 (polyclonal, lapin, 1:150), PDK3 (monoclonal, souris, 1:500), tous provenant de Sigma-Aldrich Co. (St. Louis, Missouri, USA). La détection a été obtenue avec le système Envision G/2/AP (K5355 ; Dako, Danemark). méthode de l’anti-phosphatase alcaline (APAAP), et la couleur a été développée avec le chromogène rouge permanent. Les lames ont été contre-colorées avec de l’hématoxyline, puis montées à l’aide d’un milieu de montage miscible à l’eau. La méthode APAAP a été utilisée pour éviter toute interférence de la pigmentation de la mélanine avec l’analyse immunohistochimique. Le tissu de l’estomac humain a été utilisé comme contrôle négatif (omission de l’anticorps primaire) et positif.
Évaluation immunohistochimique
Trois observateurs (J.M.L., H.P. et R.C.) ont évalué l’immunoréactivité des cellules tumorales sans connaître les données cliniques des cas. L’immunoréactivité des tissus adjacents non tumoraux a été utilisée comme contrôle interne. Un score IHC a été établi pour PDK1 – 3, et résulte de la multiplication du score d’intensité de la coloration (négatif = 0, faible = 1, modéré = 2 et fort = 3) et du score d’extension de l’immunoréactivité des cellules tumorales (0 – 5% = 0, 6 – 25% = 1, 26 – 50% = 2, 51 – 75% = 3, 76 – 100% = 4). Les scores IHC ont ensuite été classés comme négatifs/faibles (valeur du score £ 4) et modérés/élevés (valeur du score > 4). Des analyses des cascades MAPK et PI3KAKT-mTOR dans une partie de la série ont été réalisées précédemment [13] et les résultats ont été utilisés dans cette étude.
Extraction de l’ADN et analyse des mutations
L’extraction de l’ADN des tumeurs de moins de 5 mm a été réalisée après microdissection à l’aide du système PALM MicroLaser (PALM, Allemagne) et du kit Quiamp DNA micro (Quiagen, Hilden). Dans les tumeurs de plus de 5 mm, l’extraction de l’ADN a été effectuée par dissection manuelle de sections entières de 10 µm de tissu inclus en paraffine à l’aide du mini kit Invisorb spin tissue (Invitek, Berlin). Les fragments BRAF exon 15 et NRAS exon 2 ont été amplifiés par réaction en chaîne par polymérase (PCR) en utilisant des amorces décrites précédemment [45]. L’ADN génomique (25 – 100 ng) a été amplifié par PCR en utilisant les conditions de cycle suivantes : 35 s à 94°C, 40 s à 58°C pour BRAF et 57°Cpour NRAS, et 45 s à 72°Cpour 40 cycles.
| Caractéristiques clinico-pathologiques | |
| Nombre de cas (n) | 120 |
| Âge médian (± SD) | 61.5 ± 17.0 |
| Sexe (n [%]) | |
| Femme | 68 (56.7) |
| Homme | 52 (43.3) |
| Exposition au soleil (n [%]) | |
| Absent | 27 (22.7) |
| Intermittente | 72 (60.5) |
| Chronique | 20 (16.8) |
| Sous-type histologique (n [%]) | |
| LMM | 16 (13.3 |
| ALM | 24 (20.0) |
| NM | 19 (15.8) |
| SSM | 61 (50.8) |
| Épaisseur médiane (mm) | 3.7 (0 — 70) |
| Ulcération épidermique (n [%]) | |
| Absente | 79 (65.8) |
| Présente | 41 (34.2) |
| Taux de mitose (n [%]) | |
| < 1/mm2 | 45 (37.5) |
| ≥1/mm2 | 75 (62.5 |
| pT (n [%]) | |
| ≤ pT2 | 62 (51.7) |
| >pT2 | 58 (48.3) |
Tous les produits PCR ont été purifiés et directement séquencés sur un séquenceur automatique ABI Prism 3130 xl (Perkin-Elmer, Foster City, CA) en utilisant le kit de séquençage ABI Prism Dye Terminator Cycle (Perkin-Elmer). La réaction de séquençage a été effectuée dans le sens direct, et une amplification PCR indépendante, dans le sens direct et inverse, a été réalisée sur les échantillons suspectés d’être porteurs de mutations. Là encore, l’analyse des mutations BRAF et NRAS dans une partie de la série a été réalisée précédemment [13] et les résultats ont été utilisés dans cette étude.
Lignées cellulaires et conditions de culture
Deux lignées cellulaires ont été utilisées dans ce travail, la lignée cellulaire de mélanome cutané A375 qui héberge BRAFV600E et la lignée cellulaire de mélanome cutané Mewo qui héberge BRAFwt. Les deux lignées cellulaires ont été testées pour la présence de mycoplasmes.
L’A375 a été maintenue dans un milieu RPMI (Gibco/BRL – Invitrogen) et la Mewo dans un milieu DMEM (Gibco/BRL – Invitrogen). Tous les milieux ont été supplémentés avec 10% de sérum bovin fœtal, 100 U/ml de pénicilline et 100 µg/ml de streptomycine. Les lignées cellulaires ont été maintenues dans une atmosphère humidifiée (5%CO2) à 37°C.
Traitement des lignées cellulaires de mélanome avec du DCA
Le DCAsodique, acheté chez Sigma-Aldrich (St. Louis, MO, EUA), a été dissous dans du dH2Oet ajouté au milieu de culture et utilisé pour un traitement de 24 et 48 heures. Les cellules de mélanome incubées avec un milieu de culture complété par du dH2Oont servi de contrôle.
Test de viabilité cellulaire
Les effets du DCA sur la croissance des lignées cellulaires de mélanome ont été analysés par le test PrestoBlue (PB). Les cellules ont été ensemencées dans des plaques à 96 puits à une densité de 5 103 dans 200 µl de milieu. Après 24 h, le milieu a été remplacé par un milieu contenant 5, 20, 40, 60 mM de DCA. Les cellules ont été incubées pendant 24 et 48 h, lavées avec du PBS (pH 7,4) et testées pour la croissance cellulaire en utilisant du PB selon les instructions du fabricant. Pendant l’incubation avec les cellules, le réactif PB est modifié par l’environnement réducteur des cellules viables et devient hautement fluorescent. La fluorescence a été mesurée à l’aide d’un lecteur de microplaques (Synergy HT Multi-Mode Microplate Reader, BioTek Instruments Inc., Winooski, VT, USA) à des longueurs d’onde d’excitation et d’émission de 560 et 590 nm, respectivement. L’absorbance des puits contenant le milieu de culture et les cellules tumorales a été utilisée comme contrôle et chaque condition expérimentale a été évaluée en triplicata et répétée en double. En comparant la fluorescence/absorbance mesurée des puits contenant du DCA avec les mesures des puits contenant des cellules non traitées, il a été possible de générer des profils dose-réponse et de déterminer les valeurs IC50 (la concentration qui inhibe la survie à 50 %), en utilisant GraphPadPrism5.0 (GraphPad Software, Inc., La Jolla, CA).
Quantification du glucose et du lactate
Pour mesurer les niveaux de glucose et de lactate, les cellules de mélanome ont été placées dans des plaques à 6 puits à une densité finale de 2 105 cellules/puits et incubées à 37°C pendant 24 h. Les cellules ont ensuite été traitées avec 35 mM de DCA. Comme témoins, les cellules ont été incubées avec le composé véhicule (dH2O). Après 24 et 48 h de traitement, le milieu de culture a été collecté. Les niveaux de glucose présents dans le milieu conditionné de la culture ont été quantifiés à l’aide du kit Glucose GOD/PAP (Roche Applied Sciences) et soustraits aux niveaux initiaux (0 h). Le lactate a été quantifié de manière similaire, en utilisant le test colorimétrique enzymatique LO-POD (Spinreact, Sant Esteve de Bas, Espagne).
Analyse du cycle cellulaire et de l’apoptose
Pour le profil du cycle cellulaire et l’analyse de l’apoptose, les cellules de mélanome ont été placées dans des plaques à 6 puits à une densité finale de 1 105 cellules/puits et incubées à 37°C pendant 24 h. Les cellules ont ensuite été traitées avec du DCA à 35 mM pendant 24 et 48 h de traitement. Comme témoins, les cellules ont été incubées avec le composé véhicule (dH2O). Pour l’analyse du cycle cellulaire, les cellules ont ensuite été récoltées et fixées pendant une nuit dans de l’éthanol à 70 % glacé. Ensuite, les cellules ont été remises en suspension dans du PBS avec 0,1 mg/ml de RNase A et 5 µg/ml d’iodure de propidium, avant d’être analysées. Pour mesurer l’apoptose, les cellules ont été récoltées et les niveaux d’apoptose ont été analysés par cytométrie en flux à l’aide du kit d’apoptose FITC Annexin-V (Clontech Laboratories, Inc., Saint-Germainen-Laye, France) conformément aux instructions du fabricant. L’analyse par cytométrie en flux du contenu en ADN cellulaire et de l’externalisation de la phosphatidylsérine a été réalisée avec un cytomètre en flux (BD Accuri C6), en traçant au moins 20 000 événements par échantillon. Les données ont été analysées à l’aide du logiciel FlowJo 7.6.5 (Tree Star, Inc., Ashland, USA).
Analyse Western blot et anticorps
Les cellules ont été lysées pendant 15 minutes à 4°C à l’aide du tampon RIPA (1 % de NP-40 dans 150 mM NaCl, 50 mM Tris (pH 7,5), 2 mM EDTA) contenant des inhibiteurs de phosphatase et de protéase. Les protéines ont été quantifiées à l’aide d’un test Bradford modifié (Biorad). Les échantillons de protéines (50 µg) ont été séparés dans des gels SDS/ PAGE à 10% et électroblotés sur une membrane Hybond ECL (Amersham Biosciences). Nous avons utilisé les anticorps primaires suivants : PDH et pPDH Ser293, provenant d’Abcam ; PDK1 (Sigma-Aldrich), PDK2 (Sigma-Aldrich), HIF1-α (Transduction Laboratories) et les effecteurs de la voie mTOR pS6 Ser235/236, s6, p-4EBP1 Thr37/46, 4EBP1, tous provenant de Cell Signaling Technology. Les anticorps secondaires ont été conjugués à la peroxydase (Santa Cruz Biotechnology) et visualisés par la solution de détection ECL. Les membranes ont été re-colorées avec un anticorps polyclonal de chèvre anti-actine (Santa Cruz Biotechnology) pour le contrôle du chargement des protéines. Toutes les expériences et les quantifications (à l’aide du logiciel d’analyse 1-D Quantity One de Bio-Rad (version 4.6.6)) ont été réalisées en triplicata.
Analyse statistique
L’analyse statistique a été réalisée à l’aide de STAT VIEW-J 5.0 (SAS Institute, Inc., Cary, NC). La relation entre le niveau d’expression moyen (score) des marqueurs immunohistochimiques et les paramètres clinico-pathologiques a été évaluée par ANOVA. Le cas échéant, des corrections pour comparaisons multiples ont été effectuées à l’aide des tests post hoc de Bonferroni et de Tamhane. La corrélation entre le score d’immunoréactivité des différents marqueurs a été évaluée à l’aide du test exact de Fisher. Les données des expériences sur les lignées cellulaires ont été analysées par le test t de Student non apparié à deux bandes. La méthode Kaplan-Meier et le test log-rank ont été utilisés pour évaluer les données de survie des mélanomes. Des analyses univariées et multivariées ont été réalisées pour déterminer la valeur pronostique des covariables concernant la SG et la SSM à l’aide du modèle de régression de Cox. La SG et la SSM ont été calculées à partir du moment du diagnostic jusqu’au décès dû à la maladie ou aux métastases, respectivement, ou censurées au moment du dernier suivi ou du décès non lié à la maladie. Une valeur p < 0,05 a été considérée comme statistiquement significative.
Résultats
Expression des PDK dans les lésions de CM et de naevus
Tant dans les lésions de CM que de naevus, les isoformes PDK 1, 2 et 3 étaient exprimées non seulement dans les mélanocytes/mélanomes, mais aussi dans les kératinocytes et dans les cellules des glandes sébacées et des follicules pileux. Les niveaux d’expression de toutes les isoformes PDK analysées étaient positivement corrélés entre elles, suggérant une expression concomitante de ces protéines (tableau 2).
| Protéine | Niveau d’expression moyen (± SD) | Corrélation (valeur p) | Corrélation (valeur p) | Corrélation (valeur p) |
| PDK1 | 4.8 ± 3.5 | < 0.01* | < 0.01§ | |
| PDK2 | 4.6 ± 3.6 | < 0.01† | ||
| PDK3 | 7.0 ± 3.2 |
*Entre PDK1 et PDK2 ; † entre PDK2 et PDK3 ; § entre PDK1 et PDK3.
Expression immunohistochimique de PDK1
Une coloration cytoplasmique de PDK1 a été observée dans 88 % des CM et 76 % des naevus (figure 2A). Un score de coloration négatif/faible a été observé dans 52 et 76% et un score de coloration modéré/élevé a été observé dans 48 et 24% des mélanomes et des naevus, respectivement. Par conséquent, les CM présentaient des niveaux moyens d’expression de PDK1 significativement plus élevés que les nævus (p = 0,03 ; Figure 2B). En ce qui concerne les sous-types histologiques de CM, LMM a affiché des niveaux d’expression de PDK1 plus élevés que NM (p = 0,04) et ALM (p = 0,04). Aucune association n’a été trouvée entre l’expression de PDK1 et le type d’exposition au soleil. En ce qui concerne les facteurs pronostiques, PDK1 était plus exprimé dans les tumeurs £ 1 mm d’épaisseur (p = 0,02). Des niveaux d’expression plus élevés de PDK1 étaient associés à des stades tumoraux plus bas (p = 0,04), mais pas à la survie globale et sans maladie des patients. Nous avons également évalué si l’expression des isoformes de PDK était liée aux voies MAPK et PI3K-AKT-mTOR. Aucune association n’a été trouvée entre l’expression de PDK1 et les mutations BRAF/NRAS ou avec l’activation de la voie MAPK (évaluée par l’expression de pERK). L’expression de PDK1 était positivement corrélée avec l’expression des protéines de la cascade PI3K-AKT-mTOR, à savoir mTOR et 4EBP1 (p = 0,02 et p = 0,04, respectivement).
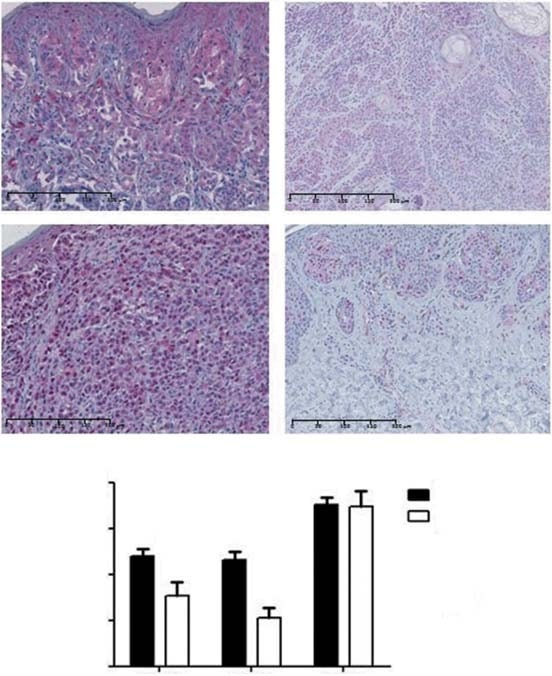
Expression immunohistochimique de PDK2
PDK2 a montré une expression à la fois nucléaire et cytoplasmique dans 87% des MC et 83% des naevus (Figure 2A). Un score de coloration négatif/faible a été observé dans 58 et 89% et un score de coloration modéré/élevé dans 42 et 11% des mélanomes et des naevus, respectivement. Le niveau d’expression moyen de PDK2 était plus élevé dans les CM par rapport aux naevus (p < 0,01 ; Figure 2B). En ce qui concerne les sous-types histologiques de CM, les SSM présentaient des niveaux d’expression de PDK2 plus élevés que les ALM (p = 0,02). Nous n’avons pas trouvé d’association entre le type d’exposition au soleil et l’expression de PDK2. Les stades tumoraux inférieurs ont montré des niveaux significativement plus élevés de PDK2 (p = 0,02), bien qu’aucune association n’ait été trouvée entre l’expression de cette isoforme et les principaux facteurs de pronostic du CM, ni avec la survie globale et sans maladie des patients. L’expression de PDK2 n’était pas associée au statut mutationnel BRAF/NRAS ou à l’activation de la voie MAPK. Une corrélation positive significative a été trouvée entre l’expression de PDK2 et l’expression d’AKT de la cascade PI3K-AKT-mTOR (p = 0,03).
Expression immunohistochimique de PDK3
PDK3 a montré une expression cytoplasmique dans tous les CM et naevus. Un score de coloration négatif/faible a été observé dans 28 et 22% et un score de coloration modéré/élevé a été observé dans 72 et 78% des CM et des naevus, respectivement. Le niveau d’expression de l’isoforme PDK3 était similaire entre les CM et les naevus (Figure 2B). Il n’y avait pas de différences significatives en comparant le niveau moyen d’expression de PDK3 entre les sous-types de CM et le type d’exposition au soleil. Les tumeurs avec une épaisseur £ 1 mm et celles avec des stades inférieurs ont montré des niveaux plus élevés de PDK3 (p = 0.01 et p < 0.01, respectivement). L’ulcération, le taux de mitose et la survie globale et sans maladie des patients n’étaient pas significativement associés à l’expression de PDK3. Aucune association n’a été trouvée entre l’expression de PDK3 et les mutations BRAF/NRAS ni l’activation de la cascade MAPK. En ce qui concerne la voie PI3K-AKT-mTOR, l’expression de PDK3 était positivement corrélée à l’expression totale de AKT, mTOR et 4EBP1 (p < 0,01, p < 0,01 et p = 0,04, respectivement). En raison de l’expression générale de PDK3 dans tous les CM et les naevus, l’analyse in vitro ultérieure n’a pas inclus cette isoforme de PDK.
Effet du traitement au DCA sur la viabilité des lignées cellulaires de mélanome
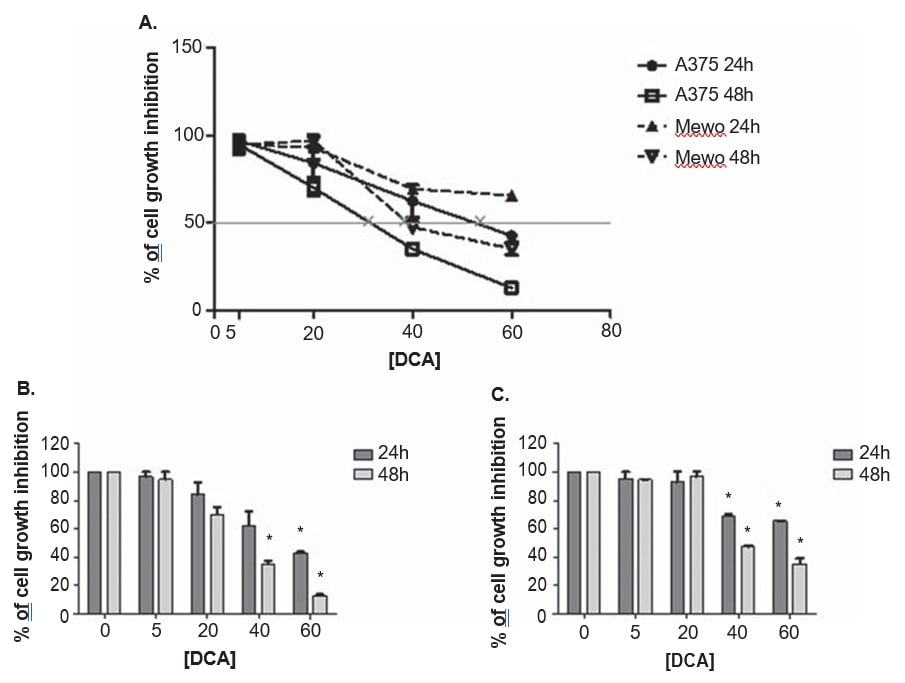
Les lignées cellulaires de mélanome A375 et Mewo ont été exposées à des concentrations croissantes de DCA afin d’établir l’effet sur la viabilité cellulaire, en utilisant le test PrestoBlue. Le DCA a réduit la viabilité des deux lignées cellulaires de manière dose-dépendante après 24 et 48 heures de traitement, bien que l’A375 ait été légèrement plus sensible au traitement par le DCA, avec une inhibition de la croissance plus importante par rapport à la lignée cellulaire Mewo (Figure 3). L’effet du DCA sur la viabilité des cellules A375 a été observé après un traitement à 5 mM et, dans la lignée cellulaire Mewo, il était plus intense après un traitement à 20 mM, avec des valeurs d’inhibition comprises entre 3 et 87 % pour l’A375 et entre 5 et 65 % pour la Mewo. Les valeurs de la CI50 ont été estimées à 33 ± 5,5 mM pour l’A375 et 39,9 ± 2,2 mM pour la lignée cellulaire Mewo, après 48 h de traitement au DCA (Figure 3).
Effet du traitement au DCA sur la consommation de glucose et la production de lactate des lignées cellulaires de mélanome
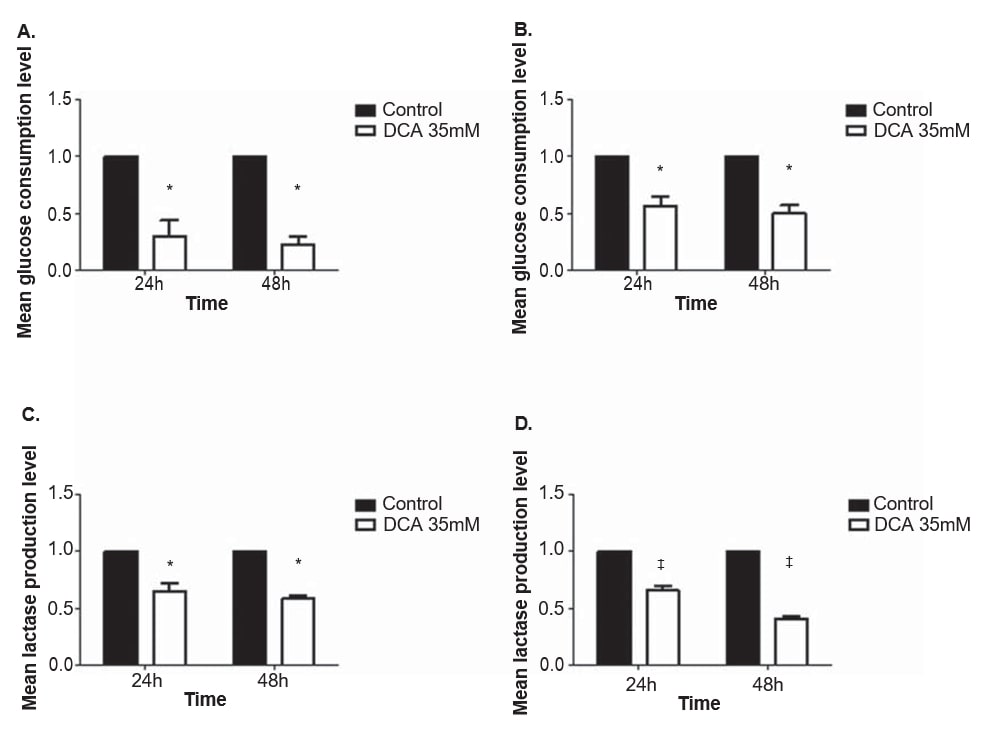
Pour évaluer l’effet du DCA sur le métabolisme des lignées cellulaires de mélanome, les niveaux de glucose et de lactate ont été quantifiés dans le milieu de culture après 24 et 48 h de traitement avec 35 mM de DCA, une concentration proche de la CI50 pour les deux lignées cellulaires. La consommation de glucose et la production de lactate ont diminué dans les deux lignées cellulaires de mélanome après traitement au DCA (Figure 4). Les lignées cellulaires A375 et Mewo ont présenté une diminution significative du niveau de consommation de glucose après 24 et 48 heures de traitement au DCA (p = 0,01 et p < 0,01, respectivement). Le niveau de production de lactate a également diminué de manière significative après 24 et 48 heures dans les lignées cellulaires A375 (p = 0,01 et p < 0,01, respectivement) et Mewo (p < 0,01).
Effet du traitement au DCA sur les effecteurs des voies PDK et mTOR, et sur l’expression de HIF1-α dans les lignées cellulaires de mélanome
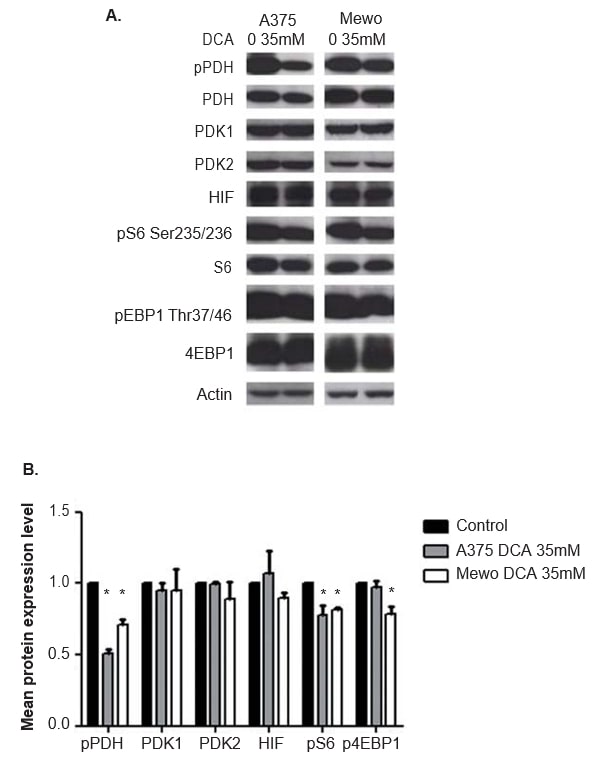
L’efficacité du traitement au DCA dans l’inhibition de l’activité PDK a été évaluée en analysant l’expression de l’effecteur en aval phosphorylé PDH, par western blot. 24 heures après le traitement au DCA, on a observé une inhibition significative de la phosphorylation de la PDH dans les deux lignées cellulaires (p < 0,01 ; figure 5). L’expression de PDK1 et PDK2 n’a pas été modifiée par le traitement au DCA (Figure 5), comme prévu. L’expression des deux effecteurs en aval et indicateurs de l’activation de la voie mTOR, S6 et 4EBP1, a également été évaluée. Bien que le traitement au DCA n’ait pas modifié l’expression de S6 et de 4EBP1, le niveau d’expression des formes phosphorylées de ces deux protéines a diminué dans les cellules A375 (p = 0,03 et non significatif, respectivement) et Mewo (p < 0,01 et p = 0,01, respectivement ; Figure 5). Comme le DCA peut affecter les niveaux de HIF1-α, l’expression de cette protéine a également été évaluée, et aucun changement n’a été observé après le traitement au DCA (Figure 5). Des résultats similaires ont été obtenus 48 h après le traitement au DCA, qui inhibe significativement la phosphorylation de la PDH dans les deux lignées cellulaires (p <0,01). Le niveau d’expression des formes phosphorylées de S6 et 4EBP1 a été significativement diminué dans l’A375 (p = 0,01 et p = 0,02, respectivement) et dans le Mewo (p = 0,01 et non significatif, respectivement). L’expression de PDK1, PDK2 et HIF1-α n’a pas été modifiée (données non présentées).
Effet du traitement au DCA sur le cycle cellulaire et l’apoptose des lignées cellulaires de mélanome
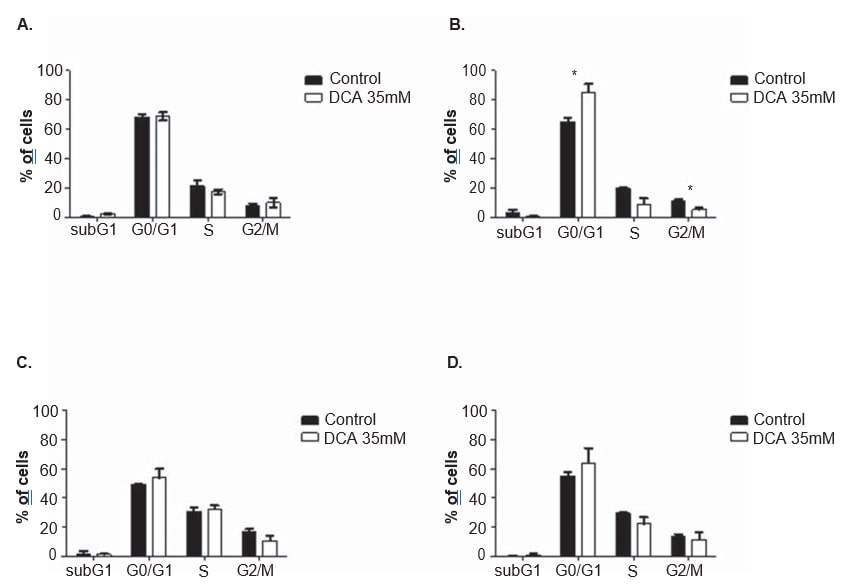
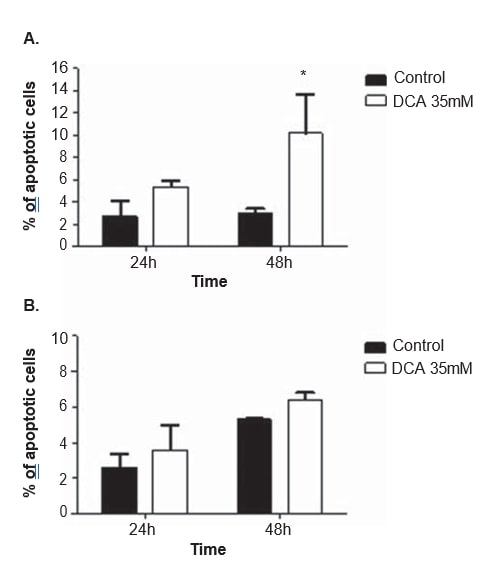
Pour clarifier le mécanisme d’action du DCA, une analyse du cycle cellulaire et des mesures de l’apoptose ont été réalisées dans les deux lignées cellulaires, après 24 et 48 h de traitement avec 35 mM de DCA. Le cycle cellulaire a été analysé avec de l’iodure de propidium. Après 48 h de traitement au DCA, la lignée cellulaire A375 a présenté une augmentation significative du pourcentage de cellules en phase G0/G1 du cycle cellulaire (de 65,0 ± 4,9 % observés dans les cellules non traitées à 85,2 ± 10,2 % dans les cellules traitées au DCA ; p = 0,04), une diminution significative de la phase G2/M (de 11.3 ± 1,3 % observé dans les cellules non traitées à 5,2 ± 2,2 % dans les cellules traitées au DCA ; p = 0,01) et une diminution, n’atteignant pas le seuil de signification statistique, de la phase S (de 19,7 ± 1,8 % observé dans les cellules non traitées à 9,1 ± 6,7 % dans les cellules traitées au DCA ; p = 0,06 ; Figure 6). La même tendance a été observée dans la lignée cellulaire Mewo 48 h après le traitement au DCA (augmentation du pourcentage de cellules en G0/G1, et diminution en S et G2/M) sans toutefois atteindre une signification statistique (Figure 6). Une analyse par cytométrie en flux a été effectuée après coloration à l’Annexin V/iodure de propidium. Une augmentation significative du nombre de cellules apoptotiques dans la lignée cellulaire A375 après 48 h de traitement au DCA a été observée (3,0 ± 0,5 % dans les cellules non traitées à 16,9 ± 4,6 % dans les cellules traitées au DCA ; p < 0,01 ; figure 7). Des augmentations non significatives du pourcentage de cellules apoptotiques ont été observées dans la lignée cellulaire A375 après 24 h et dans la lignée cellulaire Mewo après 24 h et 48 h de traitement au DCA.
Discussion
Dans ce travail, nous avons établi, pour la première fois, que le CM surexprime les protéines PDK1 et PDK2. Les PDKs sont des protéines clés, régulées par HIF1-α, conduisant les cellules malignes dans le phénotype de la glycolyse aérobie, une caractéristique du cancer [20,22,31].
L’effet Warburg est considéré comme un facteur de résistance aux chimiothérapies conventionnelles [21]. La surexpression de la PDK trouvée dans le CM nous a conduit à évaluer l’effet du traitement au DCA dans les lignées cellulaires de mélanome. Nous avons observé que le traitement au DCA diminue la consommation de glucose et la production de lactate dans les lignées cellulaires de mélanome, ce qui est cohérent avec l’effet du DCA dans le passage de la glycolyse aérobie à la phosphorylation oxydative précédemment signalé dans les cellules cancéreuses [41]. Ce changement dans le métabolisme du glucose résulte probablement de la diminution significative observée dans l’expression de la forme phosphorylée de la PDH, ce qui confirme également l’efficacité du traitement au DCA. Il est important de mentionner que la lecture correcte de l’effet du DCA est le niveau de PDH phosphorylée, puisqu’en inhibant l’activité de la PDK et non son expression, le DCA stimule la déphosphorylation de la PDH (Figure 1).
Nous avons observé une diminution dose-dépendante de la viabilité cellulaire après traitement au DCA dans les deux lignées cellulaires de mélanome. De même, une diminution de la prolifération, par arrêt G0/G1, ainsi qu’une augmentation de l’apoptose ont été observées dans les deux lignées cellulaires, mais seulement de manière statistiquement significative dans la lignée A375. Le fait que la concentration de DCA choisie dans la présente étude (35 mM) soit légèrement inférieure à la CI50 pour la lignée cellulaire Mewo peut expliquer l’effet moins prononcé observé dans ces cellules.
La relation entre le DCA et le HIF-1a dans le cancer n’est pas complètement comprise, car le DCA augmente la consommation d’oxygène des tissus et la production de ROS, ce qui devrait entraîner une régulation à la hausse ou une stabilisation du HIF-1a, respectivement [46,47]. A l’inverse, certains auteurs ont rapporté que le DCA conduisait à une diminution de l’expression de HIF1-α [25,48,49]. Dans notre travail, aucune altération n’a été observée dans le niveau de HIF1-α après le traitement par DCA de lignées cellulaires de mélanome, ce qui concorde avec Shahrzad et al. qui ont rapporté que l’expression de HIF1-α ne diminue qu’après le traitement par DCA dans des conditions hypoxiques [49].
Pour surmonter la sénescence induite par le mutant BRAF et évoluer vers des phénotypes plus agressifs, le CM active probablement d’autres cascades de signalisation, comme la voie mTOR [12]. Cette voie contribue à l’effet Warburg en favorisant l’activité de HIF1-α, qui à son tour peut augmenter l’expression de PDK [23,31]. Nous avons précédemment rapporté une activation complète de cette voie dans le CM [13]. Dans cette étude, nous avons observé une expression concomitante de PDK 1, 2 et 3 et des effecteurs de la voie mTOR dans les CMs. L’expression des isoformes de PDK dans les CM est corrélée positivement avec l’expression de mTOR, ainsi qu’avec les effecteurs en amont et en aval de cette voie : AKT et 4EBP1. De plus, nous avons observé une diminution significative de pS6 et p4EBP1 dans les lignées cellulaires A375 et Mewo après le traitement au DCA.
Les naevi partagent certaines altérations génétiques avec les CM, car tous deux présentent une fréquence élevée de mutations BRAF [10]. Bien qu’à un niveau beaucoup plus faible, l’expression de PDK a également été observée dans les naevus. Nous avons évalué si le statut mutationnel BRAF et l’activation en aval de ERK pouvaient être associés à une expression plus élevée de PDK. Nous n’avons pas trouvé d’association dans les naevus ni dans les CM. La lignée cellulaire A375 porte la mutation BRAFV600E et la lignée cellulaire Mewo est de type sauvage pour ce gène, et la réponse des deux lignées cellulaires au traitement DCA était similaire, malgré une intensité variable. En fait, il a déjà été signalé que le statut mutationnel de BRAF ne modifie pas la sensibilité au traitement par DCA [50]. Ces résultats contrastent avec ceux que nous avons obtenus précédemment en utilisant des inhibiteurs de la voie mTOR dans plusieurs lignées cellulaires dérivées de mélanomes, où nous avons constaté une plus grande sensibilité au traitement par RAD001 dans les lignées cellulaires CM portant la mutation BRAFV600E [51].
Les isoformes PDK étaient exprimées dans le cytoplasme des cellules de mélanome, ce qui confirme la fonction mitochondriale bien connue de ces isoformes [33]. En ce qui concerne PDK2, une expression nucléaire a été observée dans la majorité des cas de CM. Cette localisation nucléaire a également été rapportée pour PDK1 dans les HNSCC [37]. En l’état actuel des connaissances, le rôle de PDK dans le noyau reste à clarifier mais, récemment, une fonction nucléaire a été avancée pour PDH, l’effecteur en aval de PDK. Sutendra et al. ont rapporté que PDH est impliqué dans la formation nucléaire d’acétyl-CoA [52]. En réponse à une stimulation par un facteur de croissance, la PDH et sa phosphatase se déplacent de la mitochondrie, où elle est inhibée par la PDK, vers le noyau, mais une localisation nucléaire de la PDK n’a pas été identifiée [52]. Pourtant, il existe des preuves que certaines enzymes glycolytiques sont engagées dans d’autres fonctions cellulaires que la glycolyse, notamment dans la régulation transcriptionnelle [53]. Des études supplémentaires sont nécessaires pour clarifier si la PDK joue également une fonction nucléaire.
Dans le carcinome des cellules rénales, l’expression de PDK1 a été signalée comme diminuant au cours de la progression tumorale [39]. De manière concordante, dans notre travail, le PDK s’est avéré être plus exprimé dans les stades tumoraux inférieurs, sans influencer la survie globale et sans maladie des patients atteints de mélanome. En revanche, la surexpression de la PDK a été identifiée comme un marqueur de la progression tumorale, du mauvais pronostic et de la récidive dans le HNSCC et le cancer gastrique [37,40,54]. Bien que notre objectif n’était pas d’explorer le rôle pronostique des PDK dans le CM, nos résultats suggèrent que les PDK dans le CM pourraient jouer un rôle plus actif dans le développement du mélanome que dans sa progression. Nous sommes conscients que notre série est principalement composée de CM primaires et que d’autres études sur le mélanome avancé (métastatique) seront nécessaires pour clarifier cette question.
Notre objectif dans cette étude était d’explorer les biomarqueurs pour de nouvelles approches thérapeutiques chez les patients atteints de CM. Nos résultats suggèrent une utilité potentielle du DCA dans la gestion des patients atteints de CM, en accord avec Abildgaard et al. qui ont rapporté une diminution des niveaux d’ATP et de la croissance du mélanome en utilisant un traitement au DCA [50]. Ces auteurs ont également rapporté une combinaison synergique entre le DCA et un inhibiteur de BRAF, le vemurafenib [50]. De plus, nos résultats montrent une expression concomitante des effecteurs de la voie PDK et mTOR dans le CM et une régulation négative de l’activité de la voie mTOR par le traitement au DCA. Compte tenu des résultats de Hong et al. qui ont rapporté un effet synergique de la combinaison du DCA avec un inhibiteur de S6K1 (effecteur de la voie mTOR) [55], il est tentant de spéculer qu’un effet thérapeutique synergique peut être atteint dans la CM en utilisant la combinaison du DCA avec un inhibiteur direct de mTOR.
Conclusion
Nous rapportons pour la première fois la surexpression de PDK1 et 2 dans les CM en comparaison avec les naevus. L’expression des PDKs s’est avérée être associée à l’expression des effecteurs de la voie mTOR et non liée au statut mutationnel de BRAF. Le traitement au DCA entraîne une modification du métabolisme, une régulation négative de la prolifération, une augmentation de l’apoptose et une diminution de l’activation de la voie mTOR dans les lignées cellulaires de mélanome. En prenant tous ces résultats en compte, nous concluons que l’expression de la PDK peut jouer un rôle dans le développement du mélanome et que son inhibition par le DCA seul ou en combinaison avec des inhibiteurs directs de mTOR peut être bénéfique aux patients atteints de CM.
Remerciements
Nous sommes reconnaissants à tous les patients qui ont participé à cette étude ainsi qu’aux médecins qui ont fourni des informations cliniques, pathologiques et de suivi. Nous remercions le Dr Madalena Pinto, de CEQUIMED, Faculté de pharmacie, Université de Porto, Portugal, qui nous a aimablement fourni la lignée cellulaire de mélanome cutané A375, et le Dr Marc Mareel, du Département de radiothérapie et de médecine nucléaire, Hôpital universitaire de Gand, Belgique, qui nous a aimablement fourni la lignée cellulaire de mélanome cutané Mewo. Nous remercions Gabriela Almeida pour ses conseils techniques utiles concernant le test PB. Nous remercions également le professeur Manuel Sobrinho Simões pour la lecture critique de ce manuscrit. H Pópulo et R Caldas ont contribué à parts égales à ce travail.
Déclaration d’intérêt
Cette étude a été soutenue par la Fondation portugaise pour la science et la technologie par le biais d’une subvention post-doc à HP (Ref. : SFRH/BPD/85249/2012). Un financement supplémentaire a été obtenu dans le cadre du projet « Microenvironnement, métabolisme et cancer » qui a été partiellement soutenu par Programa Operacional Regional do Norte (ON.2 – O Novo Norte) dans le cadre du Quadro de Referência Estratégico Nacional (QREN) et du Fundo Europeu de Desenvolvimento Regional (FEDER). L’IPATIMUP intègre l’unité de recherche i3S, qui est partiellement soutenue par la FCT, la Fondation portugaise pour la science et la technologie. Ce travail a été financé par des fonds FEDER à travers le Programme Opérationnel pour les Facteurs de Compétitivité – COMPETE et des fonds nationaux à travers la FCT, dans le cadre des projets « PEst-C/SAU/LA0003/2013 » Les auteurs ne révèlent aucun conflit d’intérêt potentiel. Les auteurs n’ont aucune autre affiliation pertinente ou participation financière avec une organisation ou une entité ayant un intérêt financier ou un conflit financier avec le sujet ou les matériaux discutés dans le manuscrit en dehors de ceux divulgués.
RÉFÉRENCES
1 Siegel R, Ma J, Zou Z, et al. Statistiques sur le cancer, 2014. CA Cancer J Clin 2014;64(1):9-29
2 Nikolaou V, Stratigos AJ. Tendances émergentes dans l’épidémiologie du mélanome. Br J Dermatol 2014;170(1):11-19
3 Smoller BR. Critères histologiques pour le diagnostic du mélanome malin cutané primaire. Mod Catholic 2006;19(Suppl 2):S34-40
4 Populo H, Soares P, Lopes JM. Insights into melanoma : targeting the mTOR pathway for therapeutics. Expert Opin Ther Targets 2012;16(7):689-705
5 Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27(36):6199-206
6 Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol 2008;84(2):289-306
7 Bertolotto C. Melanoma : from melanocyte to genetic alterations and clinical options. Scientifica 2013;2013:22
8 Populo H, Boaventura P, Vinagre J, et al. Mutations du promoteur TERT dans le cancer de la peau : les effets de l’exposition au soleil et de l’irradiation X. J Invest Dermatol 2014;134(8):2251-7
9 Elder DE. Les précurseurs du mélanome et leurs mimiques : les nævus de sites particuliers. Modern pathology Inc 2006;19(Suppl 2):S4-20
10 Kumar R, Angelini S, Snellman E, et al. Les mutations BRAF sont des événements somatiques communs
11 dans les naevus mélanocytaires. J Invest Dermatol 2004;122(2):342-8 Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436(7051):720-4
12 Vredeveld LC, Possik PA, Smit MA, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012;26(10):1055-69
13 Populo H, Soares P, Faustino A, et al. L’activation de la voie mTOR dans le mélanome cutané est associée à des caractéristiques de mauvais pronostic. Pigment Cell Melanoma Res 2011;24(1):254-7
14 Olszanski AJ. Rôles actuels et futurs de la thérapie ciblée et de l’immunothérapie dans le mélanome avancé. J manag Care Spec Pharm 2014;20(4):346-56
15 Hodi FS, O’Day SJ, McDermott DF, et al. Amélioration de la survie avec l’ipilimumab chez les patients atteints de mélanome métastatique. N Engl J Med 2010;363(8):711-23
16 Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507-16
17 Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367(2):107-14
18 Ballantyne AD, Garnock-Jones KP. Dabrafenib : première approbation mondiale. Médicaments 2013;73(12):1367-76
19Disponible à partir de: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm412861.htm
20 Gillies RJ, Robey I, Gatenby RA. Causes et conséquences de l’augmentation du métabolisme du glucose des cancers. J Nucl Med 2008;49(Suppl 2):24S-42S
21 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532
22 Semenza GL. HIF-1 médiateur des réponses métaboliques à l’hypoxie intratumorale et aux mutations oncogènes. J Clin Invest 2013;123(9):3664-71
23 Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851-64
24 Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines : beyond the Warburg effect. J Biol Chem 2011;286(49):42626-34
25 Kluza J, Corazao-Rozas P, Touil Y, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res 2012;72(19):5035-47
26 Baudy AR, Dogan T, Flores-Mercado JE, et al. La TEP-FDG est un bon biomarqueur de la réponse précoce et de la résistance acquise dans les mélanomes mutants BRAFV600 traités par le vémurafénib et l’inhibiteur MEK GDC-0973. Recherche EJNMMI 2012;2(1):22
27 Hall A, Meyle KD, Lange MK, et al. La phosphorylation oxydative dysfonctionnelle rend les cellules de mélanome malin dépendantes de la glycolyse dirigée par l’oncogène (V600E)BRAF. Oncotarget 2013;4(4):584-99
28 Kumar SM, Yu H, Edwards R, et al. Le mutant V600E BRAF augmente l’expression du facteur 1 alpha inductible à l’hypoxie dans le mélanome. Cancer Res 2007;67(7):3177-84
29 Kuphal S, Winklmeier A, Warnecke C, et al. Constitutive HIF-1 activity in malignant melanoma. Eur J Cancer 2010;46(6):1159-69
30 Slominski A, Kim TK, Brozyna AA, et al. The role of melanogenesis in regulation of melanoma behavior : Melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways. Archives de biochimie et de biophysique 2014;563:79-93
31 Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3(3):177-85
32 Lu CW, Lin SC, Chen KF, et al. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008;283(42):28106-14
33 Patel MS, Korotchkina LG. Régulation du complexe pyruvate déshydrogénase. Biochem Soc Trans 2006;34(Pt 2):217-22
34 Bowker-Kinley MM, Davis WI, Wu P, et al. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6
35 Papandreou I, Goliasova T, Denko NC. Les médicaments anticancéreux qui ciblent le métabolisme : le dichloroacétate est-il le nouveau paradigme ? Int J Cancer 2011;128(5):1001-8
36 Zhang S, Hulver MW, McMillan RP, et al. Le rôle pivot des pyruvate déshydrogénase kinases dans la flexibilité métabolique. Nutrition & métabolisme 2014;11(1):10
37 Wigfield SM, Winter SC, Giatromanolaki A, et al. PDK-1 régule la production de lactate en hypoxie et est associé à un mauvais pronostic dans le cancer squameux de la tête et du cou. Br J Cancer 2008;98(12):1975-84
38 Lu CW, Lin SC, Chien CW, et al. La surexpression de la pyruvate déshydrogénase kinase 3 augmente la résistance aux médicaments et la récidive précoce dans le cancer du côlon. Am J Pathol 2011;179(3):1405-14
39 Baumunk D, Reichelt U, Hildebrandt J, et al. Paramètres d’expression des gènes de la voie métabolique pyruvate déshydrogénase kinase-1 (PDK-1) et DJ-1/PARK7 dans le carcinome rénal (RCC). World J Urol 2013;31(5):1191-6
40 Hur H, Xuan Y, Kim YB, et al. Expression de la pyruvate déshydrogénase kinase-1 dans le cancer gastrique comme cible thérapeutique potentielle. Int J Oncol 2013;42(1):44-54
41 Bonnet S, Archer SL, Allalunis-Turner J, et al. Un axe mitochondrie-canal K+ est supprimé dans le cancer et sa normalisation favorise l’apoptose et inhibe la croissance du cancer. Cancer Cell 2007;11(1):37-51
42 Kankotia S, Stacpoole PW. Dichloroacétate et cancer : Un nouveau foyer pour un médicament orphelin ? Biochimica et biophysica acta 2014;1846(2):617-29
43 Sutendra G, Dromparis P, Kinnaird A, et al. L’activation mitochondriale par l’inhibition de PDKII supprime la signalisation HIF1a et l’angiogenèse dans le cancer. Oncogene 2013;32(13):1638-50
44 Michelakis ED, Sutendra G, Dromparis P, et al. Modulation métabolique du glioblastome avec le dichloroacétate. Sci Transl Med 2010;2(31):31ra34
45 Castro P, Rebocho AP, Soares RJ, et al. PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 2006;91(1):213-20
46 Sun W, Zhou S, Chang SS, et al. Les mutations mitochondriales contribuent à l’accumulation de HIF1alpha via l’augmentation des espèces réactives de l’oxygène et la régulation à la hausse de la pyruvate déshydrogénase kinase 2 dans le carcinome épidermoïde de la tête et du cou. Clin Cancer Res 2009;15(2):476-84
47 Cairns RA, Bennewith KL, Graves EE, et al. Pharmacologically increased tumor hypoxia can be measured by 18F-Fluoroazomycin arabinoside positron emission tomography and enhance tumor response to hypoxic cytotoxin PR-104. Clin Cancer Res 2009;15(23):7170-4
48 Sun RC, Board PG, Blackburn AC. Cibler le métabolisme avec le trioxyde d’arsenic et le dichloroacétate dans les cellules du cancer du sein. Mol Cancer 2011;10:142
49 Shahrzad S, Lacombe K, Adamcic U, et al. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;297(1):75-83
50 Abildgaard C, Dahl C, Basse AL, et al. La modulation bioénergétique avec le dichloroacétate réduit la croissance des cellules de mélanome et potentialise leur réponse à l’inhibition de BRAFV600E. J Transl Med 2014;12:247
51 Populo H, Tavares S, Faustino A, et al. Les mutations GNAQ et BRAF montrent une activation différentielle de la voie mTOR dans les cellules humaines transformées. Peer J 2013;1:e104
52 Sutendra G, Kinnaird A, Dromparis P, et al. Un complexe nucléaire de pyruvate déshydrogénase est important pour la génération d’acétyl-CoA et l’acétylation des histones. Cell 2014;158(1):84-97
53 Kim JW, Dang CV. Rôles à multiples facettes des enzymes glycolytiques. Trends Biochem Sci 2005;30(3):142-50
54 Xuan Y, Hur H, Ham IH, et al. Le dichloroacétate atténue la résistance au 5-fluorouracile induite par l’hypoxie dans le cancer gastrique par la régulation du métabolisme du glucose. Exp Cell Res 2014;321(2):219-30
55 Hong SE, Shin KS, Lee YH, et al. L’inhibition de S6K1 améliore la mort cellulaire induite par le dichloroacétate. J Cancer Res Clin Oncol 2014.