Kasirajan Ayyanathan1,2,*,#, Shailaja Kesaraju1,#, Ken Dawson-Scully1,2, Herbert Weissbach1
1 Center for Molecular Biology and Biotechnology, Charles E. Schmidt College of Science, Florida Atlantic University, Jupiter, Florida, Estados Unidos de América,
2 Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Ratón, Florida, Estados Unidos de América
*E-mail: [email protected]
#Theseauthors contributed equally to this work.
Recibido: 12 de agosto de 2011
Aceptado: 4 de junio de 2012
Publicado: 1 de julio de
Resumen
El sulindaco es un fármaco antiinflamatorio no esteroideo aprobado por la FDA con actividades anticancerígenas documentadas. Nuestros estudios recientes mostraron que el sulindaco aumentó selectivamente la muerte de las células cancerosas expuestas a agentes oxidantes a través de la producción de especies reactivas de oxígeno (ROS) que resulta en la disfunción mitocondrial. Este efecto del sulindac y del estrés oxidativo en las células cancerosas podría estar relacionado con el defecto en la respiración de las células cancerosas, descrito por primera vez por Warburg hace 50 años, conocido como efecto Warburg. Postulamos que el sulindac podría potenciar la eliminación selectiva de células cancerosas cuando se combina con cualquier compuesto que altere la respiración mitocondrial. Para probar esta hipótesis hemos utilizado dicloroacetato (DCA), que se sabe que desplaza el metabolismo del piruvato de la formación de ácido láctico a la respiración. Cabría esperar que el DCA, dado que estimula el metabolismo aeróbico, pudiera estresar la respiración mitocondrial en las células cancerosas, lo que daría lugar a una mayor destrucción en presencia de sulindac. En este estudio, hemos demostrado que la combinación de sulindac y DCA potencia la destrucción selectiva de las células cancerosas A549 y SCC25 en las condiciones utilizadas. Como se predijo, el mecanismo de destrucción implica la producción de ROS, la disfunción mitocondrial, la señalización JNK y la muerte por apoptosis. Nuestros resultados sugieren que la combinación de fármacos sulindac-DCA puede proporcionar una terapia eficaz contra el cáncer.
Citación: Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H (2012) La combinación de sulindac y dicloroacetato mata las células cancerosas a través del daño oxidativo
. PLoS ONE 7(7): e39949. doi:10.1371/journal.pone.0039949
Editor: Joseph Alan Bauer, Fundación de Investigación Bauer, Estados Unidos de América
2012 Ayyanathan et al. Este es un artículo de acceso abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons, que permite a
su uso, distribución y reproducción sin restricciones en cualquier medio, siempre que se cite al autor original y la fuente.
Financiación: Se agradece la ayuda financiera de los Institutos Nacionales de Salud a KA (subvención 5K01CA95620) y HW (subvención R15 CA122001) y del Estado de Florida a HW (subvención SURECAG R94007) para llevar a cabo este trabajo. Los financiadores no intervinieron en el diseño del estudio, la recopilación y el análisis de los datos, la decisión de publicar o la preparación del manuscrito.
Competing Interests: Los autores han declarado que no tienen intereses contrapuestos.
INTRODUCCIÓN
El sulindaco es un fármaco antiinflamatorio no esteroideo (AINE) aprobado por la FDA, que también ha demostrado tener actividad anticancerígena [1-6]. Estudios recientes de nuestro laboratorio han demostrado que las líneas celulares de cáncer RKO, A549 y SCC25 mostraban sensibilidad a una combinación de sulindac y un agente oxidante, como TBHP o H2O2 [7]. Los datos indicaban que el efecto del sulindac no estaba relacionado con su actividad como AINE, sino que el sulindac hacía que las células cancerosas fueran más sensibles al estrés oxidativo, lo que provocaba disfunción mitocondrial y pérdida de viabilidad. En cambio, las células normales no mostraron una mayor destrucción en condiciones similares [7]. En los últimos 10 años ha habido informes dispersos sobre la mejora de la eliminación del cáncer utilizando sulindac en combinación con una variedad de compuestos, incluyendo trióxido de arsénico, bortezomib, difluorometilornitina (DFMO) y ácido hidroxámico suberoilanílido (SAHA) [8-14]. Aunque estos compuestos tienen diferentes sitios de acción, un mecanismo común para la mejora de la destrucción de la combinación sulindac/fármaco podría implicar el daño oxidativo, como se demostró claramente en nuestros estudios anteriores utilizando sulindac y un agente oxidante [7], [15]. De hecho, las ERO han sido implicadas en los estudios que utilizan sulindac en combinación con trióxido de arsénico, bortezomib y SAHA [10], [12], [14].
Nuestros resultados anteriores sugerían que la mayor destrucción de células cancerosas por la combinación de sulindac y un agente oxidante podría deberse a un defecto en la respiración de las células cancerosas, como describió por primera vez Warburg hace más de 50 años [16], quien observó que las células cancerosas favorecen la glucólisis, y no la respiración, para obtener energía, a diferencia de las células normales. Algunas células cancerosas obtenían hasta el 50% de su energía de la glucólisis, mientras que en las células normales la glucólisis representaba menos del 5% de las necesidades energéticas [16]. Para obtener más pruebas de las posibles funciones de la respiración alterada y las ERO en la muerte de las células cancerosas por sulindac y estrés oxidativo, iniciamos estudios con ácido dicloroacético sódico (DCA). El DCA es un candidato ideal, ya que se sabe que inhibe una quinasa que regula a la baja la actividad de la piruvato deshidrogenasa, lo que provoca un desplazamiento del metabolismo del piruvato desde la formación de ácido láctico hacia la respiración [17], [18]. El DCA se ha utilizado clínicamente para tratar a pacientes con acidosis láctica [19] y, gracias a sus propiedades bioquímicas, también se ha probado como agente anticancerígeno. Bonnet et al. 2007 han demostrado que el DCA invierte el efecto Warburg en las células cancerosas redirigiendo el metabolismo de las células cancerosas de la glucólisis a la fosforilación oxidativa. En estos estudios previos se demostró que el DCA aumenta los niveles de ROS del complejo I. Esto a su vez desencadena una «remodelación» del metabolismo mitocondrial (reduce ΔΨm, abre el poro de transición mitocondrial) en las células cancerosas empujándolas hacia la apoptosis. Además, varios estudios recientes han verificado que el DCA puede aumentar los niveles de ROS en las células cancerosas y despolarizar la membrana de las mitocondrias en líneas celulares de pulmón, endometrio y glioblastoma, provocando la apoptosis tanto in vitro como in vivo [18], [20-22]. Resulta interesante observar que, en las condiciones utilizadas, el DCA no parece afectar significativamente al metabolismo mitocondrial ni a la viabilidad de las células normales [18], [23].
Basándonos en nuestras observaciones previas sobre el efecto anticancerígeno de sulindac y un agente oxidante que afectaba al metabolismo mitocondrial [7], postulamos que la combinación de sulindac y DCA podría potenciar sinérgicamente la eliminación del cáncer y tener un importante valor terapéutico. En el presente estudio hemos examinado el efecto del uso de sulindac en combinación con DCA sobre la viabilidad de las líneas celulares de cáncer A549 y SCC25. También hemos estudiado el papel de la función mitocondrial y la apoptosis en la eliminación del cáncer observada con esta combinación de fármacos.
Materiales y métodos
Materiales
Sulindac, N-acetilcisteína y Tiron se adquirieron a Sigma (St.Louis, MO). La sal sódica de DCA se obtuvo de Acros Organics (Geel, Bélgica). H2DCFDA y JC-1 se adquirieron a Molecular Probes (Eugene, OR). El reactivo de ensayo MTS y el kit Deadend Tunel se obtuvieron de Promega (Madison, WI). El kit de fraccionamiento de citosol/mitocondrias y el kit CBA077 InnoCyte™ Flow Cytometric Cytochrome c Release eran de Calbiochem, Gibbstown, NJ. Todos los medios de cultivo celular, el suero bovino fetal y otros suplementos como penicilina/estreptomicina, glutamina, etc. se adquirieron a American Type Culture Collection (ATCC; Rockville, MD).
Cultivo celular
Se adquirieron a ATCC (Rockville, MD)unalínea celular de carcinoma pulmonar de células no pequeñas (CPCNP), A549, la línea celular de pulmón humano normal, MRC-5, y una línea de carcinoma de células escamosas derivado de la lengua, SCC25, y se mantuvieron en medio F12-K suplementado con suero bovino fetal al 10%, 2 mM de glutamina, 100 UI/ml de penicilina y 100 µg/ml de estreptomicina en un incubador humidificado con un 5% deCO2 a 37°C. Los queratinocitos epidérmicos humanos normales se obtuvieron de Promocell GmbH (Heidelberg, Alemania) y se mantuvieron en el medio de cultivo recomendado. Para los experimentos se utilizaron células normales no inmortalizadas de paso temprano.
Ensayo de viabilidad celular
Las células cancerosas A549 y las normales de pulmón se sembraron a 3×103 células por pocillo, mientras que las células cancerosas SCC25 y los queratinocitos normales se sembraron a 7,5×103 células por pocillo en una placa de 96 pocillos. Las células se cultivaron durante 18-20 horas, el medio se desechó en condiciones asépticas y se sustituyó por medio de cultivo fresco que contenía las combinaciones de fármacos indicadas. Cuando se indicó, se utilizaron 500 µM de sulindac con las células A549 cancerosas y normales de pulmón y 100 µM de sulindac con las células SCC25 cancerosas y normales de queratinocitos. Las placas se incubaron durante 48 horas a 37°C en un incubadorcon un 5% deCO2. Se desechó el medio de cultivo y las células se lavaron a fondo en 1× PBS. La viabilidad celular se determinó mediante el ensayo CellTiter 96 Aqueous One Cell Proliferation Assay (Promega) siguiendo las instrucciones del fabricante. El ensayo utiliza un compuesto de tetrazolio que se convierte en formazán hidrosoluble por la acción de las deshidrogenasas celulares presentes en las células metabólicamente activas [24]. El formazán se cuantificó midiendo la absorbancia a 490 nm mediante un lector colorimétrico de placas de microtitulación (SpectraMax Plus; Molecular Devices). La absorbancia de fondo se restó de cada muestra.
Medición intracelular de ROS
Las líneas celulares de cáncer A549 y SCC25 se sembraron como se ha indicado anteriormente. Tras el tratamiento farmacológico de 48 horas, las células se incubaron con 50 µM de diacetato de diclorodihidrofluoresceína (H2DCFDA, Molecular Probes) en medio libre de indicador durante 30 min a 37°C. Las células se enjuagaron con PBS y los niveles de ROS se visualizaron mediante microscopía de fluorescencia. Las imágenes se capturaron utilizando el software Qcapture y se procesaron en Adobe photoshop. El análisis de las imágenes se realizó con el software Slidebook. Los datos obtenidos de un experimento representativo se utilizaron para la cuantificación de células DCF-positivas medidas por la fluorescencia verde debida al DCF oxidado.
Tinción JC-1 para monitorizar el potencial de membrana mitocondrial
El potencial de membranamitocondrialse determinó utilizando el colorante JC-1 (Molecular Probes). Las líneas celulares de cáncer A549 y SCC25 se sembraron como se ha indicado anteriormente. Tras el tratamiento farmacológico de 48 horas, las células se incubaron con 5 ng/ml de colorante JC-1 en medio libre de indicadores durante 30 min a 37°C. Las células se enjuagaron con PBS y se visualizaron mediante microscopía de fluorescencia. Las mitocondrias normales captan activamente el colorante JC-1 de forma dependiente del potencial y forman agregados J, lo que produce una fluorescencia roja. La interrupción y posterior pérdida del potencial de membrana mitocondrial conduce a un aumento de la fluorescencia verde en el citosol debido al JC-1 monomérico, que se determina siguiendo la aparición de fluorescencia verde utilizando un filtro FITC (microscopio invertido Zeiss-Axiovert 40 CFL). La captura de imágenes, el procesamiento y el análisis se realizaron como se ha indicado anteriormente. Los datos obtenidos de un experimento representativo se utilizaron para la cuantificación de células positivas al verde JC-1.
Effect of ROS Scavengers on Cell Viability in the Presence of Sulindac and DCA
Las líneas celulares de cáncer A549 y SCC25 se sembraron como se ha descrito anteriormente. Para eliminar las ERO, se añadieron 2 mM de N-acetilcisteína (NAC) o 2 mM de Tiron (sal disódica del ácido 4,5-dihidroxi-1,3-bencenodisulfónico) junto con sulindac y DCA durante 48 h a 37°C. La viabilidad celular se monitorizó mediante el ensayo MTS y el análisis estadístico se realizó como se ha mencionado anteriormente.
Tinción TUNEL para monitorizar las células sometidas a apoptosis
El ensayoTUNELse realizó en placas de 96 pocillos utilizando el kit de ensayo colorimétrico TUNEL DeadEnd (Promega) siguiendo el protocolo del fabricante. Las líneas celulares de cáncer A549 y SCC25 se sembraron como se ha indicado anteriormente y se trataron durante 48 horas sin fármaco, con sulindac, con DCA o con una combinación de fármacos. Tras el tratamiento farmacológico, las células se fijaron con formalina y se permeabilizaron con Triton X-100 al 0,2% en PBS. Las células se incubaron con desoxinucleotidil transferasa terminal recombinante (TdT) y nucleótidos biotinilados. Las peroxidasas endógenas se bloquearon con H2O2 al 0,3% antes de la incubación con peroxidasa de rábano picante-estreptavidina (HRP-estreptavidina) que se une a los nucleótidos biotinilados incorporados en los extremos mellados presentes en las células sometidas a apoptosis. Las células marcadas con HRP-estreptavidina se detectaron mediante peróxido de hidrógeno y diaminobencidina (DAB). Las células que muestran una tinción nuclear marrón oscura son indicativas de apoptosis.
Análisis Western Blot
Lascélulas se cultivaron hasta un 70% de confluencia, se trataron con los fármacos especificados durante los periodos indicados y se aislaron las fracciones citosólicas utilizando el kit de fraccionamiento de citosol/mitocondrias (Calbiochem, Gibbstown, NJ) siguiendo el protocolo del fabricante. Brevemente, las células se cosecharon en diferentes puntos temporales y luego se centrifugaron a 600×g durante 5 min a 4°C. Las células sedimentadas se suspendieron en el tampón suministrado y se incubaron durante 10 minutos en hielo. A continuación, las células se homogeneizaron con un separador de vidrio y el homogeneizado se centrifugó a 700×g durante 10 minutos a 4°C para sedimentar los núcleos y los restos celulares. El sobrenadante se centrifugó a 10, 000×g durante 30 min a 4°C para obtener el pellet mitocondrial y el sobrenadante se consideró la fracción citosólica. La concentración de proteínas se determinó mediante un ensayo estándar de Bradford.
Se cargaron 60 microgramos de proteína total y se separaron en un gel NuPage Bis-Tris al 4-12% (Invitrogen, Eugene, OR) y se transfirieron a una membrana de PVDF que se sondeó con los anticuerpos primarios. Los anticuerpos primarios, JNK, pJNK, citocromo c y PARP (Cell Signaling Technology, Danvers, MA), se utilizaron en dilución 1∶1000. la β-actina (Santa Cruz Biotechnologies, Santa Cruz, California) se utilizó a una dilución de 1∶4000. Se utilizaron anticuerpos secundarios conjugados con peroxidasa de rábano picante y las bandas se visualizaron mediante un método de quimioluminiscencia mejorada (GE Healthcare, Piscataway, NJ).
Ensayode laddering de ADN basado en PCR mediada por ligadurapara monitorizar el grado de apoptosis de las células
Para confirmar el grado de apoptosis, se realizó un ensayo de laddering de ADN nucleosómico basado en PCR mediada por ligadura tal y como se describe [25]. Las líneas celulares de cáncer A549 y SCC25 se sembraron con 5×104 y 1×105 células por pocillo en placas de 35 mm. Las células cancerosas A549 se trataron durante 48 horas con a) ningún fármaco, b) 500 µM de sulindac, c) 20 mM de DCA y d) 500 µM de sulindac más 20 mM de DCA. Del mismo modo, las células cancerosas SCC25 se trataron con las cuatro combinaciones diferentes de fármacos mencionadas, excepto que el sulindac y el DCA se utilizaron a concentraciones de 100 µm y 10 mM, respectivamente. Tras el tratamiento, se extrajo el ADN celular total y se ligó al adaptador construido a partir de 27-mer 5′-GACGTCGACGTCGTACACGTGTCGACT-3′ y 12-mer 5′- AGTCGACACGTGTAC-3′. Tras la ligación, el ADN se calentó para liberar el 12-mer, se llenó con Taq polimerasa, se sometió a PCR semicuantitativa y se analizó en un gel de agarosa al 1,2% junto con marcadores de tamaño.
Localizaciónin situdel citocromo c mediante inmunofluorescencia
La localización intracelular del citocromo c se monitorizó mediante inmunofluorescencia utilizando el kit de liberación citométrica del citocromo c CBA077 InnoCyte™ Flow Cytometric Cytochrome c
acuerdo con las instrucciones del fabricante. Brevemente, las células SCC25 se sembraron a 3,5×105 células por placa de fondo de cristal de 35 mm y se trataron con los fármacos indicados durante 15 h. Las células se enjuagaron en 5 ml de PBS 1× y se permeabilizaron en hielo durante 10 min en 300 µl del tampón suministrado. Las células se fijaron a temperatura ambiente durante 20 minutos en 500 µl de paraformaldehído al 4%. Tras lavar y bloquear, las células se incubaron con 250 µl de anticuerpo anti-citocromo c (dilución 1∶500) durante 1 hora a temperatura ambiente. Tras el lavado, las células se incubaron con 300 µl de FITC-IgG (dilución 1∶300) durante 1 hora a temperatura ambiente. Por último, las células se tiñeron con 300 µl de DAPI (1 mg/ml) durante 10 minutos a temperatura ambiente. Las células se visualizaron con un microscopio fluorescente invertido Olympus. Las imágenes se capturaron y procesaron como se ha mencionado anteriormente. Se analizaron varios campos y se obtuvieron micrografías representativas que mostraban los patrones de localización del citocromo c en cada condición de tratamiento. Los valores cuantitativos se presentan en el texto.
Análisis estadístico y determinación de índices de combinación
Los datos se presentan como media ± SEM para los ensayos de viabilidad celular. Para el análisis estadístico, se utilizó el programa estadístico Minitab para realizar la prueba t de Student y los valores con p<0,05 se consideraron estadísticamente significativos. Para determinar el efecto sinérgico de sulindac y DCA en las líneas celulares de cáncer A549 y SCC25, se realizó un análisis cuantitativo de la relación dosis-efecto para determinar los índices de combinación [26]. Tanto el sulindac como el DCA se ensayaron solos en las células A549 y SCC25 a las concentraciones indicadas. Para las células A549, se mantuvo una relación de 1∶50 para las combinaciones de fármacos sulindac:DCA desde 0,2 mM:10 mM hasta 1 mM:50 mM, respectivamente. Para las células SCC25, se mantuvo un índice de 1∶100 para las combinaciones de fármacos sulindac:DCA que oscilaban entre 0,05 mM:5 mM y 0,3 mM:30 mM, respectivamente. Nuestros resultados experimentales y los valores determinados del índice de combinación se incluyen en el texto.
Resultados
El sulindacy el DCAprovocan una mayor destrucción de las célulascancerosas A549y SCC25, pero no de las células normales
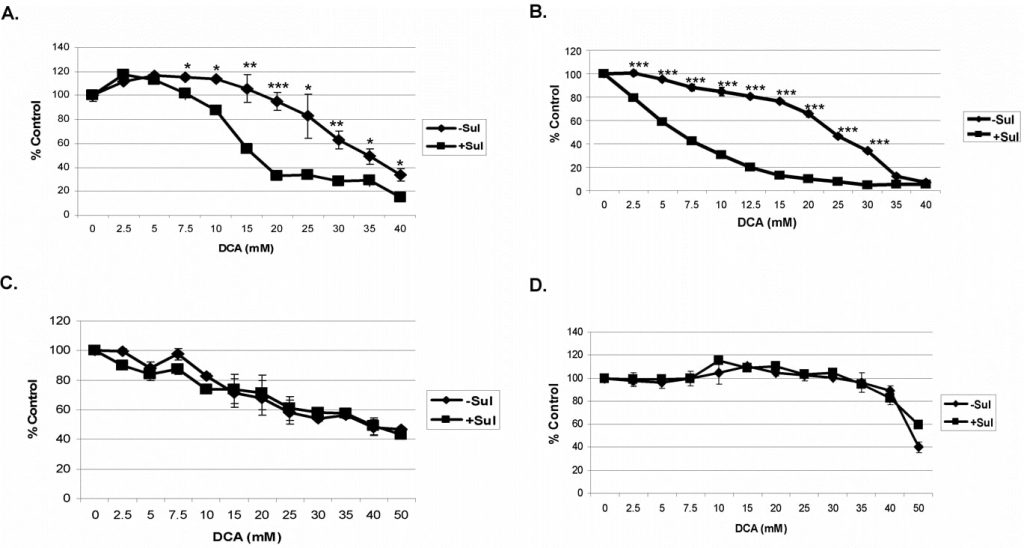
Para estos estudios probamos la combinación de sulindac y DCA en las células cancerosas A549 y SCC25. Las células se incubaron con cada compuesto solo o en combinación durante 48 horas antes de analizar su viabilidad (ver Métodos). Una curva dosis-respuesta de sulindac en estas condiciones indicó que las células cancerosas A549 y SCC25 pueden tolerar una concentración máxima de 500 µM y 100 µM de sulindac, respectivamente, sin mostrar ninguna muerte significativa (datos no mostrados), y estas concentraciones se utilizaron en todos los estudios. El DCA, cuando se añadió, se utilizó en concentraciones de 0-40 mM, como se indica. Utilizamos estas concentraciones basándonos en informes anteriores, que indicaban que se requieren más de 5 mM para causar disfunción mitocondrial en experimentos in vitro [27]. Como se muestra en la Figura 1A, el DCA solo (sin sulindac) es algo tóxico para las células cancerosas A549, especialmente por encima de concentraciones de 20 mM, pero en presencia de sulindac se produce una mayor destrucción de estas células a concentraciones de DCA superiores a 5 mM. En el caso de las células cancerosas SCC25, se observó cierta pérdida de viabilidad celular con DCA solo, incluso a concentraciones de DCA inferiores a 10 mM (Figura 1B). Sin embargo, en presencia de sulindac se produjo de nuevo un marcado aumento de la muerte celular que fue claramente evidente entre concentraciones de DCA de 2-10 mM. Anteriormente demostramos que la combinación de sulindac y un agente oxidante era selectiva para las células cancerosas y no potenciaba la muerte de células normales [7]. El sulindac y el DCA tampoco potenciaron la muerte de células normales de pulmón y piel en las condiciones experimentales utilizadas, como se muestra en las Figuras 1C y D. Cabe señalar que las células MRC-5 (normales de pulmón) son especialmente sensibles al DCA, como se informó anteriormente [28], por razones que se desconocen.
Para verificar que se producía un efecto sinérgico cuando se utilizaba la combinación de fármacos, determinamos los índices de combinación realizando un análisis cuantitativo de la relación dosis-efecto [26] en dos líneas celulares de cáncer diferentes (Figura S1). Los índices de combinación fueron de 0,84 para las células cancerosas A549 y de 0,73 para las células cancerosas SCC25, respectivamente. Un valor inferior a 1,00 indica un efecto sinérgico de eliminación del cáncer (Figura S2).
El efecto del sulindac no se debe a su actividad como AINE
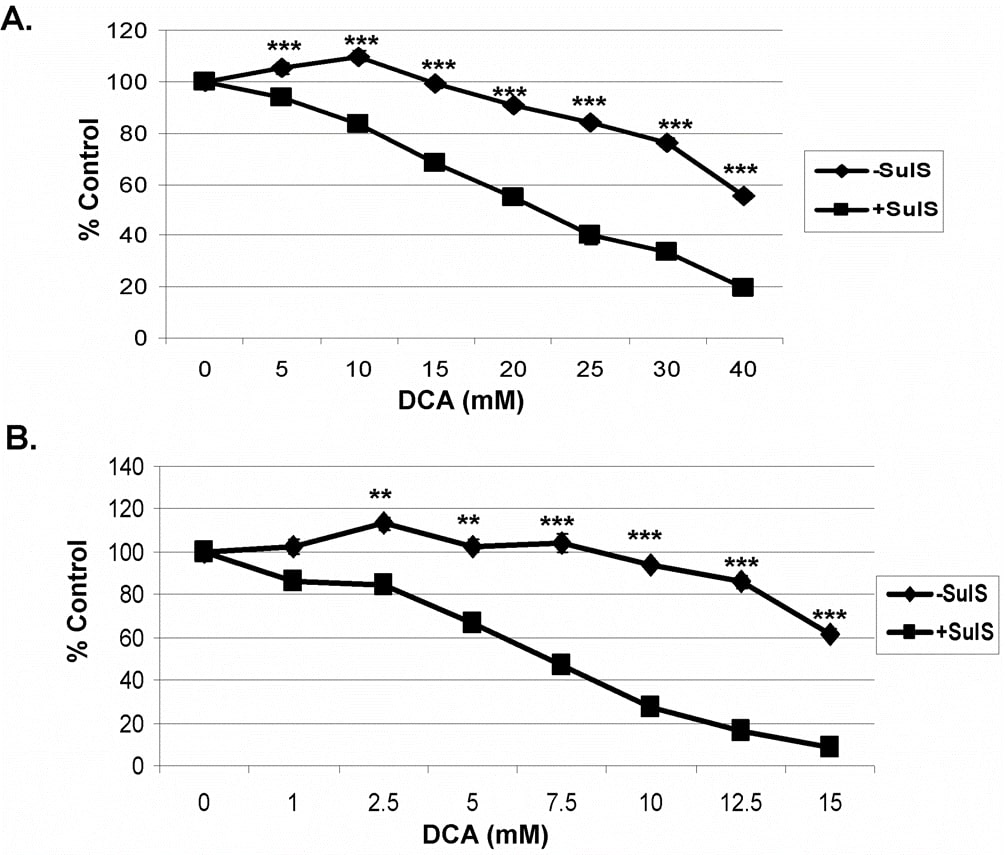
En estudios anteriores en los que se utilizó sulindac y un agente oxidante se demostró que la eliminación selectiva y potenciada de células cancerosas por sulindac y un agente oxidante no estaba relacionada con la conocida capacidad como AINE del sulindac. Para determinar el papel de la inhibición de la COX se puede utilizar un metabolito del sulindac, la sulfona de sulindac, ya que no inhibe la COX 1 o 2 [7], [29]. Como se muestra en la Figura 2, utilizando células cancerosas A549 (A) y SCC25 (B), la combinación de sulindac sulfona y DCA mostró un efecto letal similar al observado anteriormente con sulindac. Estos resultados indican que la mejora del efecto anticancerígeno del sulindac en presencia de DCA no está relacionada con su conocida actividad antiinflamatoria.
La combinación de sulindac y DCA genera ROS
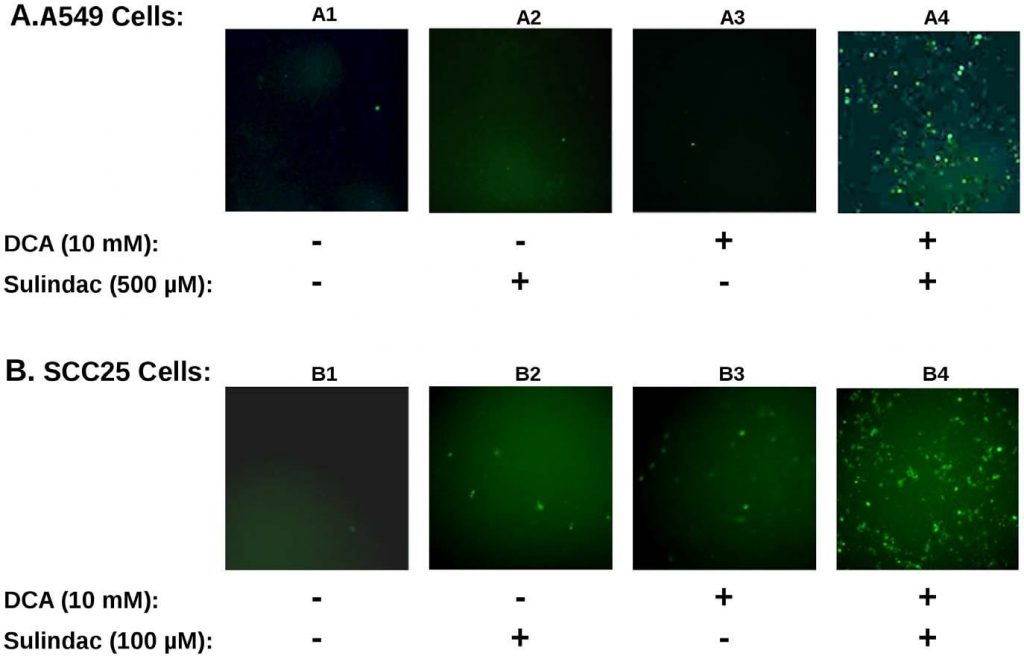
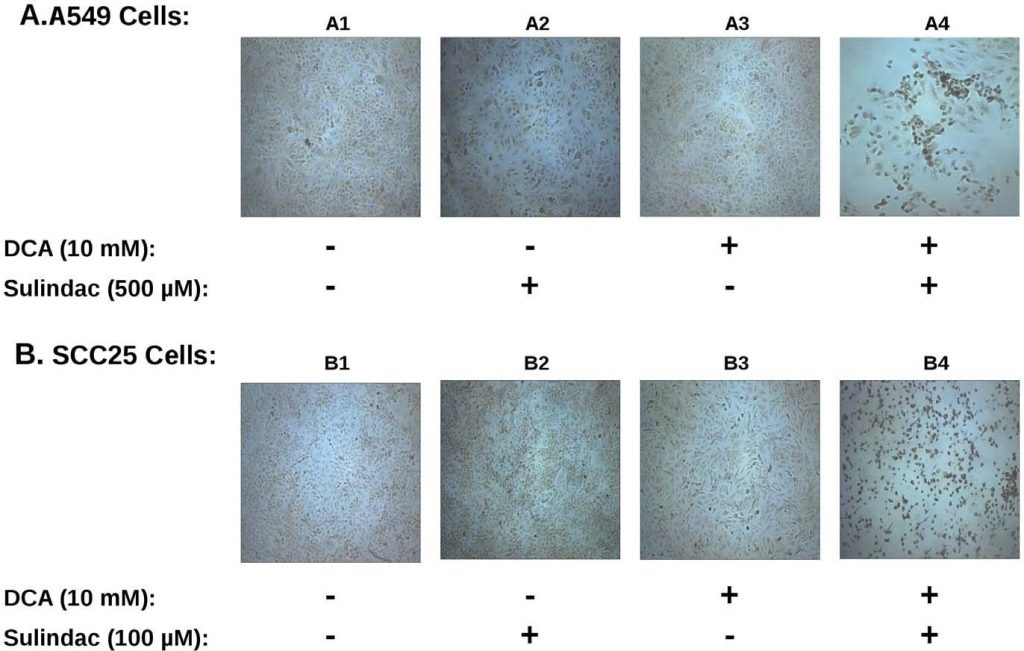
El efecto sinérgico sobre la viabilidad observado con sulindac y dicloroacetato con las células cancerosas A549 y SCC25 es sorprendentemente similar al de estudios anteriores en los que se utilizó la combinación de sulindac y TBHP [7]. Para determinar si la producción de ROS estaba implicada en la muerte selectiva observada en los presentes estudios, se determinó la producción de ROS, utilizando el colorante indicador H2DCFDA(ver Métodos), en las líneas celulares cancerosas expuestas a sulindac y DCA. Los resultados se resumen en la Figura 3. La Figura 3A muestra los resultados con las células cancerosas A549. De los resultados representados en la Figura 3A se desprende que las células cancerosas A549 no tratadas (panel A1), o las células tratadas sólo con sulindac (panel A2), o sólo con DCA (panel A3), muestran sólo unas pocas células teñidas positivamente. Sin embargo, cuando las células se expusieron tanto a sulindac como a DCA (panel A4), se observa un gran aumento de células teñidas positivamente para ROS (fluorescencia verde), lo que demuestra que la presencia tanto de sulindac como de DCA da lugar a la generación de niveles significativos de ROS. Como se muestra en la Figura 3B, se observan resultados similares con las células cancerosas SCC25. El sulindac o el DCA por sí solos dan lugar a un pequeño aumento de las células productoras de ROS (paneles B2 y B3), pero se observa de nuevo un gran aumento de la producción de ROS cuando se añaden ambos fármacos (panel B4). La cuantificación utilizando células SCC25 muestra que el número de células DCF-positivas (ver Métodos) es de 9-10× más cuando las células se tratan con sulindac y DCA en comparación con cada uno de los fármacos por separado (ver Figura S3A). De estos resultados y de estudios anteriores se desprende que la producción de ROS puede ser una característica común en el aumento de la destrucción de células cancerosas cuando se utiliza sulindac en combinación con compuestos que afectan a la función mitocondrial.
Sulindac en combinación con DCA provoca una pérdida del potencial de membrana mitocondrial
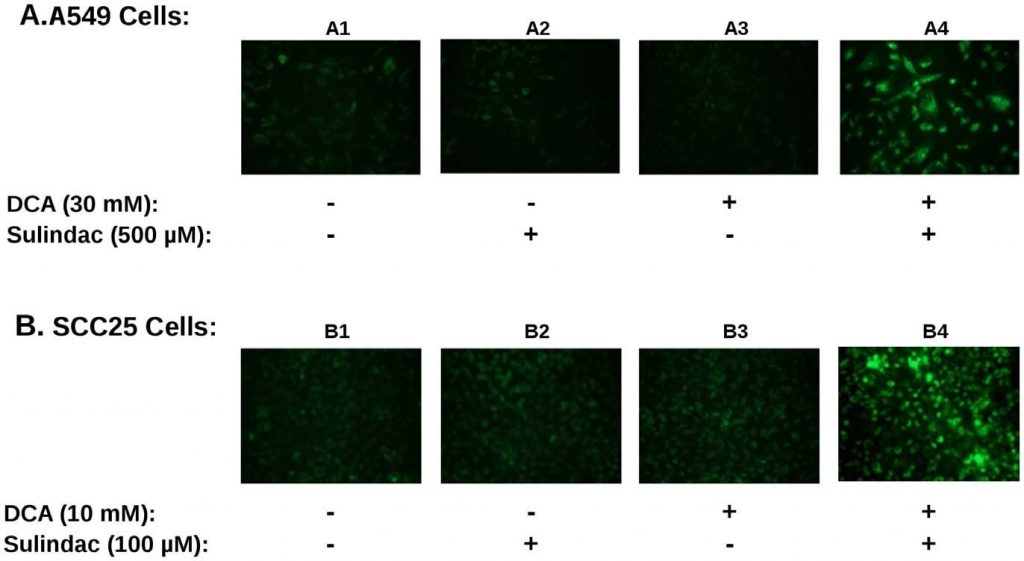
Si la producción de ROS está implicada en el efecto letal potenciado por sulindac/DCA, cabría esperar que la producción de ROS por la combinación de fármacos afectara a la función mitocondrial. Para determinar esto, se midió el potencial de membrana mitocondrial utilizando la tinción JC-1 como se describe en Métodos. Una pérdida de potencial de membrana se indica mediante un aumento de la fluorescencia verde, como se describe en Métodos. En la Figura 4 se resume un resultado típico. Las células cancerosas A549 y SCC25 se expusieron a sulindac y DCA solos o combinados durante 48 horas y se tiñeron con JC-1 para monitorizar el potencial de membrana mitocondrial. La Figura 4A muestra los resultados obtenidos con la línea celular A549. En ausencia de cualquier fármaco, las mitocondrias aparecen intactas y mantienen su potencial de membrana como indica la escasa fluorescencia verde (panel A1). En presencia de sulindac solo (panel A2) o DCA solo (panel A3) se produce un pequeño aumento de la fluorescencia verde, lo que indica cierta pérdida de potencial de membrana mitocondrial. Sin embargo, cuando están presentes tanto el sulindac como el DCA, se produce una pérdida notable del potencial de membrana mitocondrial, evidenciada por un gran aumento de la fluorescencia verde (panel A4). Observamos el mismo patrón cuando se analizaron varios campos independientes mediante microscopía de fluorescencia. La Figura 4B muestra resultados similares con las células cancerosas SCC25. Una vez más, sólo se observó una pérdida significativa del potencial de membrana mitocondrial cuando las células se expusieron tanto a sulindac como a DCA (panel B4). La cuantificación del efecto se muestra en la Figura S3B. Puede observarse que el porcentaje de células positivas al verde JC1 cuando se utilizó la combinación de fármacos es 3-4 veces superior al observado con cualquiera de los fármacos por separado.
Las EROestán implicadas en la destrucción de las células cancerosas por la combinación de sulindac y DCA
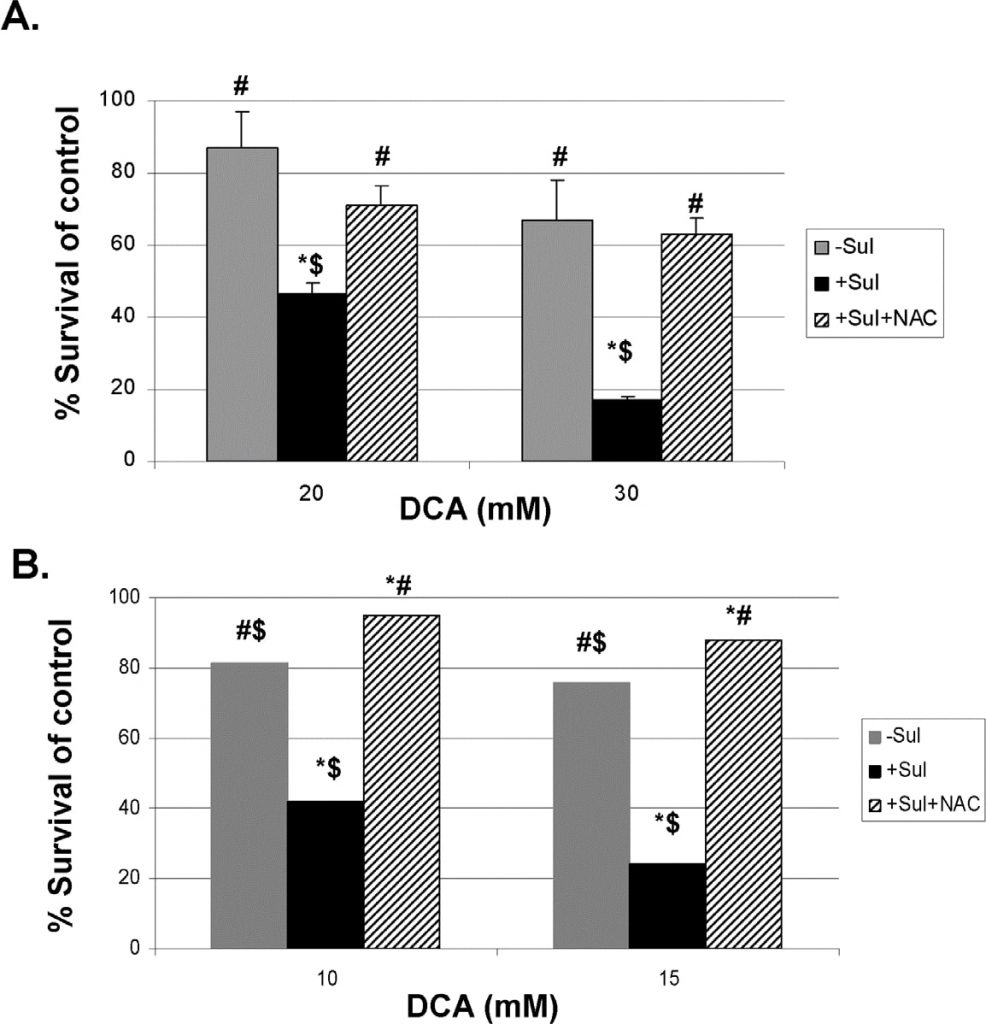
Para proporcionar pruebas más directas de que las ERO producidas están implicadas en la destrucción mejorada de las células cancerosas por sulindac y DCA, hemos utilizado dos eliminadores de ERO conocidos, N-acetilcisteína (NAC) y Tiron (ver Métodos). Los resultados utilizando NAC se muestran en la Figura 5. La Figura 5, panel A, muestra que tanto a 20 como a 30 mM de DCA, la mayor destrucción de células cancerosas A549 observada en presencia de sulindac, se evita en gran medida si NAC (2 mM) está presente durante las 48 horas de incubación. Se observan resultados muy similares con las células cancerosas SCC25, como se muestra en la Figura 5, panel B. Se obtuvieron resultados comparables cuando se utilizó Tiron en lugar de NAC (Figura S4).
Sulindac and DCA Killing of Cancer Cells Involves Apoptotic Death
Los resultados anteriores (Figuras 3, 4, 5) muestran que el aumento de la muerte de las líneas celulares de cáncer implica la disfunción mitocondrial, lo que sugiere que la muerte celular observada es a través de la apoptosis. Estudios anteriores han indicado que el sulindac y sus derivados son fármacos proapoptóticos [5], [6]. También hay informes de que el DCA puede causar la muerte celular por apoptosis [20], [23]. Para determinar si la muerte de las células cancerosas por la combinación de estos dos fármacos, mediada por ROS, implica muerte apoptótica, realizamos la tinción TUNEL para medir la apoptosis (ver Métodos). Para los experimentos de tinción TUNEL se realizaron múltiples réplicas con sulindac y DCA solos, o en combinación. Un resultado típico se ilustra en la Figura 6, donde los paneles superiores (Figura 6A, paneles A1-A4) representan los resultados con las células cancerosas A549 y los paneles inferiores (Figura 6B, paneles B1-B4) representan los resultados con las células cancerosas SCC25. Cuando las células se trataron sin fármaco, sólo con sulindac o sólo con DCA (Figura 6, paneles A1-A3 y B1-B3), sólo se observaron unas pocas células TUNEL positivas. Sin embargo, cuando las células se expusieron tanto a sulindac como a DCA, se produce un aumento significativo de las células apoptóticas TUNEL-positivas (Figura 6, paneles A4 y B4), lo que indica una gran inducción de la apoptosis. Para verificar los resultados de TUNEL, también se utilizó un ensayo de ADN laddering mediado por PCR más sensible para monitorizar la apoptosis [25]. Los resultados también mostraron la presencia de una fuerte escalera nucleosómica enriquecida sólo cuando se utilizaron sulindac y DCA en combinación (Figura S5; carriles 4 y 8), lo que apoya firmemente los datos del ensayo TUNEL.
Sulindac and DCA Killing Involves Proapoptotic JNK Signaling
De las conocidas proteínas quinasas activadas por mitógenos (MAP quinasas), la quinasa inducida por estrés, c-Jun N-terminal quinasa (JNK/SAPK) ha sido directamente implicada en la muerte celular apoptótica [11]. Por lo tanto, investigamos el papel de la señalización JNK en la apoptosis mediada por sulindac-DCA utilizando SP600125, un inhibidor específico de JNK (JNKI) y estos resultados se presentan en la Figura 7A. Como se ha mostrado anteriormente, las células SCC25 tratadas con sulindac mostraron una mayor muerte en presencia de concentraciones crecientes de DCA. Sin embargo, cuando estas células se incubaron con sulindac junto con SP600125, se evitó en gran medida la muerte celular mediada por sulindac-DCA. Estos resultados indican la participación de la señalización proapoptótica mediada por JNK en la muerte celular mediada por sulindac-DCA.
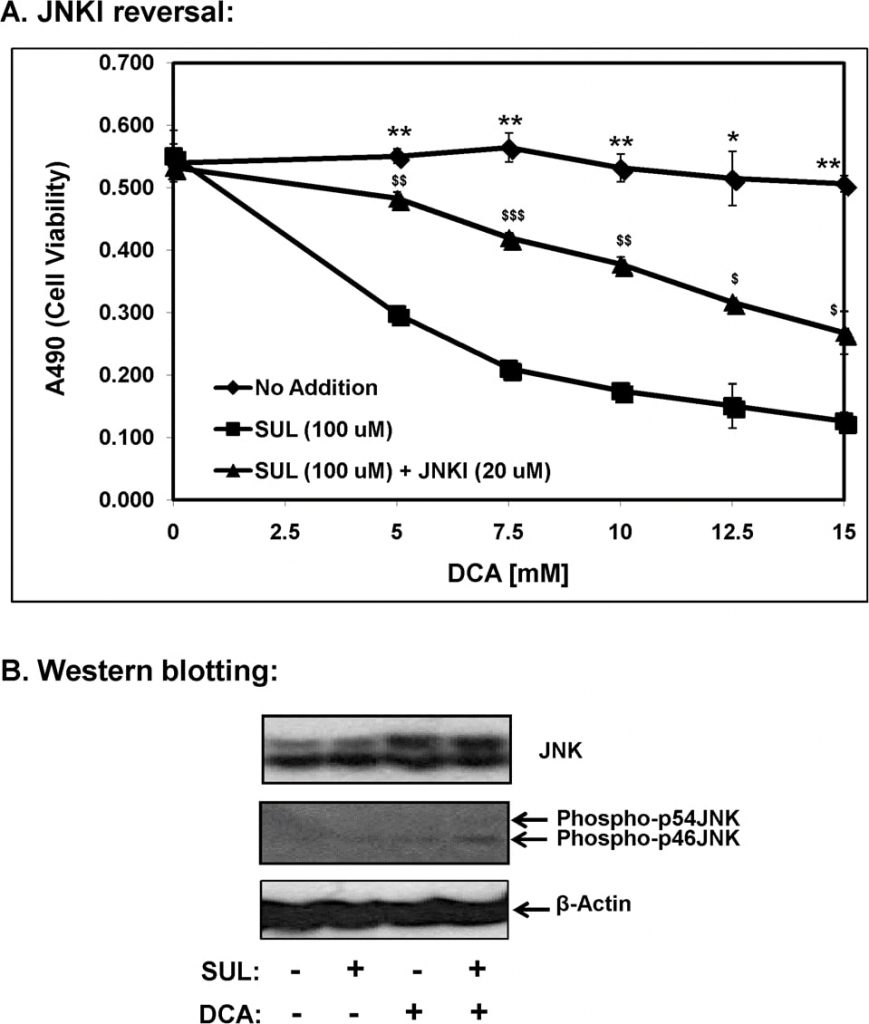
Mediante análisis western blot, también determinamos que la combinación de sulindac y DCA aumentaba significativamente los niveles de fosfo-JNK en fracciones citosólicas 12 h después de que las células fueran expuestas a sulindac y DCA (Figura 7B). Se observó un aumento de los niveles de JNK total (bandas de proteína a 46 y 54 kDa) cuando las células se trataron con DCA solo, así como cuando las células se trataron con la combinación de sulindac y DCA. Cabe señalar que tanto las isoformas fosfo-p46JNK como fosfo-p54JNK fueron inducidas por la combinación del tratamiento con sulindac y DCA, aunque el aumento de fosfo-p46JNK fue más significativo (Figura 7B).
Existe un conjunto de pruebas que sugieren que JNK inicia la liberación de factores inductores de la apoptosis desde las mitocondrias, como el citocromo c, que conducen a la escisión de las caspasas y la PARP (poli(ADP-ribosa) polimerasa) [30], [31]. Los estudios también han demostrado que durante la apoptosis, el citocromo c liberado de las mitocondrias al citoplasma acaba entrando en el núcleo [32]. Nuestros resultados indicaron que la máxima activación de JNK se produjo alrededor de 12 h después de la exposición a sulindac y DCA. Esto parece resultar en la translocación del citocromo c al citoplasma y la escisión de PARP 18 h después del tratamiento inicial con sulindac y DCA (Figura S6A). Como control positivo para estos experimentos, tratamos las células con 100 µM de etopósido, un agente inductor de la apoptosis. Bajo el tratamiento combinado de sulindac y DCA, puede observarse un aumento de la fluorescencia nuclear en la mayoría de las células que están experimentando activamente la apoptosis (Figura S6B).
El análisis detallado de los datos experimentales de inmunofluorescencia de células enteras reveló que ∼94% de las células no tratadas con sulindac ni con DCA mostraban fluorescencia mitocondrial del citocromo c punteada, con poca tinción difusa en el citoplasma o en los núcleos. Por el contrario, tras el tratamiento con sulindac, el 81% de las células mostraron fluorescencia citoplasmática difusa y distinta, y muy poca fluorescencia nuclear. Tras el tratamiento con DCA, ∼83% de las células mostraron una fluorescencia citoplasmática difusa y distinta, y <5% de las células mostraron una fuerte fluorescencia nuclear. Sin embargo, cuando las células fueron tratadas con sulindac y DCA, ∼72% de las células mostraron fluorescencia nuclear y citoplasmática y ∼11% de las células mostraron fluorescencia nuclear fuerte. Estos resultados sugieren que el citocromo c liberado de las mitocondrias puede iniciar la vía apoptótica intrínseca que funciona en la destrucción del cáncer mediada por sulindac y DCA.
Discusión
El presente estudio es una extensión de nuestro trabajo anterior, que demostró que el sulindac hacía que las células cancerosas, pero no las normales, fueran más sensibles al estrés oxidativo [7]. En estos experimentos previos, el sulindac se preincubó con las células durante 24-48 horas y después se retiró el sulindac antes de exponer las células al TBHP o al H2O2 durante 2 horas. De los experimentos anteriores se desprendía que el pretratamiento con sulindac hacía que las células cancerosas fueran mucho más sensibles al agente oxidante, lo que provocaba un gran aumento de ROS y la pérdida de la función mitocondrial [7].
Basándose en estos resultados, parecía razonable que el sulindac, en combinación con compuestos que afectaban a la respiración mitocondrial, produjera un aumento selectivo de la destrucción de células cancerosas, pero no de células normales. En el presente estudio, en el que se utilizaron las líneas celulares de cáncer A549 y SCC25, la combinación de sulindac y DCA aumentó la destrucción de estas líneas celulares de cáncer, pero no de las células normales de pulmón o piel. Nuestros resultados sobre las cantidades de DCA necesarias en células enteras concuerdan con lo comunicado anteriormente [28], [33], [34]. En nuestro sistema, la IC50 para DCA con células SCC25 es de 23 mM y para células A549 es de 35 mM. El IC50 para queratinocitos normales es >50 mM y para células pulmonares normales (MRC5) es ∼40 mM. Los resultados también indicaron que la muerte de las células cancerosas observada implica la producción de ROS, la activación de JNK y la apoptosis iniciada por las mitocondrias. Con respecto a la falta de efecto sobre las células normales, se ha demostrado que el sulindac protege a las células pulmonares normales contra el daño oxidativo resultante de la exposición al TBHP [7] y también hemos informado recientemente de que el sulindac puede proteger a las células cardíacas contra el daño oxidativo resultante de la isquemia/reperfusión a través de un mecanismo de preacondicionamiento [35].
Hasta donde sabemos, en la actualidad existen al menos 8 compuestos, incluidos nuestros estudios con TBHP, H2O2 y DCA, que han demostrado una eliminación selectiva y mejorada del cáncer en presencia de sulindac [7]–[9], [12]–[15]. Aunque se conocen sus objetivos metabólicos dentro de la célula, y son diferentes, es bastante probable que todos ellos, directa o indirectamente, causen la muerte celular en presencia de sulindac a través de un mecanismo que implica una alteración en la respiración mitocondrial y la producción de ROS [10], [12], [14]. Parece probable que cuando se encuentra un fármaco que, en combinación con sulindac, mata selectivamente las células cancerosas, pero no las normales, el mecanismo de muerte implique un estrés oxidativo que lleve a una disfunción mitocondrial. La alteración de la respiración puede ser un factor común en estos experimentos con combinaciones de sulindac y fármacos, y los resultados actuales con DCA apoyan esta opinión. Es muy posible que el efecto del sulindac esté relacionado con las observaciones realizadas hace más de 50 años por Warburg, quien observó que las células normales prefieren la respiración para obtener su energía, mientras que las células cancerosas prefieren la glucólisis, debido a un defecto en la cadena respiratoria [16]. Esta diferencia básica en la respiración mitocondrial entre células normales y cancerosas puede hacer que las células cancerosas sean más sensibles al estrés oxidativo [36], [37]. Parece que el sulindac puede amplificar esta diferencia fundamental en la bioquímica de las células normales y cancerosas. Aunque todavía no está claro cómo el sulindac sensibiliza a las células cancerosas a los fármacos que afectan a la respiración mitocondrial, se está investigando activamente. Spitz y colaboradores [38], en estudios sobre la privación de glucosa en células cancerosas, han llegado a una conclusión similar respecto a las diferencias en el metabolismo entre células normales y cancerosas. En línea con estos resultados, otro estudio reciente ha demostrado que la inhibición farmacológica de la lactato deshidrogenasa podría provocar la muerte selectiva del cáncer [39]. En estos últimos estudios se demostró que el aumento de la destrucción selectiva de las células cancerosas también implicaba la producción de ROS, y el efecto observado se atribuyó a un proceso respiratorio alterado en las células cancerosas.
Cabe señalar que la combinación de sulindac con un agente oxidante o fármacos que pueden afectar a la función mitocondrial ya se ha probado clínicamente. Meyskens et al. 2008 demostraron que la combinación de sulindac con DFMO tenía un efecto significativo sobre la recurrencia de pólipos de colon y la aparición de cáncer de colon en un estudio clínico de 3 años [13]. Recientemente hemos informado del uso de sulindac, con H2O2, en un estudio clínico de prueba de concepto para el tratamiento tópico de las queratosis actínicas [15]. Una de las desventajas del uso de esta combinación era la necesidad de dos formulaciones tópicas, ya que los compuestos no podían almacenarse durante largos periodos sin que el H2O2 destruyera el sulindac. Además, no se puede utilizar el H2O2 para el tratamiento de tumores internos, ya que no se puede tomar por vía oral. Sin embargo, la combinación de sulindac y DCA podría administrarse como una única formulación susceptible de uso tópico, y los dos compuestos pueden utilizarse por vía oral. De hecho, durante varios años, el DCA se ha utilizado clínicamente para reducir los niveles de ácido láctico en pacientes que sufren acidosis láctica [40]–[42]. El DCA también se ha utilizado como agente anticancerígeno in vitro e in vivo utilizando varias líneas celulares de cáncer diferentes, lo que indica que el metabolismo mitocondrial en las células cancerosas podría ser una nueva diana terapéutica [18], [20], [22]. Michelakis et al., (2010) han demostrado que el tratamiento con DCA «remodela» el metabolismo mitocondrial en pacientes con glioblastoma con efectos tóxicos reversibles. Cabe destacar que tanto el sulindac como el DCA son asequibles, relativamente atóxicos y pueden tomarse por vía oral. Si la combinación tiene éxito in vivo, añadirá una nueva dimensión al tratamiento del cáncer, ya que ambos fármacos actúan sobre el metabolismo mitocondrial en múltiples tipos de cáncer [22].
En resumen, nuestros estudios con la combinación de sulindac y DCA sugieren que el sulindac hace que las células cancerosas sean más sensibles a los agentes que afectan a la respiración mitocondrial, lo que provoca estrés oxidativo y disfunción mitocondrial. Estos resultados podrían estar relacionados con el defecto de respiración en las células cancerosas, observado originalmente por Warburg [16]. Actualmente se están realizando estudios para comprender las diferencias fundamentales entre la respuesta de las células cancerosas y las células normales al sulindac y a los agentes que afectan a la función mitocondrial.
Información complementaria
Figura S1 Citotoxicidad de sulindac, DCA o la combinación de fármacos en células cancerosas A549 y SCC25
Las células cancerosas A549 y SCC25 se trataron con las concentraciones indicadas de sulindac (paneles 1 y 4) o DCA (paneles 2 y 5). En los experimentos de combinación de fármacos sulindac/DCA, se mantuvo una proporción de 1:50 (sulindac:DCA) para las células A549, con combinaciones de fármacos que iban desde 0,2 mM:10 mM hasta 1 mM:50 mM (panel 3) y se mantuvo una proporción de 1:100 (sulindac:DCA) para las células SCC25 con combinaciones de fármacos que iban desde 0,05 mM:5 mM hasta 0,3 mM:30 mM (panel 6). (TIF) Figura S2 Determinación de los índices de combinación para las células cancerosas A549 y SCC25. Los índices de combinación de fármacos se determinaron incorporando los valores de viabilidad celular obtenidos anteriormente a las ecuaciones de Chou y Talalay [26]. Véanse más detalles en el texto. (TIF) Figura S3 Cuantificación de células fluorescentes verdes DCF y JC-1 positivas. Las células SCC25 se trataron con sulindac, DCA o la combinación de fármacos durante 48 horas y se tiñeron con colorantes H2DCFDA (Fig. S3A) o JC-1 (Fig. S3B) como se menciona en Métodos. Para el control positivo, las células fueron tratadas con 200 mM de TBHP durante 2 horas y la tinción se realizó como se ha indicado anteriormente. Las células se analizaron con gran aumento utilizando el objetivo 1006 en un microscopio fluorescente invertido Olympus. Se visualizaron al menos 100 células individuales para cada condición y los porcentajes de células fluorescentes verdes DCF-positivas y JC1-positivas se presentan en formato tabular y gráfico. (TIF) Figura S4 El eliminador de ROS Tiron revierte la destrucción de células cancerosas por la combinación de sulindac y DCA. Las células cancerosas A549 y SCC25 se trataron con las concentraciones indicadas de DCA en ausencia (barra gris) o presencia de sulindac (barra negra) o presencia de sulindac y Tiron (barra rayada) durante 48 horas. La viabilidad celular se monitorizó mediante el ensayo MTS como se menciona en los Métodos. La viabilidad celular se expresa como % del control (células no tratadas con sulindac). Las barras de error son el error estándar de la media (SEM) expresado como % del valor medio de cuadruplicados de un experimento representativo. La inhibición del crecimiento de las células cancerosas se produjo de forma dependiente de la dosis durante el tratamiento combinado de DCA y sulindac (barras negras) tanto en las células cancerosas A549 (A) como en las células cancerosas SCC25 (B). Sin embargo, esta mayor destrucción se evitó cuando Tiron estaba presente junto con el tratamiento de combinación de fármacos (barras rayadas en A y B). (TIF) Figura S5 Durante la destrucción de células cancerosas por la combinación de sulindac y DCA se produce un escalamiento pronunciado del ADN nucleosómico. Las células cancerosas A549 y SCC25 se trataron con los fármacos indicados durante 48 horas. Se extrajo el ADN nucleosómico y se sometió a PCR mediada por ligación como se describe en Métodos y se analizó en un gel de agarosa al 1,2% junto con marcadores de tamaño. El carril «M» indica los marcadores de tamaño molecular. Los carriles 1-4 y 5-8 muestran los resultados obtenidos con las células cancerosas A549 y SCC25, respectivamente. Los resultados se ilustran en los carriles 1 y 5 (sin fármaco), carriles 2 y 6 (sulindac solo), carriles 3 y 7 (DCA solo), y carriles 4 y 8 (sulindac y DCA). Sólo se observó un aumento del escalamiento del ADN nucleosómico con el tratamiento combinado de sulindac y DCA (carriles 4 y 8). (TIF)
Figura S6 La combinación de sulindac y DCA conduce a la liberación de citocromo c de las mitocondrias y a la escisión de PARP
Las célulasSCC25se trataron con sulindac, DCA, combinación de fármacos o etopósido para analizar la localización intracelular del citocromo c mediante western blot e inmunofluorescencia. A. Se aislaron fracciones citosólicas a las 18 h y se determinó la presencia de citocromo c en el citoplasma y la escisión de PARP mediante western blotting. Se utilizaron los niveles de b-actina como control interno. B. La inmunofluorescencia se realizó utilizando el kit citométrico de liberación de citocromo c CBA077 InnoCyteTM Flow Cytometric Cytochrome c Release Kit siguiendo las instrucciones del fabricante. Se analizaron varios campos independientes y las micrografías representativas muestran los patrones de localización del citocromo c cuando las células se exponen a sulindac y/o DCA. Los valores cuantitativos se presentan en el texto. (TIF)
Agradecimientos
Los autores expresan su agradecimiento a David Brunell por su ayuda en la determinación de los índices de combinación y a Edna Gamliel por su asistencia en el cultivo celular.
Contribuciones de los autores
Concepción y diseño de los experimentos: KA SK KDS HW. Realización de los experimentos: KA SK. Análisis de los datos: KA SK HW. Contribución de reactivos/materiales/herramientas de análisis: KDS. Escribió el artículo: KA SK HW.
REFERENCIAS
1 Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, et al. (1996) Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res 56: 2556-2560.
2 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J Natl
3 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst 90: 1529-1536.
4 Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, et al. (1995) Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res 55: 1464-1472.
5 Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, et al. (2001) Progression and NSAID-induced apoptosis in malignant melanomas are independent of cyclooxygenase II. Melanoma Res 11: 587-599.
6 Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogénesis 22: 17-25.
7 Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, et al. (2009) Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS One 4: e5804.
8 Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, et al. (1999) Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 59: 6178-6184.
9 Jiang TT, Brown SL, Kim JH (2004) Combined effect of arsenic trioxide and sulindac sulfide in A549 human lung cancer cells in vitro. J Exp Clin Cancer Res 23: 259-262.
10 Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, et al. (2006) Synergistic induction of apoptosis by sulindac and arsenic trioxide in human lung cancer A549 cells via reactive oxygen species-dependent down-regulation of survivin. Biochem Pharmacol 72: 1228-1236.
11 Jin HO, Seo SK, Woo SH, Lee HC, Kim ES, et al. (2008) A combination of sulindac and arsenic trioxide synergistically induces apoptosis in human lung cancer H1299 cells via c-Jun NH2-terminal kinase-dependent Bcl-xL phosphorylation. Cáncer de pulmón 61: 317-327.
12 Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, et al. (2005) Sulindac enhances the proteasome inhibitor bortezomib-mediated oxidative stress and anticancer activity. Clin Cancer Res 11: 5248-5256.
13 Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, et al. (2008) Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila Pa) 1: 32-38.
14 Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, et al. (2008) Combined effects of sulindac and suberoylanilide hydroxamic acid on apoptosis induction in human lung cancer cells. Mol Pharmacol 73: 1005-1012.
15 Resnick L, Rabinovitz H, Binninger D, Marchetti M, Weissbach H (2009) Topical sulindac combined with hydrogen peroxide in the treatment of actinic keratoses. J Drugs Dermatol 8: 29-32.
16 Warburg O (1956) Sobre el origen de las células cancerosas. Science 123: 309-314.
17 Whitehouse S, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141: 761-774.
18 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37-51.
19 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60: 1478-1487.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109: 394-402.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68: 1223-1231.
<span id=»22″ class=»referencess blue-text «22 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, et al. (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2: 31ra34.
23 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989-994.
24 Cory AH, Owen TC, Barltrop JA, Cory JG (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3: 207-212.
<span id=»25″ class=»referencess blue-text «25 Staley K, Blaschke AJ, Chun J (1997) Apoptotic DNA fragmentation is detected by a semi-quantitative ligation-mediated PCR of blunt DNA ends. Cell Death Differ 4: 66-75.
26 Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27-55.
<span id=»27″ class=»referencess blue-text «27 Papandreou I, Goliasova T, Denko NC (2010) Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer 128: 1001-1008.
28 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, et al. (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127: 2510-2519.
29 Babbar N, Ignatenko NA, Casero RA Jr, Gerner EW (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J Biol Chem 278: 47762-47775.
30 Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haikel Y, et al. (2011) Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogénesis 32: 1268-1278.
31 Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, et al. (2007) c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 14: 1001-1010.
<span id=»32″ class=»referencess blue-text «32 Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, et al. (2004) Nuclear translocation of cytochrome c during apoptosis. J Biol Chem 279: 24911-24914.
33 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, et al. (2011) Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 67: 647-655.
34 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 102: 1746-1752.
35 Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac confers high level ischemic protection to the heart through late preconditioning mechanisms. Proc Natl Acad Sci U S A 106: 19611-19616.
<span id=»36″ class=»referencess blue-text «36 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18: 54-61.
37 Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029-1033.
<span id=»38″ class=»referencess blue-text «38 Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, et al. (2005) Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280: 4254-4263.
<span id=»39″ class=»referencess blue-text «39 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107: 2037-2042.
<span id=»40″ class=»referencess blue-text «49 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI (1983) Treatment of lactic acidosis with dichloroacetate. N Engl J Med 309: 390-396.
41 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77: 535-541.
42 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, et al. (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121: e1223-1228.
Contenido relacionado: