Helena Populo1,2, Regina Caldas1,2,3, Jose Manuel Lopes1,2,4,5, Joana Pardal5, Valdemar Maximo1,2,4 & Paula Soares†,2,4
† Institut für Forschung und Innovation im Gesundheitswesen (Instituto de Investigacao e Inovacao em Saude), Universität Porto, Porto, Portugal1
Institut für Molekularpathologie und Immunologie der Universität Porto (IPATIMUP), Universität Porto, Porto, Portugal
Tel: +22 557 0700; Fax: +22 557 0799; E-mail:
[email protected] Institut für Forschung und Innovation im Gesundheitswesen (Instituto de Investigacao e Inovac¸aoem Saude), Universität Porto, Porto, Portugal
3 Medizinische Fakultät, Universität Porto, Porto, Portugal
4 Abteilung für Pathologie und Onkologie, Medizinische Fakultät, Universität Porto, Porto, Portugal
5 Dienst für anatomische Pathologie Hospital Sao Joao, Porto, Portugal
Online veröffentlicht: 14. Mai 2015
Zusammenfassung
Zielsetzung: Wir wollten prüfen, ob Dichloracetat (DCA), das die Pyruvat-Dehydrogenase-Kinase (PDK) hemmt und die Stoffwechselverschiebung der Krebszellen von der Glykolyse zur oxidativen Phosphorylierung umkehrt, als vielversprechendes Medikament für die Therapie von Patienten mit kutanem Melanom (CM) in Betracht kommt.
Forschungsdesign und Methoden: Wir untersuchten das Expressionsprofil von PDK 1, 2 und 3 in einer Reihe von Melanomproben, um festzustellen, ob Melanomtumore die DCA-Targets exprimieren, ob diese Expression mit der Aktivierung wichtiger Signalkaskaden für die Melanomagenese korreliert und auch mit der Prognose von Melanompatienten. Außerdem haben wir die Empfindlichkeit von Melanomzelllinien gegenüber der DCA-Behandlung ermittelt, indem wir ihre Stoffwechselveränderungen, ihre Proliferation und ihr Überleben untersucht haben.
Ergebnisse: Wir stellten fest, dass sowohl PDK 1 als auch PDK 2 Isoformen in CM im Vergleich zu Nävi überexprimiert sind, wobei diese Expression mit der Expression der Effektoren des mTOR-Stoffwechselweges verbunden und unabhängig vom BRAF-Mutationsstatus ist. Mit DCA behandelte Melanomzelllinien zeigten eine Verschiebung des Stoffwechsels, d. h. einen Rückgang des Glukoseverbrauchs und der Laktatproduktion, eine Herunterregulierung der Proliferation, einen Anstieg der Apoptose und einen Rückgang der Aktivierung des mTOR-Signalwegs.
Schlussfolgerung: Unsere Ergebnisse deuten darauf hin, dass die PDK-Expression bei der Melanomentwicklung eine Rolle spielen kann und dass DCA für die CM-Therapie nützlich sein kann, allein oder in Kombination mit mTOR-Inhibitoren.
Schlüsselwörter: Dichloracetat, Melanom, Stoffwechsel, mTOR, Pyruvat-Dehydrogenase-Kinase
© 2015 Informa UK, Ltd. ISSN 1472-8222, e-ISSN 1744-7631
EINLEITUNG
Das kutane Melanom (CM) ist ein sehr aggressives Malignom, und obwohl es die am wenigsten verbreitete Hautkrebsart ist, ist es für die Mehrzahl der hautkrebsbedingten Todesfälle verantwortlich. Da die Inzidenz des Melanoms zunimmt, ist es derzeit die wahrscheinlichste invasive Krebserkrankung, die vor dem 50 [1,2]. Die Exposition gegenüber UV-Strahlung gilt als Hauptrisikofaktor für die Melanomagenese [2]. CM kann in verschiedene histologische Subtypen eingeteilt werden, wobei das oberflächlich streuende Melanom (SSM) am häufigsten vorkommt, gefolgt vom nodulären Melanom (NM), dem Lentigo maligna Melanom (LMM) und dem akralen lentiginösen Melanom (ALM). SSM und NM entstehen in der Haut mit intermittierender Sonnenexposition, während LMM in chronisch sonnengeschädigter Haut auftritt und ALM auf Haut ohne Sonnenexposition beschränkt ist. Diese histologische Klassifizierung hat keinen prognostischen Wert [3,4]. Bei der Stadieneinteilung von CM werden Tumordicke, Ulzeration, Mitoserate, Knotenbefall und das Vorhandensein von Metastasen berücksichtigt [5]. Glücklicherweise werden die meisten Fälle von CM in einem frühen Stadium diagnostiziert, und die 5-Jahres-Überlebensrate liegt bei 98 % [1]. Bei Patienten mit metastasiertem Melanom beträgt die mediane Überlebenszeit jedoch nur 8 bis 9 Monate [5].
CM ist ein sehr heterogener Tumor und viele Zellsignalwege sind bei der Melanomagenese dereguliert [6,7]. Der MAPK-Signalweg ist bei der Mehrheit der CM konstitutiv aktiviert. NRAS-Mutationen wurden bei 10-20 % der CM festgestellt, wobei NRASQ61K/R die häufigste Mutation ist. BRAF-Mutationen, insbesondere die BRAFV600E-Mutation, wurden in 40 bis 60 % der Fälle festgestellt. Unsere Gruppe beobachtete einen Zusammenhang zwischen dem Vorhandensein von BRAF-Mutationen und den kürzlich beschriebenen Mutationen des TERT-Promotors bei CM [8]. BRAFV600E-Mutationen und in geringerem Maße NRASQ61K/R-Mutationen wurden auch in fast 80 % der Nävi (gutartige melanozytäre Läsionen, die in 25 % der Fälle als Vorläufer von CM angesehen werden) festgestellt, was darauf hindeutet, dass die Aktivierung des MAPK-Signalwegs für die Melanomentwicklung zwar notwendig, aber nicht ausreichend ist [7,9,10]. Tatsächlich wurde BRAFV600E mit onkogen-induzierter Seneszenz in Verbindung gebracht und kann daher bei vielen Nävi zu einem Wachstumsstillstand führen [11]. Die Aktivierung des PI3K-AKTmTOR-Signalwegs kann diesen Seneszenz-Phänotyp überwinden und die Tumorprogression fördern [12]. Unsere Gruppe hat die Bedeutung des PI3K-AKT-mTOR-Signalwegs für die Aggressivität von CM bestätigt, indem sie seine Aktivierung mit dem Vorhandensein von BRAF-Mutationen und schlechteren prognostischen Merkmalen in Verbindung gebracht hat. Die Überexpression von mTOR-Signalweg-Effektoren wurde mit einer höheren Mitoserate, einem höheren Clark-Level, einer größeren Tumordicke und dem Vorhandensein von Hautulzerationen in Verbindung gebracht [13].
Vor 2011 beruhten die Behandlungsmöglichkeiten für Patienten mit fortgeschrittener CM hauptsächlich auf der konventionellen Chemotherapie, die geringe Ansprechraten und geringe Auswirkungen auf das Gesamtüberleben der Patienten hatte. Seitdem wurden fünf neue Medikamente für CM-Patienten im Stadium IV zugelassen [14]. Ipilimumab, ein Anti-CTLA4-Antikörper, zeigte deutliche Verbesserungen beim Gesamtüberleben der Patienten, war aber aufgrund seiner unerwünschten Wirkungen nicht für alle Patienten geeignet [15]. Vemurafenib und Dabrafenib, selektive Inhibitoren von BRAFV600E, und Trametinib, ein MEK-Inhibitor, zielen alle auf den MAPK-Signalweg ab und erzielten Ansprechraten von mehr als 50 %, die sogar besser waren als die von Ipilimumab. Aufgrund der Entwicklung von Resistenzen übersteigt ihre Verbesserung des OS jedoch nicht 7 bis 8 Monate [16-18]. Kürzlich wurde ein weiterer Immunmodulator, Pembrolizumab, ein Antikörper gegen den programmierten Todesrezeptor-1 (PD-1), für die Behandlung von Patienten mit inoperablem oder metastasiertem Melanom und Fortschreiten der Erkrankung nach Ipilimumab oder, bei positiver BRAFV600-Mutation, einem BRAF-Inhibitor zugelassen [19]. Klinische Studien mit Medikamenten, die auf den PI3K-AKT-mTOR-Signalweg abzielen, sind im Gange [4,14], doch sind neue therapeutische Ansätze erforderlich.
Der Stoffwechsel der Krebszellen unterscheidet sich von dem der normalen Zellen. In normalen Zellen kann Glukose je nachO2-Verfügbarkeit unter hypoxischen Bedingungen durch Glykolyse teilweise zu Laktat verstoffwechselt oder in Gegenwart vonO2 durch mitochondriale oxidative Phosphorylierung, einen effizienteren energetischen Prozess, vollständig zuCO2 oxidiert werden. Krebszellen hingegen verstoffwechseln die meiste Glukose zu Laktat, unabhängig von ihrerO2-Versorgung, dem so genannten Warburg-Effekt [20,21]. Die Entstehung dieses glykolytischen Phänotyps in Krebszellen ist noch nicht vollständig geklärt, doch wurde dem Transkriptionsfaktor Hypoxie-induzierbarer Faktor 1a (HIF1-α) eine wesentliche Rolle zuerkannt [22]. Es ist bekannt, dass das HIF1-α-Protein unter Hypoxie stabilisiert wird, aber auch onkogene Signalwege, wie der MAPK- und der mTOR-Signalweg, scheinen seine Aktivierung bei Krebs zu vermitteln (Abbildung 1) [22,23]. Vieles deutet darauf hin, dass CM diesen glykolytischen Phänotyp annimmt, was durch FDGPET-Scans von Patienten bestätigt werden kann [24-27]. Tatsächlich scheint HIF1-α bei CM überexprimiert zu sein, da die Haut eine mild-hypoxische Umgebung darstellt und die Produktion von Melanin indirekt die HIF1-α-Expression durch ROS-Produktion stimuliert [28-30]. Allerdings muss die zytoplasmatische Glykolyse von der oxidativen Phosphorylierung in den Mitochondrien abgekoppelt werden, damit der größte Teil des Pyruvats in Laktat umgewandelt werden kann. Dieser letzte Prozess wird durch die Pyruvat-Dehydrogenase-Kinase (PDK) angetrieben, ein Enzym, das durch HIF1-α hochreguliert wird (Abbildung 1) [31,32].
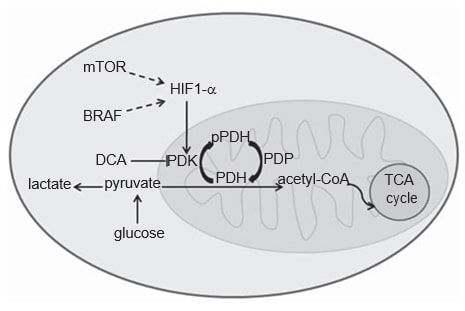
PDK ist ein Element des mitochondrialen Pyruvatdehydrogenase-Komplexes (PDC). Der PDC besteht aus dem Enzym Pyruvatdehydrogenase (PDH) und seinen regulatorischen Proteinen: PDK, das durch Phosphorylierung der PDH als Inhibitor wirkt, und Pyruvatdehydrogenase-Phosphatase, die die PDH durch Dephosphorylierung aktiviert [33]. Die dephosphorylierte PDH katalysiert die oxidative Decarboxylierung von Pyruvat zu Acetyl-CoA,CO2 und NADH (H+ ), wodurch der glykolytische Weg mit dem oxidativen Weg des Tricarbonsäurezyklus verbunden wird (Abbildung 1) [33]. Es gibt vier Isoformen der PDK (1, 2, 3, 4), die sich in ihrer intrinsischen Aktivität, ihrer Gewebeverteilung und ihrer Empfindlichkeit gegenüber dem selektiven Inhibitor Dichloracetat (DCA) unterscheiden [33-35]. Obwohl PDK1 als einzige in der Lage ist, an alle PDH-Phosphorylierungsstellen zu binden, scheint PDK3 die aktivste Isoform zu sein [33]. PDK2 reagiert am empfindlichsten auf die Hemmung durch DCA und wird ubiquitär in verschiedenen menschlichen Geweben exprimiert, während die anderen Isoformen eher gewebespezifisch sind [33,34]. PDK4 ist im Wesentlichen mit der physiologischen metabolischen Flexibilität verbunden und ist die einzige Isoform, die nicht durch HIF1-α hochreguliert wird [32,36]. Die Expression der PDK-Isoformen wurde bei verschiedenen Krebsarten untersucht, insbesondere bei Plattenepithelkarzinomen des Kopfes und Halses (HNSCC) [37]dickdarmkrebs [38]nierenzellkarzinom [39] und Magenkrebs [40].
DCA wurde zur Behandlung von Erkrankungen eingesetzt, die mit Laktatazidose im Zusammenhang mit mitochondrialer Dysfunktion einhergehen, wie z. B. angeborene mitochondriale Erkrankungen [35]. Bonnet et al. berichteten, dass DCA bei nicht-kleinzelligen Lungen-, Glioblastom- und Brustkrebszellen die Apoptose auslöst und das Zellwachstum verringert, indem es die Glukoseoxidation, die Depolarisierung der Mitochondrienmembran und die ROS-Produktion fördert [41]. Diese Wirkungen wurden in verschiedenen Krebsmodellen wiederholt und scheinen bei Krebszellen selektiv zu sein [35,41,42]. Darüber hinaus ist DCA auch in der Lage, die Angiogenese durch indirekte HIF1-α-Hemmung zu unterdrücken [43]. All diese Daten in Verbindung mit dem bekannten Sicherheitsprofil beim Menschen belegen, dass DCA ein vielversprechendes Medikament für die Krebstherapie ist. Eine klinische Studie zur Bewertung der Wirkung von DCA bei Glioblastom-Patienten wurde bereits durchgeführt
Das kutane Melanom (CM) ist ein sehr aggressives Malignom, und obwohl es die am wenigsten verbreitete Form von Hautkrebs ist, ist es für die Mehrzahl der hautkrebsbedingten Todesfälle verantwortlich. Da die Inzidenz des CM steigt, ist es derzeit die wahrscheinlichste invasive Krebserkrankung, die vor dem 50 [1,2]. Die Exposition gegenüber UV-Strahlung gilt als Hauptrisikofaktor für die Melanomagenese [2]. CM kann in verschiedene histologische Subtypen eingeteilt werden, wobei das oberflächlich streuende Melanom (SSM) am häufigsten vorkommt, gefolgt vom nodulären Melanom (NM), dem Lentigo maligna Melanom (LMM) und dem akralen lentiginösen Melanom (ALM). SSM und NM entstehen in der Haut mit intermittierender Sonnenexposition, während LMM in chronisch sonnengeschädigter Haut auftritt und ALM auf Haut ohne Sonnenexposition beschränkt ist. Diese histologische Klassifizierung hat keinen prognostischen Wert [3,4]. Bei der Stadieneinteilung von CM werden Tumordicke, Ulzeration, Mitoserate, Knotenbefall und das Vorhandensein von Metastasen berücksichtigt [5]. Glücklicherweise werden die meisten Fälle von CM in einem frühen Stadium diagnostiziert, wobei die 5-Jahres-Überlebensrate 98 % beträgt [1]. Bei Patienten mit metastasiertem Melanom beträgt die mediane Überlebenszeit jedoch nur 8 bis 9 Monate [5]. CM ist ein sehr heterogener Tumor und viele Zellsignalwege sind bei der Melanomagenese dereguliert [6,7]. Der MAPK-Signalweg ist in der Mehrzahl der CM konstitutiv aktiviert. NRAS-Mutationen wurden bei 10 bis 20 % der CMs festgestellt, wobei NRASQ61K/R die häufigste Mutation ist. BRAF-Mutationen, insbesondere die BRAFV600E-Mutation, wurden in 40 bis 60 % der Fälle festgestellt. Unsere Gruppe beobachtete einen Zusammenhang zwischen dem Vorhandensein von BRAF-Mutationen und den kürzlich beschriebenen Mutationen des TERT-Promotors bei CM [8]. BRAFV600E-Mutationen und in geringerem Maße NRASQ61K/R-Mutationen wurden auch in fast 80 % der Nävi (gutartige melanozytäre Läsionen, die in 25 % der Fälle als Vorläufer von CM angesehen werden) festgestellt, was darauf hindeutet, dass die Aktivierung des MAPK-Signalwegs für die Melanomentwicklung zwar notwendig, aber nicht ausreichend ist [7,9,10]. Tatsächlich wurde BRAFV600E mit onkogen-induzierter Seneszenz in Verbindung gebracht und kann daher bei vielen Nävi zu einem Wachstumsstillstand führen [11]. Die Aktivierung des PI3K-AKTmTOR-Signalwegs kann diesen Seneszenz-Phänotyp überwinden und die Tumorprogression fördern [12]. Unsere Gruppe hat die Bedeutung des PI3K-AKT-mTOR-Signalwegs für die Aggressivität von CM bestätigt, indem sie seine Aktivierung mit dem Vorhandensein von BRAF-Mutationen und schlechteren prognostischen Merkmalen in Verbindung gebracht hat. Die Überexpression von mTOR-Signalweg-Effektoren wurde mit einer höheren Mitoserate, einem höheren Clark-Level, einer größeren Tumordicke und dem Vorhandensein von Hautulzerationen in Verbindung gebracht [13].
Vor 2011 beruhten die Behandlungsmöglichkeiten für Patienten mit fortgeschrittener CM hauptsächlich auf der konventionellen Chemotherapie, die geringe Ansprechraten und geringe Auswirkungen auf das Gesamtüberleben der Patienten hatte. Seitdem wurden fünf neue Medikamente für CM-Patienten im Stadium IV zugelassen [14]. Ipilimumab, ein Anti-CTLA4-Antikörper, zeigte deutliche Verbesserungen beim Gesamtüberleben der Patienten, war aber aufgrund seiner unerwünschten Wirkungen nicht für alle Patienten geeignet [15]. Vemurafenib und Dabrafenib, selektive Inhibitoren von BRAFV600E, und Trametinib, ein MEK-Inhibitor, zielen alle auf den MAPK-Signalweg ab und erzielten Ansprechraten von mehr als 50 %, die sogar besser waren als die von Ipilimumab. Aufgrund der Entwicklung von Resistenzen übersteigt ihre Verbesserung des OS jedoch nicht 7 bis 8 Monate [16-18]. Kürzlich wurde ein weiterer Immunmodulator, Pembrolizumab, ein Antikörper gegen den programmierten Todesrezeptor-1 (PD-1), für die Behandlung von Patienten mit inoperablem oder metastasiertem Melanom und Fortschreiten der Erkrankung nach Ipilimumab oder, bei positiver BRAFV600-Mutation, einem BRAF-Inhibitor zugelassen [19]. Klinische Studien mit Arzneimitteln, die auf den PI3K-AKT-mTOR-Signalweg abzielen, sind im Gange [4,14]dennoch sind neue therapeutische Ansätze erforderlich. Der Stoffwechsel von Krebszellen unterscheidet sich von dem normaler Zellen. In normalen Zellen kann Glukose je nachO2-Verfügbarkeit unter hypoxischen Bedingungen durch Glykolyse teilweise zu Laktat verstoffwechselt werden, oder sie kann in Gegenwart vonO2 durch mitochondriale oxidative Phosphorylierung, einem effizienteren energetischen Prozess, vollständig zuCO2 oxidiert werden. Krebszellen hingegen verstoffwechseln die meiste Glukose zu Laktat, unabhängig von ihrerO2-Versorgung, dem so genannten Warburg-Effekt [20,21]. Die Entstehung dieses glykolytischen Phänotyps in Krebszellen ist noch nicht vollständig geklärt, jedoch wurde eine wesentliche Rolle für den Transkriptionsfaktor Hypoxie-induzierbarer Faktor 1a (HIF1-α) erkannt [22]. Es ist bekannt, dass das HIF1-α-Protein unter Hypoxie stabilisiert wird, aber auch onkogene Signalwege, wie der MAPK- und der mTOR-Signalweg, scheinen seine Aktivierung bei Krebs zu vermitteln (Abbildung 1) [22,23]. Vieles deutet darauf hin, dass CM diesen glykolytischen Phänotyp annimmt, was durch FDGPET-Scans von Patienten bestätigt werden kann [24-27]. Tatsächlich scheint HIF1-α bei CM überexprimiert zu sein, da die Haut eine mild-hypoxische Umgebung darstellt und die Produktion von Melanin die HIF1-α-Expression durch die ROS-Produktion indirekt stimuliert [28-30]. Allerdings muss die zytoplasmatische Glykolyse von der oxidativen Phosphorylierung in den Mitochondrien abgekoppelt werden, damit der größte Teil des Pyruvats in Laktat umgewandelt werden kann. Dieser letzte Prozess wird durch die Pyruvat-Dehydrogenase-Kinase (PDK) angetrieben, ein Enzym, das durch HIF1-α hochreguliert wird (Abbildung 1) [31,32]. PDK ist ein Element des mitochondrialen Pyruvatdehydrogenase-Komplexes (PDC). Der PDC besteht aus dem Enzym Pyruvatdehydrogenase (PDH) und seinen regulatorischen Proteinen: PDK, das durch Phosphorylierung der PDH als Inhibitor wirkt, und Pyruvatdehydrogenase-Phosphatase, die die PDH durch Dephosphorylierung aktiviert [33]. Die dephosphorylierte PDH katalysiert die oxidative Decarboxylierung von Pyruvat zu Acetyl-CoA,CO2 und NADH (H+ ), wodurch der glykolytische Weg mit dem oxidativen Weg des Tricarbonsäurezyklus verbunden wird (Abbildung 1) [33]. Es gibt vier Isoformen der PDK (1, 2, 3, 4), die sich in ihrer intrinsischen Aktivität, ihrer Gewebeverteilung und ihrer Empfindlichkeit gegenüber dem selektiven Inhibitor Dichloracetat (DCA) unterscheiden [33-35]. Obwohl PDK1 die einzige Isoform ist, die an alle PDH-Phosphorylierungsstellen binden kann, scheint PDK3 die aktivste Isoform zu sein [33]. PDK2 reagiert am empfindlichsten auf die Hemmung durch DCA und wird ubiquitär in verschiedenen menschlichen Geweben exprimiert, während die anderen Isoformen eher gewebespezifisch sind [33,34]. PDK4 ist im Wesentlichen mit der physiologischen metabolischen Flexibilität verbunden und ist die einzige Isoform, die nicht durch HIF1-α hochreguliert wird [32,36]. Die Expression der PDK-Isoformen wurde bei verschiedenen Krebsarten untersucht, insbesondere bei Plattenepithelkarzinomen des Kopfes und Halses (HNSCC) [37]dickdarmkrebs [38]nierenzellkarzinom [39] und Magenkrebs [40]. DCA wurde zur Behandlung von Erkrankungen eingesetzt, die mit Laktatazidose im Zusammenhang mit mitochondrialer Dysfunktion einhergehen, wie z. B. angeborene mitochondriale Erkrankungen [35]. Bonnet et al. berichteten, dass DCA bei nicht-kleinzelligen Lungen-, Glioblastom- und Brustkrebszellen die Apoptose auslöst und das Zellwachstum verringert, indem es die Glukoseoxidation, die Depolarisierung der Mitochondrienmembran und die ROS-Produktion fördert [41]. Diese Wirkungen wurden in verschiedenen Krebsmodellen wiederholt und scheinen in Krebszellen selektiv zu sein [35,41,42]. Darüber hinaus ist DCA auch in der Lage, die Angiogenese durch indirekte Hemmung von HIF1-α zu unterdrücken [43]. All diese Daten in Verbindung mit dem bekannten Sicherheitsprofil beim Menschen belegen, dass DCA ein vielversprechendes Medikament für die Krebstherapie ist. Es wurde bereits eine klinische Studie zur Bewertung der Wirkung von DCA bei Glioblastom-Patienten durchgeführt, die bei vier von fünf Patienten vielversprechende Ergebnisse hinsichtlich der Steigerung der Apoptose und der Verringerung der Angiogenese zeigte [44].
In der vorliegenden Studie wurde die Expression der PDK 1 – 3 Isoformen in CM untersucht und ihr Zusammenhang mit der Expression der wichtigsten Signalwegkaskaden für die Melanomagenese und mit der Prognose von Melanompatienten bewertet. Außerdem wurden In-vitro-Studien durchgeführt, um die Auswirkungen der DCA-Behandlung auf den Stoffwechsel, die Proliferation und das Überleben von Melanomzelllinien mit unterschiedlichen genetischen Profilen zu bewerten.
Materialien und Methoden
Auswahl der Proben, klinisch-pathologische und prognostische Parameter
Formalin-fixierte, in Paraffin eingebettete Gewebe von 120 CM-Fällen und von 22 melanozytären Nävi (12 Compound-Nävi und 10 Spitz-Nävi) wurden vom Dienst für anatomische Pathologie des Krankenhauses S. Joa˜o, Porto, und des Krankenhauses S. Marcos, Braga, entnommen. Klinisch-pathologische (Tabelle 1) und Follow-up-Daten wurden aus den Patientenakten und den Onkologieregistern des Krankenhauses S. Joa˜o und des Krankenhauses S. Marcos sowie aus dem RORENO (Onkologieregister der Region Nord) entnommen. Alle Fälle wurden gemäß der 7. Auflage des AJCC überarbeitet und eingestuft [5]. Die Nachbeobachtungsdaten umfassen die Rezidive und Metastasen (DFS; n = 108) und die Anzahl der Todesfälle aufgrund des Melanoms (krankheitsspezifische Mortalität; OS; n = 118). Die mittlere Nachbeobachtungszeit der Patienten für das DFS betrug 51 Monate (SE ± 3,59, Spanne 1-195) und für das OS 55 Monate (SE ± 3,48, Spanne 1 – 207). Diese Arbeit wurde von der lokalen Ethik-Kommission genehmigt und stand im Einklang mit den nationalen ethischen Regeln.
Immunhistochemische Analyse
Die Färbung der untersuchten Proteine erfolgte an 3µm-Paraffinschnitten repräsentativer Tumorbereiche, die auf Poly-L-Lysin-beschichtete Objektträger aufgezogen wurden. Die Schnitte wurden deparaffiniert und rehydriert, gefolgt von einem Mikrowellen-Antigen-Retrieval-Verfahren mit 10 mM Natriumcitratpuffer pH 6,0 mit 1 mM EDTA pH 9,0 (PDK2) bzw. EDTA-Puffer pH 9,0 (PDK1 und PDK3). Die Schnitte wurden über Nacht bei 4 °C in einer befeuchteten Kammer mit den primären Antikörpern PDK1 (polyklonal, Kaninchen, 1:50), PDK2 (polyklonal, Kaninchen, 1:150), PDK3 (monoklonal, Maus, 1:500), alle von Sigma-Aldrich Co. (St. Louis, Missouri, USA), inkubiert. Der Nachweis erfolgte mit dem Envision G/2 System/AP (K5355; Dako, Dänemark). alkalische Phosphatase-Anti-Alkalische-Phosphatase-Methode (APAAP), und die Farbe wurde mit permanentem roten Chromogen entwickelt. Die Objektträger wurden mit Hämatoxylin gegengefärbt und anschließend mit einem wassermischbaren Einbettungsmedium aufgezogen. Die APAAP-Methode wurde angewandt, um zu vermeiden, dass die Melaninpigmentierung die immunhistochemische Analyse beeinträchtigt. Menschliches Magengewebe wurde als Negativ- (Weglassen des Primärantikörpers) und Positivkontrolle verwendet.
Immunhistochemische Auswertung
Drei Beobachter (J.M.L., H.P. und R.C.) bewerteten die Immunreaktivität der Tumorzellen ohne Kenntnis der klinischen Daten der Fälle. Die Immunreaktivität des angrenzenden Nicht-Tumorgewebes wurde als interne Kontrolle verwendet. Für PDK1 – 3 wurde ein IHC-Score festgelegt, der sich aus der Multiplikation des Intensitätsscores der Färbung (negativ = 0, schwach = 1, mäßig = 2 und stark = 3) und des Ausdehnungsscores der Immunreaktivität der Tumorzellen (0 – 5% = 0, 6 – 25% = 1, 26 – 50% = 2, 51 – 75% = 3, 76 – 100% = 4) ergibt. Die IHC-Scores wurden dann als negativ/gering (Score-Wert £ 4) und mäßig/hoch (Score-Wert > 4) eingestuft. Analysen der MAPK- und der PI3KAKT-mTOR-Kaskaden in einem Teil der Serie wurden bereits früher durchgeführt [13] und die Ergebnisse wurden in dieser Studie verwendet.
DNA-Extraktion und Mutationsanalyse
Die DNA-Extraktion aus Tumoren, die kleiner als 5 mm sind, erfolgte nach Mikrodissektion mit PALM MicroLaser Systems (PALM, Deutschland) und unter Verwendung des Quiamp DNA micro kit (Quiagen, Hilden). Bei Tumoren, die größer als 5 mm waren, erfolgte die DNA-Extraktion durch manuelle Präparation von 10 µm großen Schnitten aus in Paraffin eingebettetem Gewebe mit dem Invisorb spin tissue mini kit (Invitek, Berlin). BRAF-Exon 15- und NRAS-Exon 2-Fragmente wurden durch Polymerase-Kettenreaktion (PCR) mit zuvor beschriebenen Primern amplifiziert [45]. Genomische DNA (25 – 100 ng) wurde mittels PCR unter den folgenden Bedingungen amplifiziert: 35 s bei 94°C, 40 s bei 58°C für BRAF und 57°C für NRAS, und 45 s bei 72°C für 40 Zyklen.
| Klinisch-pathologische Merkmale | |
| Anzahl der Fälle (n) | 120 |
| Medianes Alter (± SD) | 61.5 ± 17.0 |
| Geschlecht (n [%]) | |
| Weiblich | 68 (56.7) |
| Männlich | 52 (43.3) |
| Sonnenexposition (n [%]) | |
| Abwesend | 27 (22.7) |
| Intermittierend | 72 (60.5) |
| Chronisch | 20 (16.8) |
| Histologischer Subtyp (n [%]) | |
| LMM | 16 (13.3 |
| ALM | 24 (20.0) |
| NM | 19 (15.8) |
| SSM | 61 (50.8) |
| Mediane Dicke (mm) | 3.7 (0 — 70) |
| Epidermisgeschwüre (n [%]) | |
| Abwesend | 79 (65.8) |
| Vorhanden | 41 (34.2) |
| Mitoserate (n [%]) | |
| < 1/mm2 | 45 (37.5) |
| ≥1/mm2 | 75 (62.5 |
| pT (n [%]) | |
| ≤ pT2 | 62 (51.7) |
| >pT2 | 58 (48.3) |
Alle PCR-Produkte wurden gereinigt und direkt auf einem ABI Prism 3130 xl Automatic sequencer (Perkin-Elmer, Foster City, CA) mit dem ABI Prism Dye Terminator Cycle sequencing Kit (Perkin-Elmer) sequenziert. Die Sequenzierungsreaktion wurde in Vorwärtsrichtung durchgeführt, und bei mutationsverdächtigen Proben wurde eine unabhängige PCR-Amplifikation in Vorwärts- und Rückwärtsrichtung durchgeführt. Auch hier wurde die BRAF- und NRAS-Mutationsanalyse in einem Teil der Serie bereits durchgeführt [13] und die Ergebnisse wurden in dieser Studie verwendet.
Zelllinien und Kulturbedingungen
In dieser Arbeit wurden zwei Zelllinien verwendet, die A375 Hautmelanom-Zelllinie, die BRAFV600E trägt, und die Mewo Hautmelanom-Zelllinie, die BRAFwt trägt. Beide Zelllinien wurden auf das Vorhandensein von Mykoplasmen getestet.
A375 wurde in RPMI-Medium (Gibco/BRL – Invitrogen) und Mewo in DMEM-Medium (Gibco/BRL – Invitrogen) gehalten. Alle Medien wurden mit 10% fötalem Rinderserum, 100 U/ml Penicillin und 100 µg/ml Streptomycin ergänzt. Die Zelllinien wurden in einer befeuchteten Atmosphäre (5%CO2) bei 37°C gehalten.
Behandlung von Melanomzelllinien mit DCA
Natrium-DCA, erworben von Sigma-Aldrich (St. Louis, MO, EUA), wurde in dH2O aufgelöst und dem Kulturmedium zugegeben und für die 24- und 48-stündige Behandlung verwendet. Melanomzellen, die mit einem mit dH2O ergänzten Nährmedium bebrütet wurden, dienten als Kontrolle.
Untersuchung der Zelllebensfähigkeit
Die Auswirkungen von DCA auf das Wachstum von Melanomzelllinien wurden mit dem PrestoBlue (PB)-Test analysiert. Die Zellen wurden in 96-Well-Platten in einer Dichte von 5 103 in 200 µl Medium ausgesät. Nach 24 Stunden wurde das Medium durch ein Medium mit 5, 20, 40 und 60 mM DCA ersetzt. Die Zellen wurden 24 und 48 Stunden lang inkubiert, mit PBS (pH 7,4) gewaschen und mit PB gemäß den Anweisungen des Herstellers auf Zellwachstum getestet. Während der Inkubation mit den Zellen wird das PB-Reagenz durch die reduzierende Umgebung der lebensfähigen Zellen modifiziert und fluoresziert stark. Die Fluoreszenz wurde mit einem Mikroplattenlesegerät (Synergy HT Multi-Mode Microplate Reader, BioTek Instruments Inc., Winooski, VT, USA) bei Anregungs- und Emissionswellenlängen von 560 bzw. 590 nm gemessen. Die Absorption der Vertiefungen, die das Kulturmedium und die Tumorzellen enthielten, diente als Kontrolle, und jede Versuchsbedingung wurde in dreifacher Ausfertigung ausgewertet und in zweifacher Ausfertigung wiederholt. Durch den Vergleich der gemessenen Fluoreszenz/Absorption der DCA-haltigen Vertiefungen mit den Messungen der Vertiefungen, die unbehandelte Zellen enthielten, konnten mit GraphPadPrism5.0 (GraphPad Software, Inc., La Jolla, CA) Dosis-Wirkungs-Profile erstellt und IC50-Werte (die Konzentration, die das Überleben zu 50 % hemmt) bestimmt werden.
Quantifizierung von Glukose und Laktat
Für die Messung des Glukose- und Laktatspiegels wurden Melanomzellen in 6-Well-Platten mit einer Enddichte von 2 105 Zellen/Well plattiert und 24 Stunden lang bei 37 °C inkubiert. Die Zellen wurden dann mit 35 mM DCA behandelt. Zur Kontrolle wurden die Zellen mit der Vehikelverbindung (dH2O) inkubiert. Nach 24 und 48 Stunden Behandlung wurde das Kulturmedium abgenommen. Der Glukosegehalt im konditionierten Kulturmedium wurde mit dem Glucose GOD/PAP Kit (Roche Applied Sciences) quantifiziert und von den Ausgangswerten (0 h) abgezogen. Laktat wurde auf ähnliche Weise mit dem enzymatischen kolorimetrischen LO-POD-Assay (Spinreact, Sant Esteve de Bas, Spanien) quantifiziert.
Zellzyklus- und Apoptose-Analyse
Für die Zellzyklus- und Apoptose-Analyse wurden Melanomzellen in 6-Well-Platten mit einer Enddichte von 1 105 Zellen/Well ausplattiert und 24 Stunden lang bei 37 °C inkubiert. Die Zellen wurden dann 24 und 48 Stunden lang mit 35 mM DCA behandelt. Als Kontrollen wurden die Zellen mit der Vehikelverbindung (dH2O) inkubiert. Für die Zellzyklusanalyse wurden die Zellen dann geerntet und über Nacht in eiskaltem 70%igem Ethanol fixiert. Danach wurden die Zellen vor der Analyse in PBS mit 0,1 mg/ml RNase A und 5 µg/ml Propidiumiodid resuspendiert. Für die Apoptosemessungen wurden die Zellen geerntet und der Apoptosegrad mittels Durchflusszytometrie unter Verwendung des Annexin-V FITC Apoptose-Kits (Clontech Laboratories, Inc., Saint-Germainen-Laye, Frankreich) gemäß den Anweisungen des Herstellers analysiert. Die durchflusszytometrische Analyse des zellulären DNA-Gehalts und der Phosphatidylserin-Externalisierung wurde mit einem Durchflusszytometer (BD Accuri C6) durchgeführt, wobei mindestens 20.000 Ereignisse pro Probe aufgezeichnet wurden. Die Daten wurden mit der Software FlowJo 7.6.5 (Tree Star, Inc., Ashland, USA) ausgewertet.
Western-Blot-Analyse und Antikörper
Die Zellen wurden 15 Minuten lang bei 4 °C mit RIPA-Puffer (1% NP-40 in 150 mM NaCl, 50 mM Tris (pH 7,5), 2 mM EDTA), der Phosphatase- und Proteaseinhibitoren enthält, lysiert. Die Proteine wurden mit einem modifizierten Bradford-Assay (Biorad) quantifiziert. Die Proteinproben (50 µg) wurden in 10%igen SDS/ PAGE-Gelen aufgetrennt und auf eine Hybond ECL-Membran (Amersham Biosciences) geblottet. Wir verwendeten die folgenden primären Antikörper: PDH und pPDH Ser293, von Abcam; PDK1 (Sigma-Aldrich), PDK2 (Sigma-Aldrich), HIF1-α (Transduction Laboratories) und die mTOR-Weg-Effektoren pS6 Ser235/236, s6, p-4EBP1 Thr37/46, 4EBP1, alle von Cell Signaling Technology. Die sekundären Antikörper wurden mit Peroxidase (Santa Cruz Biotechnology) konjugiert und mit der ECL-Detektionslösung sichtbar gemacht. Die Membranen wurden mit einem polyklonalen Ziegen-Antiactin-Antikörper (Santa Cruz Biotechnology) als Protein-Ladekontrolle nachgefärbt. Alle Experimente und Quantifizierungen (mit der Bio-Rad Quantity One 1-D Analysis Software (Version 4.6.6)) wurden in dreifacher Ausführung durchgeführt.
Statistische Analyse
Die statistische Analyse wurde mit STAT VIEW-J 5.0 (SAS Institute, Inc., Cary, NC) durchgeführt. Die Beziehung zwischen dem durchschnittlichen Expressionsniveau (Score) der immunhistochemischen Marker und den klinisch-pathologischen Parametern wurde mittels ANOVA bewertet. Gegebenenfalls wurden Korrekturen für Mehrfachvergleiche mit den Post-hoc-Tests von Bonferroni und Tamhane durchgeführt. Die Korrelation zwischen den Immunreaktivitätswerten der verschiedenen Marker wurde mit dem exakten Test von Fisher bewertet. Die Daten aus den Zelllinienexperimenten wurden mit dem zweiseitigen ungepaarten Student’s t-Test analysiert. Zur Auswertung der Melanom-Überlebensdaten wurden die Kaplan-Meier-Methode und der Log-rank-Test verwendet. Univariate und multivariate Analysen wurden durchgeführt, um den prognostischen Wert von Kovariaten in Bezug auf OS und DFS mithilfe des Cox-Regressionsmodells zu bestimmen. OS und DFS wurden vom Zeitpunkt der Diagnose bis zum Tod aufgrund der Erkrankung bzw. der Metastasierung berechnet oder zum Zeitpunkt der letzten Nachuntersuchung bzw. des Todes ohne Zusammenhang mit der Erkrankung zensiert. Ein p-Wert < 0,05 wurde als statistisch signifikant angesehen.
Ergebnisse
Expression von PDKs in CM und Nävi
Sowohl in CM als auch in Nävi-Läsionen wurden die PDK-Isoformen 1, 2 und 3 nicht nur in Melanozyten/Melanomzellen, sondern auch in Keratinozyten und in den Zellen der Talgdrüsen und Haarfollikel exprimiert. Die Expressionsniveaus aller untersuchten PDK-Isoformen waren untereinander positiv korreliert, was auf eine gleichzeitige Expression dieser Proteine schließen lässt (Tabelle 2).
| Protein | Mittleres Expressionsniveau (± SD) | Korrelation (p-Wert) | Korrelation (p-Wert) | Korrelation (p-Wert) |
| PDK1 | 4.8 ± 3.5 | < 0.01* | < 0.01§ | |
| PDK2 | 4.6 ± 3.6 | < 0.01† | ||
| PDK3 | 7.0 ± 3.2 |
*Zwischen PDK1 und PDK2; † zwischen PDK2 und PDK3; § zwischen PDK1 und PDK3.
Immunhistochemische PDK1-Expression
Zytoplasmatische PDK1-Färbung wurde in 88 % der CM und 76 % der Nävi beobachtet (Abbildung 2A). Negative/schwache Färbung wurde bei 52 bzw. 76 % und mäßige/hohe Färbung bei 48 bzw. 24 % der Melanome und Nävi beobachtet. Daher wiesen CM im Durchschnitt eine signifikant höhere PDK1-Expression auf als Nävi (p = 0,03; Abbildung 2B). Was die histologischen Subtypen von CM betrifft, so wiesen LMM höhere PDK1-Expressionswerte auf als NM (p = 0,04) und ALM (p = 0,04). Es wurde kein Zusammenhang zwischen der Expression von PDK1 und der Art der Sonnenexposition festgestellt. Was die prognostischen Faktoren anbelangt, so wurde PDK1 in Tumoren mit einer Dicke von £ 1 mm stärker exprimiert (p = 0,02). Höhere PDK1-Expressionswerte waren mit niedrigeren Tumorstadien assoziiert (p = 0,04), jedoch nicht mit dem Gesamtüberleben und dem krankheitsfreien Überleben der Patienten. Wir untersuchten auch, ob die Expression von PDK-Isoformen mit dem MAPK- und dem PI3K-AKT-mTOR-Signalweg zusammenhängt. Es wurde kein Zusammenhang zwischen der Expression von PDK1 und BRAF/NRAS-Mutationen oder mit der Aktivierung des MAPK-Signalwegs (bewertet durch die pERK-Expression) festgestellt. Die PDK1-Expression war positiv korreliert mit der Expression von Proteinen der PI3K-AKT-mTOR-Kaskade, nämlich mTOR und 4EBP1 (p = 0,02 bzw. p = 0,04).
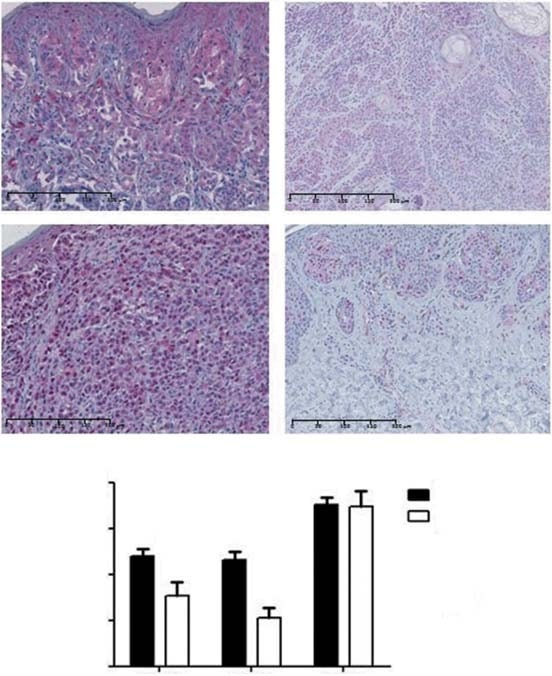
PDK2 immunhistochemische Expression
PDK2 wurde sowohl nukleär als auch zytoplasmatisch in 87 % der CM und 83 % der Nävi exprimiert (Abbildung 2A). Ein negativer/geringer Färbegrad wurde bei 58 bzw. 89 % und ein mittlerer/hoher Färbegrad bei 42 bzw. 11 % der Melanome bzw. Nävi festgestellt. Die mittlere Expressionsrate von PDK2 war bei CM höher als bei Nävi (p < 0,01; Abbildung 2B). Was die histologischen Subtypen von CM betrifft, so wiesen SSM eine höhere PDK2-Expression auf als ALM (p = 0,02). Wir konnten keinen Zusammenhang zwischen der Art der Sonnenexposition und der PDK2-Expression feststellen. In niedrigeren Tumorstadien war die PDK2-Expression signifikant höher (p = 0,02), obwohl kein Zusammenhang zwischen der Expression dieser Isoform und den wichtigsten prognostischen Faktoren von CM sowie mit dem Gesamtüberleben und dem krankheitsfreien Überleben der Patienten festgestellt wurde. Die PDK2-Expression stand weder mit dem BRAF/NRAS-Mutationsstatus noch mit der Aktivierung des MAPK-Signalwegs in Zusammenhang. Eine signifikante positive Korrelation wurde zwischen der PDK2-Expression und der AKT-Expression der PI3K-AKT-mTOR-Kaskade festgestellt (p = 0,03).
PDK3 immunhistochemische Expression
PDK3 zeigte eine zytoplasmatische Expression in allen CM und Nävi. Ein negativer/geringer Färbegrad wurde bei 28 bzw. 22 % und ein mittlerer/hoher Färbegrad bei 72 bzw. 78 % der CM und Nävi beobachtet. Der Grad der Expression der PDK3-Isoform war bei CM und Nävi ähnlich (Abbildung 2B). Beim Vergleich der mittleren PDK3-Expression zwischen CM-Subtypen und der Art der Sonnenexposition ergaben sich keine signifikanten Unterschiede. Tumore mit einer Dicke von £ 1 mm und solche mit niedrigeren Stadien wiesen höhere PDK3-Werte auf (p = 0,01 bzw. p < 0,01). Ulzeration, Mitoserate sowie Gesamtüberleben und krankheitsfreies Überleben der Patienten waren nicht signifikant mit der PDK3-Expression verbunden. Es wurde kein Zusammenhang zwischen der Expression von PDK3 und BRAF/NRAS-Mutationen oder der Aktivierung der MAPK-Kaskade festgestellt. Was den PI3K-AKT-mTOR-Signalweg betrifft, so war die PDK3-Expression positiv mit der gesamten AKT-, mTOR- und 4EBP1-Expression korreliert (p < 0,01, p < 0,01 bzw. p = 0,04). Aufgrund der allgemeinen Expression von PDK3 in allen CM und Nävi wurde diese PDK-Isoform bei der anschließenden In-vitro-Analyse nicht berücksichtigt.
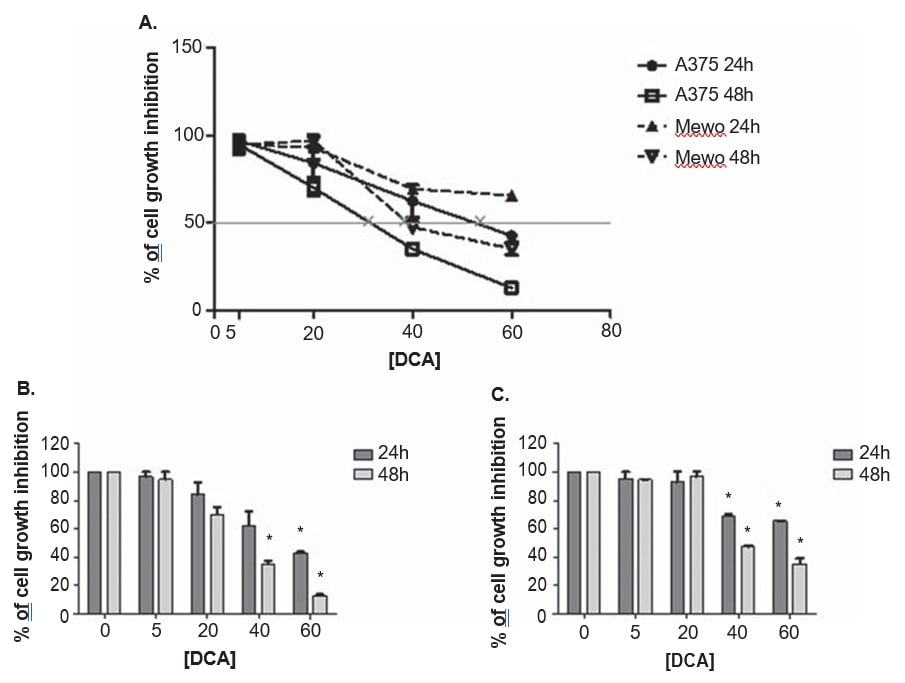
Die Auswirkung der DCA-Behandlung auf die Lebensfähigkeit der Melanom-Zelllinien
Die Melanom-Zelllinien A375 und Mewo wurden steigenden Konzentrationen von DCA ausgesetzt, um die Auswirkung auf die Lebensfähigkeit der Zellen mit Hilfe des PrestoBlue-Tests zu ermitteln. DCA verringerte die Lebensfähigkeit beider Zelllinien in dosisabhängiger Weise nach 24- und 48-stündiger Behandlung, wobei A375 etwas empfindlicher auf die DCA-Behandlung reagierte und eine höhere Wachstumshemmung aufwies als die Mewo-Zelllinie (Abbildung 3). Die Wirkung von DCA auf die Lebensfähigkeit der A375-Zellen wurde nach einer Behandlung mit 5 mM beobachtet und war bei der Mewo-Zelllinie nach einer Behandlung mit 20 mM intensiver, wobei die Hemmungswerte zwischen 3 und 87 % bei A375 und 5 bis 65 % bei Mewo lagen. Die IC50-Werte wurden auf 33 ± 5,5 mM für A375 und 39,9 ± 2,2 mM für die Mewo-Zelllinie nach 48 Stunden DCA-Behandlung geschätzt (Abbildung 3).
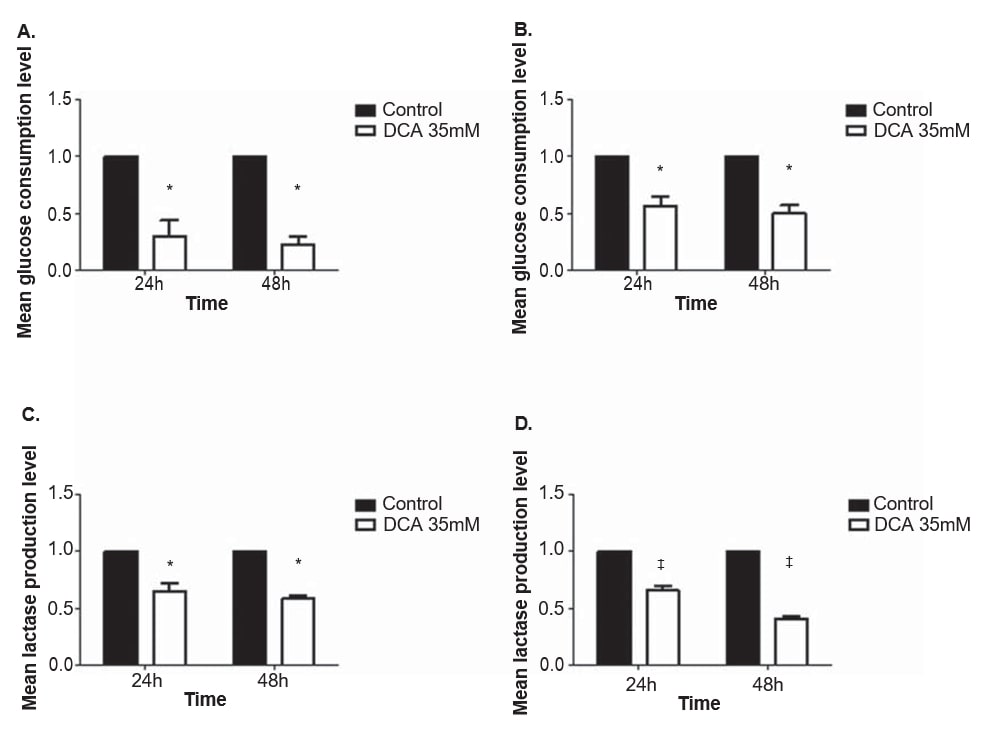
Auswirkung der DCA-Behandlung auf den Glukoseverbrauch und die Laktatproduktion von Melanomzelllinien
Um die Auswirkung von DCA auf den Stoffwechsel von Melanomzelllinien zu bewerten, wurden die Glukose- und Laktatwerte im Kulturmedium nach 24- und 48-stündiger Behandlung mit 35 mM DCA quantifiziert, einer Konzentration nahe der IC50 für beide Zelllinien. Der Verbrauch von Glukose und die Produktion von Laktat nahmen bei beiden Melanomzelllinien nach der Behandlung mit DCA ab (Abbildung 4). Die Zelllinien A375 und Mewo zeigten sowohl nach 24 als auch nach 48 Stunden DCA-Behandlung einen signifikanten Rückgang des Glukoseverbrauchs (p = 0,01 bzw. p < 0,01). Auch die Laktatproduktion war nach 24 und 48 Stunden in den Zelllinien A375 (p = 0,01 bzw. p < 0,01) und Mewo (p < 0,01) signifikant verringert.
Wirkung der DCA-Behandlung auf die PDK- und mTOR-Weg-Effektoren sowie auf die HIF1-α-Expression in Melanom-Zelllinien
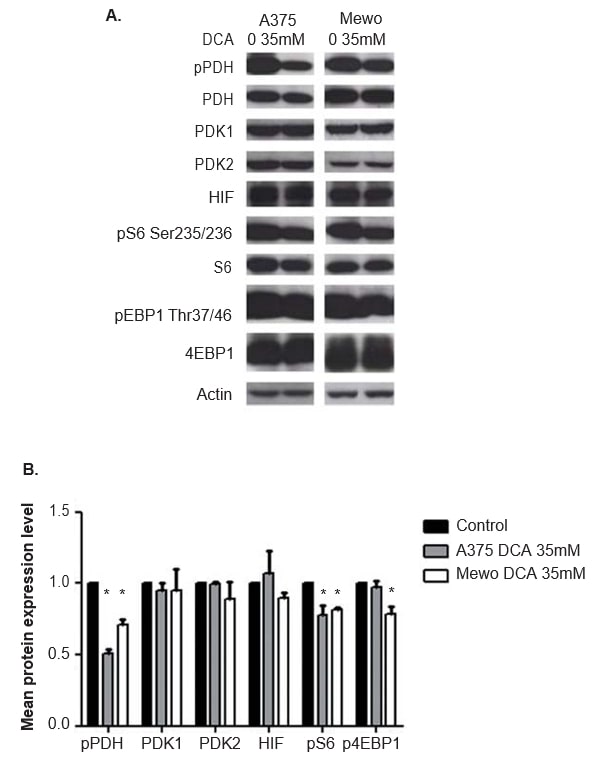
Die Wirksamkeit der DCA-Behandlung bei der Hemmung der PDK-Aktivität wurde durch Analyse der Expression des phosphorylierten nachgeschalteten Effektors PDH mittels Western Blot bewertet. 24 Stunden nach der DCA-Behandlung wurde eine signifikante Hemmung der Phosphorylierung von PDH in beiden Zelllinien beobachtet (p < 0,01; Abbildung 5). Die Expression von PDK1 und PDK2 wurde durch die DCA-Behandlung erwartungsgemäß nicht verändert (Abbildung 5). Die Expression der beiden nachgeschalteten Effektoren und Indikatoren für die Aktivierung des mTOR-Weges, S6 und 4EBP1, wurde ebenfalls untersucht. Obwohl die DCA-Behandlung die Expression von S6 und 4EBP1 nicht veränderte, war das Expressionsniveau der phosphorylierten Formen beider Proteine in A375 (p = 0,03 bzw. nicht signifikant) und in Mewo (p < 0,01 bzw. p = 0,01; Abbildung 5) verringert. Da DCA die HIF1-α-Konzentration beeinflussen kann, wurde auch die Expression dieses Proteins untersucht, und es wurde keine Veränderung nach der DCA-Behandlung beobachtet (Abbildung 5). Ähnliche Ergebnisse wurden 48 Stunden nach der DCA-Behandlung erzielt, die die Phosphorylierung von PDH in beiden Zelllinien signifikant hemmt (p <0,01). Die Expression der phosphorylierten Formen von S6 und 4EBP1 war in A375 (p = 0,01 bzw. p = 0,02) und in Mewo (p = 0,01 bzw. nicht signifikant) deutlich verringert. Die Expression von PDK1, PDK2 und HIF1-α war nicht verändert (Daten nicht gezeigt).
Auswirkung der DCA-Behandlung auf den Zellzyklus und die Apoptose von Melanomzelllinien
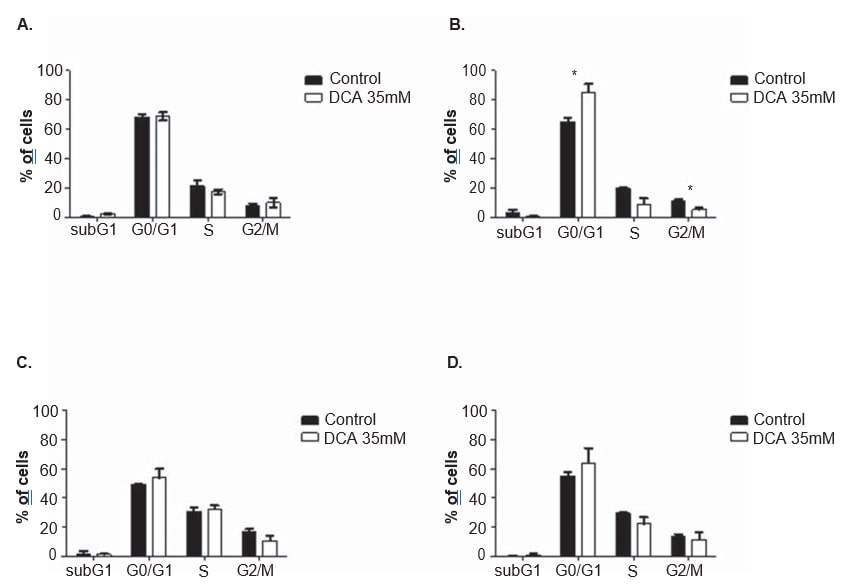
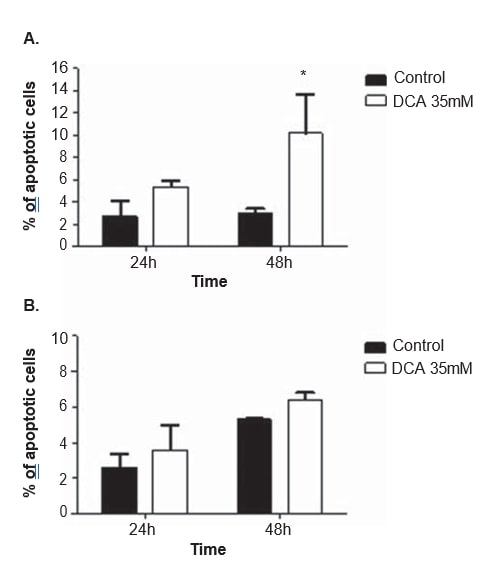
Um den Wirkmechanismus von DCA zu klären, wurden bei beiden Zelllinien nach 24 und 48 Stunden Behandlung mit 35 mM DCA Zellzyklusanalysen und Apoptosemessungen durchgeführt. Der Zellzyklus wurde mit Propidiumjodid analysiert. Nach 48 Stunden DCA-Behandlung zeigte die A375-Zelllinie einen signifikanten Anstieg des Prozentsatzes der Zellen in der G0/G1-Phase des Zellzyklus (von 65,0 ± 4,9 % in den nicht behandelten Zellen auf 85,2 ± 10,2 % in den DCA-behandelten Zellen; p = 0,04), einen signifikanten Rückgang in G2/M (von 11.3 ± 1,3 % in unbehandelten Zellen auf 5,2 ± 2,2 % in DCA-behandelten Zellen; p = 0,01) und eine Abnahme in der S-Phase (von 19,7 ± 1,8 % in unbehandelten Zellen auf 9,1 ± 6,7 % in DCA-behandelten Zellen; p = 0,06; Abbildung 6), die jedoch nicht die Schwelle der statistischen Signifikanz erreicht. Die gleiche Tendenz wurde bei der Mewo-Zelllinie 48 Stunden nach der DCA-Behandlung beobachtet (Zunahme des Prozentsatzes der Zellen in G0/G1 und Abnahme der S- und G2/M-Phase), obwohl sie keine statistische Signifikanz erreichte (Abbildung 6). Nach der Annexin V/Propidiumjodid-Färbung wurde eine Durchflusszytometrie-Analyse durchgeführt. Es wurde ein signifikanter Anstieg der Zahl der apoptotischen Zellen in der A375-Zelllinie nach 48 Stunden DCA-Behandlung beobachtet (3,0 ± 0,5 % in nicht behandelten Zellen bis 16,9 ± 4,6 % in DCA-behandelten Zellen; p < 0,01; Abbildung 7). Ein nicht signifikanter Anstieg des Prozentsatzes apoptotischer Zellen wurde in der A375-Zelllinie nach 24 Stunden und in der Mewo-Zelllinie nach 24- und 48-stündiger Behandlung mit DCA beobachtet.
Diskussion
In dieser Arbeit haben wir zum ersten Mal festgestellt, dass CM die Proteine PDK1 und PDK2 überexprimiert. PDKs sind Schlüsselproteine, die durch HIF1-α reguliert werden und bösartige Zellen in den Phänotyp der aeroben Glykolyse treiben, ein Markenzeichen von Krebs [20,22,31].
Der Warburg-Effekt gilt als ein Resistenzfaktor gegenüber herkömmlichen Chemotherapien [21]. Die in CM festgestellte Überexpression von PDK veranlasste uns, die Wirkung einer DCA-Behandlung in Melanomzelllinien zu untersuchen. Wir stellten fest, dass die DCA-Behandlung den Glukoseverbrauch und die Laktatproduktion in Melanomzelllinien senkt, was mit der Wirkung von DCA auf die Verlagerung von aerober Glykolyse zu oxidativer Phosphorylierung übereinstimmt, die zuvor in Krebszellen berichtet wurde [41]. Diese Verlagerung des Glukosestoffwechsels ist wahrscheinlich auf den beobachteten signifikanten Rückgang der Expression der phosphorylierten Form der PDH zurückzuführen, was ebenfalls die Wirksamkeit der DCA-Behandlung bestätigt. Es ist wichtig, darauf hinzuweisen, dass die korrekte Messgröße für die Wirkung von DCA der Gehalt an phosphorylierter PDH ist, da DCA durch die Hemmung der PDK-Aktivität und nicht der PDH-Expression die Dephosphorylierung von PDH stimuliert (Abbildung 1).
Wir beobachteten eine dosisabhängige Abnahme der Zelllebensfähigkeit nach DCA-Behandlung bei beiden Melanom-Zelllinien. Außerdem wurde bei beiden Zelllinien eine Herabregulierung der Proliferation durch G0/G1-Arrest sowie eine Zunahme der Apoptose beobachtet, die jedoch nur bei der Zelllinie A375 statistisch signifikant war. Die Tatsache, dass die in der vorliegenden Studie gewählte DCA-Konzentration (35 mM) geringfügig unter der IC50 für die Mewo-Zelllinie liegt, könnte die weniger ausgeprägte Wirkung erklären, die bei diesen Zellen beobachtet wurde.
Die Beziehung zwischen DCA und HIF-1a bei Krebs ist nicht vollständig geklärt, da DCA den Sauerstoffverbrauch und die ROS-Produktion im Gewebe erhöht, was zu einer Hochregulierung bzw. Stabilisierung von HIF-1a führen sollte [46,47]. Umgekehrt berichteten einige Autoren, dass DCA zu einem Rückgang der HIF1-α-Expression führt [25,48,49]. In unserer Arbeit wurde keine Veränderung des HIF1-α-Spiegels nach DCA-Behandlung von Melanomzelllinien beobachtet, was mit Shahrzad et al. übereinstimmt, die berichteten, dass die HIF1-α-Expression nach DCA-Behandlung nur unter hypoxischen Bedingungen abnimmt [49].
Um die durch die BRAF-Mutation ausgelöste Seneszenz zu überwinden und zu einem aggressiveren Phänotyp überzugehen, aktiviert CM wahrscheinlich andere Signalkaskaden, wie den mTOR-Signalweg [12]. Dieser Weg trägt zum Warburg-Effekt bei, indem er die HIF1-α-Aktivität fördert, die wiederum die PDK-Expression erhöhen kann [23,31]. Wir haben bereits über eine vollständige Aktivierung dieses Signalwegs bei CM berichtet [13]. In dieser Studie beobachteten wir eine gleichzeitige Expression von PDK 1, 2 und 3 und der mTOR-Weg-Effektoren in CM. Die Expression der PDK-Isoformen in CM korrelierte positiv mit der Expression von mTOR sowie mit vor- und nachgeschalteten Effektoren dieses Weges: AKT und 4EBP1. Darüber hinaus beobachteten wir eine signifikante Abnahme von pS6 und p4EBP1 in den Zelllinien A375 und Mewo nach DCA-Behandlung.
Naevi haben einige genetische Veränderungen mit CM gemeinsam, da beide eine hohe Frequenz von BRAF-Mutationen aufweisen [10]. Die PDK-Expression wurde auch bei Nävi beobachtet, wenn auch in viel geringerem Ausmaß. Wir untersuchten, ob der BRAF-Mutationsstatus und die nachgeschaltete ERK-Aktivierung mit einer höheren PDK-Expression verbunden sein könnten. Wir konnten weder bei Nävi noch bei CM einen Zusammenhang feststellen. Die A375-Zelllinie weist die BRAFV600E-Mutation auf, während die Mewo-Zelllinie ein Wildtyp für dieses Gen ist, und die Reaktion beider Zelllinien auf die DCA-Behandlung war trotz unterschiedlicher Intensität ähnlich. In der Tat wurde bereits berichtet, dass der BRAF-Mutationsstatus die Empfindlichkeit gegenüber einer DCA-Behandlung nicht verändert [50]. Diese Ergebnisse stehen im Gegensatz zu unseren früheren Erkenntnissen über die Verwendung von mTOR-Signalweg-Inhibitoren bei verschiedenen Melanom-Zelllinien, bei denen wir eine höhere Empfindlichkeit gegenüber der Behandlung mit RAD001 bei den CM-Zelllinien mit BRAFV600E-Mutation festgestellt haben [51].
Die PDK-Isoformen wurden im Zytoplasma der Melanomzellen exprimiert, was die bekannte mitochondriale Funktion dieser Isoformen bestätigt [33]. Bei PDK2 wurde in der Mehrzahl der CM-Fälle eine nukleäre Expression beobachtet. Diese nukleäre Lokalisation wurde auch für PDK1 in HNSCC berichtet [37]. Soweit wir wissen, ist die Rolle der PDK im Zellkern noch nicht geklärt, aber vor kurzem wurde eine nukleäre Funktion für PDH, den nachgeschalteten PDK-Effektor, vermutet. Sutendra et al. berichteten, dass PDH an der nukleären Bildung von Acetyl-CoA beteiligt ist [52]. Als Reaktion auf die Stimulierung durch einen Wachstumsfaktor verlagern sich PDH und ihre Phosphatase von den Mitochondrien, wo sie von PDK gehemmt werden, in den Zellkern, aber ein nukleärer Ort für PDK wurde nicht identifiziert [52]. Es gibt jedoch Hinweise darauf, dass einige glykolytische Enzyme neben der Glykolyse noch andere zelluläre Funktionen erfüllen, insbesondere die Transkriptionsregulation [53]. Weitere Studien sind notwendig, um zu klären, ob PDK auch eine nukleare Funktion hat.
Beim Nierenzellkarzinom wurde berichtet, dass die PDK1-Expression mit der Tumorprogression abnimmt [39]. In Übereinstimmung damit wurde in unserer Arbeit festgestellt, dass PDK in niedrigeren Tumorstadien stärker exprimiert wird, ohne das allgemeine und das krankheitsfreie Überleben der Melanompatienten zu beeinflussen. Im Gegensatz dazu wurde die Überexpression von PDK als Marker für Tumorprogression, schlechte Prognose und Wiederauftreten bei HNSCC und Magenkrebs identifiziert [37,40,54]. Obwohl es nicht unser Ziel war, die prognostische Rolle von PDK bei CM zu untersuchen, deuten unsere Ergebnisse darauf hin, dass PDKs in CM eine aktivere Rolle bei der Melanomentwicklung als bei der Melanomprogression spielen könnten. Wir sind uns bewusst, dass unsere Serie hauptsächlich aus primären CM besteht und dass weitere Studien bei fortgeschrittenen (metastasierten) Melanomen erforderlich sind, um diese Frage zu klären.
Unser Ziel in dieser Studie war es, Biomarker für neue therapeutische Ansätze bei CM-Patienten zu untersuchen. Unsere Ergebnisse deuten auf einen potenziellen Nutzen von DCA bei der Behandlung von CM-Patienten hin, was im Einklang mit Abildgaard et al. steht, die über einen Rückgang der ATP-Werte und des Melanomwachstums unter DCA-Behandlung berichteten [50]. Diese Autoren berichteten auch über eine synergistische Kombination zwischen DCA und einem BRAF-Inhibitor, Vemurafenib [50]. Darüber hinaus zeigen unsere Ergebnisse eine gleichzeitige Expression von PDK- und mTOR-Weg-Effektoren in CM und eine Herunterregulierung der mTOR-Weg-Aktivität durch DCA-Behandlung. In Anbetracht der Ergebnisse von Hong et al., die über einen synergistischen Effekt der Kombination von DCA mit einem S6K1-Inhibitor (mTOR-Stoffwechselweg-Effektor) [55]ist es verlockend zu spekulieren, dass ein synergistischer therapeutischer Effekt bei CM durch die Kombination von DCA mit einem direkten mTOR-Inhibitor erzielt werden könnte.
Schlussfolgerung
Wir berichten zum ersten Mal über die Überexpression von PDK1 und 2 in CM im Vergleich zu Nävi. Es wurde festgestellt, dass die Expression von PDKs mit der Expression von Effektoren des mTOR-Stoffwechselweges assoziiert ist und nicht mit dem BRAF-Mutationsstatus in Zusammenhang steht. Die DCA-Behandlung führt zu einer Verschiebung des Stoffwechsels, einer Herunterregulierung der Proliferation, einer erhöhten Apoptose und einer verringerten Aktivierung des mTOR-Signalwegs in Melanomzelllinien. Aus all diesen Ergebnissen schließen wir, dass die PDK-Expression bei der Melanomentwicklung eine Rolle spielen könnte und dass ihre Hemmung durch DCA allein oder in Kombination mit direkten mTOR-Inhibitoren CM-Patienten zugute kommen könnte.
Danksagung
Wir danken allen Patienten, die an dieser Studie teilgenommen haben, sowie den Ärzten, die klinische und pathologische Daten sowie Informationen zur Nachsorge zur Verfügung gestellt haben. Wir danken Dr. Madalena Pinto vom CEQUIMED, Fakultät für Pharmazie, Universität Porto, Portugal, die uns freundlicherweise die A375 Hautmelanom-Zelllinie zur Verfügung gestellt hat, und Dr. Marc Mareel von der Abteilung für Strahlentherapie und Nuklearmedizin, Universitätsklinikum Gent, Belgien, der uns freundlicherweise die Mewo Hautmelanom-Zelllinie zur Verfügung gestellt hat. Wir danken Gabriela Almeida für die hilfreichen technischen Ratschläge bezüglich des PB-Tests. Wir danken auch Prof. Manuel Sobrinho Simões für das kritische Lesen dieses Manuskripts. H. Pópulo und R. Caldas haben gleichermaßen zu dieser Arbeit beigetragen.
Erklärung zu den Interessen
Diese Studie wurde von der Portugiesischen Stiftung für Wissenschaft und Technologie durch ein Post-Doc-Stipendium für HP unterstützt (Ref.: SFRH/BPD/85249/2012). Weitere Mittel stammen aus dem Projekt „Microenvironment, metabolism and cancer“ (Mikroumgebung, Stoffwechsel und Krebs), das teilweise vom Programa Operacional Regional do Norte (ON.2 – O Novo Norte) im Rahmen des Quadro de Referência Estratégico Nacional (QREN) und dem Fundo Europeu de Desenvolvimento Regional (FEDER) unterstützt wurde. IPATIMUP umfasst die i3S-Forschungseinheit, die teilweise von der FCT, der portugiesischen Stiftung für Wissenschaft und Technologie, unterstützt wird. Diese Arbeit wurde aus Mitteln des FEDER über das Operationelle Programm für Wettbewerbsfaktoren – COMPETE und aus nationalen Mitteln über die FCT im Rahmen des Projekts „PEst-C/SAU/LA0003/2013“ finanziert Die Autoren legen keine potenziellen Interessenkonflikte offen. Die Autoren haben keine anderen relevanten Verbindungen oder finanziellen Beteiligungen zu Organisationen oder Einrichtungen, die ein finanzielles Interesse an oder einen finanziellen Konflikt mit dem im Manuskript behandelten Thema oder Material haben, abgesehen von den offengelegten Verbindungen.
REFERENZEN
1 Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin 2014;64(1):9-29
2 Nikolaou V, Stratigos AJ. Emerging trends in the epidemiology of melanoma. Br J Dermatol 2014;170(1):11-19
3 Smoller BR. Histologische Kriterien für die Diagnose des primären kutanen malignen Melanoms. Mod Pathol 2006;19(Suppl 2):S34-40
4 Populo H, Soares P, Lopes JM. Insights into melanoma: Targeting the mTOR pathway for therapeutics. Expert Opin Ther Targets 2012;16(7):689-705
5 Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27(36):6199-206
6 Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol 2008;84(2):289-306
7 Bertolotto C. Melanoma: from melanocyte to genetic alterations and clinical options. Scientifica 2013;2013:22
8 Populo H, Boaventura P, Vinagre J, et al. TERT-Promotor-Mutationen bei Hautkrebs: die Auswirkungen von Sonnenexposition und Röntgenbestrahlung. J Invest Dermatol 2014;134(8):2251-7
9 Elder DE. Vorstufen des Melanoms und ihre Nachahmer: Nävi an besonderen Stellen. Modern Pathology Inc 2006;19(Suppl 2):S4-20
10 Kumar R, Angelini S, Snellman E, et al. BRAF-Mutationen sind häufige somatische Ereignisse
11 bei melanozytären Nävi. J Invest Dermatol 2004;122(2):342-8 Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-assoziierter seneszenzähnlicher Zellzyklus-Stillstand bei menschlichen Nävi. Natur 2005;436(7051):720-4
12 Vredeveld LC, Possik PA, Smit MA, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012;26(10):1055-69
13 Populo H, Soares P, Faustino A, et al. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res 2011;24(1):254-7
14 Olszanski AJ. Aktuelle und zukünftige Rolle der zielgerichteten Therapie und Immuntherapie beim fortgeschrittenen Melanom. J Manag Care Spec Pharm 2014;20(4):346-56
15 Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711-23
16 Chapman PB, Hauschild A, Robert C, et al. Verbessertes Überleben mit Vemurafenib bei Melanom mit BRAF V600E-Mutation. N Engl J Med 2011;364(26):2507-16
17 Flaherty KT, Robert C, Hersey P, et al. Verbessertes Überleben mit MEK-Inhibition bei BRAF-mutiertem Melanom. N Engl J Med 2012;367(2):107-14
18 Ballantyne AD, Garnock-Jones KP. Dabrafenib: first global approval. Drugs 2013;73(12):1367-76
19Availablefrom:http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm412861.htm
20 Gillies RJ, Robey I, Gatenby RA. Ursachen und Folgen eines erhöhten Glukosestoffwechsels bei Krebserkrankungen. J Nucl Med 2008;49(Suppl 2):24S-42S
21 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532
22 Semenza GL. HIF-1 vermittelt metabolische Reaktionen auf intratumorale Hypoxie und onkogene Mutationen. J Clin Invest 2013;123(9):3664-71
23 Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851-64
24 Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 2011;286(49):42626-34
25 Kluza J, Corazao-Rozas P, Touil Y, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res 2012;72(19):5035-47
26 Baudy AR, Dogan T, Flores-Mercado JE, et al. FDG-PET ist ein guter Biomarker sowohl für frühes Ansprechen als auch für erworbene Resistenz bei BRAFV600-mutierten Melanomen, die mit Vemurafenib und dem MEK-Inhibitor GDC-0973 behandelt werden. EJNMMI research 2012;2(1):22
27 Hall A, Meyle KD, Lange MK, et al. Dysfunktionale oxidative Phosphorylierung macht maligne Melanomzellen süchtig nach Glykolyse, angetrieben durch das (V600E)BRAF-Onkogen. Oncotarget 2013;4(4):584-99
28 Kumar SM, Yu H, Edwards R, et al. Mutant V600E BRAF erhöht die Expression des Hypoxie-induzierbaren Faktors-1alpha in Melanomen. Krebsforschung 2007;67(7):3177-84
29 Kuphal S, Winklmeier A, Warnecke C, et al. Konstitutive HIF-1-Aktivität in malignen Melanomen. Eur J Cancer 2010;46(6):1159-69
30 Slominski A, Kim TK, Brozyna AA, et al. The role of melanogenesis in regulation of melanoma behavior: Melanogenese führt zur Stimulation der HIF-1alpha-Expression und HIF-abhängiger Begleitwege. Archives of Biochemistry and Biophysics 2014;563:79-93
31 Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Zell Metab 2006;3(3):177-85
32 Lu CW, Lin SC, Chen KF, et al. Induktion von Pyruvat-Dehydrogenase-Kinase-3 durch Hypoxie-induzierbaren Faktor-1 fördert Stoffwechselumstellung und Arzneimittelresistenz. J Biol Chem 2008;283(42):28106-14
33 Patel MS, Korotchkina LG. Die Regulierung des Pyruvat-Dehydrogenase-Komplexes. Biochem Soc Trans 2006;34(Pt 2):217-22
34 Bowker-Kinley MM, Davis WI, Wu P, et al. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6
35 Papandreou I, Goliasova T, Denko NC. Krebsmedikamente, die auf den Stoffwechsel abzielen: Ist Dichloracetat das neue Paradigma? Int J Cancer 2011;128(5):1001-8
36 Zhang S, Hulver MW, McMillan RP, et al. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutrition & Metabolism 2014;11(1):10
37 Wigfield SM, Winter SC, Giatromanolaki A, et al. PDK-1 reguliert die Laktatproduktion bei Hypoxie und ist mit einer schlechten Prognose bei Plattenepithelkarzinomen des Kopfes und Halses verbunden. Br J Cancer 2008;98(12):1975-84
38 Lu CW, Lin SC, Chien CW, et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol 2011;179(3):1405-14
39 Baumunk D, Reichelt U, Hildebrandt J, et al. Expressionsparameter der Stoffwechselweggene Pyruvat-Dehydrogenase-Kinase-1 (PDK-1) und DJ-1/PARK7 beim Nierenzellkarzinom (RCC). World J Urol 2013;31(5):1191-6
40 Hur H, Xuan Y, Kim YB, et al. Expression von Pyruvat-Dehydrogenase-Kinase-1 in Magenkrebs als potenzielles therapeutisches Ziel. Int J Oncol 2013;42(1):44-54
41 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 2007;11(1):37-51
42 Kankotia S, Stacpoole PW. Dichloroacetat und Krebs: Neue Heimat für ein Orphan Drug? Biochimica et biophysica acta 2014;1846(2):617-29
43 Sutendra G, Dromparis P, Kinnaird A, et al. Mitochondriale Aktivierung durch Hemmung von PDKII unterdrückt HIF1a-Signalisierung und Angiogenese bei Krebs. Oncogene 2013;32(13):1638-50
44 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2(31):31ra34
45 Castro P, Rebocho AP, Soares RJ, et al. PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 2006;91(1):213-20
46 Sun W, Zhou S, Chang SS, et al. Mitochondriale Mutationen tragen über erhöhte reaktive Sauerstoffspezies und hochregulierte Pyruvat-Dehydrogenase-Kinase 2 beim Plattenepithelkarzinom des Kopfes und Halses zur Akkumulation von HIF1alpha bei. Clin Cancer Res 2009;15(2):476-84
47 Cairns RA, Bennewith KL, Graves EE, et al. Pharmakologisch erhöhte Tumorhypoxie kann durch 18F-Fluoroazomycin-Arabinosid-Positronenemissionstomographie gemessen werden und verstärkt die Tumorantwort auf das hypoxische Zytotoxin PR-104. Clin Cancer Res 2009;15(23):7170-4
48 Sun RC, Board PG, Blackburn AC. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol Cancer 2011;10:142
49 Shahrzad S, Lacombe K, Adamcic U, et al. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 2010;297(1):75-83
50 Abildgaard C, Dahl C, Basse AL, et al. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition. J Transl Med 2014;12:247
51 Populo H, Tavares S, Faustino A, et al. GNAQ- und BRAF-Mutationen zeigen eine unterschiedliche Aktivierung des mTOR-Signalwegs in menschlichen transformierten Zellen. Peer J 2013;1:e104
52 Sutendra G, Kinnaird A, Dromparis P, et al. Ein nukleärer Pyruvatdehydrogenase-Komplex ist wichtig für die Bildung von Acetyl-CoA und die Histon-Acetylierung. Cell 2014;158(1):84-97
53 Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 2005;30(3):142-50
54 Xuan Y, Hur H, Ham IH, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res 2014;321(2):219-30
55 Hong SE, Shin KS, Lee YH, et al. Inhibition of S6K1 enhances dichloroacetate-induced cell death. J Cancer Res Clin Oncol 2014.