Helena Populo1,2, Regina Caldas1,2,3, Jose Manuel Lopes1,2,4,5, Joana Pardal5, Valdemar Maximo1,2,4 & Paula Soares†,2,4
† Instituto de Investigación e Innovación en Salud (Instituto de Investigacao e Inovacao em Saude), Universidad de Oporto, Oporto, Portugal
1 Instituto de Patología Molecular e Inmunología de la Universidad de Oporto (IPATIMUP), Universidad de Oporto, Oporto, Portugal
Tel: +22 557 0700; Fax: +22 557 0799; E-mail: [email protected]
2 Instituto de Investigacao e Inovac¸aoem Saude, Universidad de Oporto, Oporto, Portugal
3 Facultad de Medicina, Universidad de Oporto, Oporto, Portugal
4 Departamento de Patología y Oncología, Facultad de Medicina, Universidad de Oporto, Oporto, Portugal
5 Servicio de Anatomía Patológica Hospital Sao Joao, Oporto, Portugal
Publicado en línea: 14 de mayo de 2015
Resumen
Objetivo: Nos propusimos verificar si existen evidencias para considerar el dicloroacetato (DCA), que inhibe la piruvato deshidrogenasa quinasa (PDK) y revierte el cambio metabólico de las células cancerosas de la glucólisis a la fosforilación oxidativa, como un fármaco prometedor para la terapia de pacientes con melanoma cutáneo (MC).
Diseño y métodos de investigación: Evaluamos el perfil de expresión de PDK 1, 2 y 3 en una serie de muestras de melanoma, para verificar si los tumores de melanoma expresan las dianas de DCA, si esta expresión se correlaciona con la activación de cascadas de señalización importantes para la melanomagénesis y también con el pronóstico de los pacientes con melanoma. También establecimos la sensibilidad de las líneas celulares de melanoma al tratamiento con DCA, evaluando sus alteraciones metabólicas, proliferación y supervivencia.
Resultados: Observamos que tanto la isoforma PDK 1 como la 2 están sobreexpresadas en el CM en comparación con los nevos, estando esta expresión asociada a la expresión de los efectores de la vía mTOR e independiente del estado mutacional de BRAF. Las líneas celulares de melanoma tratadas con DCA mostraron un cambio en el metabolismo, es decir, una disminución del consumo de glucosa y de la producción de lactato, una regulación a la baja de la proliferación, un aumento de la apoptosis y una disminución de la activación de la vía mTOR.
Conclusiones: Nuestros resultados sugieren que la expresión de PDK puede desempeñar un papel en el desarrollo del melanoma y que el DCA puede ser útil para la terapia del CM, solo o en combinación con inhibidores de mTOR.
Palabras clave: dicloroacetato, melanoma, metabolismo, mTOR, piruvato deshidrogenasa cinasa
2015 Informa UK, Ltd. ISSN 1472-8222, e-ISSN 1744-7631
INTRODUCCIÓN
El melanoma cutáneo (MC) es una neoplasia maligna muy agresiva y, a pesar de ser el tipo de cáncer de piel menos frecuente, es responsable de la mayoría de las muertes relacionadas con el cáncer de piel. Dado que la incidencia del MC está aumentando, en la actualidad es el cáncer invasivo con mayor probabilidad de desarrollarse antes de los 50 años en el sexo masculino [1,2]. La exposición a la radiación UV se considera el principal factor de riesgo para la melanomagénesis [2]. El MC puede clasificarse en diferentes subtipos histológicos, siendo el más frecuente el melanoma de extensión superficial (MSE), seguido del melanoma nodular (MN), el melanoma lentigo maligno (MLM) y el melanoma lentiginoso acral (MLA). El SSM y el NM surgen en la piel con exposición solar intermitente, mientras que el LMM se produce en la piel dañada por el sol de forma crónica y el ALM se limita a la piel sin exposición solar. Esta clasificación histológica no tiene valor pronóstico [3,4]. La estadificación del CM tiene en cuenta el grosor del tumor, la ulceración, la tasa mitótica, la afectación ganglionar y la presencia de metástasis [5]. Afortunadamente, la mayoría de los casos de CM se diagnostican en una fase temprana, con una tasa de supervivencia a 5 años que alcanza el 98% [1]. Sin embargo, en los pacientes con melanoma metastásico, la supervivencia media es de sólo 8-9 meses [5].
El MC es un tumor muy heterogéneo y muchas vías de señalización celular están desreguladas en la melanomagénesis [6,7]. La vía MAPK se activa de forma constitutiva en la mayoría de los CM. Se han descrito mutaciones de NRAS en el 10-20% de los CM, siendo NRASQ61K/R la más común. Las mutaciones BRAF, y en particular la mutación BRAFV600E, se identificaron en el 40 – 60% de los casos. Nuestro grupo observó una asociación entre la presencia de mutaciones BRAF y las recientemente descritas mutaciones del promotor TERT en CM [8]. Las mutaciones en BRAFV600E y, en menor medida, en NRASQ61K/R también se observaron en casi el 80% de los nevos (lesiones melanocíticas benignas que se consideran precursoras del CM en el 25% de los casos), lo que indica que la activación de la vía MAPK puede ser necesaria pero no suficiente para el desarrollo del melanoma [7,9,10]. De hecho, BRAFV600E se ha relacionado con la senescencia inducida por oncogenes y, por tanto, puede conducir, en muchos nevos, a una condición de detención del crecimiento [11]. La activación de la vía PI3K-AKTmTOR puede superar este fenotipo de senescencia e impulsar la progresión tumoral [12]. Nuestro grupo ha confirmado la importancia de la vía PI3K-AKT-mTOR en la agresividad del CM al asociar su activación con la presencia de mutaciones en BRAF y peores características pronósticas. De hecho, la sobreexpresión de los efectores de la vía mTOR se asoció con una mayor tasa mitótica, un mayor nivel de Clark, un mayor grosor tumoral y la presencia de ulceración cutánea [13].
Antes de 2011, las opciones de tratamiento para los pacientes con CM avanzado se basaban principalmente en la quimioterapia convencional, que tenía bajas tasas de respuesta y efectos menores en la supervivencia global (SG) de los pacientes. Desde entonces, se han aprobado cinco nuevos fármacos para pacientes con CM en estadio IV [14]. El ipilimumab, un anticuerpo anti-CTLA4, reveló mejoras notables en la SG de los pacientes, pero sus efectos adversos lo convirtieron en no apto para todos los pacientes [15]. El vemurafenib y el dabrafenib, inhibidores selectivos de BRAFV600E, y el trametinib, un inhibidor de MEK, se dirigen todos a la vía MAPK y se notificaron tasas de respuesta superiores al 50%, incluso mejores que las del ipilimumab. Sin embargo, sus mejoras en la SG no superan los 7-8 meses debido al desarrollo de resistencia [16-18]. Recientemente, se aprobó otro inmunomodulador, el pembrolizumab, un anticuerpo contra el receptor-1 de la muerte programada (PD-1), para el tratamiento de pacientes con melanoma irresecable o metastásico y progresión de la enfermedad tras ipilimumab o, si la mutación BRAFV600 es positiva, un inhibidor de BRAF [19]. Se están llevando a cabo ensayos clínicos con fármacos dirigidos a la vía PI3K-AKT-mTOR [4,14], pero se necesitan nuevos enfoques terapéuticos.
El metabolismo de las células cancerosas difiere del de las células normales. En las células normales, dependiendo de la disponibilidad de O2, la glucosa puede ser parcialmente metabolizada a lactato a través de la glucólisis en condiciones de hipoxia, o puede ser totalmente oxidada aCO2 en presencia de O2, a través de la fosforilación oxidativa mitocondrial, un proceso energético más eficiente. Las células cancerosas, por el contrario, metabolizan la mayor parte de la glucosa a lactato, independientemente de su aporte de O2, el llamado efecto Warburg [20,21]. La adquisición de este fenotipo glucolítico en las células cancerosas no se conoce del todo, pero se ha reconocido un papel esencial para el factor de transcripción factor 1a inducible por hipoxia (HIF1-α) [22]. Se sabe que la proteína HIF1-α se estabiliza en condiciones de hipoxia, pero las vías oncogénicas, como las vías MAPK y mTOR, también parecen mediar en su activación en el cáncer (Figura 1) [22,23]. Existen pruebas sólidas que indican que el CM adquiere este fenotipo glucolítico, lo que puede confirmarse mediante la exploración FDGPET de los pacientes [24-27]. De hecho, HIF1-α parece estar sobreexpresado en el CM, ya que la piel es un entorno hipóxico leve y la producción de melanina estimula indirectamente la expresión de HIF1-α mediante la producción de ROS [28-30]. Sin embargo, la glucólisis citoplasmática debe desacoplarse de la fosforilación oxidativa mitocondrial, para que la mayor parte del piruvato pueda convertirse en lactato. Este último proceso está dirigido por la piruvato deshidrogenasa cinasa (PDK), una enzima regulada por HIF1-α (Figura 1) [31,32].
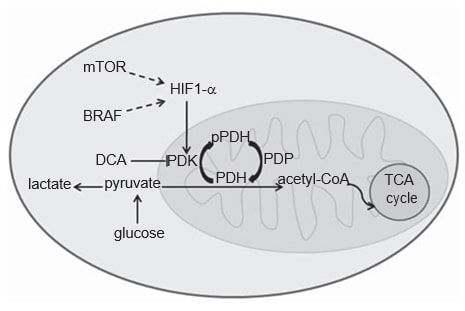
La PDK es un elemento del complejo piruvato deshidrogenasa mitocondrial (PDC). El PDC está formado por la enzima piruvato deshidrogenasa (PDH) y sus proteínas reguladoras: PDK que al fosforilar la PDH actúa como inhibidor, y la piruvato deshidrogenasa fosfatasa que activa la PDH por desfosforilación [33]. La PDH desfosforilada cataliza la descarboxilación oxidativa del piruvato en acetil-CoA,CO2 y NADH (H+ ), uniendo así la vía glucolítica con la vía oxidativa del ciclo del ácido tricarboxílico (Figura 1) [33]. Existen cuatro isoformas de PDK (1, 2, 3, 4) que difieren en su actividad intrínseca, distribución tisular y sensibilidad a su fármaco inhibidor selectivo, el dicloroacetato (DCA) [33-35]. Aunque la PDK1 es la única capaz de unirse a todos los sitios de fosforilación de la PDH, la PDK3 parece ser la isoforma más activa [33]. La PDK2 es la más sensible a la inhibición por DCA y se expresa de forma ubicua en diferentes tejidos humanos, mientras que las otras son más específicas de tejido [33,34]. PDK4 está esencialmente relacionada con la flexibilidad metabólica fisiológica y es la única isoforma no regulada por HIF1-α [32,36]. La expresión de las isoformas PDK se ha evaluado en varios tipos de cáncer, como el carcinoma de células escamosas de cabeza y cuello (HNSCC) [37], el cáncer de colon [38], el carcinoma de células renales [39] y el cáncer gástrico [40].
El DCA se ha utilizado en el tratamiento de afecciones asociadas con la acidosis láctica en el contexto de la disfunción mitocondrial, como las enfermedades mitocondriales congénitas [35]. En células de cáncer de pulmón no microcítico, glioblastoma y mama, Bonnet et al. informaron de que el DCA induce la apoptosis y disminuye el crecimiento celular, al promover la oxidación de la glucosa, la despolarización de la membrana mitocondrial y la producción de ROS [41]. Estos efectos se han replicado en diferentes modelos de cáncer y parecen ser selectivos en células cancerosas [35,41,42]. Además, el DCA también es capaz de suprimir la angiogénesis a través de la inhibición indirecta de HIF1-α [43]. Todos estos datos, combinados con el conocido perfil de seguridad en humanos, demuestran que el DCA es un fármaco prometedor para la terapia del cáncer. Ya se ha realizado un ensayo clínico para evaluar el efecto del DCA en pacientes con glioblastoma
El melanoma cutáneo (MC) es una neoplasia muy agresiva y, a pesar de ser el tipo menos frecuente de cáncer de piel, es responsable de la mayoría de las muertes relacionadas con este tipo de cáncer. Dado que la incidencia del MC está aumentando, en la actualidad es el cáncer invasivo con mayor probabilidad de desarrollarse antes de los 50 años en el sexo masculino [1,2]. La exposición a la radiación UV se considera el principal factor de riesgo para la melanomagénesis [2]. El MC puede clasificarse en diferentes subtipos histológicos, siendo el más frecuente el melanoma de extensión superficial (MSE), seguido del melanoma nodular (MN), el melanoma lentigo maligno (MLM) y el melanoma lentiginoso acral (MLA). El SSM y el NM surgen en la piel con exposición solar intermitente, mientras que el LMM se produce en la piel dañada por el sol de forma crónica y el ALM se limita a la piel sin exposición solar. Esta clasificación histológica no tiene valor pronóstico [3,4]. La estadificación del CM tiene en cuenta el grosor del tumor, la ulceración, la tasa mitótica, la afectación ganglionar y la presencia de metástasis [5]. Afortunadamente, la mayoría de los casos de CM se diagnostican en una fase temprana, con una tasa de supervivencia a 5 años que alcanza el 98% [1]. Sin embargo, en los pacientes con melanoma metastásico, la supervivencia media es de sólo 8-9 meses [5]. El MC es un tumor muy heterogéneo y muchas vías de señalización celular están desreguladas en la melanomagénesis [6,7]. La vía MAPK se activa de forma constitutiva en la mayoría de los CM. Se han descrito mutaciones de NRAS en el 10-20% de los CM, siendo NRASQ61K/R la más común. Las mutaciones BRAF, y en particular la mutación BRAFV600E, se identificaron en el 40 – 60% de los casos. Nuestro grupo observó una asociación entre la presencia de mutaciones BRAF y las recientemente descritas mutaciones del promotor TERT en CM [8]. Las mutaciones en BRAFV600E y, en menor medida, en NRASQ61K/R también se observaron en casi el 80% de los nevos (lesiones melanocíticas benignas que se consideran precursoras del CM en el 25% de los casos), lo que indica que la activación de la vía MAPK puede ser necesaria pero no suficiente para el desarrollo del melanoma [7,9,10]. De hecho, BRAFV600E se ha relacionado con la senescencia inducida por oncogenes y, por tanto, puede conducir, en muchos nevos, a una condición de detención del crecimiento [11]. La activación de la vía PI3K-AKTmTOR puede superar este fenotipo de senescencia e impulsar la progresión tumoral [12]. Nuestro grupo ha confirmado la importancia de la vía PI3K-AKT-mTOR en la agresividad del CM al asociar su activación con la presencia de mutaciones en BRAF y peores características pronósticas. De hecho, la sobreexpresión de los efectores de la vía mTOR se asoció con una mayor tasa mitótica, un mayor nivel de Clark, un mayor grosor tumoral y la presencia de ulceración cutánea [13].
Antes de 2011, las opciones de tratamiento para los pacientes con CM avanzado se basaban principalmente en la quimioterapia convencional, que tenía bajas tasas de respuesta y efectos menores en la supervivencia global (SG) de los pacientes. Desde entonces, se han aprobado cinco nuevos fármacos para pacientes con CM en estadio IV [14]. El ipilimumab, un anticuerpo anti-CTLA4, reveló mejoras notables en la SG de los pacientes, pero sus efectos adversos lo convirtieron en no apto para todos los pacientes [15]. El vemurafenib y el dabrafenib, inhibidores selectivos de BRAFV600E, y el trametinib, un inhibidor de MEK, se dirigen a la vía MAPK y, según los informes, logran tasas de respuesta superiores al 50%, incluso mejores que las del ipilimumab. Sin embargo, sus mejoras en la SG no superan los 7-8 meses debido al desarrollo de resistencia [16-18]. Recientemente, se aprobó otro inmunomodulador, el pembrolizumab, un anticuerpo contra el receptor-1 de la muerte programada (PD-1), para el tratamiento de pacientes con melanoma irresecable o metastásico y progresión de la enfermedad tras ipilimumab o, si la mutación BRAFV600 es positiva, un inhibidor de BRAF [19]. Se están realizando ensayos clínicos con fármacos dirigidos a la vía PI3K-AKT-mTOR [4,14], pero se necesitan nuevos enfoques terapéuticos. El metabolismo de las células cancerosas difiere del de las células normales. En las células normales, dependiendo de la disponibilidad de O2, la glucosa puede ser parcialmente metabolizada a lactato a través de la glucólisis en condiciones de hipoxia, o puede ser totalmente oxidada aCO2 en presencia de O2, a través de la fosforilación oxidativa mitocondrial, un proceso energético más eficiente. Las células cancerosas, por el contrario, metabolizan la mayor parte de la glucosa a lactato, independientemente de su aporte de O2, el llamado efecto Warburg [20,21]. La adquisición de este fenotipo glucolítico en las células cancerosas no se conoce del todo, pero se ha reconocido un papel esencial para el factor de transcripción factor 1a inducible por hipoxia (HIF1-α) [22]. Se sabe que la proteína HIF1-α se estabiliza en condiciones de hipoxia, pero las vías oncogénicas, como las vías MAPK y mTOR, también parecen mediar en su activación en el cáncer (Figura 1) [22,23]. Existen pruebas sólidas que indican que el CM adquiere este fenotipo glucolítico, lo que puede confirmarse mediante la exploración FDGPET de los pacientes [24-27]. De hecho, HIF1-α parece estar sobreexpresado en el CM, ya que la piel es un entorno hipóxico leve y la producción de melanina estimula indirectamente la expresión de HIF1-α mediante la producción de ROS [28-30]. Sin embargo, la glucólisis citoplasmática debe desacoplarse de la fosforilación oxidativa mitocondrial, para que la mayor parte del piruvato pueda convertirse en lactato. Este último proceso está dirigido por la piruvato deshidrogenasa cinasa (PDK), una enzima regulada por HIF1-α (Figura 1) [31,32]. La PDK es un elemento del complejo piruvato deshidrogenasa mitocondrial (PDC). El PDC comprende la enzima piruvato deshidrogenasa (PDH) y sus proteínas reguladoras: PDK que al fosforilar la PDH actúa como inhibidor, y la piruvato deshidrogenasa fosfatasa que activa la PDH por desfosforilación [33]. La PDH desfosforilada cataliza la descarboxilación oxidativa del piruvato en acetil-CoA,CO2 y NADH (H+ ), uniendo así la vía glucolítica con la vía oxidativa del ciclo del ácido tricarboxílico (Figura 1) [33]. Existen cuatro isoformas de PDK (1, 2, 3, 4) que difieren en su actividad intrínseca, distribución tisular y sensibilidad a su fármaco inhibidor selectivo, el dicloroacetato (DCA) [33-35]. Aunque la PDK1 es la única capaz de unirse a todos los sitios de fosforilación de la PDH, la PDK3 parece ser la isoforma más activa [33]. La PDK2 es la más sensible a la inhibición por DCA y se expresa de forma ubicua en diferentes tejidos humanos, mientras que las otras son más específicas de tejido [33,34]. PDK4 está esencialmente relacionada con la flexibilidad metabólica fisiológica y es la única isoforma no regulada por HIF1-α [32,36]. La expresión de las isoformas PDK se ha evaluado en varios tipos de cáncer, concretamente en el carcinoma de células escamosas de cabeza y cuello (HNSCC) [37], el cáncer de colon [38], el carcinoma de células renales [39] y el cáncer gástrico [40]. El DCA se ha utilizado en el tratamiento de afecciones asociadas con la acidosis láctica en el contexto de la disfunción mitocondrial, como las enfermedades mitocondriales congénitas [35]. En células de cáncer de pulmón no microcítico, glioblastoma y mama, Bonnet et al. informaron de que el DCA induce la apoptosis y disminuye el crecimiento celular, al promover la oxidación de la glucosa, la despolarización de la membrana mitocondrial y la producción de ROS [41]. Estos efectos se han replicado en diferentes modelos de cáncer y parecen ser selectivos en células cancerosas [35,41,42]. Además, el DCA también es capaz de suprimir la angiogénesis a través de la inhibición indirecta de HIF1-α [43]. Todos estos datos, combinados con el conocido perfil de seguridad en humanos, demuestran que el DCA es un fármaco prometedor para la terapia del cáncer. Ya se realizó un ensayo clínico para evaluar el efecto del DCA en pacientes con glioblastoma, en el que se observó un aumento de la apoptosis y una disminución de la angiogénesis, con resultados prometedores en cuatro de cinco pacientes [44].
En el presente estudio, se evaluó la expresión de las isoformas PDK 1 – 3 en el CM y su asociación con la expresión de las principales cascadas de vías de señalización para la melanomagénesis y con el pronóstico de los pacientes con melanoma. Asimismo, se realizaron estudios in vitro para evaluar el efecto del tratamiento con DCA en el metabolismo, la proliferación y la supervivencia de líneas celulares de melanoma con diferentes perfiles genéticos.
Materiales y métodos
Selección de muestras, parámetros clínico-patológicos y pronósticos
Se recuperaron tejidos fijados en formol e incluidos en parafina de 120 casos de MC y de 22 nevos melanocíticos (12 nevos compuestos y 10 nevos de Spitz) del Servicio de Anatomía Patológica del Hospital S. Joa˜o de Oporto y del Hospital S. Marcos de Braga. Los datos clínico-patológicos (Tabla 1) y de seguimiento se obtuvieron de las historias clínicas de los pacientes y de los Registros de Oncología del Hospital S. Joa˜o y del Hospital S. Marcos, y del RORENO (Registro de Oncología de la Región Norte). Todos los casos fueron revisados y estadificados según la 7ª edición de la AJCC [5]. Los datos de seguimiento incluyen las recurrencias y metástasis (DFS; n = 108) y el número de muertes debidas al melanoma (mortalidad específica de la enfermedad; OS; n = 118). El tiempo medio de seguimiento de los pacientes para la SSE fue de 51 meses (SE ± 3,59, rango 1-195) y para la SG fue de 55 meses (SE ± 3,48, rango 1 – 207). Este trabajo fue aprobado por el Comité Ético Local y se ajustó a las normas éticas nacionales.
Análisis inmunohistoquímico
La tinción para las proteínas analizadas se realizó en secciones de parafina de 3 µm de áreas tumorales representativas, montadas en portaobjetos recubiertos de poli-L-lisina. Las secciones se desparafinaron y rehidrataron, seguidas de un procedimiento de recuperación de antígenos por microondas con tampón citrato sódico 10 mM pH 6,0 con 1 mM EDTA pH 9,0 (PDK2), o tampón EDTA pH 9,0 (PDK1 y PDK3). Las secciones se incubaron durante la noche a 4 °C en una cámara humidificada con los anticuerpos primarios PDK1 (policlonal, conejo, 1:50), PDK2 (policlonal, conejo, 1:150), PDK3 (monoclonal, ratón, 1:500), todos ellos de Sigma-Aldrich Co. (St. Louis, Missouri, EE.UU.). La detección se obtuvo con el sistema Envision G/2 System/AP (K5355; Dako, Dinamarca). método antifosfatasa alcalina (APAAP), y el color se reveló con cromógeno rojo permanente. Los portaobjetos se teñían con hematoxilina y se montaban con un medio de montaje miscible en agua. El método APAAP se utilizó para evitar interferencias de la pigmentación de melanina en el análisis inmunohistoquímico. Se utilizó tejido de estómago humano como control negativo (omisión del anticuerpo primario) y positivo.
Evaluación inmunohistoquímica
Tres observadores (J.M.L., H.P. y R.C.) evaluaron la inmunorreactividad de las células tumorales sin conocer ningún dato clínico de los casos. La inmunorreactividad del tejido adyacente no tumoral se utilizó como control interno. Se estableció una puntuación IHC para PDK1 – 3, que resulta de la multiplicación de la puntuación de intensidad de la tinción (negativa = 0, débil = 1, moderada = 2 y fuerte = 3) y la puntuación de extensión de la inmunorreactividad de las células tumorales (0 – 5% = 0, 6 – 25% = 1, 26 – 50% = 2, 51 – 75% = 3, 76 – 100% = 4). A continuación, las puntuaciones IHC se clasificaron como negativas/bajas (valor de puntuación £ 4) y moderadas/altas (valor de puntuación > 4). Los análisis de las cascadas MAPK y PI3KAKT-mTOR en parte de la serie se han realizado previamente [13] y los resultados se utilizaron en este estudio.
Extracción de ADN y análisis de mutaciones
La extracción de ADN de tumores menores de 5 mm se realizó tras microdisección con PALM MicroLaser Systems (PALM, Alemania) y utilizando el kit Quiamp DNA micro (Quiagen, Hilden). En los tumores de más de 5 mm, la extracción de ADN se realizó mediante disección manual de secciones enteras de 10 µm de tejido embebido en parafina utilizando el kit Invisorb spin tissue mini (Invitek, Berlín). Los fragmentos del exón 15 de BRAF y del exón 2 de NRAS se amplificaron mediante reacción en cadena de la polimerasa (PCR) utilizando cebadores descritos previamente [45]. El ADN genómico (25 – 100 ng) se amplificó mediante PCR utilizando las siguientes condiciones de ciclado: 35 s a 94°C, 40 s a 58°C para BRAF y 57°Cpara NRAS, y 45 s a 72°Cdurante 40 ciclos.
| Características clínico-patológicas | |
| Número de casos (n) | 120 |
| Edad mediana (± DE) | 61.5 ± 17.0 |
| Sexo (n [%]) | |
| Mujer | 68 (56.7) |
| Varón | 52 (43.3) |
| Exposición al sol (n [%]) | |
| Ausente | 27 (22.7) |
| Intermitente | 72 (60.5) |
| Crónica | 20 (16.8) |
| Subtipo histológico (n [%]) | |
| LMM | 16 (13.3 |
| ALM | 24 (20.0) |
| NM | 19 (15.8) |
| SSM | 61 (50.8) |
| Grosor medio (mm) | 3.7 (0 — 70) |
| Ulceración epidérmica (n [%]) | |
| Ausente | 79 (65.8) |
| Presente | 41 (34.2) |
| Tasa mitótica (n [%]) | |
| < 1/mm2 | 45 (37.5) |
| ≥1/mm2 | 75 (62.5 |
| pT (n [%]) | |
| ≤ pT2 | 62 (51.7) |
| >pT2 | 58 (48.3) |
Todos los productos de PCR se purificaron y secuenciaron directamente en un secuenciador automático ABI Prism 3130 xl (Perkin-Elmer, Foster City, CA) utilizando el kit de secuenciación ABI Prism Dye Terminator Cycle (Perkin-Elmer). La reacción de secuenciación se realizó en la dirección directa, y se llevó a cabo una amplificación PCR independiente, en la dirección directa e inversa, en las muestras sospechosas de portar mutaciones. Una vez más, el análisis mutacional de BRAF y NRAS en parte de la serie se ha realizado previamente [13] y los resultados se utilizaron en este estudio.
Líneas celulares y condiciones de cultivo
En este trabajo se utilizaron dos líneas celulares, la línea celular A375 de melanoma cutáneo que alberga BRAFV600E y la línea celular Mewo de melanoma cutáneo que alberga BRAFwt. Se comprobó la presencia de micoplasma en ambas líneas celulares.
A375 se mantuvo en medio RPMI (Gibco/BRL – Invitrogen) y Mewo en medio DMEM (Gibco/BRL – Invitrogen). Todos los medios se complementaron con un 10% de suero bovino fetal, 100 U/ml de penicilina y 100 µg/ml de estreptomicina. Las líneas celulares se mantuvieron en una atmósfera humidificada (5%CO2) a 37°C.
Tratamiento de las líneas celulares de melanoma con DCA
ElDCAsódico, adquirido a Sigma-Aldrich (St. Louis, MO, EUA), se disolvió en dH2Oy se añadió al medio de cultivo, utilizándose para el tratamiento de 24 y 48 h. Las células de melanoma incubadas con medio de cultivo suplementado con dH2Osirvieron de control.
Ensayo de viabilidad celular
Los efectos del DCA en el crecimiento de las líneas celulares de melanoma se analizaron mediante el ensayo PrestoBlue (PB). Las células se sembraron en placas de 96 pocillos a una densidad de 5 103 en 200 µl de medio. Tras 24 h, se sustituyó el medio por otro que contenía 5, 20, 40, 60 mM de DCA. Las células se incubaron durante 24 y 48 h, se lavaron con PBS (pH 7,4) y se analizó el crecimiento celular utilizando PB según las instrucciones del fabricante. Durante la incubación con las células, el reactivo PB se modifica por el entorno reductor de las células viables y se vuelve altamente fluorescente. La fluorescencia se midió con un lector de microplacas (Synergy HT Multi-Mode Microplate Reader, BioTek Instruments Inc., Winooski, VT, EE.UU.) a longitudes de onda de excitación y emisión de 560 y 590 nm, respectivamente. La absorbancia de los pocillos que contenían medio de cultivo y células tumorales se utilizó como control y cada condición experimental se evaluó por triplicado y se repitió por duplicado. Comparando la fluorescencia/absorbancia medida de los pocillos que contenían DCA con las mediciones de los pocillos que contenían células no tratadas, fue posible generar perfiles dosis-respuesta y determinar los valores de IC50 (la concentración que inhibe la supervivencia en un 50%), utilizando GraphPadPrism5.0 (GraphPad Software, Inc., La Jolla, CA).
Cuantificación de glucosa y lactato
Para medir los niveles de glucosa y lactato, se sembraron células de melanoma en placas de 6 pocillos a una densidad final de 2.105 células/pocillo y se incubaron a 37 °C durante 24 h. A continuación, se trataron las células con 35 mM de DCA. Como controles, las células se incubaron con el compuesto vehículo (dH2O). Tras 24 y 48 h de tratamiento, se recogió el medio de cultivo. Los niveles de glucosa presentes en el medio condicionado en cultivo se cuantificaron utilizando el Kit Glucose GOD/PAP (Roche Applied Sciences) y se restaron a los niveles iniciales (0 h). El lactato se cuantificó de forma similar, utilizando el ensayo enzimático colorimétrico LO-POD (Spinreact, Sant Esteve de Bas, España).
Análisis del ciclo celular y de la apoptosis
Para el perfil del ciclo celular y el análisis de la apoptosis, se sembraron células de melanoma en placas de 6 pocillos a una densidad final de 1 105 células/pocillo y se incubaron a 37 °C durante 24 h. A continuación, se trataron las células con DCA a 35 mM durante 24 y 48 h de tratamiento. Como controles, las células se incubaron con el compuesto vehículo (dH2O). Para el análisis del ciclo celular, se cosecharon las células y se fijaron durante la noche en etanol al 70% enfriado en hielo. Después, las células se resuspendieron en PBS con 0,1 mg/ml de RNasa A y 5 µg/ml de yoduro de propidio, antes del análisis. Para medir la apoptosis, se cosecharon las células y se analizaron los niveles de apoptosis por citometría de flujo utilizando el Annexin-V FITC Apoptosis Kit (Clontech Laboratories, Inc., Saint-Germainen-Laye, Francia) siguiendo las instrucciones del fabricante. El análisis por citometría de flujo del contenido de ADN celular y la externalización de fosfatidilserina se realizó con un citómetro de flujo (BD Accuri C6), trazando al menos 20.000 eventos por muestra. Los datos se analizaron con el software FlowJo 7.6.5 (Tree Star, Inc., Ashland, EE.UU.).
Análisis Western blot y anticuerpos
Las célulasse lisaron durante 15 min a 4°C utilizando tampón RIPA (1% NP-40 en 150 mM NaCl, 50 mM Tris (pH 7,5), 2 mM EDTA) que contenía inhibidores de la fosfatasa y la proteasa. Las proteínas se cuantificaron mediante un ensayo de Bradford modificado (Biorad). Las muestras de proteínas (50 µg) se separaron en geles SDS/PAGE al 10% y se electroblotaron en una membrana Hybond ECL (Amersham Biosciences). Se utilizaron los siguientes anticuerpos primarios: PDH y pPDH Ser293, de Abcam; PDK1 (Sigma-Aldrich), PDK2 (Sigma-Aldrich), HIF1-α (Transduction Laboratories) y los efectores de la vía mTOR pS6 Ser235/236, s6, p-4EBP1 Thr37/46, 4EBP1, todos de Cell Signaling Technology. Los anticuerpos secundarios se conjugaron con peroxidasa (Santa Cruz Biotechnology) y se visualizaron con la solución de detección ECL. Las membranas se volvieron a teñir con un anticuerpo policlonal antiactina de cabra (Santa Cruz Biotechnology) para el control de carga de proteínas. Todos los experimentos y cuantificaciones (utilizando el software Bio-Rad Quantity One 1-D Analysis (versión 4.6.6)) se realizaron por triplicado.
Análisis estadístico
El análisis estadístico se realizó utilizando STAT VIEW-J 5.0 (SAS Institute, Inc., Cary, NC). La relación entre el nivel medio de expresión (puntuación) de los marcadores inmunohistoquímicos y los parámetros clínico-patológicos se evaluó mediante ANOVA. Cuando procedió, se realizaron correcciones de comparaciones múltiples mediante las pruebas post hoc de Bonferroni y Tamhane. La correlación entre la puntuación de inmunorreactividad de los distintos marcadores se evaluó mediante la prueba exacta de Fisher. Los datos de los experimentos con líneas celulares se analizaron mediante la prueba t de Student no emparejada de dos colas. Para evaluar los datos de supervivencia del melanoma se utilizaron el método de Kaplan-Meier y la prueba de rangos logarítmicos. Se realizaron análisis univariantes y multivariantes para determinar el valor pronóstico de las covariantes en relación con la SG y la SSE mediante el modelo de regresión de Cox. La SG y la SSE se calcularon desde el momento del diagnóstico hasta la muerte por enfermedad o metástasis, respectivamente, o se censuraron en el momento del último seguimiento o de la muerte no relacionada con la enfermedad. Un valor p < 0,05 se consideró estadísticamente significativo.
Resultados
Expresión de PDK en CM y nevus
Tanto en las lesiones de CM como en las de nevus, las isoformas PDK 1, 2 y 3 se expresaron no sólo en los melanocitos/células de melanoma, sino también en los queratinocitos y en las células de las glándulas sebáceas y los folículos pilosos. Los niveles de expresión de todas las isoformas PDK analizadas estaban positivamente correlacionados entre sí, lo que sugiere una expresión concomitante de estas proteínas (Tabla 2).
| Proteína | Nivel de expresión medio (± SD) | Correlación (valor p) | Correlación (valor p) | Correlación (valor p) |
| PDK1 | 4.8 ± 3.5 | < 0.01* | < 0.01§ | |
| PDK2 | 4.6 ± 3.6 | < 0.01† | ||
| PDK3 | 7.0 ± 3.2 |
*Entre PDK1 y PDK2; † entre PDK2 y PDK3; § entre PDK1 y PDK3.
Expresión inmunohistoquímica de PDK1
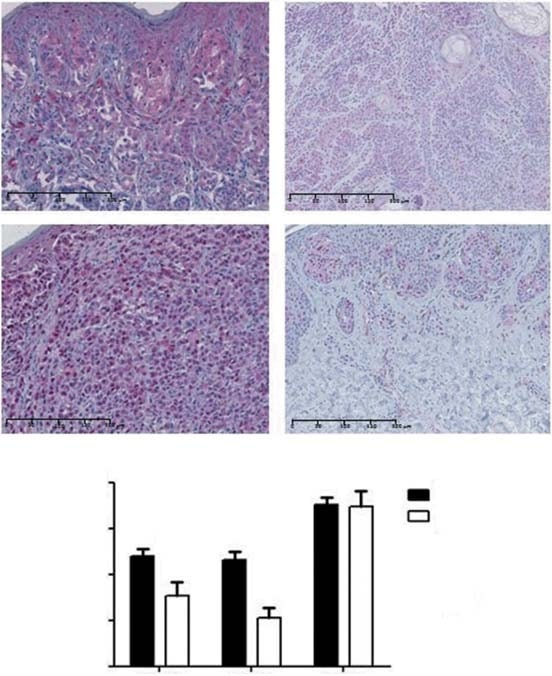
Se observó tinción citoplasmática de PDK1 en el 88% de los CM y en el 76% de los nevos (Figura 2A). Se observó una tinción negativa/baja en el 52% y el 76% y una tinción moderada/alta en el 48% y el 24% de los melanomas y nevos, respectivamente. Por lo tanto, los MC mostraron niveles medios de expresión de PDK1 significativamente más altos que los nevos (p = 0,03; Figura 2B). En cuanto a los subtipos histológicos de MC, los LMM mostraron mayores niveles de expresión de PDK1 que los NM (p = 0,04) y los ALM (p = 0,04). No se encontró asociación entre la expresión de PDK1 y el tipo de exposición solar. En cuanto a los factores pronósticos, PDK1 se expresaba más en tumores de £ 1 mm de grosor (p = 0,02). Los mayores niveles de expresión de PDK1 se asociaron con estadios tumorales más bajos (p = 0,04), pero no con la supervivencia global y libre de enfermedad de los pacientes. También evaluamos si la expresión de isoformas de PDK estaba relacionada con las vías MAPK y PI3K-AKT-mTOR. No se encontró ninguna asociación entre la expresión de PDK1 y las mutaciones BRAF/NRAS o con la activación de la vía MAPK (evaluada mediante la expresión de pERK). La expresión de PDK1 se correlacionó positivamente con la expresión de proteínas de la cascada PI3K-AKT-mTOR, concretamente mTOR y 4EBP1 (p = 0,02 y p = 0,04, respectivamente).
Expresión inmunohistoquímica de PDK2
PDK2 mostró expresión tanto nuclear como citoplasmática en el 87% de los MC y el 83% de los nevos (Figura 2A). Se observó una tinción negativa/baja en el 58% y el 89% y una tinción moderada/alta en el 42% y el 11% de los melanomas y nevos, respectivamente. El nivel medio de expresión de PDK2 fue mayor en los MC que en los nevos (p < 0,01; Figura 2B). Con respecto a los subtipos histológicos de MC, el MSS mostró niveles más altos de expresión de PDK2 que el MLA (p = 0,02). No encontramos ninguna asociación entre el tipo de exposición solar y la expresión de PDK2. Los estadios tumorales más bajos mostraron niveles significativamente más altos de PDK2 (p = 0,02), aunque no se encontró asociación entre la expresión de esta isoforma y los principales factores pronósticos del CM, ni con la supervivencia global y libre de enfermedad de los pacientes. La expresión de PDK2 no se asoció con el estado mutacional BRAF/NRAS ni con la activación de la vía MAPK. Se encontró una correlación positiva significativa entre la expresión de PDK2 y la expresión de AKT de la cascada PI3K-AKT-mTOR (p = 0,03).
Expresión inmunohistoquímica de PDK3
PDK3 mostró expresión citoplasmática en todos los CM y nevos. Se observó una tinción negativa/baja en el 28% y el 22% y una tinción moderada/alta en el 72% y el 78% de los CM y nevos, respectivamente. El nivel de expresión de la isoforma PDK3 fue similar entre el CM y los nevos (Figura 2B). No hubo diferencias significativas al comparar el nivel medio de expresión de PDK3 entre los subtipos de CM y el tipo de exposición solar. Los tumores con un grosor de £ 1 mm y aquellos con estadios inferiores mostraron niveles más altos de PDK3 (p = 0,01 y p < 0,01, respectivamente). La ulceración, la tasa mitótica y la supervivencia global y libre de enfermedad de los pacientes no se asociaron significativamente con la expresión de PDK3. No se encontró ninguna asociación entre la expresión de PDK3 y las mutaciones BRAF/NRAS ni la activación de la cascada MAPK. En cuanto a la vía PI3K-AKT-mTOR, la expresión de PDK3 se correlacionó positivamente con la expresión total de AKT, mTOR y 4EBP1 (p < 0,01, p < 0,01 y p = 0,04, respectivamente). Debido a la expresión general de PDK3 en todos los CM y nevos, el posterior análisis in vitro no incluyó esta isoforma de PDK.
Efecto del tratamiento con DCA en la viabilidad de las líneas celulares de melanoma
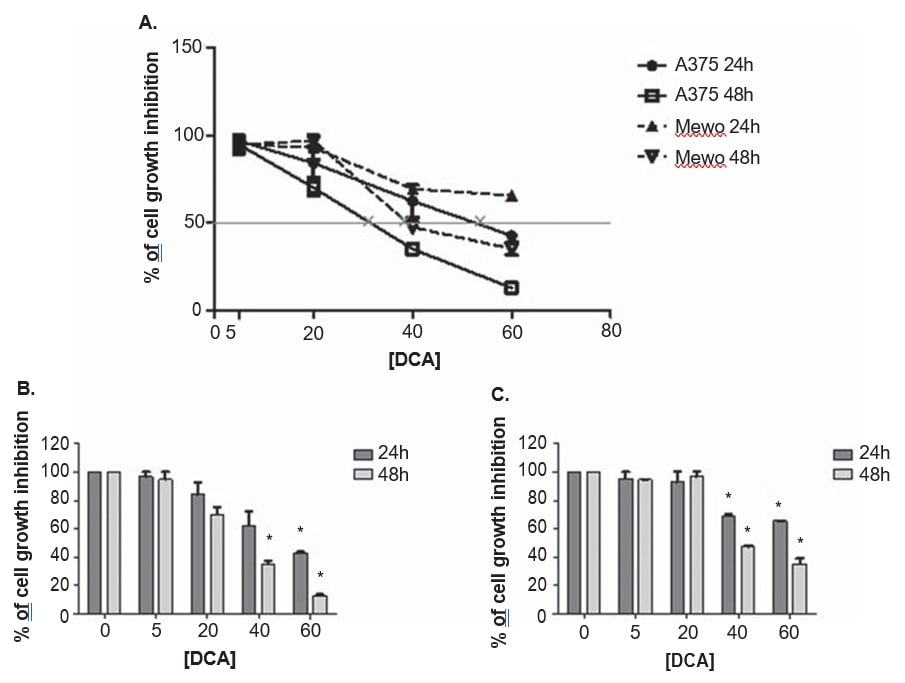
Las líneas celulares de melanoma A375 y Mewo se expusieron a concentraciones crecientes de DCA para establecer el efecto sobre la viabilidad celular, utilizando el ensayo PrestoBlue. El DCA redujo la viabilidad de ambas líneas celulares de forma dosis-dependiente tras 24 y 48 h de tratamiento, aunque A375 fue ligeramente más sensible al tratamiento con DCA, con una mayor inhibición del crecimiento en comparación con la línea celular Mewo (Figura 3). El efecto del DCA sobre la viabilidad de las células A375 se observó tras el tratamiento con 5 mM y en la línea celular Mewo fue más intenso tras el tratamiento con 20 mM, con valores de inhibición que oscilaron entre el 3 y el 87% en A375 y entre el 5 y el 65% en Mewo. Los valores de IC50 se estimaron en 33 ± 5,5 mM para A375 y 39,9 ± 2,2 mM para la línea celular Mewo, tras 48 h de tratamiento con DCA (Figura 3).
Efecto del tratamiento con DCA en los niveles de consumo de glucosa y producción de lactato de las líneas celulares de melanoma
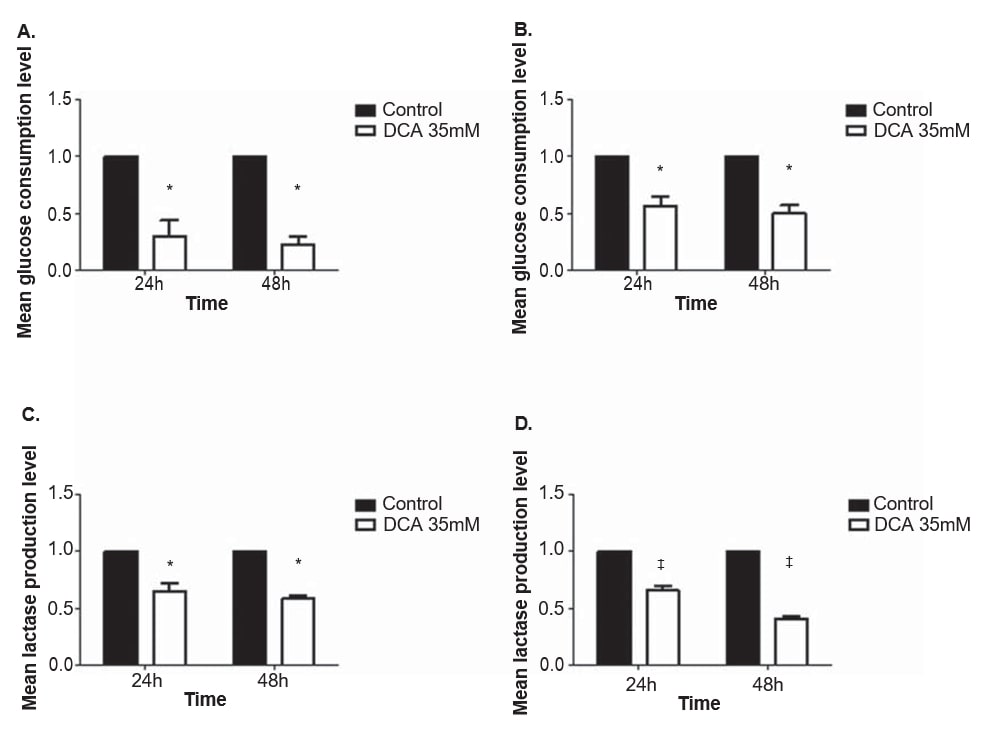
Para evaluar el efecto del DCA en el metabolismo de las líneas celulares de melanoma, se cuantificaron los niveles de glucosa y lactato en el medio de cultivo tras 24 y 48 h de tratamiento con 35 mM de DCA, una concentración cercana a la IC50 para ambas líneas celulares. El consumo de glucosa y la producción de lactato disminuyeron en ambas líneas celulares de melanoma tras el tratamiento con DCA (Figura 4). Las líneas celulares A375 y Mewo mostraron un descenso significativo en el nivel de consumo de glucosa tras 24 y 48 h de tratamiento con DCA (p = 0,01 y p < 0,01, respectivamente). El nivel de producción de lactato también disminuyó significativamente tras 24 y 48 h en las líneas celulares A375 (p = 0,01 y p < 0,01, respectivamente) y Mewo (p < 0,01).
Efecto del tratamiento con DCA en los efectores de las vías PDK y mTOR, y en la expresión de HIF1-α en líneas celulares de melanoma
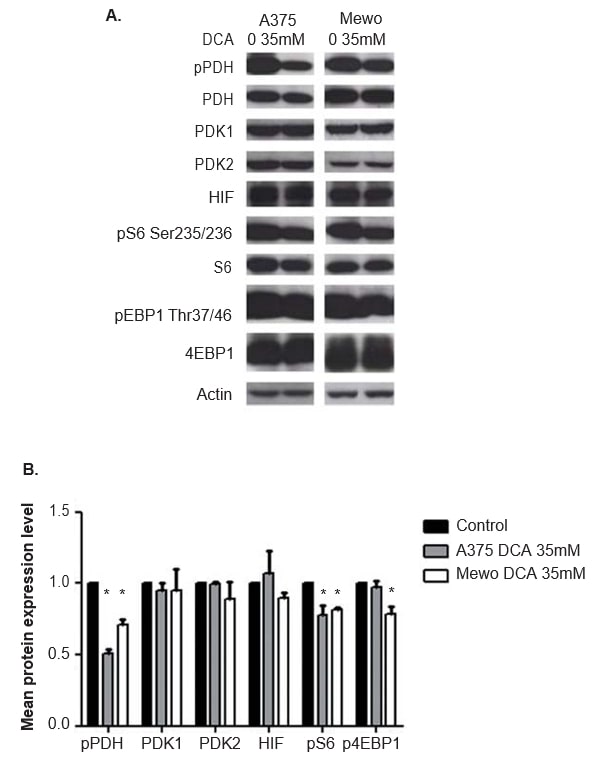
La eficacia del tratamiento con DCA en la inhibición de la actividad PDK se evaluó analizando la expresión del efector corriente abajo fosforilado PDH, mediante western blot. A las 24 h del tratamiento con DCA, se observó una inhibición significativa de la fosforilación de PDH en ambas líneas celulares (p < 0,01; Figura 5). La expresión de PDK1 y PDK2 no se vio alterada por el tratamiento con DCA (Figura 5), como era de esperar. También se evaluó la expresión de S6 y 4EBP1, dos efectores de la activación de la vía mTOR. Aunque el tratamiento con DCA no alteró la expresión de S6 y 4EBP1, el nivel de expresión de las formas fosforiladas de ambas proteínas disminuyó en A375 (p = 0,03 y no significativo, respectivamente) y en Mewo (p < 0,01 y p = 0,01, respectivamente; Figura 5). Dado que el DCA puede afectar a los niveles de HIF1-α, también se evaluó la expresión de esta proteína, y no se observaron cambios tras el tratamiento con DCA (Figura 5). Se obtuvieron resultados similares a las 48 h del tratamiento con DCA, que inhibe significativamente la fosforilación de la PDH en ambas líneas celulares (p <0,01). El nivel de expresión de las formas fosforiladas de S6 y 4EBP1 disminuyó significativamente en A375 (p = 0,01 y p = 0,02, respectivamente) y en Mewo (p = 0,01 y no significativo, respectivamente). La expresión de PDK1, PDK2 y HIF1-α no se vio alterada (datos no mostrados).
Efecto del tratamiento con DCA en el ciclo celular y la apoptosis de las líneas celulares de melanoma
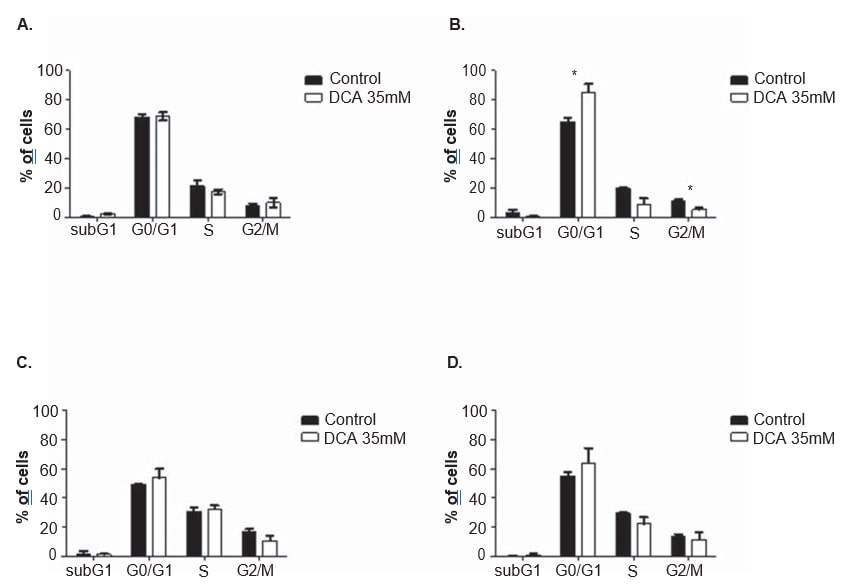
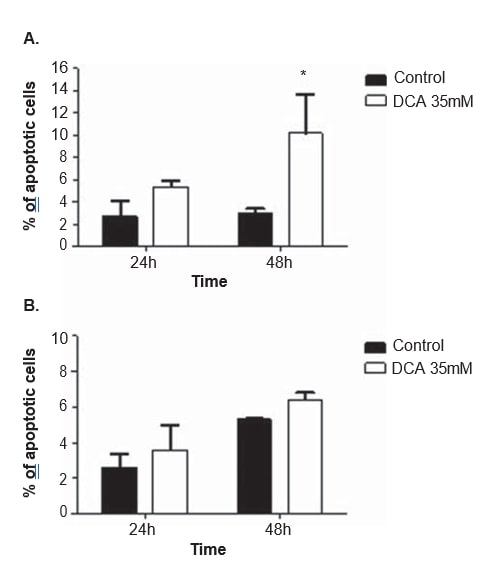
Para aclarar el mecanismo de acción del DCA, se realizaron análisis del ciclo celular y mediciones de la apoptosis en ambas líneas celulares, tras 24 y 48 h de tratamiento con 35 mM de DCA. El ciclo celular se analizó con yoduro de propidio. Tras 48 h de tratamiento con DCA, la línea celular A375 mostró un aumento significativo del porcentaje de células en la fase G0/G1 del ciclo celular (del 65,0 ± 4,9% observado en las células no tratadas al 85,2 ± 10,2% en las células tratadas con DCA; p = 0,04), una disminución significativa en G2/M (del 11.3 ± 1,3% observado en las células no tratadas a 5,2 ± 2,2% en las células tratadas con DCA; p = 0,01) y una disminución, que no alcanzó el umbral de significación estadística, en la fase S (de 19,7 ± 1,8% observado en las células no tratadas a 9,1 ± 6,7% en las células tratadas con DCA; p = 0,06; Figura 6). La misma tendencia se observó en la línea celular Mewo 48 h después del tratamiento con DCA (aumento del porcentaje de células en G0/G1, y disminución en S y G2/M) aunque sin alcanzar significación estadística (Figura 6). El análisis por citometría de flujo se llevó a cabo tras la tinción con Annexin V/yoduro de propidio. Se observó un aumento significativo del número de células apoptóticas en la línea celular A375 tras 48 h de tratamiento con DCA (3,0 ± 0,5% en las células no tratadas a 16,9 ± 4,6% en las células tratadas con DCA; p < 0,01; Figura 7). Se observaron aumentos no significativos del porcentaje de células apoptóticas en la línea celular A375 tras 24 h y en la línea celular Mewo tras 24 h y 48 h de tratamiento con DCA.
Discusión
En este trabajo hemos establecido, por primera vez, que el CM sobreexpresa las proteínas PDK1 y PDK2. Las PDKs son proteínas clave, reguladas por HIF1-α, que conducen a las células malignas al fenotipo de glucólisis aeróbica, una característica distintiva del cáncer [20,22,31].
El efecto Warburg se considera un factor de resistencia a las quimioterapias convencionales [21]. La sobreexpresión de PDK encontrada en CM nos lleva a evaluar el efecto del tratamiento con DCA en líneas celulares de melanoma. Observamos que el tratamiento con DCA disminuye el consumo de glucosa y la producción de lactato en las líneas celulares de melanoma, lo que concuerda con el efecto del DCA en el cambio de la glucólisis aeróbica a la fosforilación oxidativa previamente descrito en células cancerosas [41]. Este cambio en el metabolismo de la glucosa probablemente es el resultado de la disminución significativa observada en la expresión de la forma fosforilada de PDH, que también confirma la eficacia del tratamiento con DCA. Es relevante referir que la lectura correcta del efecto del DCA es el nivel de PDH fosforilada, ya que al inhibir la actividad de la PDK y no su expresión, el DCA estimula la desfosforilación de la PDH (Figura 1).
Observamos una disminución dosis-dependiente de la viabilidad celular tras el tratamiento con DCA en ambas líneas celulares de melanoma. Además, se observó una disminución de la proliferación, a través de la detención G0/G1, junto con un aumento de la apoptosis en ambas líneas celulares, pero sólo estadísticamente significativo en la línea celular A375. El hecho de que la concentración de DCA seleccionada en el presente estudio (35 mM) sea ligeramente inferior a la IC50 para la línea celular Mewo puede explicar el efecto menos pronunciado observado en esas células.
La relación entre el DCA y HIF-1a en el cáncer no se conoce completamente, ya que el DCA aumenta el consumo tisular de oxígeno y la producción de ROS, lo que debería conducir a la regulación al alza o a la estabilización de HIF-1a, respectivamente [46,47]. Por el contrario, algunos autores informaron de que el DCA conduce a una disminución de la expresión de HIF1-α [25,48,49]. En nuestro trabajo, no se observó ninguna alteración en el nivel de HIF1-α tras el tratamiento con DCA de las líneas celulares de melanoma, lo que concuerda con Shahrzad et al. que informaron de que la expresión de HIF1-α sólo disminuye tras el tratamiento con DCA en condiciones de hipoxia [49].
Para superar la senescencia inducida por el mutante BRAF y progresar hacia fenotipos más agresivos, el CM probablemente activa otras cascadas de señalización, como la vía mTOR [12]. Esta vía contribuye al efecto Warburg promoviendo la actividad de HIF1-α, que a su vez puede aumentar la expresión de PDK [23,31]. Anteriormente habíamos descrito una activación completa de esta vía en el CM [13]. En este estudio, observamos una expresión concomitante de PDK 1, 2 y 3 y de los efectores de la vía mTOR en CMs. La expresión de las isoformas de PDK en CM se correlacionó positivamente con la expresión de mTOR, así como con los efectores upstream y downstream de esta vía: AKT y 4EBP1. Además, observamos una disminución significativa de pS6 y p4EBP1 en las líneas celulares A375 y Mewo tras el tratamiento con DCA.
Los nevus comparten algunas alteraciones genéticas con el CM, ya que ambos presentan una alta frecuencia de mutaciones en BRAF [10]. Aunque en un nivel mucho más bajo, la expresión de PDK también se observó en los nevos. Evaluamos si el estado mutacional de BRAF y la activación descendente de ERK podrían asociarse con una mayor expresión de PDK. No encontramos ninguna asociación en los nevos ni en el CM. La línea celular A375 alberga la mutación BRAFV600E y la línea celular Mewo es de tipo salvaje para este gen, y la respuesta de ambas líneas celulares al tratamiento con DCA fue similar, a pesar de tener una intensidad variable. De hecho, ya se informó de que el estado mutacional de BRAF no altera la sensibilidad al tratamiento con DCA [50]. Estos resultados contrastan con nuestros hallazgos previos utilizando inhibidores de la vía mTOR en varias líneas celulares derivadas de melanoma, donde encontramos una mayor sensibilidad al tratamiento con RAD001 en las líneas celulares de CM portadoras de la mutación BRAFV600E [51].
Las isoformas PDK se expresaron en el citoplasma de las células de melanoma, lo que apoya la conocida función mitocondrial de dichas isoformas [33]. En cuanto a PDK2, se observó una expresión nuclear en la mayoría de los casos de MC. Esta localización nuclear también se ha descrito para PDK1 en HNSCC [37]. Por lo que sabemos, el papel de PDK en el núcleo sigue sin aclararse pero, recientemente, se ha avanzado una función nuclear para PDH, el efector aguas abajo de PDK. Sutendra et al. informaron de que la PDH está implicada en la formación nuclear de acetil-CoA [52]. En respuesta a la estimulación del factor de crecimiento, la PDH y su fosfatasa se translocan de la mitocondria, donde es inhibida por la PDK, al núcleo, pero no se identificó una localización nuclear para la PDK [52]. Sin embargo, existen pruebas de que algunas enzimas glucolíticas participan en otras funciones celulares además de la glucólisis, concretamente en la regulación transcripcional [53]. Se necesitan estudios adicionales para aclarar si la PDK también desempeña una función nuclear.
En el carcinoma de células renales, se ha descrito que la expresión de PDK1 disminuye a lo largo de la progresión tumoral [39]. Concordantemente, en nuestro trabajo, PDK se encontró más expresada en estadios tumorales más bajos, sin influir en la supervivencia global y libre de enfermedad de los pacientes con melanoma. En cambio, la sobreexpresión de PDK se identificó como un marcador de progresión tumoral, mal pronóstico y recurrencia en el HNSCC y el cáncer gástrico [37,40,54]. Aunque nuestro objetivo no era explorar el papel pronóstico de las PDK en el CM, nuestros resultados sugieren que las PDK en el CM pueden desempeñar un papel más activo en el desarrollo del melanoma que en su progresión. Somos conscientes de que nuestra serie se compone principalmente de MC primarios y de que serán necesarios más estudios en melanoma avanzado (metastásico) para aclarar esta cuestión.
Nuestro objetivo en este estudio era explorar biomarcadores para nuevos enfoques terapéuticos en pacientes con CM. Nuestros resultados sugieren una utilidad potencial del DCA en el tratamiento de pacientes con MC, en línea con Abildgaard et al. que informaron de una disminución de los niveles de ATP y del crecimiento del melanoma mediante el tratamiento con DCA [50]. Estos autores también informaron de una combinación sinérgica entre el DCA y un inhibidor de BRAF, el vemurafenib [50]. Además, nuestros resultados muestran una expresión concomitante de los efectores de las vías PDK y mTOR en el CM y una regulación a la baja de la actividad de la vía mTOR mediante el tratamiento con DCA. Teniendo en cuenta los resultados de Hong et al., que informaron de un efecto sinérgico de la combinación de DCA con un inhibidor de S6K1 (efector de la vía mTOR) [55], resulta tentador especular con la posibilidad de conseguir un efecto terapéutico sinérgico en el CM utilizando la combinación de DCA con un inhibidor directo de mTOR.
Conclusión
Informamos por primera vez de la sobreexpresión de PDK1 y 2 en el CM en comparación con los nevos. La expresión de PDKs se encontró asociada a la expresión de efectores de la vía mTOR y no relacionada con el estado mutacional de BRAF. El tratamiento con DCA produce un cambio en el metabolismo, una regulación a la baja de la proliferación, un aumento de la apoptosis y una disminución de la activación de la vía mTOR en las líneas celulares de melanoma. Tomando todos estos resultados en conjunto, concluimos que la expresión de PDK puede desempeñar un papel en el desarrollo del melanoma y que su inhibición mediante DCA solo o en combinación con inhibidores directos de mTOR puede beneficiar a los pacientes con CM.
Agradecimientos
Agradecemos a todos los pacientes que participaron en este estudio, así como a los médicos que proporcionaron información clínica, patológica y de seguimiento. Agradecemos a la Dra. Madalena Pinto, del CEQUIMED, Facultad de Farmacia, Universidad de Oporto, Portugal, que amablemente nos proporcionó la línea celular de melanoma cutáneo A375, y al Dr. Marc Mareel, del Departamento de Radioterapia y Medicina Nuclear, Hospital Universitario de Gante, Bélgica, que amablemente nos proporcionó la línea celular de melanoma cutáneo Mewo. Damos las gracias a Gabriela Almeida por sus útiles consejos técnicos en relación con el ensayo PB. También agradecemos al Prof. Manuel Sobrinho Simões la lectura crítica de este manuscrito. H Pópulo y R Caldas han contribuido a partes iguales a este trabajo.
Declaración de intereses
Este estudio fue apoyado por la Fundación Portuguesa para la Ciencia y la Tecnología a través de la beca Post-Doc a HP (Ref.: SFRH/BPD/85249/2012). Se obtuvo financiación adicional del proyecto «Microambiente, metabolismo y cáncer» que fue parcialmente apoyado por el Programa Operacional Regional do Norte (ON.2 – O Novo Norte) bajo el Quadro de Referência Estratégico Nacional (QREN) y el Fundo Europeu de Desenvolvimento Regional (FEDER). IPATIMUP integra la Unidad de Investigación i3S, que cuenta con el apoyo parcial de FCT, la Fundación Portuguesa para la Ciencia y la Tecnología. Este trabajo fue financiado por fondos FEDER a través del Programa Operativo de Factores de Competitividad – COMPETE y Fondos Nacionales a través de la FCT, bajo los proyectos «PEst-C/SAU/LA0003/2013.» Los autores declaran no tener potenciales conflictos de intereses. Los autores no tienen otras afiliaciones relevantes o participación financiera con cualquier organización o entidad con un interés financiero o conflicto financiero con el tema o los materiales discutidos en el manuscrito aparte de los revelados.
REFERENCIAS
1 Siegel R, Ma J, Zou Z, et al. Estadísticas sobre el cáncer, 2014. CA Cancer J Clin 2014;64(1):9-29
2 Nikolaou V, Stratigos AJ. Tendencias emergentes en la epidemiología del melanoma. Br J Dermatol 2014;170(1):11-19
3 Smoller BR. Criterios histológicos para el diagnóstico del melanoma maligno cutáneo primario. Mod Pathol 2006;19(Suppl 2):S34-40
4 Populo H, Soares P, Lopes JM. Insights into melanoma: targeting the mTOR pathway for therapeutics. Expert Opin Ther Targets 2012;16(7):689-705
5 Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27(36):6199-206
6 Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol 2008;84(2):289-306
7 Bertolotto C. Melanoma: del melanocito a las alteraciones genéticas y opciones clínicas. Scientifica 2013;2013:22
8 Populo H, Boaventura P, Vinagre J, et al. Mutaciones del promotor TERT en cáncer de piel: efectos de la exposición solar y la irradiación X. J Invest Dermatol 2014;134(8):2251-7
9 Elder DE. Precursores del melanoma y sus imitadores: nevos de sitios especiales. Modern pathology Inc 2006;19(Suppl 2):S4-20
10 Kumar R, Angelini S, Snellman E, et al. BRAF mutations are common somatic events
11 in melanocytic nevi. J Invest Dermatol 2004;122(2):342-8 Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436(7051):720-4
12 Vredeveld LC, Possik PA, Smit MA, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012;26(10):1055-69
13 Populo H, Soares P, Faustino A, et al. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res 2011;24(1):254-7
14 Olszanski AJ. Funciones actuales y futuras de la terapia dirigida y la inmunoterapia en el melanoma avanzado. J manag Care Spec Pharm 2014;20(4):346-56
15 Hodi FS, O’Day SJ, McDermott DF, et al. Mejora de la supervivencia con ipilimumab en pacientes con melanoma metastásico. N Engl J Med 2010;363(8):711-23
16 Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507-16
17 Flaherty KT, Robert C, Hersey P, et al. Mejora de la supervivencia con inhibición de MEK en melanoma con mutación de BRAF. N Engl J Med 2012;367(2):107-14
18 Ballantyne AD, Garnock-Jones KP. Dabrafenib: primera aprobación mundial. Drugs 2013;73(12):1367-76
19Disponible en:http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm412861.htm
20 Gillies RJ, Robey I, Gatenby RA. Causas y consecuencias del aumento del metabolismo de la glucosa de los cánceres. J Nucl Med 2008;49(Suppl 2):24S-42S
21 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532
22 Semenza GL. HIF-1 media respuestas metabólicas a la hipoxia intratumoral y mutaciones oncogénicas. J Clin Invest 2013;123(9):3664-71
23 Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851-64
24 Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 2011;286(49):42626-34
25 Kluza J, Corazao-Rozas P, Touil Y, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res 2012;72(19):5035-47
26 Baudy AR, Dogan T, Flores-Mercado JE, et al. FDG-PET is a good biomarker of both early response and acquired resistance in BRAFV600 mutant melanomas treated with vemurafenib and the MEK inhibitor GDC-0973. EJNMMI research 2012;2(1):22
27 Hall A, Meyle KD, Lange MK, et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 2013;4(4):584-99
28 Kumar SM, Yu H, Edwards R, et al. Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res 2007;67(7):3177-84
29 Kuphal S, Winklmeier A, Warnecke C, et al. Constitutive HIF-1 activity in malignant melanoma. Eur J Cancer 2010;46(6):1159-69
30 Slominski A, Kim TK, Brozyna AA, et al. El papel de la melanogénesis en la regulación del comportamiento del melanoma: Melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways. Archivos de bioquímica y biofísica 2014;563:79-93
31 Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3(3):177-85
32 Lu CW, Lin SC, Chen KF, et al. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008;283(42):28106-14
33 Patel MS, Korotchkina LG. Regulación del complejo piruvato deshidrogenasa. Biochem Soc Trans 2006;34(Pt 2):217-22
34 Bowker-Kinley MM, Davis WI, Wu P, et al. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6
35 Papandreou I, Goliasova T, Denko NC. Fármacos anticancerígenos dirigidos al metabolismo: ¿Es el dicloroacetato el nuevo paradigma? Int J Cancer 2011;128(5):1001-8
36 Zhang S, Hulver MW, McMillan RP, et al. El papel fundamental de las quinasas piruvato deshidrogenasa en la flexibilidad metabólica. Nutrición y metabolismo 2014;11(1):10
37 Wigfield SM, Winter SC, Giatromanolaki A, et al. PDK-1 regula la producción de lactato en hipoxia y se asocia con un mal pronóstico en el cáncer escamoso de cabeza y cuello. Br J Cancer 2008;98(12):1975-84
38 Lu CW, Lin SC, Chien CW, et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol 2011;179(3):1405-14
39 Baumunk D, Reichelt U, Hildebrandt J, et al. Parámetros de expresión de los genes de la vía metabólica piruvato deshidrogenasa quinasa-1 (PDK-1) y DJ-1/PARK7 en el carcinoma de células renales (CCR). World J Urol 2013;31(5):1191-6
40 Hur H, Xuan Y, Kim YB, et al. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int J Oncol 2013;42(1):44-54
41 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11(1):37-51
42 Kankotia S, Stacpoole PW. Dicloroacetato y cáncer: ¿Nuevo hogar para un medicamento huérfano? Biochimica et biophysica acta 2014;1846(2):617-29
43 Sutendra G, Dromparis P, Kinnaird A, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013;32(13):1638-50
44 Michelakis ED, Sutendra G, Dromparis P, et al. Modulación metabólica del glioblastoma con dicloroacetato. Sci Transl Med 2010;2(31):31ra34
45 Castro P, Rebocho AP, Soares RJ, et al. PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 2006;91(1):213-20
46 Sun W, Zhou S, Chang SS, et al. Las mutaciones mitocondriales contribuyen a la acumulación de HIF1 alfa a través del aumento de especies reactivas de oxígeno y la regulación al alza de la piruvato deshidrogenasa quinasa 2 en el carcinoma de células escamosas de cabeza y cuello. Clin Cancer Res 2009;15(2):476-84
47 Cairns RA, Bennewith KL, Graves EE, et al. Pharmacologically increased tumor hypoxia can be measured by 18F-Fluoroazomycin arabinoside positron emission tomography and enhances tumour response to hypoxic cytotoxin PR-104. Clin Cancer Res 2009;15(23):7170-4
48 Sun RC, Board PG, Blackburn AC. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol Cancer 2011;10:142
49 Shahrzad S, Lacombe K, Adamcic U, et al. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;297(1):75-83
50 Abildgaard C, Dahl C, Basse AL, et al. La modulación bioenergética con dicloroacetato reduce el crecimiento de células de melanoma y potencia su respuesta a la inhibición de BRAFV600E. J Transl Med 2014;12:247
51 Populo H, Tavares S, Faustino A, et al. Las mutaciones GNAQ y BRAF muestran una activación diferencial de la vía mTOR en células humanas transformadas. Peer J 2013;1:e104
52 Sutendra G, Kinnaird A, Dromparis P, et al. Un complejo nuclear de piruvato deshidrogenasa es importante para la generación de acetil-CoA y la acetilación de histonas. Cell 2014;158(1):84-97
53 Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 2005;30(3):142-50
54 Xuan Y, Hur H, Ham IH, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res 2014;321(2):219-30
55 Hong SE, Shin KS, Lee YH, et al. La inhibición de S6K1 mejora la muerte celular inducida por dicloroacetato. J Cancer Res Clin Oncol 2014.