Kasirajan Ayyanathan1,2,*,#, Shailaja Kesaraju1,#, Ken Dawson-Scully1,2, Herbert Weissbach1
1 Centrum voor Moleculaire Biologie en Biotechnologie, Charles E. Schmidt College of Science, Florida Atlantic University, Jupiter, Florida, Verenigde Staten van Amerika,
2 Afdeling Biologische Wetenschappen, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, Florida, Verenigde Staten van Amerika
*E-mail: [email protected]
#Dezeauteurs hebben in gelijke mate bijgedragen aan dit werk.
Ontvangen: 12 augustus 2011
Geaccepteerd: 4 juni 2012
Gepubliceerd: 1 juli 2012
Abstract
Sulindac is een FDA-goedgekeurd niet-steroïdaal anti-inflammatoir geneesmiddel met gedocumenteerde antikanker activiteiten. Onze recente studies toonden aan dat sulindac selectief het doden van kankercellen blootgesteld aan oxiderende middelen bevorderde via productie van reactieve zuurstofspecies (ROS) resulterend in mitochondriale disfunctie. Dit effect van sulindac en oxidatieve stress op kankercellen zou verband kunnen houden met het defect in de ademhaling van kankercellen, voor het eerst beschreven door Warburg 50 jaar geleden, bekend als het Warburg-effect. Wij stelden dat sulindac het selectief doden van kankercellen zou kunnen versterken wanneer het gecombineerd wordt met een verbinding die de mitochondriale ademhaling verandert. Om deze hypothese te testen hebben wij dichlooracetaat (DCA) gebruikt, waarvan bekend is dat het pyruvaatmetabolisme verschuift van melkzuurvorming naar ademhaling. Men zou kunnen verwachten dat DCA, omdat het de aërobe stofwisseling stimuleert, de mitochondriale ademhaling in kankercellen zou kunnen belasten, wat zou resulteren in een verhoogde doding in aanwezigheid van sulindac. In deze studie hebben wij aangetoond dat de combinatie van sulindac en DCA de selectieve doding van A549- en SCC25-kankercellen onder de gebruikte omstandigheden versterkt. Zoals voorspeld omvat het moordmechanisme ROS-productie, mitochondriale disfunctie, JNK-signalering en dood door apoptose. Onze resultaten suggereren dat de combinatie van sulindac en DCA een effectieve kankertherapie kan bieden.
Citatie: Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H (2012) Combinatie van sulindac en dichlooracetaat doodt kankercellen via oxidatieve
schade. PLoS ONE 7(7): e39949. doi:10.1371/journal.pone.0039949
Editor: Joseph Alan Bauer, Bauer Research Foundation, Verenigde Staten van Amerika
© 2012 Ayyanathan et al. Dit is een open-access artikel verspreid onder de voorwaarden van de Creative Commons Attribution License, die
onbeperkt gebruik, distributie en reproductie in elk medium toestaat, mits de oorspronkelijke auteur en bron worden gecrediteerd.
Financiering: Financiële steun van de National Institutes of Health aan KA (grant 5K01CA95620) en HW (grant R15 CA122001) en van de staat Florida aan HW (SURECAG grant R94007) om dit werk uit te voeren wordt dankbaar erkend. De financiers hadden geen rol in de studieopzet, gegevensverzameling en -analyse, beslissing tot publicatie of voorbereiding van het manuscript.
Concurrerende belangen: De auteurs hebben verklaard dat er geen concurrerende belangen bestaan.
INLEIDING
Sulindac is een door de FDA goedgekeurd niet-steroïde anti-inflammatoir geneesmiddel (NSAID), waarvan ook is aangetoond dat het kanker bestrijdt [1-6]. Recente studies uit ons laboratorium hebben aangetoond dat RKO, A549 en SCC25 kankercellijnen gevoelig waren voor een combinatie van sulindac en een oxidatiemiddel, zoals TBHP of H2O2 [7]. De gegevens wezen erop dat het sulindac-effect geen verband hield met de NSAID-activiteit ervan, maar dat sulindac kankercellen gevoeliger maakte voor oxidatieve stress, wat resulteerde in mitochondriale disfunctie en verlies van levensvatbaarheid. Normale cellen daarentegen vertoonden geen verhoogde doding onder vergelijkbare omstandigheden [7]. In de afgelopen 10 jaar zijn er verspreid meldingen geweest van verbeterde kankerdoding met behulp van sulindac in combinatie met verschillende verbindingen, waaronder arseentrioxide, bortezomib, difluormethylornithine (DFMO) en suberoylanilide hydroxaminezuur (SAHA) [8-14]. Hoewel deze verbindingen verschillende werkingsplaatsen hebben, zou een gemeenschappelijk mechanisme voor de verbeterde doding door de combinatie sulindac/drug een rol kunnen spelen bij oxidatieve schade, zoals duidelijk werd aangetoond in onze eerdere studies met sulindac en een oxidatiemiddel [7], [15]. In feite zijn ROS betrokken bij studies waarin sulindac werd gebruikt in combinatie met arseentrioxide, bortezomib en SAHA [10], [12], [14].
Onze eerdere resultaten suggereerden dat de verhoogde doding van kankercellen door de combinatie van sulindac en een oxiderende stof te wijten zou kunnen zijn aan een defect in de ademhaling bij kankercellen, zoals voor het eerst beschreven door Warburg meer dan 50 jaar geleden [16], die opmerkte dat kankercellen de voorkeur geven aan glycolyse, en niet aan ademhaling, om energie te verkrijgen, in tegenstelling tot normale cellen. Sommige kankercellen halen tot 50% van hun energie uit de glycolyse, terwijl de glycolyse in normale cellen minder dan 5% van de energiebehoefte uitmaakt [16]. Om verder bewijs te verkrijgen voor de mogelijke rol van veranderde ademhaling en ROS bij het doden van kankercellen door sulindac en oxidatieve stress, zijn wij begonnen met studies met natriumdichloorazijnzuur (DCA). DCA is een ideale kandidaat, omdat bekend is dat het een kinase remt dat de activiteit van pyruvaatdehydrogenase vermindert, waardoor het pyruvaatmetabolisme verschuift van melkzuurvorming naar ademhaling [17], [18]. DCA is klinisch gebruikt voor de behandeling van patiënten met melkzuuracidose [19], en op grond van zijn biochemische eigenschappen is DCA ook getest als middel tegen kanker. Bonnet et al. 2007 hebben aangetoond dat DCA het Warburg-effect in kankercellen omkeert door het metabolisme van kankercellen om te leiden van glycolyse naar oxidatieve fosforylering. In deze eerdere studies werd aangetoond dat DCA de niveaus van ROS van complex I verhoogt. Dit leidt op zijn beurt tot “remodeling” van het mitochondriaal metabolisme (vermindert ΔΨm, opent de mitochondriale overgangspoort) in kankercellen, waardoor deze in de richting van apoptose worden geduwd. Bovendien hebben verschillende recente studies aangetoond dat DCA het ROS-niveau in kankercellen kan verhogen en het mitochondriummembraan kan depolariseren in long-, endometriale en glioblastoma-cellijnen, wat resulteert in apoptose, zowel in vitro als in vivo [18], [20-22]. Interessant was de observatie dat DCA onder de gebruikte omstandigheden het mitochondriale metabolisme of de levensvatbaarheid in normale cellen niet significant leek te beïnvloeden [ 18], [23].
Op grond van onze eerdere waarnemingen over het kankerdodend effect van sulindac en een oxidatiemiddel dat het mitochondriaal metabolisme beïnvloedde [7], stelden wij dat de combinatie van sulindac en DCA synergetisch de kankerdoding zou kunnen versterken en belangrijke therapeutische waarde zou kunnen hebben. In de huidige studie hebben wij het effect onderzocht van het gebruik van sulindac in combinatie met DCA op de levensvatbaarheid van A549 en SCC25 kankercellijnen. Wij hebben ook de rol van de mitochondriale functie en apoptose bij de kankerdoding door deze combinatie onderzocht.
Materialen en methoden
Materialen
Sulindac, N-acetylcysteïne en Tiron werden gekocht van Sigma (St.Louis, MO). Natriumzout van DCA werd verkregen van Acros Organics (Geel, België). H2DCFDA en JC-1 werden aangekocht bij Molecular Probes (Eugene, OR). MTS assay reagens en Deadend Tunel Kit werden verkregen van Promega (Madison, WI). Cytosol/mitochondriën-fractioneringskit en de CBA077 InnoCyte™ Flow Cytometric Cytochrome c Release kit waren afkomstig van Calbiochem, Gibbstown, NJ. Alle celkweekmedia, foetaal runderserum en andere supplementen zoals penicilline/streptomycine, glutamine, enz. werden gekocht bij American Type Culture Collection (ATCC; Rockville, MD).
Celcultuur
Eenniet-kleincellige longcarcinoomcellijn (NSCLC), A549, de normale menselijke longcellijn, MRC-5, en een van tong afgeleide plaveiselcelcarcinoomlijn, SCC25, werden gekocht bij ATCC (Rockville, MD) en onderhouden in F12-K-medium aangevuld met 10% foetaal runderserum, 2 mM glutamine, 100 IE/ml penicilline en 100 µg/ml streptomycine in een bevochtigde, 5%CO2-incubator bij 37°C. Normale menselijke epidermale keratinocyten werden verkregen van Promocell GmbH (Heidelberg, Duitsland) en onderhouden in het aanbevolen kweekmedium. Voor de experimenten werden vroege, niet-geïmmortaliseerde normale cellen gebruikt.
Levensvatbaarheidsonderzoek van de cellen
De A549 kankercellen en normale longcellen werden uitgezet op 3×103 cellen per well, terwijl SCC25 kankercellen en normale keratinocyten werden uitgezet op 7,5×103 cellen per well in een 96-wells plaat. De cellen werden 18-20 uur gekweekt, het medium werd onder aseptische omstandigheden weggegooid en vervangen door vers kweekmedium met de aangegeven combinaties van geneesmiddelen. Waar aangegeven werd 500 µM sulindac gebruikt met de A549 kanker- en longnormale cellen en 100 µM sulindac met de SCC25 kanker- en normale keratinocytencellen. De platen werden gedurende 48 uur geïncubeerd bij 37°C in een incubator met 5%CO2. Het kweekmedium werd weggegooid en de cellen werden grondig gespoeld in 1× PBS. De levensvatbaarheid van de cellen werd bepaald met behulp van de CellTiter 96 Aqueous One Cell Proliferation Assay (Promega) volgens de instructies van de fabrikant. De assay maakt gebruik van een tetrazoliumverbinding die wordt omgezet in een in water oplosbaar formazan door de werking van cellulaire dehydrogenases die aanwezig zijn in de metabolisch actieve cellen [24]. Het formazan werd gekwantificeerd door de absorptie bij 490 nm te meten met een colorimetrische microtiterplaatlezer (SpectraMax Plus; Molecular Devices). De achtergrondabsorptie werd van elk monster afgetrokken.
Intracellulaire meting van ROS
De A549 en SCC25 kankercellijnen werden uitgezet zoals hierboven beschreven. Na 48 uur behandeling met geneesmiddelen werden de cellen gedurende 30 minuten bij 37°C geïncubeerd met 50 µM dichlorodihydrofluoresceïne-diacetaat (H2DCFDA, Molecular Probes) in indicatorvrij medium. De cellen werden gespoeld met PBS en de ROS-niveaus werden gevisualiseerd met fluorescentiemicroscopie. De beelden werden vastgelegd met de Qcapture software en verwerkt in Adobe photoshop. Beeldanalyse werd gedaan met de slidebook software. De gegevens van een representatief experiment werden gebruikt voor de kwantificering van DCF-positieve cellen, gemeten aan de hand van de groene fluorescentie als gevolg van geoxideerd DCF.
JC-1 kleuring om het mitochondriale membraanpotentieel te controleren
Het mitochondrialemembraanpotentieel werd bepaald met behulp van de JC-1 kleurstof (Molecular Probes). De A549 en SCC25 kankercellijnen werden uitgezet zoals hierboven beschreven. Na 48 uur behandeling met geneesmiddelen werden de cellen gedurende 30 minuten bij 37°C geïncubeerd met 5 ng/ml JC-1 kleurstof in indicatorvrij medium. De cellen werden gespoeld met PBS en gevisualiseerd met fluorescentiemicroscopie. Normale mitochondriën nemen actief JC-1 kleurstof op in een potentiaalafhankelijke manier en vormen J-aggregaten, wat een rode fluorescentie geeft. Verstoring en vervolgens verlies van mitochondriale membraanpotentiaal leidt tot verhoogde groene fluorescentie in het cytosol als gevolg van monomere JC-1, die wordt bepaald door het verschijnen van groene fluorescentie te volgen met behulp van een FITC-filter (Zeiss inverted microscope-Axiovert 40 CFL). Beeldopname, verwerking en analyse werden uitgevoerd zoals hierboven beschreven. De gegevens van een representatief experiment werden gebruikt voor de kwantificering van JC-1-groen positieve cellen.
Effect van ROS-vangers op de levensvatbaarheid van de cellen in aanwezigheid van sulindac en DCA
De A549 en SCC25 kankercellijnen werden uitgezet zoals hierboven beschreven. Om ROS weg te vangen werd 2 mM N-acetylcysteïne (NAC) of 2 mM Tiron (4,5-dihydroxy-1,3-benzeendisulfonzuur dinatriumzout) toegevoegd samen met sulindac en DCA gedurende 48 uur bij 37°C. De levensvatbaarheid van de cellen werd gecontroleerd met de MTS-test en de statistische analyse werd uitgevoerd zoals hierboven vermeld.
TUNEL-kleuring om cellen die apoptose ondergaan te controleren
TUNEL-testwerd uitgevoerd in 96 wells platen met behulp van de DeadEnd colorimetric tunel assay kit (Promega) volgens het protocol van de fabrikant. De A549 en SCC25 kankercellijnen werden uitgezet zoals hierboven beschreven en gedurende 48 uur behandeld met geen geneesmiddel, sulindac, DCA of een combinatie van geneesmiddelen. Na de behandeling met geneesmiddelen werden de cellen gefixeerd met formaline en gepermeabiliseerd met 0,2% Triton X-100 in PBS. De cellen werden geïncubeerd met recombinant terminal deoxynucleotidyl transferase (TdT) en gebiotinyleerde nucleotiden. Endogene peroxidases werden geblokkeerd met 0,3% H2O2 voorafgaand aan de incubatie met mierikswortelperoxidase-streptavidine (HRP-streptavidine) dat bindt aan de gebiotinyleerde nucleotiden die zijn opgenomen in de geknipte uiteinden die aanwezig zijn in cellen die apoptose ondergaan. Met HRP-streptavidine gelabelde cellen werden gedetecteerd met waterstofperoxide en diaminobenzidine (DAB). Cellen met donkerbruine kernkleuring wijzen op apoptose.
Western Blot Analyse
Cellenwerden gekweekt tot 70% confluentie, behandeld met gespecificeerde geneesmiddelen voor de aangegeven duur, en cytosolische fracties werden geïsoleerd met behulp van de cytosol/mitochondria-fractioneringskit (Calbiochem, Gibbstown, NJ) volgens het protocol van de fabrikant. Kort gezegd werden de cellen op verschillende tijdstippen geoogst en vervolgens bij 600×g gedurende 5 minuten bij 4°C gecentrifugeerd. De gepelleteerde cellen werden gesuspendeerd in de bijgeleverde buffer en gedurende 10 minuten geïncubeerd op ijs. De cellen werden vervolgens gehomogeniseerd met een glazen douncer en het homogenaat werd gecentrifugeerd bij 700×g gedurende 10 minuten bij 4°C om kernen en celresten af te zetten. Het supernatant werd gesponnen bij 10.000×g gedurende 30 minuten bij 4°C om de mitochondriale pellet te verkrijgen en het supernatant werd beschouwd als de cytosolische fractie. De eiwitconcentratie werd bepaald met een standaard Bradford-test.
Zestig microgram totaal eiwit werd geladen en gescheiden op een 4-12% NuPage Bis-Tris-gel (Invitrogen, Eugene, OR) en overgebracht op een PVDF-membraan dat werd onderzocht met de primaire antilichamen. De primaire antilichamen JNK, pJNK, cytochroom c en PARP (Cell Signaling Technology, Danvers, MA) werden gebruikt in een verdunning van 1∶1000. β-actine (Santa Cruz Biotechnologies, Santa Cruz, Californië) werd gebruikt in een verdunning van 1∶4000. Er werden mierikswortelperoxidasegeconjugeerde secundaire antilichamen gebruikt en de banden werden gevisualiseerd met een versterkte chemiluminescentiemethode (GE Healthcare, Piscataway, NJ).
Ligatie-gemedieerde PCR op basis van DNA-laddering assay om de omvang van cellen die apoptose ondergaan te controleren
Om de mate van apoptose te bevestigen werd ligatie-gemedieerde PCR op basis van nucleosomaal DNA laddering assay uitgevoerd zoals beschreven [25]. De A549 en SCC25 kankercellijnen werden uitgezet bij 5×104 en 1×105 cellen per well in schaaltjes van 35 mm. De A549 kankercellen werden gedurende 48 uur behandeld met a) geen geneesmiddel, b) 500 µM sulindac, c) 20 mM DCA, en d) 500 µM sulindac plus 20 mM DCA. Evenzo werden SCC25-kankercellen behandeld met de bovengenoemde vier verschillende combinaties van geneesmiddelen, behalve dat sulindac en DCA werden gebruikt in concentraties van respectievelijk 100 µm en 10 mM. Na de behandeling werd totaal cellulair DNA geëxtraheerd, geligeerd aan de adaptor opgebouwd uit 27-mer 5′-GACGTCGACGTCGTACGTGACT-3′ en 12-mer 5′- AGTCGACGTAC-3′. Na de ligatie werd het DNA verwarmd om het 12-mer vrij te maken, gevuld met Taq polymerase, onderworpen aan semi-kwantitatieve PCR, en geanalyseerd op een 1,2% agarose gel samen met grootte-markers.
In situ lokalisatie van cytochroom c door immunofluorescentie
Intracellulaire locatie van cytochroom c werd gecontroleerd door immunofluorescentie met behulp van de CBA077 InnoCyte™ Flow Cytometric Cytochrome c Release Kit volgens de instructies van de fabrikant. Kortom, de SCC25-cellen werden uitgezet op 3,5×105 cellen per 35 mm glasbodemschotel en gedurende 15 uur behandeld met de aangegeven geneesmiddelen. De cellen werden gespoeld in 5 ml 1× PBS en gedurende 10 minuten op ijs gepermeabiliseerd in 300 µl bijgeleverde buffer. De cellen werden gedurende 20 minuten bij kamertemperatuur gefixeerd in 500 µl 4% paraformaldehyde. Na het wassen en blokkeren werden de cellen geïncubeerd met 250 µl anti-cytochroom c antilichaam (1∶500 verdunning) gedurende 1 uur bij kamertemperatuur. Na het wassen werden de cellen geïncubeerd met 300 µl FITC-IgG (verdunning 1∶300) gedurende 1 uur bij kamertemperatuur. Ten slotte werden de cellen gekleurd met 300 µl DAPI (1 mg/ml) gedurende 10 minuten bij kamertemperatuur. De cellen werden gevisualiseerd met een Olympus omgekeerde fluorescentiemicroscoop. Beelden werden gemaakt en verwerkt zoals hierboven vermeld. Verschillende velden werden geanalyseerd en representatieve microfoto’s met de lokalisatiepatronen van cytochroom c onder elke behandelingsconditie werden verkregen. Kwantitatieve waarden worden in de tekst gepresenteerd.
Statistische analyse en bepaling van combinatie-indices
De gegevens worden gepresenteerd als gemiddelde ± SEM voor de levensvatbaarheidstests van de cellen. Voor de statistische analyse werd de statistische software Minitab gebruikt om de Student’s t-test uit te voeren en waarden met p<0,05 werden als statistisch significant beschouwd. Om het synergetisch effect van sulindac en DCA op A549 en SCC25 kankercellijnen vast te stellen, werd een kwantitatieve analyse van de dosis-effectrelatie uitgevoerd om de combinatie-indices te bepalen [26]. Zowel sulindac als DCA werden afzonderlijk getest op A549 en SCC25 cellen in de aangegeven concentraties. Voor A549 cellen werd een ratio van 1∶50 aangehouden voor de sulindac:DCA geneesmiddelcombinaties van respectievelijk 0,2 mM:10 mM tot 1 mM:50 mM. Voor SCC25-cellen werd een ratio van 1∶100 aangehouden voor de sulindac:DCA-geneesmiddelcombinaties van respectievelijk 0,05 mM:5 mM tot 0,3 mM:30 mM. Onze experimentele resultaten en de vastgestelde combinatie-indexwaarden zijn opgenomen in de tekst.
Resultaten
Sulindac en DCA veroorzaken een verhoogde doding van A549 en SCC25 kankercellen, maar niet van normale cellen
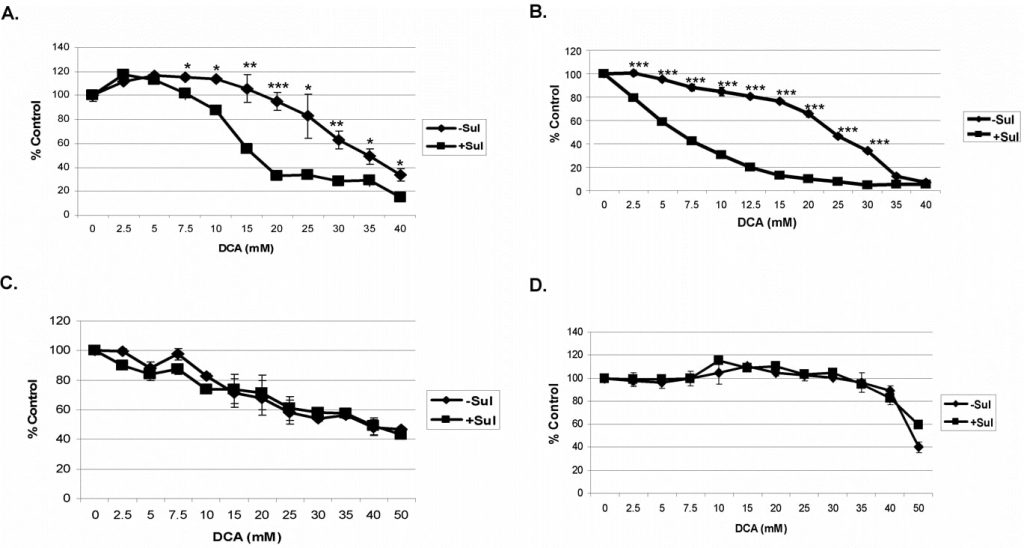
Voor deze studies hebben we de combinatie van sulindac en DCA getest op A549 en SCC25 kankercellen. De cellen werden geïncubeerd met elke verbinding alleen of in combinatie gedurende 48 uur voordat de levensvatbaarheid werd bepaald (zie Methoden). Een sulindac dosis-responscurve onder deze omstandigheden gaf aan dat A549 en SCC25 kankercellen een maximale concentratie van respectievelijk 500 µM en 100 µM sulindac kunnen verdragen zonder significante doding (gegevens niet getoond), en deze concentraties werden in alle studies gebruikt. DCA, indien toegevoegd, werd gebruikt in concentraties van 0-40 mM, zoals aangegeven. Wij gebruikten deze concentraties op basis van eerdere rapporten, die aangaven dat meer dan 5 mM nodig is om mitochondriale disfunctie te veroorzaken in in vitro experimenten [27]. Zoals getoond in figuur 1A, is DCA alleen (zonder sulindac) enigszins toxisch voor A549 kankercellen, vooral boven concentraties van 20 mM, maar in aanwezigheid van sulindac worden deze cellen beter gedood bij DCA-concentraties boven 5 mM. In het geval van de SCC25 kankercellen werd zelfs bij DCA-concentraties onder 10 mM enig verlies aan levensvatbaarheid van de cellen met DCA alleen waargenomen (figuur 1B). In aanwezigheid van sulindac was er echter opnieuw een duidelijke toename van de celdood die duidelijk zichtbaar was tussen DCA-concentraties van 2-10 mM. Eerder toonden wij aan dat de combinatie van sulindac en een oxidatiemiddel selectief was voor kankercellen en het doden van normale cellen niet versterkte [ 7]. Sulindac en DCA versterkten ook niet het doden van normale long- en huidcellen onder de gebruikte experimentele omstandigheden, zoals getoond in figuren 1C en D. Opgemerkt moet worden dat de MRC-5 (normale long) cellen bijzonder gevoelig zijn voor DCA, zoals eerder gemeld [28], om redenen die niet bekend zijn.
Om na te gaan of er sprake was van een synergetisch effect bij gebruik van de geneesmiddelencombinatie, bepaalden wij de combinatie-indices door een kwantitatieve analyse van de dosis-effectrelatie [26] uit te voeren op twee verschillende kankercellijnen (figuur S1). De combinatie-indices waren respectievelijk 0,84 voor de A549 en 0,73 voor de SCC25 kankercellen. Een waarde van minder dan 1,00 wijst op een synergetisch kankerdodend effect (figuur S2).
Het sulindac-effect is niet het gevolg van zijn NSAID-activiteit
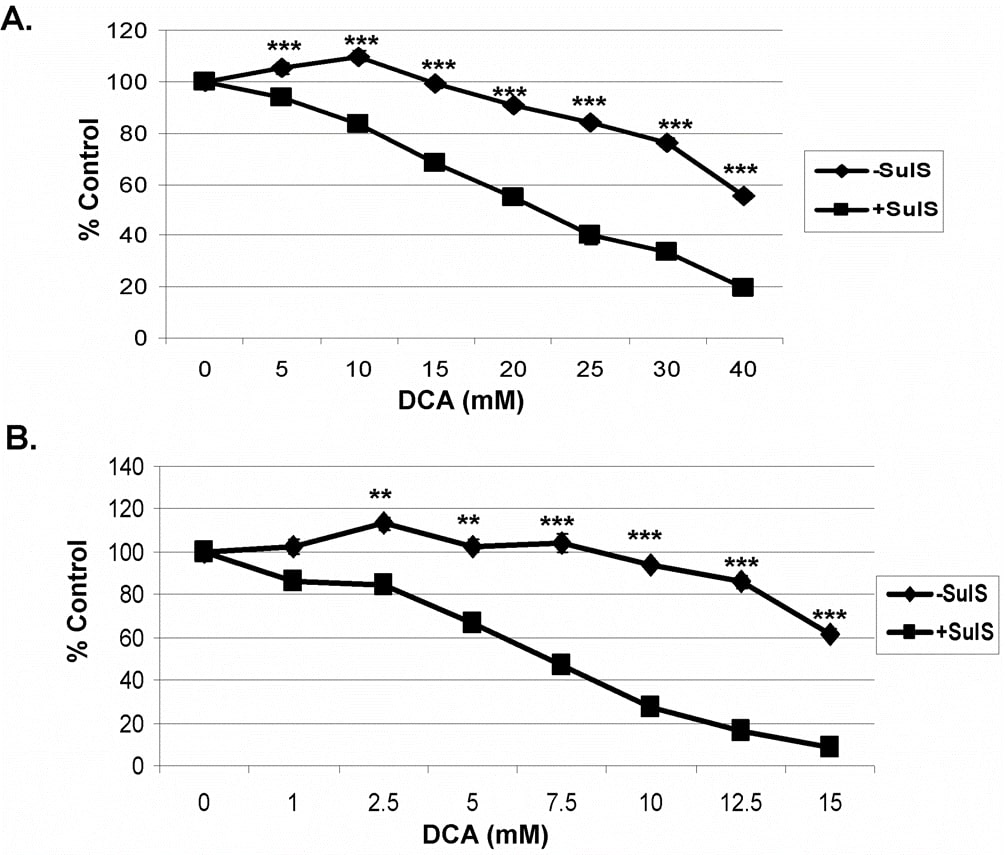
In eerdere studies met sulindac en een oxidatiemiddel werd aangetoond dat de versterkte en selectieve doding van kankercellen door sulindac en een oxidatiemiddel geen verband hield met het bekende NSAID-vermogen van sulindac. Om de rol van COX-remming te bepalen kan een sulindac-metaboliet, sulindac-sulfon, worden gebruikt, aangezien deze COX 1 of 2 niet remt [7], [29]. Zoals getoond in figuur 2, toonde de combinatie van sulindac-sulfon en DCA bij gebruik van zowel A549 (A) als SCC25 (B) kankercellen een vergelijkbaar dodend effect als hierboven gezien met sulindac. Deze resultaten wijzen erop dat het versterkte kankerdodende effect van sulindac in aanwezigheid van DCA geen verband houdt met de bekende ontstekingsremmende werking ervan.
De combinatie van sulindac en DCA genereert ROS
Het met sulindac en dichlooracetaat waargenomen synergetisch effect op de levensvatbaarheid bij zowel A549 als SCC25 kankercellen is opvallend vergelijkbaar met eerdere studies waarbij de combinatie van sulindac en TBHP
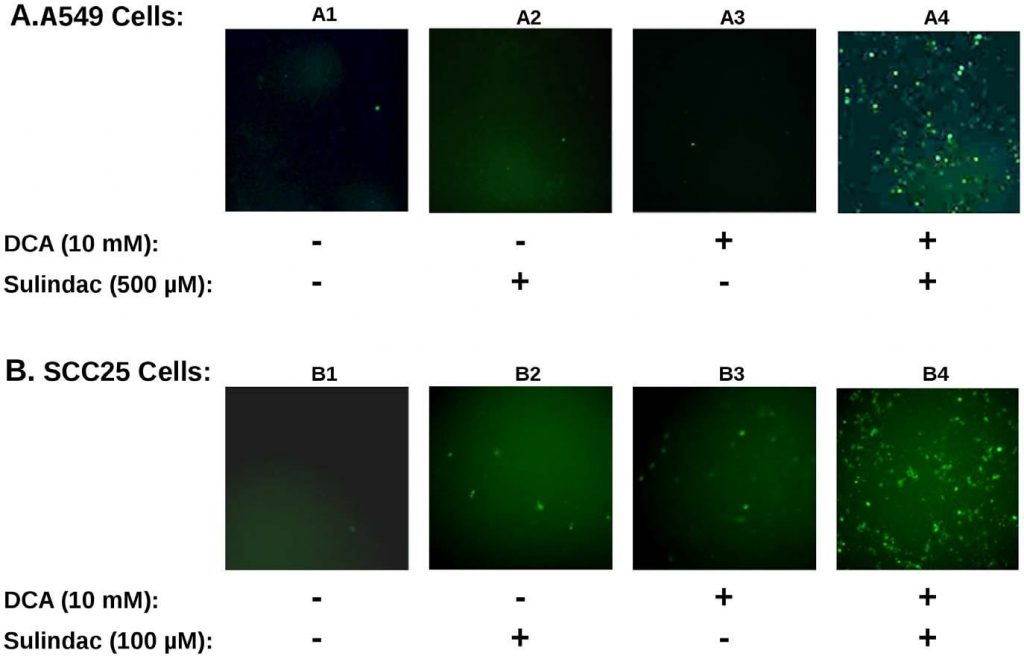
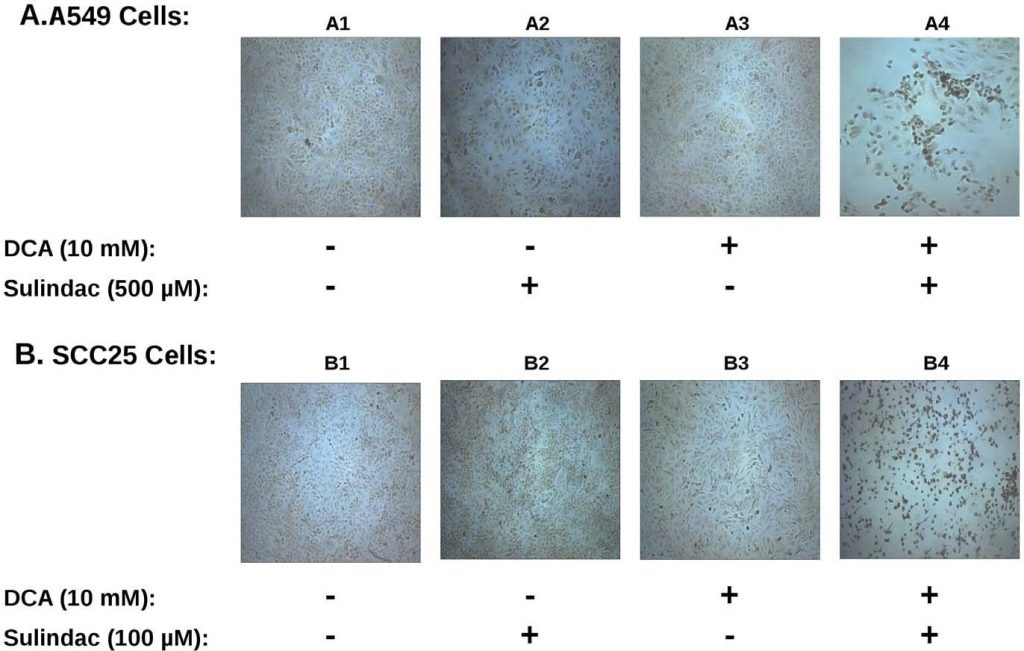
[7]. Om na te gaan of de productie van ROS betrokken was bij de selectieve doding die in deze studies werd waargenomen, werd de productie van ROS, met behulp van de indicator kleurstofH2DCFDA(zie Methoden), bepaald in de kankercellijnen die aan sulindac en DCA werden blootgesteld. De resultaten zijn samengevat in figuur 3. Figuur 3A toont de resultaten met A549 kankercellen. Uit de resultaten in figuur 3A blijkt dat onbehandelde A549 kankercellen (paneel A1), of cellen die alleen met sulindac (paneel A2) of alleen met DCA (paneel A3) zijn behandeld, slechts enkele positief gekleurde cellen vertonen. Wanneer de cellen echter werden blootgesteld aan zowel sulindac als DCA (paneel A4), wordt een grote toename van positief gekleurde cellen voor ROS (groene fluorescentie) gezien, waaruit blijkt dat de aanwezigheid van zowel sulindac als DCA resulteert in het genereren van aanzienlijke hoeveelheden ROS. Zoals blijkt uit figuur 3B worden soortgelijke resultaten gezien met de SCC25-kankercellen. Sulindac of DCA alleen resulteren in een kleine toename van ROS producerende cellen (panelen B2 en B3), maar een grote toename van de ROS-productie wordt opnieuw waargenomen wanneer beide geneesmiddelen worden toegevoegd (paneel B4). Kwantificering met SCC25-cellen laat zien dat het aantal DCF-positieve cellen (zie Methoden) 9-10× hoger is wanneer de cellen worden behandeld met sulindac en DCA in vergelijking met elk van de geneesmiddelen alleen (zie figuur S3A). Uit deze resultaten en eerdere studies blijkt dat ROS-productie een gemeenschappelijk kenmerk kan zijn van de versterkte doding van kankercellen wanneer sulindac wordt gebruikt in combinatie met verbindingen die de mitochondriale functie beïnvloeden.
Sulindac in combinatie met DCA resulteert in een verlies aan mitochondriaal membraanpotentieel
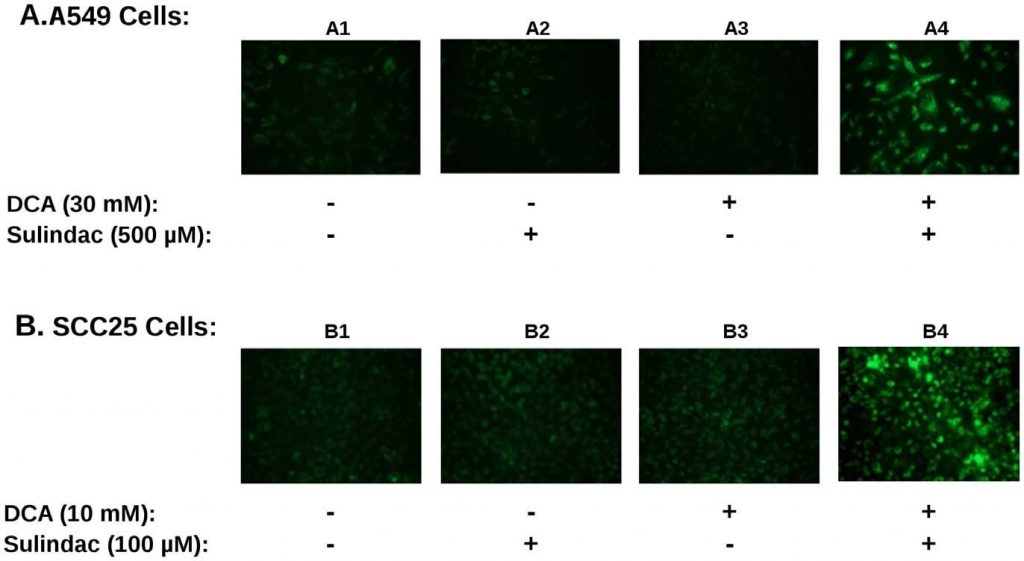
Als ROS-productie betrokken is bij het versterkte dodingseffect van sulindac/DCA zou men verwachten dat de productie van ROS door de combinatie van geneesmiddelen de mitochondriale functie zou beïnvloeden. Om dit te bepalen werd de mitochondriale membraanpotentiaal gemeten met behulp van JC-1 kleuring, zoals beschreven in Methoden. Een verlies aan membraanpotentiaal wordt aangegeven door een toename van de groene fluorescentie, zoals beschreven in de Methoden. Een typisch resultaat is samengevat in figuur 4. Zowel A549 als SCC25 kankercellen werden gedurende 48 uur alleen of in combinatie blootgesteld aan sulindac en DCA en gekleurd met JC-1 om het mitochondriale membraanpotentieel te controleren. Figuur 4A toont de resultaten met de A549 kankercellijn. In afwezigheid van enig geneesmiddel lijken de mitochondriën intact en behouden zij hun membraanpotentiaal, zoals blijkt uit de geringe groene fluorescentie (paneel A1). In aanwezigheid van sulindac alleen (paneel A2) of DCA alleen (paneel A3) is er een kleine toename van de groene fluorescentie, hetgeen wijst op enig verlies van mitochondriaal membraanpotentiaal. Wanneer echter zowel sulindac als DCA aanwezig zijn, is er een opvallend verlies van mitochondriaal membraanpotentiaal, zoals blijkt uit een grote toename van de groene fluorescentie (paneel A4). Wij namen hetzelfde patroon waar toen verschillende onafhankelijke velden met fluorescentiemicroscopie werden geanalyseerd. Figuur 4B toont soortgelijke resultaten met de SCC25-kankercellen. Opnieuw werd een significant verlies van mitochondriaal membraanpotentieel alleen gezien wanneer de cellen werden blootgesteld aan zowel sulindac als DCA (paneel B4). Kwantificering van het effect wordt getoond in figuur S3B. Te zien is dat het percentage JC1-groenpositieve cellen bij gebruik van de geneesmiddelencombinatie 3-4× zo hoog is als bij beide geneesmiddelen alleen.
ROS zijn betrokken bij het doden van kankercellen door de combinatie van sulindac en DCA
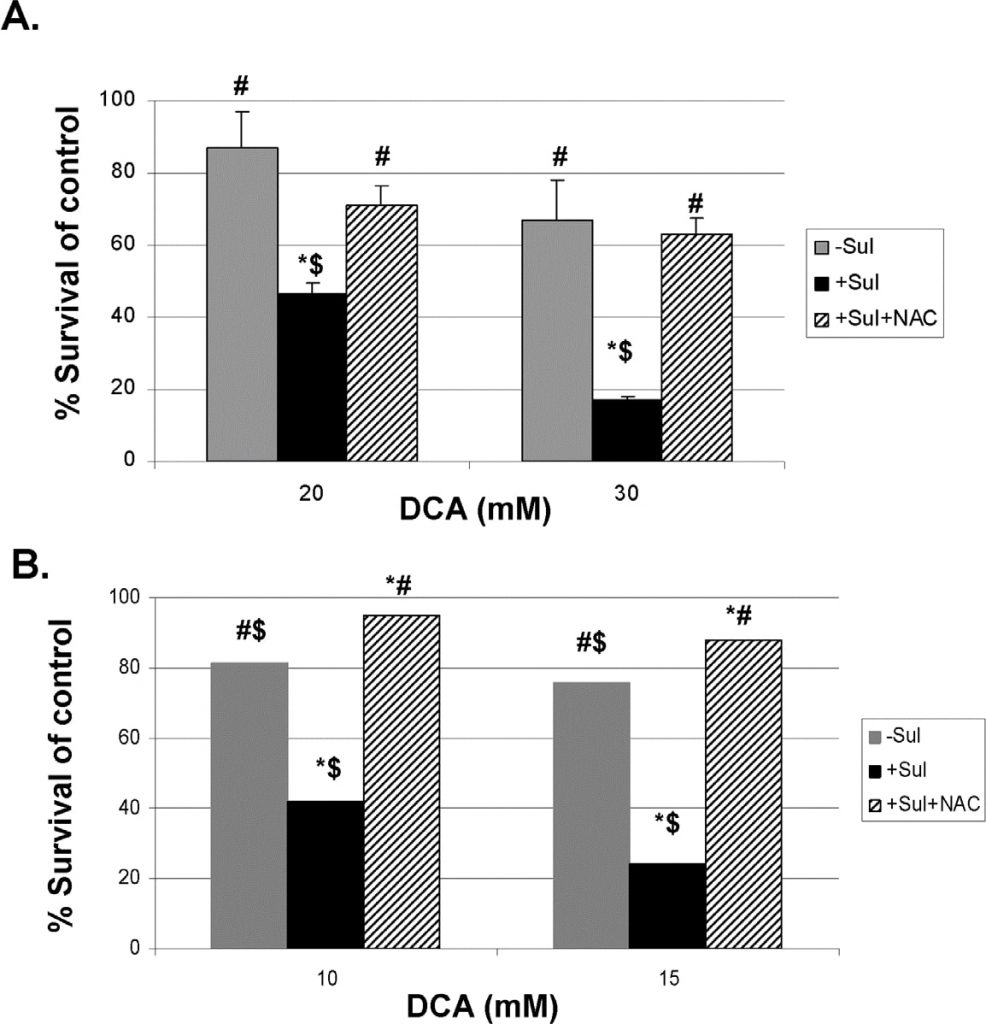
Om meer direct bewijs te leveren dat de geproduceerde ROS betrokken zijn bij het versterkte doden van de kankercellen door sulindac en DCA, hebben we twee bekende ROS-vangers gebruikt, N-acetylcysteïne (NAC) en Tiron (zie Methoden). De resultaten met NAC worden getoond in figuur 5. Figuur 5, paneel A, toont aan dat bij zowel 20 als 30 mM DCA, de versterkte doding van A549 kankercellen, waargenomen in aanwezigheid van sulindac, grotendeels wordt voorkomen als NAC (2 mM) aanwezig is tijdens de 48 uur durende incubaties. Zeer vergelijkbare resultaten worden gezien met de SCC25 kankercellen zoals getoond in figuur 5, paneel B. Vergelijkbare resultaten werden verkregen wanneer Tiron werd gebruikt in plaats van NAC (figuur S4).
Sulindac en DCA doden kankercellen via apoptose
De bovenstaande resultaten (figuren 3, 4 en 5) laten zien dat de versterkte doding van de kankercellijnen gepaard gaat met mitochondriale disfunctie, hetgeen suggereert dat de waargenomen celdood via apoptose verloopt. Eerdere studies hebben aangetoond dat sulindac en zijn derivaten proapoptotische geneesmiddelen zijn [5], [6]. Er zijn ook meldingen dat DCA celdood door apoptose kan veroorzaken [20], [23]. Om te bepalen of het doden van de kankercellen door de combinatie van deze twee geneesmiddelen, gemedieerd door ROS, gepaard gaat met apoptose, hebben we TUNEL-kleuring uitgevoerd om apoptose te meten (zie Methoden). Meerdere herhalingen werden getest voor sulindac en DCA alleen, of in combinatie, voor de TUNEL-kleuringsexperimenten. Een typisch resultaat is weergegeven in figuur 6, waarbij de bovenste panelen (figuur 6A, panelen A1-A4) de resultaten met de A549 kankercellen weergeven en de onderste panelen (figuur 6B, panelen B1-B4) de resultaten met de SCC25 kankercellen. Wanneer de cellen werden behandeld met geen geneesmiddel, sulindac alleen of DCA alleen (figuur 6, panelen A1-A3 en B1-B3), worden slechts enkele TUNEL-positieve cellen waargenomen. Wanneer de cellen echter werden blootgesteld aan zowel sulindac als DCA, is er een significante toename van TUNEL-positieve apoptotische cellen (figuur 6, panelen A4 en B4), hetgeen wijst op een grote inductie van apoptose. Ter verificatie van de TUNEL-resultaten werd ook een gevoeligere ligatie-gemedieerde PCR-gebaseerde DNA laddering assay gebruikt om apoptose te controleren [25]. De resultaten toonden ook de aanwezigheid van een verrijkte sterke nucleosomale ladder alleen wanneer zowel sulindac als DCA in combinatie werden gebruikt (figuur S5; lanes 4 en 8), hetgeen de gegevens van de TUNEL-assay sterk ondersteunt.
Sulindac en DCA Killing Betrekt Proapoptotic JNK Signaling
Van de bekende mitogen activated protein kinases (MAP kinases) is het stress-geïnduceerde kinase, c-Jun N-terminal kinase (JNK/SAPK) direct betrokken bij apoptotische celdood [11]. Daarom onderzochten wij de rol van JNK signalering in sulindac-DCA gemedieerde apoptose door gebruik te maken van SP600125, een JNK-specifieke remmer (JNKI) en deze resultaten zijn weergegeven in figuur 7A. Zoals hierboven getoond, vertoonden SCC25-cellen die met sulindac waren behandeld een versterkte sterfte in aanwezigheid van toenemende DCA-concentraties. Wanneer deze cellen echter werden geïncubeerd met sulindac samen met SP600125, werd sulindac-DCA gemedieerde celdood grotendeels voorkomen. Deze resultaten wijzen op de deelname van JNK-gemedieerde pro-apoptotische signalering in de door sulindac-DCA gemedieerde celdood.
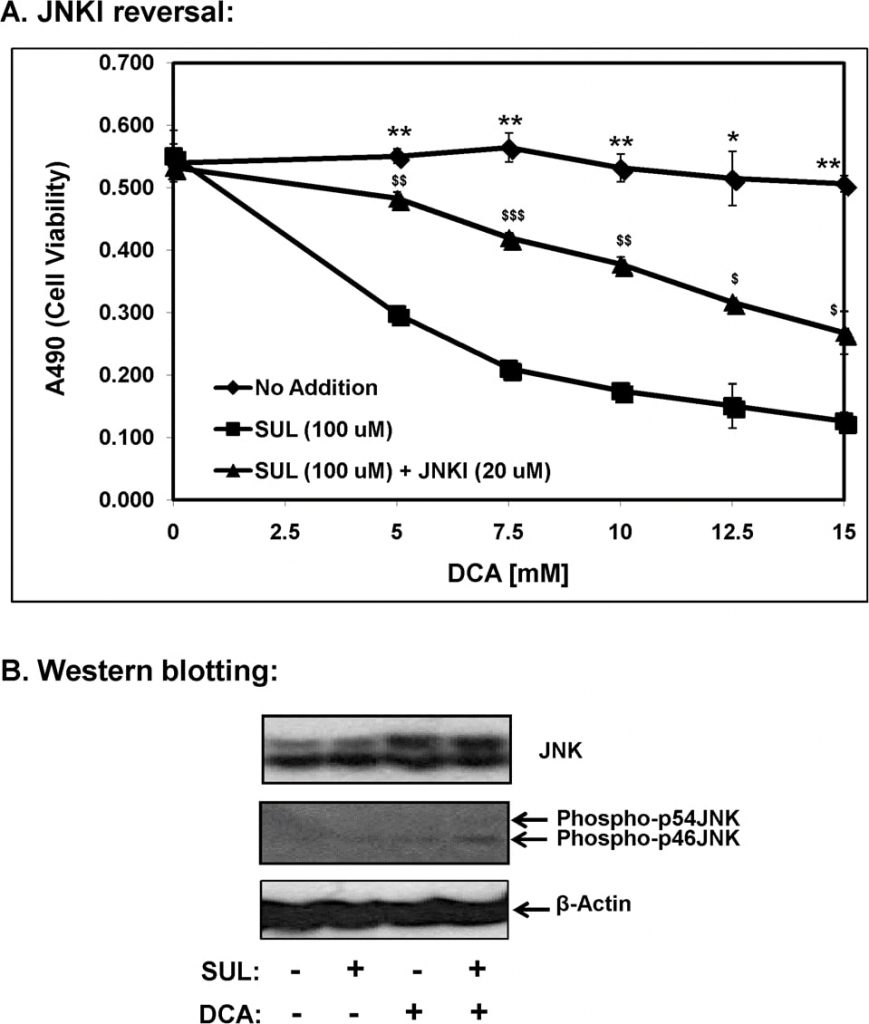
Door middel van western blot analyse werd ook vastgesteld dat de combinatie van sulindac en DCA de niveaus van fosfo-JNK in cytosolische fracties aanzienlijk verhoogde 12 uur nadat de cellen waren blootgesteld aan sulindac en DCA (figuur 7B). Een toename van de totale hoeveelheid JNK (eiwitbanden op 46 en 54 kDa) werd gezien wanneer de cellen alleen met DCA werden behandeld en wanneer de cellen werden behandeld met de combinatie van sulindac en DCA. Opgemerkt zij dat zowel fosfo-p46JNK als fosfo-p54JNK isovormen werden geïnduceerd door de combinatie van behandeling met sulindac en DCA, hoewel de toename van fosfo-p46JNK significanter was (figuur 7B).
Er zijn aanwijzingen dat JNK het vrijkomen van apoptose inducerende factoren uit mitochondria, zoals cytochroom c, in gang zet, die leiden tot splitsing van caspases en PARP (poly(ADP-ribose) polymerase) [30], [31]. Studies hebben ook aangetoond dat tijdens apoptose het cytochroom c dat uit de mitochondriën in het cytoplasma vrijkomt, uiteindelijk in de kern terechtkomt [ 32]. Onze resultaten wijzen op een maximale activering van JNK rond 12 uur na blootstelling aan sulindac en DCA. Dit lijkt te resulteren in de translocatie van cytochroom c naar het cytoplasma en splitsing van PARP 18 uur na de eerste behandeling met sulindac en DCA (figuur S6A). Als positieve controle voor deze experimenten behandelden wij de cellen met 100 µM etoposide, een apoptose-inducerend middel. Onder de combinatiebehandeling met sulindac en DCA kan een verhoogde nucleaire fluorescentie worden waargenomen in een meerderheid van de cellen die actief apoptose ondergaan (figuur S6B).
Gedetailleerde analyse van de experimentele immunofluorescentiegegevens van hele cellen toonde aan dat ∼94% van de cellen die noch met sulindac, noch met DCA waren behandeld, punctuele, mitochondriale cytochroom c-fluorescentie vertoonden met weinig diffuse kleuring in het cytoplasma of in de kernen. Na behandeling met sulindac daarentegen vertoonde 81% van de cellen een diffuse, duidelijke cytoplasmatische fluorescentie en zeer weinig nucleaire fluorescentie. Na DCA-behandeling vertoonde ∼83% van de cellen een diffuse, duidelijke cytoplasmatische fluorescentie, en <5% van de cellen een sterke nucleaire fluorescentie. Wanneer de cellen echter met zowel sulindac als DCA werden behandeld, vertoonde ∼72% van de cellen zowel nucleaire als cytoplasmatische fluorescentie en ∼11% van de cellen vertoonde een sterke nucleaire fluorescentie. Deze resultaten suggereren dat het vrijgekomen cytochroom c uit de mitochondriën de intrinsieke apoptotische route kan initiëren die functioneert bij de door sulindac en DCA gemedieerde kankerdoding.
Bespreking
De huidige studie is een uitbreiding van ons eerdere werk, waaruit bleek dat sulindac kankercellen, maar geen normale cellen, gevoeliger maakte voor oxidatieve stress [7]. In deze eerdere experimenten werd sulindac gedurende 24-48 uur met de cellen gepreïncubeerd, waarna de sulindac werd verwijderd voordat de cellen gedurende 2 uur werden blootgesteld aan TBHP of H2O2. Uit de eerdere experimenten bleek dat de voorbehandeling met sulindac de kankercellen veel gevoeliger maakte voor het oxidatiemiddel, wat resulteerde in een grote toename van ROS en verlies van de mitochondriale functie [7].
Het leek redelijk, op basis van deze resultaten, dat sulindac in combinatie met verbindingen die de mitochondriale ademhaling beïnvloeden, zou resulteren in een selectieve versterkte doding van kankercellen, maar niet van normale cellen. In het huidige onderzoek met A549 en SCC25 kankercellijnen bleek dat de combinatie van sulindac en DCA de doding van deze kankercellijnen versterkte, maar niet van normale long- of huidcellen. Onze resultaten over de benodigde hoeveelheden DCA in hele cellen komen overeen met wat eerder is gerapporteerd [28], [33], [34]. In ons systeem is de IC50 voor DCA bij SCC25-cellen 23 mM en bij A549-cellen 35 mM. De IC50 voor normale keratinocyten is >50 mM en voor normale longcellen (MRC5) is ∼40 mM. De resultaten wezen er ook op dat bij de waargenomen kankerceldood ROS-productie, JNK-activering en door mitochondriën geïnitieerde apoptose betrokken zijn. Wat betreft een gebrek aan effect op normale cellen, is aangetoond dat sulindac normale longcellen beschermt tegen oxidatieve schade als gevolg van blootstelling aan TBHP [7] en wij hebben onlangs ook gemeld dat sulindac hartcellen kan beschermen tegen oxidatieve schade als gevolg van ischemie/reperfusie via een preconditioneringsmechanisme [35].
Voor zover wij weten zijn er nu ten minste 8 verbindingen, waaronder onze studies met TBHP, H2O2 en DCA, die een verbeterde en selectieve kankerdoding hebben laten zien in aanwezigheid van sulindac [7]–[9], [12]–[15]. Hoewel hun metabolische doelen binnen de cel bekend en verschillend zijn, is het zeer waarschijnlijk dat ze allemaal, direct of indirect, celdood veroorzaken in aanwezigheid van sulindac via een mechanisme dat een verandering in de mitochondriale ademhaling en ROS-productie inhoudt [10], [12], [14]. Het lijkt waarschijnlijk dat wanneer men een geneesmiddel vindt dat in combinatie met sulindac selectief kankercellen doodt, maar geen normale cellen, het moordmechanisme oxidatieve stress inhoudt die leidt tot mitochondriale disfunctie. Een veranderde ademhaling kan een gemeenschappelijke factor zijn in deze experimenten met combinaties van sulindac en geneesmiddelen en de huidige resultaten met DCA ondersteunen deze zienswijze. Het is goed mogelijk dat het sulindac effect verband houdt met de waarnemingen die meer dan 50 jaar geleden werden gedaan door Warburg, die opmerkte dat normale cellen de voorkeur geven aan ademhaling om hun energie te verkrijgen, terwijl kankercellen de voorkeur geven aan glycolyse, als gevolg van een defect in de ademhalingsketen [16]. Dit fundamentele verschil in mitochondriale ademhaling tussen normale en kankercellen kan kankercellen gevoeliger maken voor oxidatieve stress [36], [37]. Het lijkt erop dat sulindac dit fundamentele verschil in de biochemie van normale en kankercellen kan versterken. Hoe sulindac kankercellen gevoelig maakt voor geneesmiddelen die de mitochondriale ademhaling aantasten is nog niet duidelijk, maar wordt actief onderzocht. Spitz en medewerkers [38] zijn in studies over glucosedeprivatie van kankercellen tot een soortgelijke conclusie gekomen met betrekking tot de verschillen in metabolisme tussen normale en kankercellen. In overeenstemming met deze resultaten heeft een andere recente studie aangetoond dat farmacologische remming van lactaatdehydrogenase zou kunnen leiden tot selectieve kankerdoding [39]. In laatstgenoemd onderzoek werd aangetoond dat de versterkte selectieve doding van kankercellen ook gepaard ging met ROS-productie, en het waargenomen effect werd toegeschreven aan een veranderd ademhalingsproces in de kankercellen.
Er zij op gewezen dat de combinatie van sulindac met een oxiderende stof of geneesmiddelen die de mitochondriale functie kunnen beïnvloeden, reeds klinisch is getest. Meyskens et al. 2008 toonden aan dat de combinatie van sulindac met DFMO een significant effect had op het terugkomen van colonpoliepen en het ontstaan van colonkanker in een klinische studie van 3 jaar [13]. Wij hebben onlangs het gebruik van sulindac, met H2O2, gerapporteerd in een proof of concept klinische studie voor de topische behandeling van actinische keratosen [ 15]. Een van de nadelen van deze combinatie was de noodzaak van twee topische formuleringen, aangezien de verbindingen niet langdurig konden worden bewaard zonder vernietiging van de sulindac door de H2O2. Bovendien kan men H2O2 niet gebruiken voor de behandeling van inwendige tumoren omdat het niet oraal kan worden ingenomen. De combinatie van sulindac en DCA zou echter als één enkele formulering kunnen worden toegediend die geschikt is voor uitwendig gebruik, en de twee verbindingen kunnen oraal worden gebruikt. In feite wordt DCA al enkele jaren klinisch gebruikt om het melkzuurgehalte te verlagen bij patiënten die lijden aan melkziekte [40]–[42]. DCA is ook gebruikt als middel tegen kanker in vitro en in vivo met verschillende kankercellijnen, wat erop wijst dat het mitochondriale metabolisme in kankercellen een nieuw therapeutisch doelwit zou kunnen zijn [18], [20], [22]. Michelakis et al., (2010) hebben aangetoond dat behandeling met DCA het mitochondriaal metabolisme in glioblastoma-patiënten “remodelleert” met omkeerbare toxische effecten. Opgemerkt zij dat zowel sulindac als DCA betaalbaar en relatief niet-toxisch zijn en oraal kunnen worden ingenomen. Als de combinatie in vivo succesvol blijkt te zijn, zal zij een nieuwe dimensie toevoegen aan de behandeling van kanker, aangezien beide geneesmiddelen het mitochondriale metabolisme bij meerdere vormen van kanker aanpakken [22].
Kortom, onze studies met de combinatie van sulindac en DCA suggereren dat sulindac kankercellen selectief gevoeliger maakt voor middelen die de mitochondriale ademhaling aantasten, wat leidt tot oxidatieve stress en mitochondriale disfunctie. Deze resultaten zouden verband kunnen houden met het ademhalingsdefect in kankercellen, dat oorspronkelijk door Warburg werd waargenomen [16]. Momenteel wordt onderzoek gedaan naar de fundamentele verschillen tussen de reactie van kankercellen en normale cellen op sulindac en stoffen die de mitochondriale functie beïnvloeden.
Ondersteunende informatie
Figuur S1 Cytotoxiciteit van sulindac, DCA, of combinatie van geneesmiddelen op A549 en SCC25 kankercellen
De A549 en SCC25 kankercellen werden behandeld met de aangegeven concentraties sulindac (panelen 1 en 4) of DCA (panelen 2 en 5). In de sulindac/DCA geneesmiddelcombinatie-experimenten werd een verhouding van 1:50 (sulindac:DCA) aangehouden voor de A549-cellen, met geneesmiddelcombinaties variërend van 0,2 mM:10 mM tot 1 mM:50 mM (paneel 3) en een verhouding van 1:100 (sulindac:DCA) werd aangehouden voor de SCC25-cellen met geneesmiddelcombinaties variërend van 0,05 mM:5 mM tot 0,3 mM:30 mM (paneel 6). (TIF) Figuur S2 Bepaling van de combinatie-indices voor A549- en SCC25-kankercellen. De indices voor de combinatie van geneesmiddelen werden bepaald door de hierboven verkregen waarden voor de levensvatbaarheid van de cellen op te nemen in de vergelijkingen van Chou en Talalay [26]. Zie de tekst voor meer details. (TIF) Figuur S3 Kwantificering van DCF- en JC-1-positieve groene fluorescerende cellen. SCC25-cellen werden gedurende 48 uur behandeld met sulindac, DCA of een combinatie van geneesmiddelen en gekleurd met H2DCFDA (fig. S3A) of JC-1 (fig. S3B) kleurstoffen zoals vermeld in de methoden. Voor een positieve controle werden de cellen gedurende 2 uur behandeld met 200 mM TBHP en werd de kleuring uitgevoerd zoals hierboven vermeld. De cellen werden geanalyseerd onder sterke vergroting met een 1006 objectief in een Olympus omgekeerde fluorescentiemicroscoop. Voor elke conditie werden ten minste 100 individuele cellen gevisualiseerd en de percentages DCF-positieve en JC1-positieve groene fluorescerende cellen worden in tabel- en grafiekvorm weergegeven. (TIF) Figuur S4 De ROS-vanger Tiron keert het doden van kankercellen door de combinatie van sulindac en DCA om. De kankercellen A549 en SCC25 werden gedurende 48 uur behandeld met de aangegeven concentraties DCA in afwezigheid (grijze balk) of aanwezigheid van sulindac (zwarte balk) of aanwezigheid van sulindac en Tiron (gestreepte balk). De levensvatbaarheid van de cellen werd gecontroleerd met de MTS-test, zoals vermeld in de methoden. De levensvatbaarheid van de cellen wordt uitgedrukt als % van de controle (cellen niet behandeld met sulindac). De foutbalkjes zijn de standaardfout van het gemiddelde (SEM), uitgedrukt als % van de gemiddelde waarde van viervoudige exemplaren van een representatief experiment. Remming van kankercelgroei trad dosisafhankelijk op bij combinatiebehandeling van DCA en sulindac (zwarte balken) in zowel A549-kankercellen (A) als SCC25-kankercellen (B). Deze versterkte doding werd echter voorkomen wanneer Tiron aanwezig was samen met de combinatiebehandeling met geneesmiddelen (gestreepte balken in A en B). (TIF) Figuur S5 Uitgesproken nucleosomale DNA-laddering treedt op tijdens het doden van kankercellen door de combinatie van sulindac en DCA. De kankercellen A549 en SCC25 werden gedurende 48 uur behandeld met de aangegeven geneesmiddelen. Nucleosomaal DNA werd geëxtraheerd en onderworpen aan ligatiegemedieerde PCR zoals beschreven in Methoden en geanalyseerd op een 1,2% agarose gel samen met grootte markers. Lane ‘M’ geeft moleculaire grootte markers aan. De banen 1-4 en 5-8 tonen de resultaten die werden verkregen met respectievelijk A549-kankercellen en SCC25-kankercellen. De resultaten zijn weergegeven in lanen 1 en 5 (geen geneesmiddel), lanen 2 en 6 (alleen sulindac), lanen 3 en 7 (alleen DCA) en lanen 4 en 8 (sulindac en DCA). Een versterkte nucleosomale DNA-laddering werd alleen waargenomen bij behandeling met een combinatie van sulindac en DCA (lanen 4 en 8). (TIF)
Figuur S6 Combinatie van sulindac en DCA leidt tot afgifte van cytochroom c uit mitochondria en splitsing van PARP
SCC25-cellen werden behandeld met sulindac, DCA, een combinatie van geneesmiddelen of etoposide om de intracellulaire locatie van cytochroom c te bepalen met behulp van western blotting en immunofluorescentie. A. Cytosolische fracties werden geïsoleerd na 18 uur en de aanwezigheid van cytochroom c in het cytoplasma en de splitsing van PARP werd bepaald door western blotting. Representatieve western blots tonen de hoeveelheid cytochroom c en gekloofd PARP. b-actine niveaus werden gebruikt als een interne controle. B. Immunofluorescentie werd uitgevoerd met behulp van de CBA077 InnoCyteTM Flow Cytometric Cytochrome c Release Kit volgens de instructies van de fabrikant. Verschillende onafhankelijke velden werden geanalyseerd en de representatieve microfoto’s tonen de lokalisatiepatronen van cytochroom c bij blootstelling van de cellen aan sulindac en/of DCA. Kwantitatieve waarden worden gepresenteerd in de tekst. (TIF)
Dankbetuigingen
De auteurs bedanken David Brunell voor hulp bij het bepalen van de combinatie-indices en Edna Gamliel voor hulp bij de celkweek.
Bijdragen van de auteurs
Opzet en ontwerp van de experimenten: KA SK KDS HW. Voerde de experimenten uit: KA SK. Analyseerde de gegevens: KA SK HW. Bijgedragen reagentia/materialen/analysehulpmiddelen: KDS. Schreef het artikel: KA SK HW.
VERWIJZINGEN
1 Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, et al. (1996) Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res 56: 2556-2560.
2 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J Natl
3 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst 90: 1529-1536.
4 Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, et al. (1995) Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res 55: 1464-1472.
5 Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, et al. (2001) Progressie en NSAID-geïnduceerde apoptose in kwaadaardige melanomen zijn onafhankelijk van cyclooxygenase II. Melanoma Res 11: 587-599.
6 Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenesis 22: 17-25.
7 Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, et al. (2009) Sulindac versterkt de doding van kankercellen blootgesteld aan oxidatieve stress. PLoS One 4: e5804.
8 Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, et al. (1999) Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 59: 6178-6184.
9 Jiang TT, Brown SL, Kim JH (2004) Gecombineerd effect van arseentrioxide en sulindac sulfide in A549 menselijke longkankercellen in vitro. J Exp Clin Cancer Res 23: 259-262.
10 Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, et al. (2006) Synergistic induction of apoptosis by sulindac and arsenic trioxide in human lung cancer A549 cells via reactive oxygen species-dependent down-regulation of survivin. Biochem Pharmacol 72: 1228-1236.
11 Jin HO, Seo SK, Woo SH, Lee HC, Kim ES, et al. (2008) A combination of sulindac and arsenic trioxide synergistically induces apoptosis in human lung cancer H1299 cells via c-Jun NH2-terminal kinase-dependent Bcl-xL phosphorylation. Longkanker 61: 317-327.
12 Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, et al. (2005) Sulindac versterkt de proteasoomremmer bortezomib-gemedieerde oxidatieve stress en antikankeractiviteit. Clin Cancer Res 11: 5248-5256.
13 Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, et al. (2008) Difluoromethylornithine plus sulindac voor de preventie van sporadische colorectale adenomen: een gerandomiseerd placebogecontroleerd, dubbelblind onderzoek. Cancer Prev Res (Phila Pa) 1: 32-38.
14 Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, et al. (2008) Combined effects of sulindac and suberoylanilide hydroxamic acid on apoptosis induction in human lung cancer cells. Mol Pharmacol 73: 1005-1012.
15 Resnick L, Rabinovitz H, Binninger D, Marchetti M, Weissbach H (2009) Topical sulindac combined with hydrogen peroxide in the treatment of actinic keratoses. J Drugs Dermatol 8: 29-32.
16 Warburg O (1956) Over het ontstaan van kankercellen. Science 123: 309-314.
17 Whitehouse S, Cooper RH, Randle PJ (1974) Mechanisme van activering van pyruvaatdehydrogenase door dichlooracetaat en andere gehalogeneerde carbonzuren. Biochem J 141: 761-774.
18 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37-51.
19 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60: 1478-1487.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichlooracetaat induceert apoptose in endometriumkankercellen. Gynecol Oncol 109: 394-402.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostaat 68: 1223-1231.
<span id=”22″ class=”referencess blue-text”22 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, et al. (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2: 31ra34.
23 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989-994.
24 Cory AH, Owen TC, Barltrop JA, Cory JG (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3: 207-212.
<span id=”25″ class=”referencess blue-text”25 Staley K, Blaschke AJ, Chun J (1997) Apoptotische DNA-fragmentatie wordt gedetecteerd door een semi-kwantitatieve ligatie-gemedieerde PCR van stompe DNA-einden. Cell Death Differ 4: 66-75.
26 Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regulering 22: 27-55.
<span id=”27″ class=”referencess blue-text”27 Papandreou I, Goliasova T, Denko NC (2010) Antikankermedicijnen die het metabolisme aanpakken: is dichlooracetaat het nieuwe paradigma? Int J Cancer 128: 1001-1008.
28 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, et al. (2010) Natriumdichlooracetaat richt zich selectief op cellen met defecten in de mitochondriale ETC. Int J Cancer 127: 2510-2519.
29 Babbar N, Ignatenko NA, Casero RA Jr, Gerner EW (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J Biol Chem 278: 47762-47775.
30 Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haikel Y, et al. (2011) Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenesis 32: 1268-1278.
31 Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, et al. (2007) c-Jun N-terminal kinase medieert waterstofperoxide-geïnduceerde celdood via aanhoudende poly(ADP-ribose) polymerase-1 activering. Cell Death Differ 14: 1001-1010.
<span id=”32″ class=”referencess blue-text”32 Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, et al. (2004) Nucleaire translocatie van cytochroom c tijdens apoptose. J Biol Chem 279: 24911-24914.
33 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, et al. (2011) Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 67: 647-655.
34 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichlooracetaat induceert apoptose en celcyclusstilstand in colorectale kankercellen. Br J Cancer 102: 1746-1752.
35 Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac biedt ischemische bescherming op hoog niveau aan het hart via late preconditioneringsmechanismen. Proc Natl Acad Sci U S A 106: 19611-19616.
<span id=”36″ class=”referencess blue-text”36 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18: 54-61.
37 Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029-1033.
<span id=”38″ class=”referencess blue-text”38 Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, et al. (2005) Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280: 4254-4263.
<span id=”39″ class=”referencess blue-text”39 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107: 2037-2042.
<span id=”40″ class=”referencess blue-text”49 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI (1983) Treatment of lactic acidosis with dichloroacetate. N Engl J Med 309: 390-396.
41 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77: 535-541.
42 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, et al. (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrie 121: e1223-1228.
Gerelateerde inhoud: