Kasirajan Ayyanathan1,2,*,#, Shailaja Kesaraju1,#, Ken Dawson-Scully1,2, Herbert Weissbach1
1 Center for Molecular Biology and Biotechnology, Charles E. Schmidt College of Science, Florida Atlantic University, Jupiter, Florida, Stati Uniti d’America,
2 Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, Florida, Stati Uniti d’America
*E-mail: [email protected]
#Questiautori hanno contribuito in egual misura a questo lavoro.
Ricevuto: 12 agosto 2011
Accettato: 4 giugno 2012
Pubblicato: 1 luglio 2012
Abstract
Il sulindac è un farmaco antinfiammatorio non steroideo approvato dalla FDA con documentate attività antitumorali. I nostri recenti studi hanno dimostrato che il sulindac aumenta selettivamente l’uccisione delle cellule tumorali esposte ad agenti ossidanti attraverso la produzione di specie reattive dell’ossigeno (ROS) con conseguente disfunzione mitocondriale. Questo effetto del sulindac e dello stress ossidativo sulle cellule tumorali potrebbe essere correlato al difetto di respirazione delle cellule tumorali, descritto per la prima volta da Warburg 50 anni fa, noto come effetto Warburg. Abbiamo ipotizzato che il sulindac possa aumentare l’uccisione selettiva delle cellule tumorali se combinato con qualsiasi composto che altera la respirazione mitocondriale. Per verificare questa ipotesi abbiamo utilizzato il dicloroacetato (DCA), noto per spostare il metabolismo del piruvato dalla formazione di acido lattico alla respirazione. Ci si potrebbe aspettare che il DCA, poiché stimola il metabolismo aerobico, possa sollecitare la respirazione mitocondriale nelle cellule tumorali, con conseguente aumento dell’uccisione in presenza di sulindac. In questo studio abbiamo dimostrato che la combinazione di sulindac e DCA aumenta l’uccisione selettiva delle cellule tumorali A549 e SCC25 nelle condizioni utilizzate. Come previsto, il meccanismo di uccisione coinvolge la produzione di ROS, la disfunzione mitocondriale, la segnalazione JNK e la morte per apoptosi. I nostri risultati suggeriscono che la combinazione di farmaci sulindac-DCA può fornire una terapia antitumorale efficace.
Citazione: Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H (2012) La combinazione di sulindac e dicloroacetato uccide le cellule tumorali attraverso il danno ossidativo
. PLoS ONE 7(7): e39949. doi:10.1371/journal.pone.0039949
Editore: Joseph Alan Bauer, Bauer Research Foundation, Stati Uniti d’America
© 2012 Ayyanathan et al. Questo è un articolo ad accesso libero distribuito secondo i termini della Creative Commons Attribution License, che ne consente
l’uso, la distribuzione e la riproduzione illimitata su qualsiasi supporto, a condizione che vengano citati l’autore e la fonte originali.
Finanziamenti: Si ringrazia l’assistenza finanziaria del National Institutes of Health a KA (sovvenzione 5K01CA95620) e HW (sovvenzione R15 CA122001) e dello Stato della Florida a HW (sovvenzione SURECAG R94007) per la realizzazione di questo lavoro. I finanziatori non hanno avuto alcun ruolo nella progettazione dello studio, nella raccolta e nell’analisi dei dati, nella decisione di pubblicare o nella preparazione del manoscritto.
Interessi in competizione: Gli autori hanno dichiarato che non esistono interessi in competizione.
INTRODUZIONE
Il sulindac è un farmaco antinfiammatorio non steroideo (FSAID) approvato dalla FDA, che ha dimostrato di avere anche un’attività antitumorale [1-6]. Recenti studi condotti dal nostro laboratorio hanno dimostrato che le linee cellulari tumorali RKO, A549 e SCC25 sono risultate sensibili alla combinazione di sulindac e di un agente ossidante, come TBHP o H2O2 [7]. I dati indicano che l’effetto del sulindac non è legato alla sua attività di FANS, ma che il sulindac rende le cellule tumorali più sensibili allo stress ossidativo, con conseguente disfunzione mitocondriale e perdita di vitalità. Al contrario, le cellule normali non hanno mostrato una maggiore uccisione in condizioni simili [7]. Negli ultimi 10 anni ci sono state segnalazioni sparse di una maggiore uccisione del cancro utilizzando il sulindac in combinazione con una serie di composti, tra cui il triossido di arsenico, il bortezomib, la difluorometilornitina (DFMO) e l’acido suberoilanilide idrossamico (SAHA) [8-14]. Sebbene questi composti abbiano siti d’azione diversi, un meccanismo comune per il potenziamento dell’uccisione da parte della combinazione sulindac/farmaco potrebbe coinvolgere il danno ossidativo, come è stato chiaramente dimostrato in nostri precedenti studi che hanno utilizzato sulindac e un agente ossidante [7], [15]. Infatti, i ROS sono stati coinvolti negli studi che hanno utilizzato il sulindac in combinazione con triossido di arsenico, bortezomib e SAHA [10], [12], [14].
I nostri risultati precedenti hanno suggerito che la maggiore uccisione delle cellule tumorali da parte della combinazione di sulindac e un agente ossidante potrebbe essere dovuta a un difetto nella respirazione delle cellule tumorali, come descritto per la prima volta da Warburg più di 50 anni fa [16], il quale ha osservato che le cellule tumorali favoriscono la glicolisi, non la respirazione, per ottenere energia, a differenza delle cellule normali. Alcune cellule tumorali ottengono fino al 50% dell’energia dalla glicolisi, mentre la glicolisi nelle cellule normali rappresenta meno del 5% del fabbisogno energetico [16]. Per ottenere ulteriori prove del possibile ruolo dell’alterazione della respirazione e dei ROS nell’uccisione delle cellule tumorali da parte del sulindac e dello stress ossidativo, abbiamo avviato studi con l’acido dicloroacetico (DCA) di sodio. Il DCA è un candidato ideale in quanto è noto che inibisce una chinasi che regola l’attività della piruvato deidrogenasi, con conseguente spostamento del metabolismo del piruvato dalla formazione di acido lattico alla respirazione [17], [18]. Il DCA è stato utilizzato clinicamente per trattare i pazienti affetti da acidosi lattica [19] e, in base alle sue proprietà biochimiche, è stato testato anche come agente antitumorale. Bonnet et al. 2007 hanno dimostrato che il DCA inverte l’effetto Warburg nelle cellule tumorali reindirizzando il metabolismo delle cellule tumorali dalla glicolisi alla fosforilazione ossidativa. In questi studi precedenti è stato dimostrato che il DCA aumenta i livelli di ROS dal complesso I. Questo a sua volta innesca un “rimodellamento” del metabolismo mitocondriale (riduce il ΔΨm, apre il poro di transizione mitocondriale) nelle cellule tumorali spingendole verso l’apoptosi. Inoltre, diversi studi recenti hanno verificato che il DCA può aumentare i livelli di ROS nelle cellule tumorali e depolarizzare la membrana dei mitocondri in linee cellulari di polmone, endometrio e glioblastoma con conseguente apoptosi sia in vitro che in vivo [18], [20-22]. È interessante l’osservazione che, nelle condizioni utilizzate, il DCA non sembra influenzare significativamente il metabolismo mitocondriale o la vitalità delle cellule normali [18], [23].
Sulla base delle nostre precedenti osservazioni sull’effetto antitumorale del sulindac e di un agente ossidante che influisce sul metabolismo mitocondriale [7], abbiamo ipotizzato che la combinazione di sulindac e DCA possa aumentare sinergicamente l’effetto antitumorale e avere un importante valore terapeutico. Nel presente studio abbiamo esaminato l’effetto dell’uso del sulindac in combinazione con il DCA sulla vitalità delle linee cellulari tumorali A549 e SCC25. Abbiamo anche studiato il ruolo della funzione mitocondriale e dell’apoptosi nell’uccisione del cancro osservata con questa combinazione di farmaci.
Materiali e metodi
Materiali
Sulindac, N-acetilcisteina e Tiron sono stati acquistati da Sigma (St.Louis, MO). Il sale sodico di DCA è stato ottenuto da Acros Organics (Geel, Belgio). H2DCFDA e JC-1 sono stati acquistati da Molecular Probes (Eugene, OR). Il reagente per il saggio MTS e il kit Deadend Tunel sono stati acquistati da Promega (Madison, WI). Il kit per il frazionamento di citosol/mitocondri e il kit per il rilascio di citocromo c a flusso CBA077 InnoCyte™ sono stati forniti da Calbiochem, Gibbstown, NJ. Tutti i terreni di coltura cellulare, il siero fetale bovino e altri integratori come penicillina/streptomicina, glutammina, ecc. sono stati acquistati dall’American Type Culture Collection (ATCC; Rockville, MD).
Coltura cellulare
Unalinea cellulare di carcinoma polmonare non a piccole cellule (NSCLC), A549, una linea cellulare di polmone umano normale, MRC-5, e una linea di carcinoma a cellule squamose derivato dalla lingua, SCC25, sono state acquistate dall’ATCC (Rockville, MD) e mantenute in terreno F12-K integrato con 10% di siero bovino fetale, 2 mM di glutammina, 100 UI/ml di penicillina e 100 µg/ml di streptomicina in un incubatore umidificato al 5% diCO2 a 37°C. I cheratinociti epidermici umani normali sono stati ottenuti da Promocell GmbH (Heidelberg, Germania) e mantenuti nel terreno di coltura raccomandato. Per gli esperimenti sono state utilizzate cellule normali di primo passaggio, non immortalate.
Saggio di vitalità cellulare
Le cellule tumorali A549 e le cellule normali polmonari sono state piastrate a 3×103 cellule per pozzetto, mentre le cellule tumorali SCC25 e i cheratinociti normali sono stati piastrati a 7,5×103 cellule per pozzetto in una piastra a 96 pozzetti. Le cellule sono state coltivate per 18-20 ore, il terreno di coltura è stato scartato in condizioni asettiche e sostituito con terreno di coltura fresco contenente le combinazioni di farmaci indicate. Dove indicato, 500 µM di sulindac sono stati utilizzati con le cellule tumorali e polmonari normali A549 e 100 µM di sulindac sono stati utilizzati con le cellule tumorali e cheratinocitarie normali SCC25. Le piastre sono state incubate per 48 ore a 37°C in un incubatore al 5% diCO2. Il terreno di coltura è stato scartato e le cellule sono state accuratamente risciacquate in PBS 1×. La vitalità cellulare è stata determinata utilizzando il saggio CellTiter 96 Aqueous One Cell Proliferation Assay (Promega) secondo le istruzioni del produttore. Il saggio utilizza un composto di tetrazolio che viene convertito in formazan solubile in acqua dall’azione delle deidrogenasi cellulari presenti nelle cellule metabolicamente attive [24]. Il formazan è stato quantificato misurando l’assorbanza a 490 nm con un lettore colorimetrico per micropiastre (SpectraMax Plus; Molecular Devices). L’assorbanza di fondo è stata sottratta da ciascun campione.
Misurazione intracellulare dei ROS
Le linee cellulari tumorali A549 e SCC25 sono state piastrate come sopra. Dopo il trattamento farmacologico di 48 ore, le cellule sono state incubate con 50 µM di diclorodiidrofluoresceina diacetato (H2DCFDA, Molecular Probes) in terreno privo di indicatori per 30 minuti a 37 °C. Le cellule sono state risciacquate con PBS e i livelli di ROS sono stati visualizzati mediante microscopia a fluorescenza. Le immagini sono state acquisite con il software Qcapture ed elaborate con Adobe photoshop. L’analisi delle immagini è stata eseguita con il software slidebook. I dati ottenuti da un esperimento rappresentativo sono stati utilizzati per la quantificazione delle cellule positive al DCF, misurate dalla fluorescenza verde dovuta al DCF ossidato.
Colorazione JC-1 per monitorare il potenziale di membrana mitocondriale
Il potenziale di membranamitocondrialeè stato determinato utilizzando il colorante JC-1 (Molecular Probes). Le linee cellulari tumorali A549 e SCC25 sono state piastrate come sopra. Dopo il trattamento farmacologico di 48 ore, le cellule sono state incubate con 5 ng/ml di colorante JC-1 in terreno libero da indicatori per 30 minuti a 37°C. Le cellule sono state risciacquate con PBS e visualizzate mediante microscopia a fluorescenza. I mitocondri normali assorbono attivamente il colorante JC-1 in modo dipendente dal potenziale e formano aggregati J, che producono una fluorescenza rossa. L’interruzione e la successiva perdita del potenziale di membrana mitocondriale porta a un aumento della fluorescenza verde nel citosol dovuta alla JC-1 monomerica, che viene determinata seguendo la comparsa della fluorescenza verde con un filtro FITC (microscopio invertito Zeiss-Axiovert 40 CFL). L’acquisizione, l’elaborazione e l’analisi delle immagini sono state eseguite come sopra. I dati ottenuti da un esperimento rappresentativo sono stati utilizzati per la quantificazione delle cellule positive alla JC-1 verde.
Effetto degli scavenger dei ROS sulla vitalità cellulare in presenza di Sulindac e DCA
Le linee cellulari tumorali A549 e SCC25 sono state piastrate come descritto sopra. Per eliminare i ROS, sono stati aggiunti 2 mM di N-acetilcisteina (NAC) o 2 mM di Tiron (sale disodico dell’acido 4,5-diidrossi-1,3-benzendisolfonico) insieme a sulindac e DCA per 48 ore a 37°C. La vitalità cellulare è stata monitorata con il saggio MTS e l’analisi statistica è stata eseguita come indicato sopra.
Colorazione TUNEL per monitorare le cellule in fase di apoptosi
Il saggioTUNELè stato eseguito in piastre da 96 pozzetti utilizzando il kit DeadEnd colorimetric tunel assay (Promega) seguendo il protocollo del produttore. Le linee cellulari tumorali A549 e SCC25 sono state piastrate come sopra e trattate per 48 ore con nessun farmaco, sulindac, DCA o combinazione di farmaci. Dopo il trattamento farmacologico, le cellule sono state fissate con formalina e permeabilizzate con Triton X-100 allo 0,2% in PBS. Le cellule sono state incubate con deossinucleotidil transferasi terminale ricombinante (TdT) e nucleotidi biotinilati. Le perossidasi endogene sono state bloccate con H2O2 allo 0,3% prima dell’incubazione con perossidasi-streptavidina di rafano (HRP-streptavidina) che si lega ai nucleotidi biotinilati incorporati nelle estremità intaccate presenti nelle cellule in fase di apoptosi. Le cellule marcate con HRP-streptavidina sono state rilevate con perossido di idrogeno e diaminobenzidina (DAB). Le cellule che mostrano una colorazione nucleare marrone scuro sono indicative di apoptosi.
Analisi Western Blot
Le cellule sono state coltivate al 70% della confluenza, trattate con i farmaci specificati per la durata indicata e le frazioni citosoliche sono state isolate utilizzando il kit di frazionamento citosol/mitocondri (Calbiochem, Gibbstown, NJ) seguendo il protocollo del produttore. In breve, le cellule sono state raccolte in diversi punti temporali e poi centrifugate a 600×g per 5 minuti a 4°C. Le cellule ridotte in pellet sono state sospese nel tampone fornito e incubate per 10 minuti in ghiaccio. Le cellule sono state quindi omogeneizzate con un douncer di vetro e l’omogenato è stato centrifugato a 700×g per 10 minuti a 4°C per sedimentare nuclei e detriti cellulari. Il surnatante è stato centrifugato a 10.000×g per 30 minuti a 4°C per ottenere il pellet mitocondriale e il surnatante è stato considerato come frazione citosolica. La concentrazione di proteine è stata determinata utilizzando un saggio standard di Bradford.
Sessanta microgrammi di proteine totali sono stati caricati e separati su gel NuPage Bis-Tris al 4-12% (Invitrogen, Eugene, OR) e trasferiti su una membrana PVDF che è stata testata con gli anticorpi primari. Gli anticorpi primari, JNK, pJNK, citocromo c e PARP (Cell Signaling Technology, Danvers, MA), sono stati utilizzati alla diluizione 1∶1000. la β-actina (Santa Cruz Biotechnologies, Santa Cruz, California) è stata utilizzata alla diluizione 1∶4000. Sono stati utilizzati anticorpi secondari coniugati con perossidasi di rafano e le bande sono state visualizzate con un metodo di chemiluminescenza potenziata (GE Healthcare, Piscataway, NJ).
Saggio di DNA laddering mediato da ligation-mediated PCR per monitorare l’estensione delle cellule sottoposte ad apoptosi
Per confermare l’entità dell’apoptosi, è stato eseguito un saggio di DNA laddering nucleosomico mediato da ligation-mediated PCR come descritto [25]. Le linee cellulari tumorali A549 e SCC25 sono state piastrate a 5×104 e 1×105 cellule per pozzetto in piatti da 35 mm. Le cellule tumorali A549 sono state trattate per 48 ore con a) nessun farmaco, b) 500 µM di sulindac, c) 20 mM di DCA e d) 500 µM di sulindac più 20 mM di DCA. Allo stesso modo, le cellule tumorali SCC25 sono state trattate con le quattro diverse combinazioni di farmaci di cui sopra, tranne che per il sulindac e il DCA, utilizzati rispettivamente a concentrazioni di 100 µm e 10 mM. Dopo il trattamento, il DNA cellulare totale è stato estratto e ligato all’adattatore costruito con 27-mer 5′-GACGTCGACGTCGTACGTGTCGACT-3′ e 12-mer 5′- AGTCGACGTGTAC-3′. Dopo la legatura, il DNA è stato riscaldato per rilasciare il 12-mer, riempito con Taq polimerasi, sottoposto a PCR semiquantitativa e analizzato su un gel di agarosio all’1,2% insieme ai marcatori di dimensione.
Localizzazionein situ del citocromo c mediante immunofluorescenza
La localizzazione intracellulare del citocromo c è stata monitorata mediante immunofluorescenza utilizzando il kit InnoCyte™ Flow Cytometric Cytochrome c Release Kit CBA077 secondo le istruzioni del produttore. In breve, le cellule SCC25 sono state placcate a 3,5×105 cellule per piatto da 35 mm con fondo in vetro e trattate con i farmaci indicati per 15 ore. Le cellule sono state risciacquate in 5 ml di PBS 1× e permeabilizzate in ghiaccio per 10 minuti in 300 µl di tampone fornito. Le cellule sono state fissate a temperatura ambiente per 20 minuti in 500 µl di paraformaldeide al 4%. Dopo il lavaggio e il blocco, le cellule sono state incubate con 250 µl di anticorpo anticitocromo c (diluizione 1∶500) per 1 ora a temperatura ambiente. Dopo il lavaggio, le cellule sono state incubate con 300 µl di FITC-IgG (diluizione 1∶300) per 1 ora a temperatura ambiente. Infine, le cellule sono state colorate con 300 µl di DAPI (1 mg/ml) per 10 minuti a temperatura ambiente. Le cellule sono state visualizzate con un microscopio invertito a fluorescenza Olympus. Le immagini sono state acquisite ed elaborate come indicato sopra. Sono stati analizzati diversi campi e sono state ottenute micrografie rappresentative che mostrano i modelli di localizzazione del citocromo c in ciascuna condizione di trattamento. I valori quantitativi sono presentati nel testo.
Analisi statistica e determinazione degli indici di combinazione
I dati sono presentati come media ± SEM per i saggi di vitalità cellulare. Per l’analisi statistica è stato utilizzato il software statistico Minitab per eseguire il test t di Student e i valori con p<0,05 sono stati considerati statisticamente significativi. Per accertare l’effetto sinergico di sulindac e DCA sulle linee cellulari tumorali A549 e SCC25, è stata eseguita un’analisi quantitativa della relazione dose-effetto per determinare gli indici di combinazione [26]. Sia il sulindac che il DCA sono stati testati da soli sulle cellule A549 e SCC25 alle concentrazioni indicate. Per le cellule A549, è stato mantenuto un rapporto di 1∶50 per le combinazioni di farmaci sulindac:DCA che vanno da 0,2 mM:10 mM fino a 1 mM:50 mM, rispettivamente. Per le cellule SCC25, è stato mantenuto un rapporto di 1∶100 per le combinazioni di farmaci sulindac:DCA che vanno da 0,05 mM:5 mM fino a 0,3 mM:30 mM, rispettivamente. I nostri risultati sperimentali e i valori determinati dell’indice di combinazione sono inclusi nel testo.
Risultati
Sulindac e DCA causano una maggiore uccisione delle cellule tumorali A549 e SCC25, ma non delle cellule normali
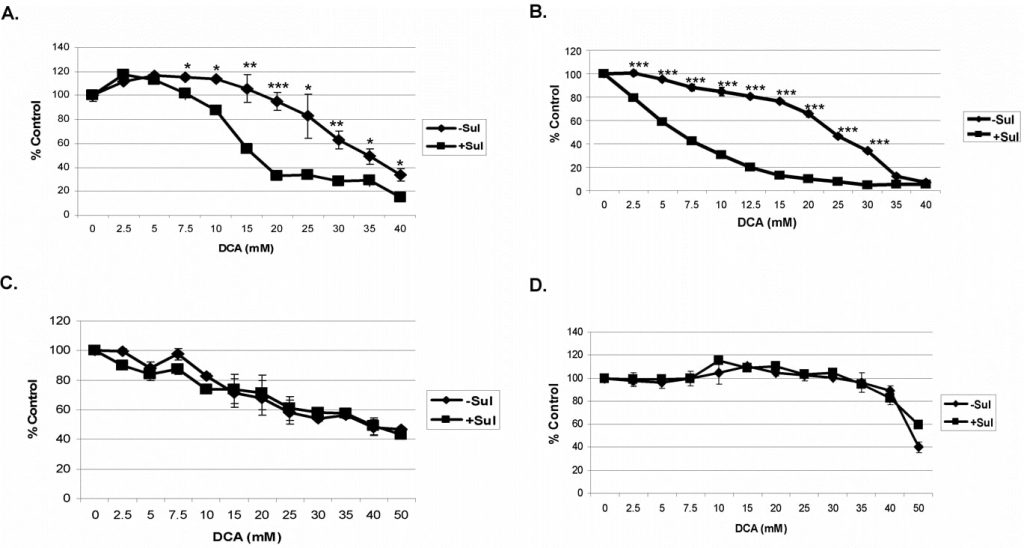
Per questi studi abbiamo testato la combinazione disulindace DCA sulle cellule tumorali A549 e SCC25. Le cellule sono state incubate con ciascun composto da solo o in combinazione per 48 ore prima di eseguire il test di vitalità (vedi Metodi). Una curva di risposta alla dose di sulindac in queste condizioni ha indicato che le cellule tumorali A549 e SCC25 possono tollerare una concentrazione massima di 500 µM e 100 µM di sulindac, rispettivamente, senza mostrare alcuna uccisione significativa (dati non mostrati), e queste concentrazioni sono state utilizzate in tutti gli studi. Il DCA, quando aggiunto, è stato utilizzato a concentrazioni comprese tra 0 e 40 mM, come indicato. Abbiamo utilizzato queste concentrazioni sulla base di precedenti rapporti, che indicavano come fossero necessari più di 5 mM per causare disfunzione mitocondriale in esperimenti in vitro [27]. Come mostrato nella Figura 1A, il DCA da solo (senza sulindac) è leggermente tossico per le cellule tumorali A549, soprattutto a concentrazioni superiori a 20 mM, ma in presenza di sulindac l’uccisione di queste cellule è maggiore a concentrazioni di DCA superiori a 5 mM. Nel caso delle cellule tumorali SCC25 è stata osservata una certa perdita di vitalità cellulare con il solo DCA anche a concentrazioni di DCA inferiori a 10 mM (Figura 1B). Tuttavia, in presenza di sulindac si è registrato un marcato aumento della morte cellulare, chiaramente evidente tra le concentrazioni di DCA di 2-10 mM. In precedenza abbiamo dimostrato che la combinazione di sulindac e di un agente ossidante era selettiva per le cellule tumorali e non aumentava l’uccisione delle cellule normali [7]. Anche il sulindac e il DCA non hanno aumentato l’uccisione delle cellule normali del polmone e della pelle nelle condizioni sperimentali utilizzate, come mostrato nelle Figure 1C e D. Va notato che le cellule MRC-5 (normali del polmone) sono particolarmente sensibili al DCA, come riportato in precedenza [28], per ragioni che non sono note.
Per verificare l’esistenza di un effetto sinergico quando è stata utilizzata la combinazione di farmaci, abbiamo determinato gli indici di combinazione eseguendo un’analisi quantitativa della relazione dose-effetto [26] su due diverse linee cellulari tumorali (FiguraS1). Gli indici di combinazione erano rispettivamente 0,84 per le cellule tumorali A549 e 0,73 per le cellule SCC25. Un valore inferiore a 1,00 indica un effetto sinergico di uccisione del cancro (Figura S2).
L’effetto del sulindac non è dovuto alla sua attività FANS
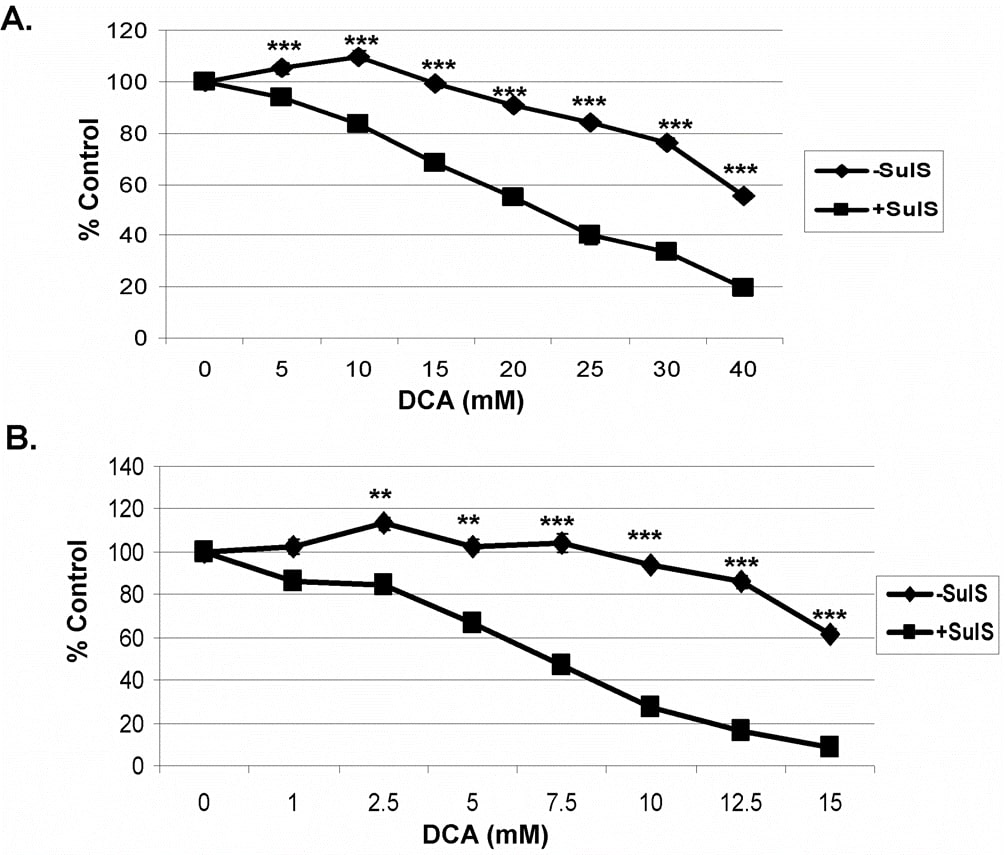
In studi precedenti condotti con il sulindac e un agente ossidante è stato dimostrato che l’uccisione potenziata e selettiva delle cellule tumorali da parte del sulindac e di un agente ossidante non era legata alla nota capacità FANS del sulindac. Per determinare il ruolo dell’inibizione della COX è possibile utilizzare un metabolita del sulindac, il sulfone del sulindac, che non inibisce la COX 1 o 2 [7], [29]. Come mostrato nella Figura 2, utilizzando cellule tumorali A549 (A) e SCC25 (B), la combinazione di sulindac solfone e DCA ha mostrato un effetto di uccisione simile a quello visto in precedenza con il sulindac. Questi risultati indicano che l’effetto cancerogeno potenziato del sulindac in presenza di DCA non è legato alla sua nota attività antinfiammatoria.
La combinazione di Sulindac e DCA genera ROS
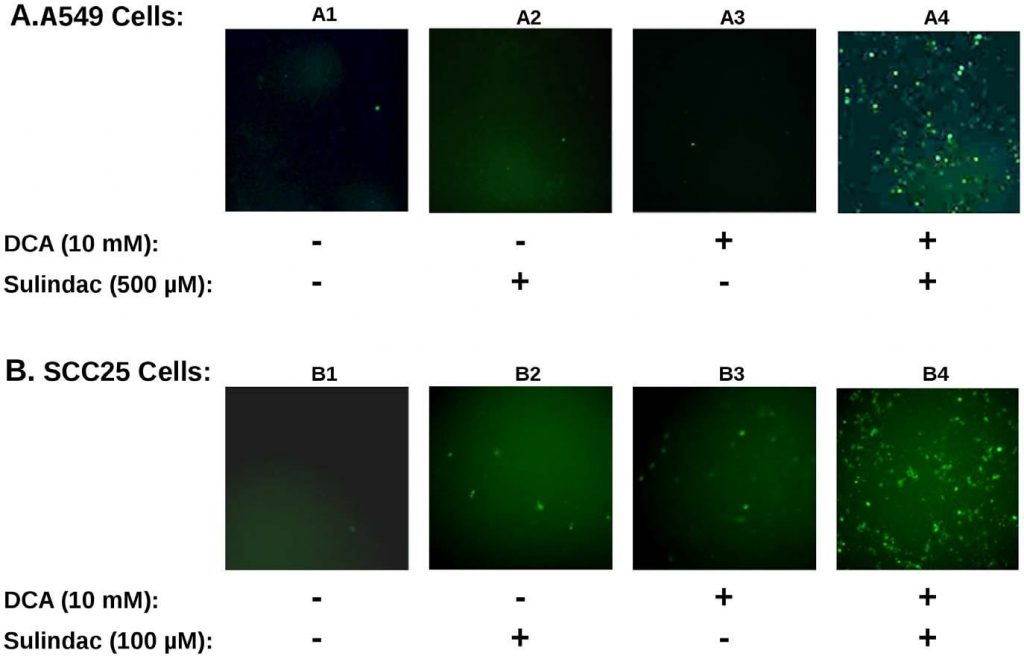
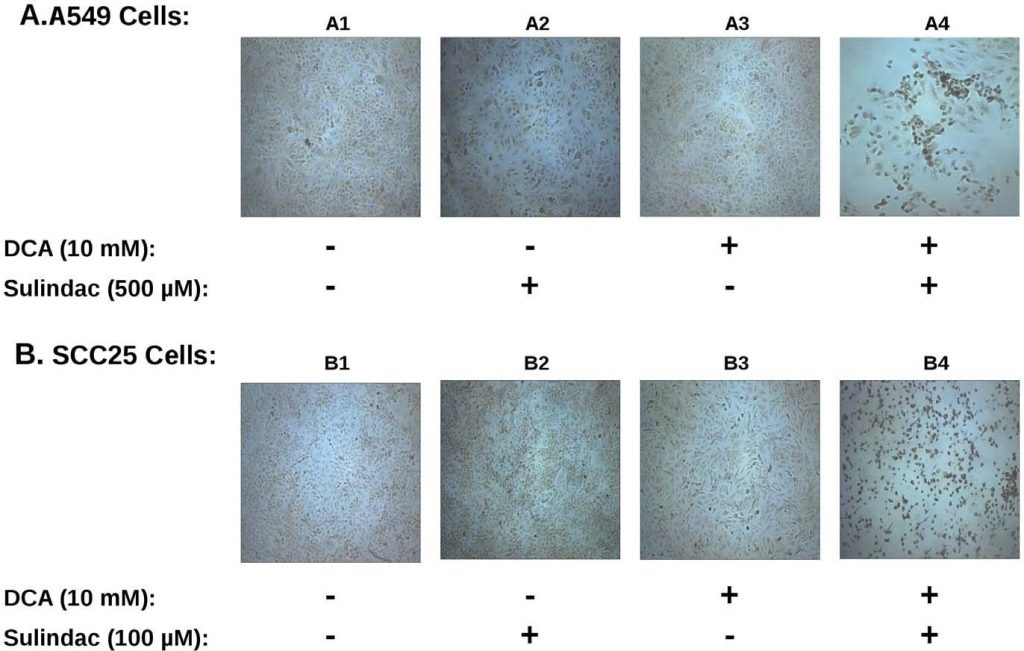
L’effetto sinergico sulla vitalità osservato con il sulindac e il dicloroacetato sia con le cellule tumorali A549 che con quelle SCC25 è sorprendentemente simile a quello di studi precedenti che utilizzavano la combinazione di sulindac e TBHP [7]. Per determinare se la produzione di ROS fosse coinvolta nell’uccisione selettiva osservata nei presenti studi, la produzione di ROS, utilizzando il colorante indicatoreH2DCFDA(vedi Metodi), è stata determinata nelle linee cellulari tumorali esposte a sulindac e DCA. I risultati sono riassunti nella Figura 3. La Figura 3A mostra i risultati ottenuti con le cellule tumorali A549. È evidente dai risultati illustrati nella Figura 3A che le cellule tumorali A549 non trattate (pannello A1) o trattate con il solo sulindac (pannello A2) o il solo DCA (pannello A3) mostrano solo poche cellule colorate positivamente. Tuttavia, quando le cellule sono state esposte sia al sulindac che al DCA (pannello A4), si osserva un forte aumento delle cellule colorate positivamente per i ROS (fluorescenza verde), a dimostrazione del fatto che la presenza di sulindac e DCA determina la generazione di livelli significativi di ROS. Come mostrato nella Figura 3B, risultati simili sono stati ottenuti con le cellule tumorali SCC25. Il sulindac o il DCA da soli determinano un piccolo aumento delle cellule che producono ROS (pannelli B2 e B3), ma si osserva nuovamente un forte aumento della produzione di ROS quando vengono aggiunti entrambi i farmaci (pannello B4). La quantificazione effettuata con le cellule SCC25 mostra che il numero di cellule DCF-positive (vedi Metodi) è 9-10 volte superiore quando le cellule sono trattate con sulindac e DCA rispetto a ciascuno dei due farmaci da solo (vedi Figura S3A). Da questi risultati e da studi precedenti emerge che la produzione di ROS può essere una caratteristica comune nella maggiore uccisione delle cellule tumorali quando il sulindac viene usato in combinazione con composti che influenzano la funzione mitocondriale.
Ilsulindac in combinazione con il DCA determina una perdita del potenziale di membrana mitocondriale
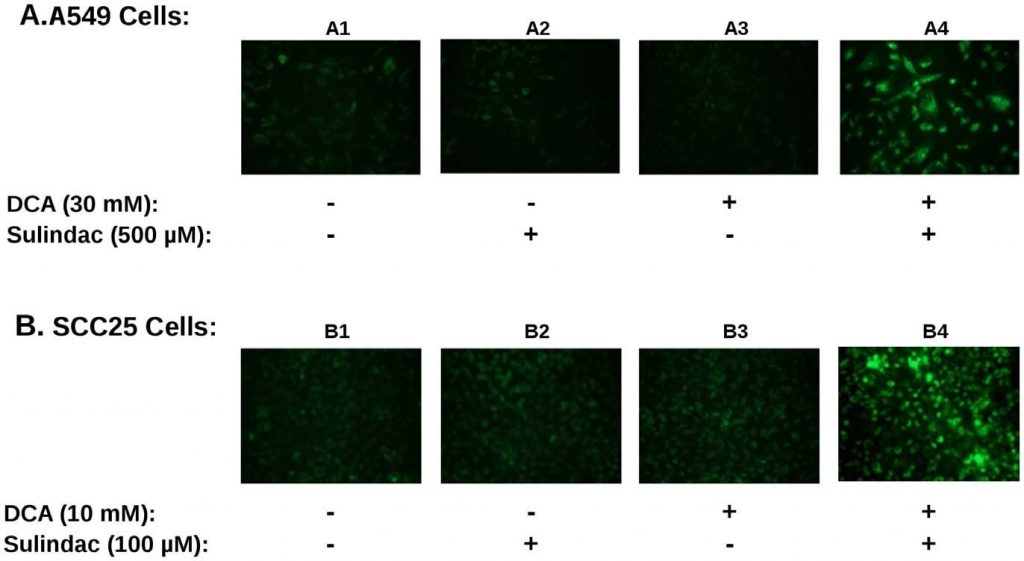
Se la produzione di ROS è coinvolta nell’effetto di uccisione potenziato dal sulindac/DCA, ci si aspetterebbe che la produzione di ROS da parte della combinazione di farmaci influisca sulla funzione mitocondriale. Per determinare ciò, il potenziale di membrana mitocondriale è stato misurato utilizzando la colorazione JC-1 come descritto nei Metodi. Una perdita di potenziale di membrana è indicata da un aumento della fluorescenza verde, come descritto nei Metodi. Un risultato tipico è riassunto nella Figura 4. Le cellule tumorali A549 e SCC25 sono state esposte a sulindac e DCA, da soli o in combinazione, per 48 ore e colorate con JC-1 per monitorare il potenziale di membrana mitocondriale. La Figura 4A mostra i risultati ottenuti con la linea cellulare A549. In assenza di farmaci, i mitocondri appaiono intatti e mantengono il loro potenziale di membrana, come indicato dalla scarsa fluorescenza verde (pannello A1). In presenza del solo sulindac (pannello A2) o del solo DCA (pannello A3) si verifica un piccolo aumento della fluorescenza verde, che indica una certa perdita del potenziale di membrana mitocondriale. Tuttavia, quando sono presenti sia il sulindac che il DCA, si verifica un’evidente perdita del potenziale di membrana mitocondriale, evidenziata da un forte aumento della fluorescenza verde (pannello A4). Abbiamo osservato lo stesso schema quando diversi campi indipendenti sono stati analizzati con la microscopia a fluorescenza. La Figura 4B mostra risultati simili con le cellule tumorali SCC25. Ancora una volta, una perdita significativa del potenziale di membrana mitocondriale è stata osservata solo quando le cellule sono state esposte sia al sulindac che al DCA (pannello B4). La quantificazione dell’effetto è riportata nella Figura S3B. Si può notare che la percentuale di cellule JC1-verdi positive quando è stata utilizzata la combinazione di farmaci è 3-4 volte superiore a quella osservata con uno dei due farmaci da soli.
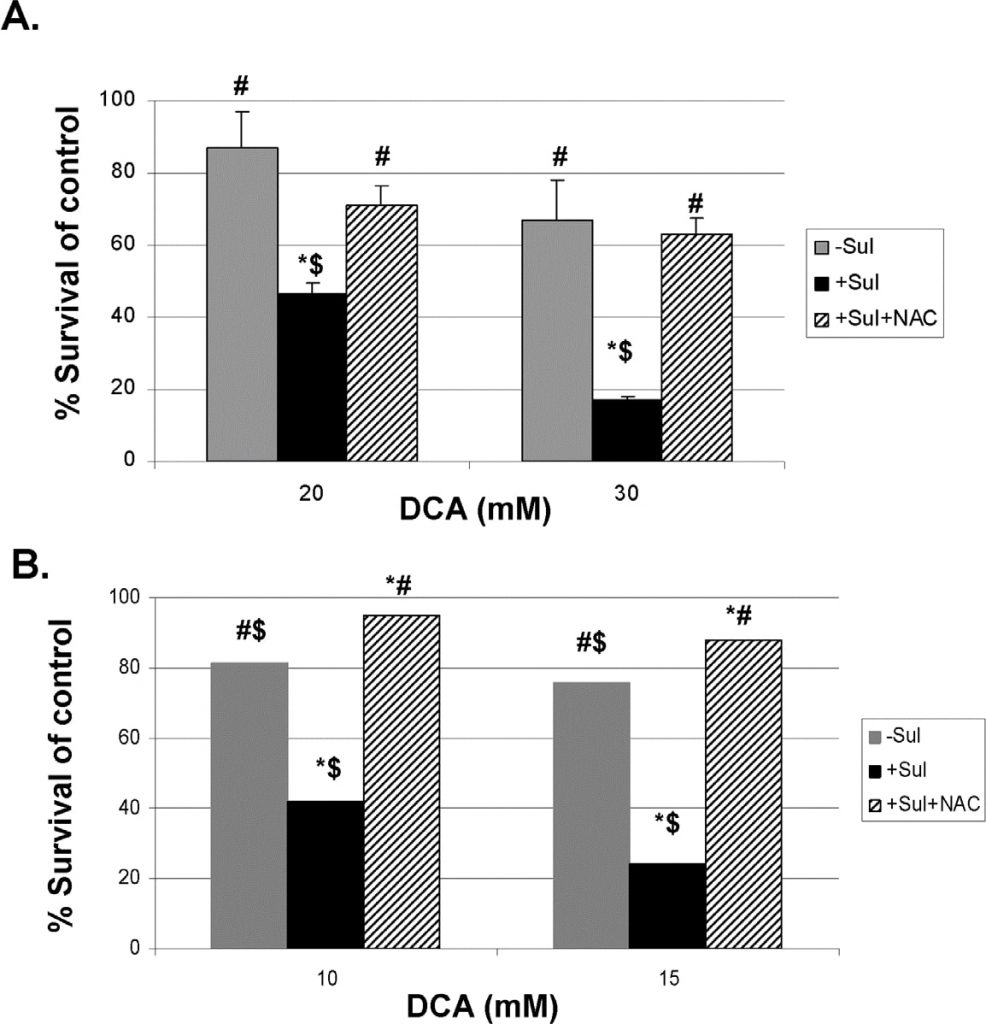
IROS sono coinvolti nell’uccisione delle cellule tumorali da parte della combinazione di sulindac e DCA
Per fornire una prova più diretta che i ROS prodotti sono coinvolti nell’uccisione potenziata delle cellule tumorali da parte di sulindac e DCA, abbiamo utilizzato due noti scavenger di ROS, N-acetilcisteina (NAC) e Tiron (vedi Metodi). I risultati ottenuti con l’uso di NAC sono illustrati nella Figura 5. La Figura 5, pannello A, mostra che, sia a 20 che a 30 mM di DCA, l’aumento dell’uccisione delle cellule tumorali A549 osservato in presenza di sulindac è in gran parte impedito dalla presenza di NAC (2 mM) durante le 48 ore di incubazione. Risultati molto simili sono stati osservati con le cellule tumorali SCC25, come mostrato nella Figura 5, pannello B. Risultati comparabili sono stati ottenuti quando è stato utilizzato il Tiron al posto della NAC (Figura S4).
L’uccisione delle cellule tumorali da parte di Sulindac e DCA coinvolge la morte apoptotica
I risultati sopra riportati (Figure 3, 4 e 5) mostrano che l’uccisione potenziata delle linee cellulari tumorali coinvolge la disfunzione mitocondriale, il che suggerisce che la morte cellulare osservata avviene tramite apoptosi. Studi precedenti hanno indicato che il sulindac e i suoi derivati sono farmaci pro-apoptotici [5], [6]. È stato anche riportato che il DCA può causare la morte cellulare per apoptosi [20], [23]. Per determinare se l’uccisione delle cellule tumorali da parte della combinazione di questi due farmaci, mediata dai ROS, comporti una morte apoptotica, abbiamo eseguito la colorazione TUNEL per misurare l’apoptosi (vedi Metodi). Per gli esperimenti di colorazione TUNEL sono state eseguite repliche multiple di sulindac e DCA da soli o in combinazione. Un risultato tipico è illustrato nella Figura 6, dove i pannelli superiori (Figura 6A, pannelli A1-A4) rappresentano i risultati con le cellule tumorali A549 e i pannelli inferiori (Figura 6B, pannelli B1-B4) rappresentano i risultati con le cellule tumorali SCC25. Quando le cellule sono state trattate con nessun farmaco, con il solo sulindac o con il solo DCA (Figura 6, pannelli A1-A3 e B1-B3), sono state osservate solo poche cellule TUNEL-positive. Tuttavia, quando le cellule sono state esposte sia al sulindac che al DCA, si è registrato un aumento significativo delle cellule apoptotiche positive alla TUNEL (Figura 6, pannelli A4 e B4), indicando una forte induzione dell’apoptosi. Per verificare i risultati di TUNEL, per monitorare l’apoptosi è stato utilizzato anche un saggio più sensibile di DNA laddering mediato da PCR [25]. I risultati hanno mostrato la presenza di un forte ladder nucleosomico solo quando sulindac e DCA sono stati usati in combinazione (Figura S5; corsie 4 e 8), il che supporta fortemente i dati del saggio TUNEL.
L’uccisione da parte di Sulindac e DCA coinvolge la segnalazione proapoptotica JNK
Tra le protein chinasi attivate da mitogeni (MAP chinasi) conosciute, la chinasi indotta dallo stress, c-Jun N-terminal kinase (JNK/SAPK), è stata direttamente implicata nella morte cellulare apoptotica [11]. Pertanto, abbiamo indagato il ruolo della segnalazione JNK nell’apoptosi mediata da sulindac-DCA utilizzando SP600125, un inibitore specifico di JNK (JNKI); i risultati sono presentati nella Figura 7A. Come mostrato in precedenza, le cellule SCC25 trattate con sulindac hanno mostrato un aumento della morte in presenza di concentrazioni crescenti di DCA. Tuttavia, quando queste cellule sono state incubate con sulindac insieme a SP600125, la morte cellulare mediata da sulindac-DCA è stata ampiamente prevenuta. Questi risultati indicano la partecipazione della segnalazione pro-apoptotica mediata da JNK nella morte cellulare mediata da sulindac-DCA.
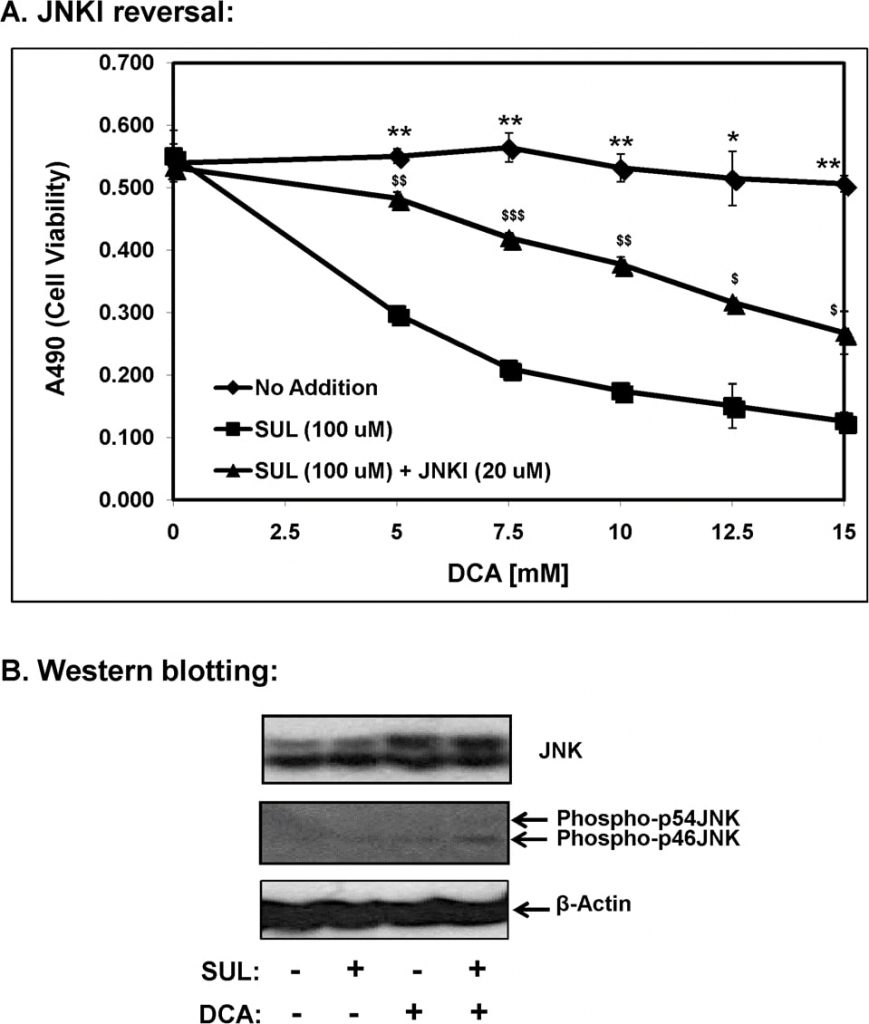
Con l’analisi western blot, abbiamo anche determinato che la combinazione di sulindac e DCA ha aumentato significativamente i livelli di fosfo-JNK nelle frazioni citosoliche 12 ore dopo che le cellule erano state esposte a sulindac e DCA (Figura 7B). Un aumento dei livelli di JNK totale (bande proteiche a 46 e 54 kDa) è stato osservato quando le cellule sono state trattate con il solo DCA e quando le cellule sono state trattate con la combinazione di sulindac e DCA. Va notato che entrambe le isoforme fosfo-p46JNK e fosfo-p54JNK sono state indotte dalla combinazione di trattamento con sulindac e DCA, sebbene l’aumento della fosfo-p46JNK sia stato più significativo (Figura 7B).
Numerose evidenze suggeriscono che JNK avvia il rilascio di fattori che inducono l’apoptosi dai mitocondri, come il citocromo c, che portano al clivaggio delle caspasi e della PARP (poli(ADP-ribosio) polimerasi) [30], [31]. Gli studi hanno anche dimostrato che durante l’apoptosi, il citocromo c rilasciato dai mitocondri nel citoplasma entra infine nel nucleo [32]. I nostri risultati indicano che l’attivazione massima di JNK si è verificata circa 12 ore dopo l’esposizione a sulindac e DCA. Questo sembra portare alla traslocazione del citocromo c nel citoplasma e al clivaggio di PARP 18 ore dopo il trattamento iniziale con sulindac e DCA (Figura S6A). Come controllo positivo per questi esperimenti abbiamo trattato le cellule con 100 µM di etoposide, un agente che induce l’apoptosi. Con il trattamento combinato di sulindac e DCA, è possibile osservare un aumento della fluorescenza nucleare nella maggior parte delle cellule che sono attivamente in fase di apoptosi (Figura S6B).
L’analisi dettagliata dei dati sperimentali dell’immunofluorescenza a cellule intere ha rivelato che il ∼94% delle cellule non trattate con sulindac o DCA mostrava una fluorescenza del citocromo c mitocondriale puntiforme, con scarsa colorazione diffusa nel citoplasma o nei nuclei. Al contrario, dopo il trattamento con sulindac, l’81% delle cellule ha mostrato una fluorescenza citoplasmatica diffusa e distinta e una fluorescenza nucleare molto ridotta. Dopo il trattamento con DCA, il ∼83% delle cellule ha mostrato una fluorescenza citoplasmatica diffusa e distinta e il <5% delle cellule ha mostrato una forte fluorescenza nucleare. Tuttavia, quando le cellule sono state trattate sia con sulindac che con DCA, il ∼72% delle cellule ha mostrato una fluorescenza sia nucleare che citoplasmatica e il ∼11% delle cellule ha mostrato una forte fluorescenza nucleare. Questi risultati suggeriscono che il citocromo c rilasciato dai mitocondri può avviare la via apoptotica intrinseca che funziona nell’uccisione del cancro mediata da sulindac e DCA.
Discussione
Il presente studio è un’estensione del nostro precedente lavoro, che ha dimostrato che il sulindac rende le cellule tumorali, ma non quelle normali, più sensibili allo stress ossidativo [7]. In questi esperimenti precedenti il sulindac è stato pre-incubato con le cellule per 24-48 ore e poi il sulindac è stato rimosso prima che le cellule fossero esposte a TBHP o H2O2 per 2 ore. Dagli esperimenti precedenti è risultato evidente che il pretrattamento con sulindac rendeva le cellule tumorali molto più sensibili all’agente ossidante, con conseguente forte aumento dei ROS e perdita della funzione mitocondriale [7].
Sulla base di questi risultati, sembrava ragionevole che il sulindac, in combinazione con composti che influenzano la respirazione mitocondriale, avrebbe portato a un’uccisione selettiva maggiore delle cellule tumorali, ma non delle cellule normali. Nel presente studio, condotto su linee cellulari tumorali A549 e SCC25, la combinazione di sulindac e DCA ha aumentato l’uccisione di queste linee cellulari tumorali, ma non di cellule normali del polmone o della pelle. I nostri risultati sulle quantità di DCA necessarie nelle cellule intere sono coerenti con quanto riportato in precedenza [28], [33], [34]. Nel nostro sistema, l’IC50 per il DCA con le cellule SCC25 è di 23 mM e per le cellule A549 è di 35 mM. L’IC50 per i cheratinociti normali è >50 mM e per le cellule polmonari normali (MRC5) è ∼40 mM. I risultati indicano inoltre che la morte delle cellule tumorali osservata coinvolge la produzione di ROS, l’attivazione di JNK e l’apoptosi avviata dai mitocondri. Per quanto riguarda la mancanza di effetti sulle cellule normali, è stato dimostrato che il sulindac protegge le cellule polmonari normali dal danno ossidativo derivante dall’esposizione al TBHP [7] e recentemente abbiamo anche riportato che il sulindac può proteggere le cellule cardiache dal danno ossidativo derivante dall’ischemia/riperfusione attraverso un meccanismo di precondizionamento [35].
A nostra conoscenza ci sono almeno 8 composti, compresi i nostri studi con TBHP, H2O2 e DCA, che hanno mostrato un’uccisione potenziata e selettiva del cancro in presenza di sulindac [7]–[9], [12]–[15]. Sebbene i loro bersagli metabolici all’interno della cellula siano noti e diversi, è abbastanza probabile che tutti, direttamente o indirettamente, causino la morte cellulare in presenza di sulindac attraverso un meccanismo che comporta un’alterazione della respirazione mitocondriale e della produzione di ROS [10], [12], [14]. Sembra probabile che quando si trova un farmaco che, in combinazione con il sulindac, uccide selettivamente le cellule tumorali, ma non le cellule normali, il meccanismo di uccisione coinvolga lo stress ossidativo che porta alla disfunzione mitocondriale. L’alterazione della respirazione potrebbe essere un fattore comune a questi esperimenti che utilizzano combinazioni di sulindac e farmaci e gli attuali risultati ottenuti con l’uso di DCA supportano questa tesi. È molto probabile che l’effetto del sulindac sia legato alle osservazioni fatte più di 50 anni fa da Warburg, il quale notò che le cellule normali preferiscono la respirazione per ottenere energia, mentre le cellule tumorali preferiscono la glicolisi, a causa di un difetto nella catena respiratoria [16]. Questa differenza di base nella respirazione mitocondriale tra cellule normali e cellule tumorali può rendere le cellule tumorali più sensibili allo stress ossidativo [36], [37]. Sembra che il sulindac possa amplificare questa differenza fondamentale nella biochimica delle cellule normali e tumorali. Il modo in cui il sulindac sensibilizza le cellule tumorali ai farmaci che influenzano la respirazione mitocondriale non è ancora chiaro, ma è oggetto di indagini attive. Spitz e collaboratori [38], in studi sulla privazione di glucosio delle cellule tumorali, sono giunti a una conclusione simile riguardo alle differenze nel metabolismo tra cellule normali e tumorali. In linea con questi risultati, un altro studio recente ha dimostrato che l’inibizione farmacologica della lattato deidrogenasi potrebbe portare a un’uccisione selettiva del cancro [39]. In questi ultimi studi è stato dimostrato che la maggiore uccisione selettiva delle cellule tumorali comportava anche la produzione di ROS e l’effetto osservato è stato attribuito a un processo respiratorio alterato nelle cellule tumorali.
Va sottolineato che la combinazione di sulindac con un agente ossidante o con farmaci che possono influenzare la funzione mitocondriale è già stata testata in ambito clinico. Meyskens et al. nel 2008 hanno dimostrato che la combinazione di sulindac con DFMO ha avuto un effetto significativo sulla recidiva dei polipi del colon e sulla comparsa del cancro del colon in uno studio clinico di 3 anni [13]. Recentemente abbiamo riportato l’uso di sulindac, con H2O2, in uno studio clinico proof of concept per il trattamento topico delle cheratosi attiniche [15]. Uno degli svantaggi dell’uso di questa combinazione era la necessità di due formulazioni topiche, poiché i composti non potevano essere conservati per lunghi periodi senza che il sulindac venisse distrutto dall’H2O2. Inoltre, non è possibile utilizzare l’H2O2 per il trattamento dei tumori interni, poiché non può essere assunto per via orale. Tuttavia, la combinazione di sulindac e DCA potrebbe essere fornita come un’unica formulazione adatta all’uso topico e i due composti possono essere utilizzati per via orale. Da diversi anni, infatti, il DCA viene utilizzato clinicamente per abbassare i livelli di acido lattico nei pazienti affetti da acidosi lattica [40]–[42]. Il DCA è stato utilizzato anche come agente antitumorale in vitro e in vivo utilizzando diverse linee cellulari tumorali, indicando che il metabolismo mitocondriale nelle cellule tumorali potrebbe essere un nuovo bersaglio terapeutico [18], [20], [22]. Michelakis et al. (2010) hanno dimostrato che il trattamento con DCA “rimodella” il metabolismo mitocondriale nei pazienti affetti da glioblastoma con effetti tossici reversibili. Va notato che sia il sulindac che il DCA sono economici, relativamente non tossici e possono essere assunti per via orale. Se la combinazione si rivelerà efficace in vivo, aggiungerà una nuova dimensione al trattamento del cancro, poiché entrambi i farmaci hanno come bersaglio il metabolismo mitocondriale in diversi tipi di cancro [22].
In sintesi, i nostri studi con la combinazione di sulindac e DCA suggeriscono che il sulindac rende selettivamente le cellule tumorali più sensibili agli agenti che influenzano la respirazione mitocondriale, con conseguente stress ossidativo e disfunzione mitocondriale. Questi risultati potrebbero essere correlati al difetto di respirazione nelle cellule tumorali, originariamente osservato da Warburg [16]. Sono attualmente in corso studi volti a comprendere le differenze fondamentali tra il modo in cui le cellule tumorali e quelle normali rispondono al sulindac e agli agenti che influenzano la funzione mitocondriale.
Informazioni di supporto
Figura S1 Citotossicità di sulindac, DCA o combinazione di farmaci su cellule tumorali A549 e SCC25
Le cellule tumorali A549 e SCC25 sono state trattate con le concentrazioni indicate di sulindac (pannelli 1 e 4) o DCA (pannelli 2 e 5). Negli esperimenti di combinazione di farmaci sulindac/DCA, è stato mantenuto un rapporto di 1:50 (sulindac:DCA) per le cellule A549, con combinazioni di farmaci che andavano da 0,2 mM:10 mM fino a 1 mM:50 mM (pannello 3) e un rapporto di 1:100 (sulindac:DCA) per le cellule SCC25 con combinazioni di farmaci che andavano da 0,05 mM:5 mM fino a 0,3 mM:30 mM (pannello 6). (TIF) Figura S2 Determinazione degli indici di combinazione per le cellule tumorali A549 e SCC25. Gli indici di combinazione dei farmaci sono stati determinati incorporando i valori di vitalità cellulare ottenuti sopra nelle equazioni di Chou e Talalay [26]. Si veda il testo per ulteriori dettagli. (TIF) Figura S3 Quantificazione delle cellule verdi fluorescenti positive a DCF e JC-1. Le cellule SCC25 sono state trattate con sulindac, DCA o una combinazione di farmaci per 48 ore e colorate con i coloranti H2DCFDA (Fig. S3A) o JC-1 (Fig. S3B) come indicato nei Metodi. Per il controllo positivo, le cellule sono state trattate con 200 mM di TBHP per 2 ore e la colorazione è stata eseguita come sopra. Le cellule sono state analizzate con ingrandimento ad alta potenza utilizzando l’obiettivo 1006 in un microscopio invertito a fluorescenza Olympus. Sono state visualizzate almeno 100 singole cellule per ogni condizione e le percentuali di cellule fluorescenti verdi DCF-positive e JC1-positive sono presentate in formato tabellare e grafico. (TIF) Figura S4 Il ROS scavenger Tiron inverte l’uccisione delle cellule tumorali da parte della combinazione di sulindac e DCA. Le cellule tumorali A549 e SCC25 sono state trattate con le concentrazioni indicate di DCA in assenza (barra grigia) o in presenza di sulindac (barra nera) o in presenza di sulindac e Tiron (barra a righe) per 48 ore. La vitalità cellulare è stata monitorata con il saggio MTS come indicato nei Metodi. La vitalità cellulare è espressa come % del controllo (cellule non trattate con sulindac). Le barre di errore rappresentano l’errore standard della media (SEM) espresso come % del valore medio di quadruplicati di un esperimento rappresentativo. L’inibizione della crescita delle cellule tumorali si è verificata in modo dipendente dalla dose durante il trattamento combinato di DCA e sulindac (barre nere) sia nelle cellule tumorali A549 (A) che in quelle SCC25 (B). Tuttavia, questo aumento dell’uccisione è stato impedito quando il Tiron è stato presente insieme al trattamento di combinazione dei farmaci (barre a righe in A e B). (TIF) Figura S5 Durante l’uccisione delle cellule tumorali da parte della combinazione di sulindac e DCA si verifica un pronunciato laddering del DNA nucleosomiale. Le cellule tumorali A549 e SCC25 sono state trattate con i farmaci indicati per 48 ore. Il DNA nucleosomiale è stato estratto e sottoposto a PCR mediata da ligazione, come descritto nei Metodi, e analizzato su un gel di agarosio all’1,2% insieme a marcatori di dimensione. La corsia “M” indica i marcatori di dimensione molecolare. Le corsie 1-4 e 5-8 illustrano i risultati ottenuti rispettivamente con cellule tumorali A549 e SCC25. I risultati sono illustrati nelle corsie 1 e 5 (nessun farmaco), nelle corsie 2 e 6 (solo sulindac), nelle corsie 3 e 7 (solo DCA) e nelle corsie 4 e 8 (sulindac e DCA). Una maggiore laddering del DNA nucleosomiale è stata osservata solo con il trattamento combinato di sulindac e DCA (corsie 4 e 8). (TIF)
Figura S6 La combinazione di sulindac e DCA porta al rilascio di citocromo c dai mitocondri e al clivaggio di PARP
Le celluleSCC25sono state trattate con sulindac, DCA, combinazione di farmaci o etoposide per analizzare la localizzazione intracellulare del citocromo c mediante western blotting e immunofluorescenza. A. Le frazioni citosoliche sono state isolate a 18 ore e la presenza di citocromo c nel citoplasma e il clivaggio di PARP sono stati determinati mediante western blotting. I western blot rappresentativi mostrano la quantità di citocromo c e di PARP clivato. I livelli di b-actina sono stati usati come controllo interno. B. L’immunofluorescenza è stata eseguita utilizzando il kit di rilascio del citocromo c a flusso InnoCyteTM CBA077, secondo le istruzioni del produttore. Sono stati analizzati diversi campi indipendenti e le micrografie rappresentative mostrano i modelli di localizzazione del citocromo c quando le cellule sono esposte a sulindac e/o DCA. I valori quantitativi sono presentati nel testo. (TIF)
Ringraziamenti
Gli autori ringraziano David Brunell per l’aiuto nella determinazione degli indici di combinazione e Edna Gamliel per l’assistenza alla coltura cellulare.
Contributi degli autori
Ideazione e progettazione degli esperimenti: KA SK KDS HW. Esecuzione degli esperimenti: KA SK. Analisi dei dati: KA SK HW. Ha contribuito con reagenti/materiali/strumenti di analisi: KDS. Ha scritto l’articolo: KA SK HW.
RIFERIMENTI
1 Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, et al. (1996) La sovraespressione della ciclossigenasi-2 e la formazione di tumori sono bloccate dal sulindac in un modello murino di poliposi adenomatosa familiare. Cancer Res 56: 2556-2560.
2 Taketo MM (1998) Inibitori della cicloossigenasi-2 nella tumorigenesi (Parte II). J Natl
3 Taketo MM (1998) Inibitori della cicloossigenasi-2 nella tumorigenesi (parte I). J Natl Cancer Inst 90: 1529-1536.
4 Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, et al. (1995) Chemioprevenzione della carcinogenesi del colon mediante sulindac, un agente antinfiammatorio non steroideo. Cancer Res 55: 1464-1472.
5 Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, et al. (2001) La progressione e l’apoptosi indotta dai FANS nei melanomi maligni sono indipendenti dalla ciclossigenasi II. Melanoma Res 11: 587-599.
6 Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Inibizione della crescita e induzione dell’apoptosi nelle cellule tumorali del colon-retto da parte degli inibitori della ciclossigenasi. Carcinogenesi 22: 17-25.
7 Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, et al. (2009) Il sulindac aumenta l’uccisione delle cellule tumorali esposte allo stress ossidativo. PLoS One 4: e5804.
8 Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, et al. (1999) Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 59: 6178-6184.
9 Jiang TT, Brown SL, Kim JH (2004) Effetto combinato di triossido di arsenico e solfuro di sulindac in cellule di cancro al polmone umano A549 in vitro. J Exp Clin Cancer Res 23: 259-262.
10 Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, et al. (2006) Synergistic induction of apoptosis by sulindac and arsenic trioxide in human lung cancer A549 cells via reactive oxygen species-dependent down-regulation of survivin. Biochem Pharmacol 72: 1228-1236.
11 Jin HO, Seo SK, Woo SH, Lee HC, Kim ES, et al. (2008) Una combinazione di sulindac e triossido di arsenico induce sinergicamente l’apoptosi nelle cellule di tumore polmonare umano H1299 attraverso la fosforilazione di Bcl-xL dipendente dalla c-Jun NH2-terminal kinase. Lung Cancer 61: 317-327.
12 Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, et al. (2005) Il sulindac potenzia l’attività antitumorale e lo stress ossidativo mediati dall’inibitore del proteasoma bortezomib. Clin Cancer Res 11: 5248-5256.
13 Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, et al. (2008) Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila Pa) 1: 32-38.
14 Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, et al. (2008) Effetti combinati del sulindac e dell’acido suberoilanilide idrossamico sull’induzione dell’apoptosi in cellule di cancro al polmone umano. Mol Pharmacol 73: 1005-1012.
15 Resnick L, Rabinovitz H, Binninger D, Marchetti M, Weissbach H (2009) Sulindac topico combinato con perossido di idrogeno nel trattamento delle cheratosi attiniche. J Drugs Dermatol 8: 29-32.
16 Warburg O (1956) Sull’origine delle cellule cancerose. Science 123: 309-314.
17 Whitehouse S, Cooper RH, Randle PJ (1974) Meccanismo di attivazione della piruvato deidrogenasi da parte del dicloroacetato e di altri acidi carbossilici alogenati. Biochem J 141: 761-774.
18 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37-51.
19 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60: 1478-1487.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109: 394-402.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Il dicloroacetato (DCA) sensibilizza in vitro alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata 68: 1223-1231.
<span id=”22″ class=”referencess blue-text “22 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, et al. (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2: 31ra34.
23 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989-994.
24 Cory AH, Owen TC, Barltrop JA, Cory JG (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3: 207-212.
<span id=”25″ class=”referencess blue-text “25 Staley K, Blaschke AJ, Chun J (1997) Apoptotic DNA fragmentation is detected by a semi-quantitative ligation-mediated PCR of blunt DNA ends. Cell Death Differ 4: 66-75.
26 Chou TC, Talalay P (1984) Analisi quantitativa delle relazioni dose-effetto: gli effetti combinati di più farmaci o inibitori enzimatici. Adv Enzyme Regul 22: 27-55.
<span id=”27″ class=”referencess blue-text “27 Papandreou I, Goliasova T, Denko NC (2010) Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer 128: 1001-1008.
28 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, et al. (2010) Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer 127: 2510-2519.
29 Babbar N, Ignatenko NA, Casero RA Jr, Gerner EW (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J Biol Chem 278: 47762-47775.
30 Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haikel Y, et al. (2011) Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenesi 32: 1268-1278.
31 Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, et al. (2007) La c-Jun N-terminal kinase media la morte cellulare indotta dal perossido di idrogeno attraverso l’attivazione sostenuta della polimerasi-1 (ADP-ribosio). Cell Death Differ 14: 1001-1010.
<span id=”32″ class=”referencess blue-text “32 Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, et al. (2004) Nuclear translocation of cytochrome c during apoptosis. J Biol Chem 279: 24911-24914.
33 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, et al. (2011) La terapia metabolicamente mirata con dicloroacetato sconfigge la citotossicità dei farmaci antitumorali standard. Cancer Chemother Pharmacol 67: 647-655.
34 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 102: 1746-1752.
35 Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac conferisce una protezione ischemica di alto livello al cuore attraverso meccanismi di precondizionamento tardivo. Proc Natl Acad Sci U S A 106: 19611-19616.
<span id=”36″ class=”referencess blue-text “36 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18: 54-61.
37 Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029-1033.
<span id=”38″ class=”referencess blue-text “38 Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, et al. (2005) Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280: 4254-4263.
<span id=”39″ class=”referencess blue-text “39 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107: 2037-2042.
<span id=”40″ class=”referencess blue-text “49 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI (1983) Treatment of lactic acidosis with dichloroacetate. N Engl J Med 309: 390-396.
41 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77: 535-541.
42 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, et al. (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatria 121: e1223-1228.
Contenuti correlati: