Kasirajan Ayyanathan1,2,*,#, Shailaja Kesaraju1,#, Ken Dawson-Scully1,2, Herbert Weissbach1
1 Center for Molecular Biology and Biotechnology, Charles E. Schmidt College of Science, Florida Atlantic University, Jupiter, Floride, États-Unis d’Amérique,
2 Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, Floride, États-Unis d’Amérique
*E-mail: [email protected]
#Cesauteurs ont contribué de manière égale à ce travail.
Reçu : 12 août 2011
Accepté : 4 juin 2012
Publié : 1 juillet 2012
Résumé
Le sulindac est un médicament anti-inflammatoire non stéroïdien approuvé par la FDA avec des activités anticancéreuses documentées. Nos études récentes ont montré que le sulindac renforçait sélectivement la destruction des cellules cancéreuses exposées à des agents oxydants via la production d’espèces réactives de l’oxygène (ERO) résultant en un dysfonctionnement mitochondrial. Cet effet du sulindac et du stress oxydatif sur les cellules cancéreuses pourrait être lié au défaut de respiration dans les cellules cancéreuses, décrit pour la première fois par Warburg il y a 50 ans, connu sous le nom d’effet Warburg. Nous avons émis l’hypothèse que le sulindac pourrait renforcer la destruction sélective des cellules cancéreuses lorsqu’il est associé à un composé qui altère la respiration mitochondriale. Pour tester cette hypothèse, nous avons utilisé le dichloroacétate (DCA), qui est connu pour déplacer le métabolisme du pyruvate de la formation d’acide lactique vers la respiration. On pourrait s’attendre à ce que le DCA, puisqu’il stimule le métabolisme aérobie, puisse stresser la respiration mitochondriale dans les cellules cancéreuses, ce qui aurait pour conséquence d’augmenter la destruction en présence de sulindac. Dans cette étude, nous avons montré que la combinaison du sulindac et du DCA augmente la destruction sélective des cellules cancéreuses A549 et SCC25 dans les conditions utilisées. Comme prévu, le mécanisme de destruction implique la production de ROS, le dysfonctionnement mitochondrial, la signalisation JNK et la mort par apoptose. Nos résultats suggèrent que l’association médicamenteuse sulindac-DCA pourrait constituer une thérapie efficace contre le cancer.
Citation : Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H (2012) La combinaison du sulindac et du dichloroacétate tue les cellules cancéreuses par le biais de dommages oxydatifs
. PLoS ONE 7(7) : e39949. doi:10.1371/journal.pone.0039949
Editeur : Joseph Alan Bauer, Fondation de recherche Bauer, États-Unis d’Amérique
© 2012 Ayyanathan et al. Il s’agit d’un article en accès libre distribué selon les termes de la licence Creative Commons Attribution, qui autorise
l’utilisation, la distribution et la reproduction sans restriction sur n’importe quel support, à condition que l’auteur original et la source soient crédités.
Financement : Nous remercions les National Institutes of Health pour leur aide financière à KA (subvention 5K01CA95620) et HW (subvention R15 CA122001) et l’État de Floride à HW (subvention SURECAG R94007) pour la réalisation de ce travail. Les financeurs n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’analyse des données, la décision de publier ou la préparation du manuscrit.
Intérêts concurrents : Les auteurs ont déclaré qu’il n’existe aucun intérêt concurrent.
INTRODUCTION
Le sulindac est un anti-inflammatoire non stéroïdien (AINS) approuvé par la FDA, dont l’activité anticancéreuse a également été démontrée [1-6]. Des études récentes de notre laboratoire ont montré que les lignées cellulaires cancéreuses RKO, A549 et SCC25 étaient sensibles à une combinaison de sulindac et d’un agent oxydant, tel que le TBHP ou H2O2 [7]. Les données indiquent que l’effet du sulindac n’est pas lié à son activité AINS, mais que le sulindac rend les cellules cancéreuses plus sensibles au stress oxydatif, ce qui entraîne un dysfonctionnement des mitochondries et une perte de viabilité. En revanche, les cellules normales n’ont pas présenté d’augmentation de la mortalité dans des conditions similaires [7]. Au cours des dix dernières années, des rapports épars ont fait état d’une meilleure élimination des cellules cancéreuses en utilisant le sulindac en association avec divers composés, dont le trioxyde d’arsenic, le bortézomib, la difluorométhylornithine (DFMO) et l’acide hydroxamique de subéroylanilide (SAHA) [8-14]. Bien que ces composés aient des sites d’action différents, un mécanisme commun pour l’amélioration de l’efficacité de l’association sulindac/médicament pourrait impliquer des dommages oxydatifs, comme l’ont clairement démontré nos études précédentes utilisant le sulindac et un agent oxydant [7], [15]. En fait, les ROS ont été impliqués dans les études utilisant le sulindac en association avec le trioxyde d’arsenic, le bortezomib et le SAHA [10], [12], [14].
Nos résultats précédents suggéraient que l’augmentation de la destruction des cellules cancéreuses par l’association du sulindac et d’un agent oxydant pouvait être due à un défaut de respiration dans les cellules cancéreuses, comme décrit pour la première fois par Warburg il y a plus de 50 ans [16], qui a noté que les cellules cancéreuses favorisaient la glycolyse, et non la respiration, pour obtenir de l’énergie, contrairement aux cellules normales. Certaines cellules cancéreuses obtiennent jusqu’à 50 % de leur énergie par la glycolyse, alors que la glycolyse dans les cellules normales représente moins de 5 % des besoins énergétiques [16]. Pour obtenir des preuves supplémentaires des rôles possibles de l’altération de la respiration et des ROS dans la destruction des cellules cancéreuses par le sulindac et le stress oxydatif, nous avons lancé des études avec de l’acide dichloroacétique (DCA) sodique. Le DCA est un candidat idéal car il est connu pour inhiber une kinase qui régule à la baisse l’activité de la pyruvate déshydrogénase, ce qui entraîne un déplacement du métabolisme du pyruvate de la formation d’acide lactique vers la respiration [17], [18]. Le DCA a été utilisé en clinique pour traiter les patients souffrant d’acidose lactique [19], et sur la base de ses propriétés biochimiques, le DCA a également été testé comme agent anticancéreux. Bonnet et al. 2007 ont montré que le DCA inverse l’effet Warburg dans les cellules cancéreuses en redirigeant le métabolisme des cellules cancéreuses de la glycolyse vers la phosphorylation oxydative. Dans ces études précédentes, il a été montré que le DCA augmente les niveaux de ROS du complexe I. Ceci déclenche à son tour un » remodelage » du métabolisme mitochondrial (réduction de ΔΨm, ouverture du pore de transition mitochondrial) dans les cellules cancéreuses, les poussant vers l’apoptose. En outre, plusieurs études récentes ont vérifié que le DCA peut augmenter les niveaux de ROS dans les cellules cancéreuses et dépolariser la membrane des mitochondries dans les lignées cellulaires de poumon, d’endomètre et de glioblastome, entraînant l’apoptose in vitro et in vivo [18], [20-22]. Il est intéressant de noter que, dans les conditions utilisées, le DCA ne semble pas affecter de manière significative le métabolisme mitochondrial ou la viabilité des cellules normales [18], [23].
Sur la base de nos observations précédentes sur l’effet anticancéreux du sulindac et d’un agent oxydant qui affecte le métabolisme mitochondrial [7], nous avons postulé que l’association du sulindac et du DCA pourrait renforcer de manière synergique l’action anticancéreuse et avoir une valeur thérapeutique importante. Dans la présente étude, nous avons examiné l’effet de l’utilisation du sulindac en association avec le DCA sur la viabilité des lignées cellulaires cancéreuses A549 et SCC25. Nous avons également étudié le rôle de la fonction mitochondriale et de l’apoptose dans la destruction du cancer observée avec cette combinaison de médicaments.
Matériel et méthodes
Matériaux
Le sulindac, la N-acétylcystéine et le Tiron ont été achetés chez Sigma (St.Louis, MO). Le sel de sodium du DCA a été obtenu auprès d’Acros Organics (Geel, Belgique). Le H2DCFDA et le JC-1 ont été achetés auprès de Molecular Probes (Eugene, OR). Le réactif de dosage MTS et le kit Deadend Tunel ont été obtenus auprès de Promega (Madison, WI). Le kit de fractionnement cytosol/mitochondries et le kit de cytochrome c release cytométrique en flux CBA077 InnoCyte™ ont été fournis par Calbiochem, Gibbstown, NJ. Tous les milieux de culture cellulaire, le sérum bovin fœtal et les autres suppléments tels que la pénicilline/streptomycine, la glutamine, etc. ont été achetés auprès de l’American Type Culture Collection (ATCC ; Rockville, MD).
Culture cellulaire
Unelignée de cellules de carcinome pulmonaire non à petites cellules (NSCLC), A549, la lignée de cellules pulmonaires humaines normales, MRC-5, et une lignée de carcinome épidermique dérivée de la langue, SCC25, ont été achetées auprès de l’ATCC (Rockville, MD) et maintenues dans un milieu F12-K complété par du sérum bovin fœtal à 10 %, de la glutamine 2 mM, de la pénicilline 100 UI/ml et de la streptomycine 100 µg/ml dans un incubateur humidifié à 5 % deCO2 à 37°C. Les kératinocytes épidermiques humains normaux ont été obtenus auprès de Promocell GmbH (Heidelberg, Allemagne) et maintenus dans le milieu de culture recommandé. Des cellules normales non immortalisées et de passage précoce ont été utilisées pour les expériences.
Test de viabilité cellulaire
Les cellules cancéreuses A549 et les cellules normales du poumon ont été placées à 3×103 cellules par puits tandis que les cellules cancéreuses SCC25 et les kératinocytes normaux ont été placés à 7,5×103 cellules par puits dans une plaque à 96 puits. Les cellules ont été cultivées pendant 18-20 heures, le milieu a été éliminé dans des conditions aseptiques et remplacé par un milieu de culture frais contenant les combinaisons de médicaments indiquées. Lorsque cela était indiqué, 500 µM de sulindac ont été utilisés avec le cancer A549 et les cellules normales du poumon et 100 µM de sulindac ont été utilisés avec le cancer SCC25 et les cellules kératinocytaires normales. Les plaques ont été incubées pendant 48 heures à 37°C dans un incubateur à 5% deCO2. Le milieu de culture a été éliminé et les cellules ont été soigneusement rincées dans du PBS 1×. La viabilité cellulaire a été déterminée à l’aide du test CellTiter 96 Aqueous One Cell Proliferation Assay (Promega) conformément aux instructions du fabricant. Le test utilise un composé de tétrazolium qui est converti en un formazan hydrosoluble par l’action des déshydrogénases cellulaires présentes dans les cellules métaboliquement actives [24]. Le formazan a été quantifié en mesurant l’absorbance à 490 nm à l’aide d’un lecteur de plaques de microtitration colorimétrique (SpectraMax Plus ; Molecular Devices). L’absorbance de fond a été soustraite de chaque échantillon.
Mesure intracellulaire des ROS
Les lignées cellulaires cancéreuses A549 et SCC25 ont été placées comme indiqué ci-dessus. Après le traitement médicamenteux de 48 heures, les cellules ont été incubées avec 50 µM de diacétate de dichlorodihydrofluorescéine (H2DCFDA, Molecular Probes) dans un milieu sans indicateur pendant 30 min à 37°C. Les cellules ont été rincées avec du PBS et les niveaux de ROS ont été visualisés par microscopie à fluorescence. Les images ont été capturées à l’aide du logiciel Qcapture et traitées dans Adobe photoshop. L’analyse des images a été réalisée à l’aide du logiciel slidebook. Les données obtenues à partir d’une expérience représentative ont été utilisées pour la quantification des cellules positives au DCF, mesurées par la fluorescence verte due au DCF oxydé.
Coloration JC-1 pour surveiller le potentiel membranaire mitochondrial
Le potentiel membranaire mitochondrial a été déterminé à l’aide du colorant JC-1 (Molecular Probes). Les lignées cellulaires cancéreuses A549 et SCC25 ont été placées comme ci-dessus. Après le traitement médicamenteux de 48 heures, les cellules ont été incubées avec 5 ng/ml de colorant JC-1 dans un milieu sans indicateur pendant 30 minutes à 37°C. Les cellules ont été rincées avec du PBS et visualisées par microscopie à fluorescence. Les mitochondries normales absorbent activement le colorant JC-1 d’une manière dépendante du potentiel et forment des agrégats J, ce qui donne une fluorescence rouge. La perturbation et la perte ultérieure du potentiel de la membrane mitochondriale entraînent une augmentation de la fluorescence verte dans le cytosol due au JC-1 monomère, qui est déterminée en suivant l’apparition de la fluorescence verte à l’aide d’un filtre FITC (microscope inversé Zeiss-Axiovert 40 CFL). La capture, le traitement et l’analyse des images ont été effectués comme ci-dessus. Les données obtenues à partir d’une expérience représentative ont été utilisées pour la quantification des cellules positives au vert JC-1.
Effet des capteurs de ROS sur la viabilité cellulaire en présence de Sulindac et de DCA
Les lignées cellulaires cancéreuses A549 et SCC25 ont été placées comme décrit ci-dessus. Pour piéger les ROS, de la N-acétylcystéine (NAC) 2 mM ou du Tiron 2 mM (sel disodique de l’acide 4,5-dihydroxy-1,3-benzènedisulfonique) ont été ajoutés au sulindac et au DCA pendant 48 heures à 37°C. La viabilité cellulaire a été contrôlée par le test MTS et l’analyse statistique a été effectuée comme mentionné ci-dessus.
Coloration TUNEL pour surveiller les cellules en cours d’apoptose
Le dosageTUNELa été réalisé dans des plaques de 96 puits à l’aide du kit de dosage colorimétrique TUNEL DeadEnd (Promega) en suivant le protocole du fabricant. Les lignées cellulaires cancéreuses A549 et SCC25 ont été placées comme indiqué ci-dessus et traitées pendant 48 heures sans médicament, avec du sulindac, du DCA ou une combinaison de médicaments. Après le traitement médicamenteux, les cellules ont été fixées avec du formol et perméabilisées avec 0,2 % de Triton X-100 dans du PBS. Les cellules ont été incubées avec de la désoxynucléotidyl transférase terminale recombinante (TdT) et des nucléotides biotinylés. Les peroxydases endogènes ont été bloquées avec 0,3 % de H2O2 avant l’incubation avec de la peroxydase de raifort-streptavidine (HRP-streptavidine) qui se lie aux nucléotides biotinylés incorporés dans les extrémités entaillées présentes dans les cellules en cours d’apoptose. Les cellules marquées à la HRP-streptavidine ont été détectées par le peroxyde d’hydrogène et la diaminobenzidine (DAB). Les cellules qui présentent une coloration nucléaire brun foncé indiquent une apoptose.
Analyse Western Blot
Les cellules ont été cultivées jusqu’à 70% de confluence, traitées avec les médicaments spécifiés pendant les durées indiquées, et les fractions cytosoliques ont été isolées à l’aide du kit de fractionnement cytosol/mitochondries (Calbiochem, Gibbstown, NJ) selon le protocole du fabricant. En bref, les cellules ont été récoltées à différents moments et ont ensuite été centrifugées à 600×g pendant 5 minutes à 4°C. Les cellules pelletées ont été mises en suspension dans le tampon fourni et incubées pendant 10 minutes sur la glace. Les cellules ont ensuite été homogénéisées à l’aide d’un videur en verre et l’homogénat a été centrifugé à 700×g pendant 10 minutes à 4°C pour sédimenter les noyaux et les débris cellulaires. Le surnageant a été centrifugé à 10 000×g pendant 30 minutes à 4°C pour obtenir le culot mitochondrial et le surnageant a été considéré comme la fraction cytosolique. La concentration en protéines a été déterminée à l’aide d’un test standard de Bradford.
Soixante microgrammes de protéines totales ont été chargés et séparés sur des gels NuPage Bis-Tris 4-12% (Invitrogen, Eugene, OR) et transférés sur une membrane PVDF qui a été sondée par les anticorps primaires. Les anticorps primaires, JNK, pJNK, cytochrome c et PARP (Cell Signaling Technology, Danvers, MA), ont été utilisés à une dilution de 1∶1000. la β-actine, (Santa Cruz Biotechnologies, Santa Cruz, Californie), a été utilisée à une dilution de 1∶4000. Des anticorps secondaires conjugués à la peroxydase de raifort ont été utilisés et les bandes ont été visualisées à l’aide d’une méthode de chimiluminescence améliorée (GE Healthcare, Piscataway, NJ).
Test d’échelonnement de l’ADN par PCR à médiation par ligature pour surveiller l’étendue des cellules subissant l’apoptose
Pour confirmer l’étendue de l’apoptose, un test d’échelonnement de l’ADN nucléosomique par PCR à médiation par ligature a été réalisé comme décrit [25]. Les lignées cellulaires cancéreuses A549 et SCC25 ont été placées à 5×104 et 1×105 cellules par puits dans des boîtes de 35 mm. Les cellules cancéreuses A549 ont été traitées pendant 48 heures avec a) aucun médicament, b) 500 µM de sulindac, c) 20 mM de DCA, et d) 500 µM de sulindac plus 20 mM de DCA. De même, les cellules cancéreuses SCC25 ont été traitées avec les quatre différentes combinaisons de médicaments mentionnées ci-dessus, sauf que le sulindac et le DCA ont été utilisés à des concentrations de 100 µm et 10 mM, respectivement. Après traitement, l’ADN cellulaire total a été extrait, ligaturé à l’adaptateur construit à partir de 27-mer 5′-GACGTCGACGTCGTACGTGTCGACT-3′ et 12-mer 5′- AGTCGACACGTGTAC-3′. Après la ligature, l’ADN a été chauffé pour libérer le 12-mer, rempli de Taq polymérase, soumis à une PCR semi-quantitative et analysé sur un gel d’agarose à 1,2 % avec des marqueurs de taille.
Localisationin situ du cytochrome c par immunofluorescence
La localisation intracellulaire du cytochrome c a été suivie par immunofluorescence à l’aide du kit de libération cytométrique du cytochrome c en flux CBA077 InnoCyte™, conformément aux instructions du fabricant. En bref, les cellules SCC25 ont été placées à 3,5×105 cellules par plat à fond de verre de 35 mm et traitées avec les médicaments indiqués pendant 15 h. Les cellules ont été rincées dans 5 ml de PBS 1× et perméabilisées sur glace pendant 10 min dans 300 µl de tampon fourni. Les cellules ont été fixées à RT pendant 20 min dans 500 µl de paraformaldéhyde à 4%. Après lavage et blocage, les cellules ont été incubées avec 250 µl d’anticorps anti-cytochrome c (dilution 1∶500) pendant 1 h à température ambiante. Après lavage, les cellules ont été incubées avec 300 µl de FITC-IgG (dilution 1∶300) pendant 1 h à température ambiante. Enfin, les cellules ont été colorées avec 300 µl de DAPI (1 mg/ml) pendant 10 min à température ambiante. Les cellules ont été visualisées à l’aide d’un microscope inversé à fluorescence Olympus. Les images ont été capturées et traitées comme mentionné ci-dessus. Plusieurs champs ont été analysés et des micrographies représentatives montrant les schémas de localisation du cytochrome c dans chaque condition de traitement ont été obtenues. Les valeurs quantitatives sont présentées dans le texte.
Analyse statistique et détermination des indices de combinaison
Les données sont présentées sous forme de moyenne ± SEM pour les tests de viabilité cellulaire. Pour l’analyse statistique, le logiciel statistique Minitab a été utilisé pour effectuer le test t de Student et les valeurs avec p<0,05 ont été considérées comme statistiquement significatives. Pour vérifier l’effet synergique du sulindac et du DCA sur les lignées cellulaires cancéreuses A549 et SCC25, une analyse quantitative de la relation dose-effet a été réalisée pour déterminer les indices de combinaison [26]. Le sulindac et le DCA ont été testés seuls sur les cellules A549 et SCC25 aux concentrations indiquées. Pour les cellules A549, un rapport de 1∶50 a été maintenu pour les combinaisons médicamenteuses sulindac:DCA allant de 0,2 mM:10 mM jusqu’à 1 mM:50 mM, respectivement. Pour les cellules SCC25, un rapport de 1∶100 a été maintenu pour les combinaisons de médicaments sulindac:DCA allant de 0,05 mM:5 mM à 0,3 mM:30 mM, respectivement. Nos résultats expérimentaux et les valeurs d’indice de combinaison déterminées sont inclus dans le texte.
Résultats
Lesulindac et le DCA entraînent une destruction accrue des cellules cancéreuses A549 et SCC25, mais pas des cellules normales
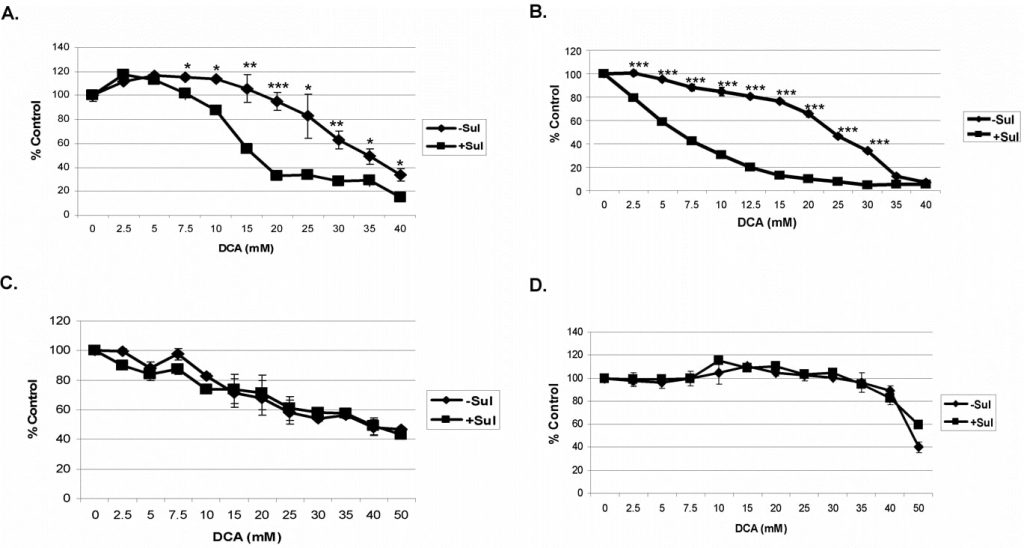
Pour ces études, nous avons testé la combinaison du sulindac et du DCA sur les cellules cancéreuses A549 et SCC25. Les cellules ont été incubées avec chaque composé seul ou en combinaison pendant 48 heures avant d’être testées pour la viabilité (voir Méthodes). Une courbe dose-réponse du sulindac dans ces conditions a indiqué que les cellules cancéreuses A549 et SCC25 peuvent tolérer une concentration maximale de 500 µM et 100 µM de sulindac, respectivement, sans présenter de destruction significative (données non présentées), et ces concentrations ont été utilisées dans toutes les études. Le DCA, lorsqu’il a été ajouté, a été utilisé à des concentrations de 0-40 mM, comme indiqué. Nous avons utilisé ces concentrations en nous basant sur des rapports précédents, qui indiquaient qu’il fallait plus de 5 mM pour provoquer un dysfonctionnement mitochondrial dans des expériences in vitro [27]. Comme le montre la figure 1A, le DCA seul (sans sulindac) est quelque peu toxique pour les cellules cancéreuses A549, surtout à des concentrations supérieures à 20 mM, mais en présence de sulindac, la destruction de ces cellules est accrue à des concentrations de DCA supérieures à 5 mM. Dans le cas des cellules cancéreuses SCC25, une certaine perte de viabilité cellulaire avec le DCA seul a été observée même à des concentrations de DCA inférieures à 10 mM (figure 1B). Cependant, en présence de sulindac, on a constaté une nouvelle fois une augmentation marquée de la mort cellulaire qui était clairement évidente entre les concentrations de DCA de 2 à 10 mM. Nous avons montré précédemment que l’association du sulindac et d’un agent oxydant était sélective pour les cellules cancéreuses et ne favorisait pas la mort des cellules normales [7]. Le sulindac et le DCA n’ont pas non plus favorisé la mort des cellules normales du poumon et de la peau dans les conditions expérimentales utilisées, comme le montrent les figures 1C et D. Il convient de noter que les cellules MRC-5 (normales du poumon) sont particulièrement sensibles au DCA, comme cela a été signalé précédemment [28], pour des raisons qui ne sont pas connues.
Pour vérifier l’existence d’un effet synergique lors de l’utilisation de l’association médicamenteuse, nous avons déterminé les indices de combinaison en effectuant une analyse quantitative de la relation dose-effet [26] sur deux lignées cellulaires cancéreuses différentes (FigureS1). Les indices de combinaison étaient respectivement de 0,84 pour les cellules cancéreuses A549 et de 0,73 pour les cellules SCC25. Une valeur inférieure à 1,00 indique un effet synergique de destruction du cancer (figure S2).
L’effet du sulindac n’est pas dû à son activité AINS
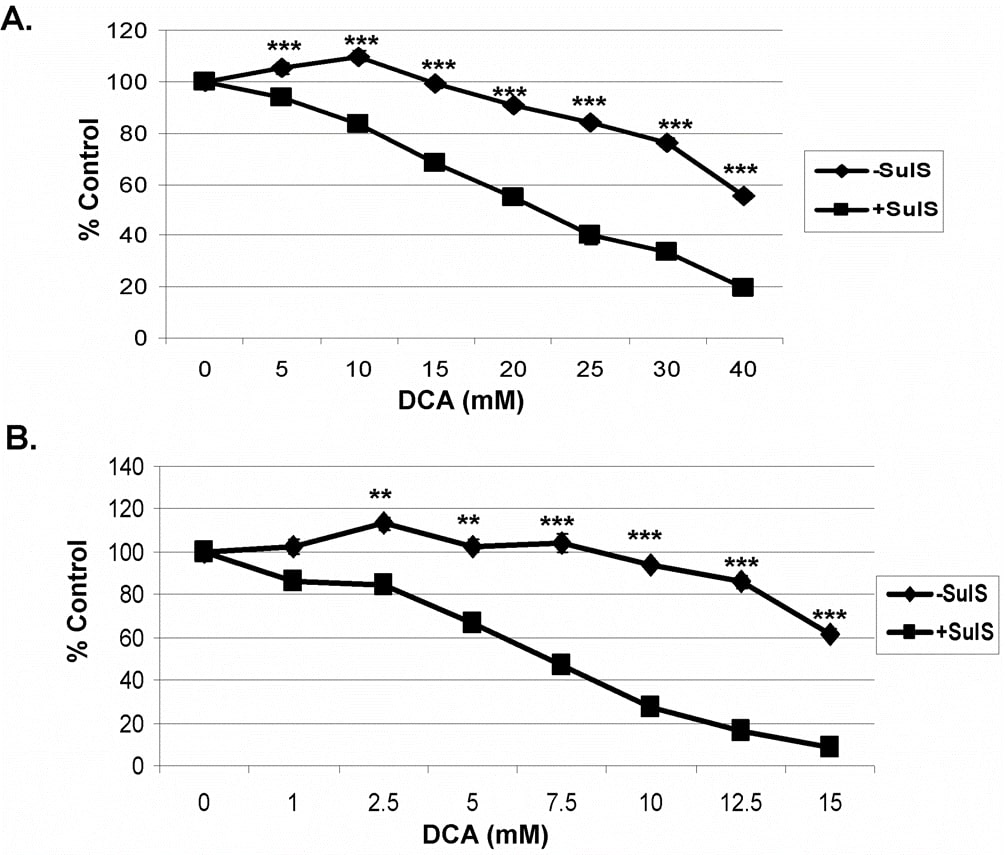
Dans des études précédentes utilisant le sulindac et un agent oxydant, il a été montré que la destruction accrue et sélective des cellules cancéreuses par le sulindac et un agent oxydant n’était pas liée à la capacité AINS connue du sulindac. Pour déterminer le rôle de l’inhibition des COX, un métabolite du sulindac, le sulindac sulfone, peut être utilisé, car il n’inhibe pas les COX 1 ou 2 [7], [29]. Comme le montre la figure 2, en utilisant les cellules cancéreuses A549 (A) et SCC25 (B), la combinaison de sulindac sulfone et de DCA a montré un effet de destruction similaire à celui observé ci-dessus avec le sulindac. Ces résultats indiquent que l’effet anticancéreux accru du sulindac en présence du DCA n’est pas lié à son activité anti-inflammatoire connue.
La combinaison du sulindac et du DCA génère des ROS
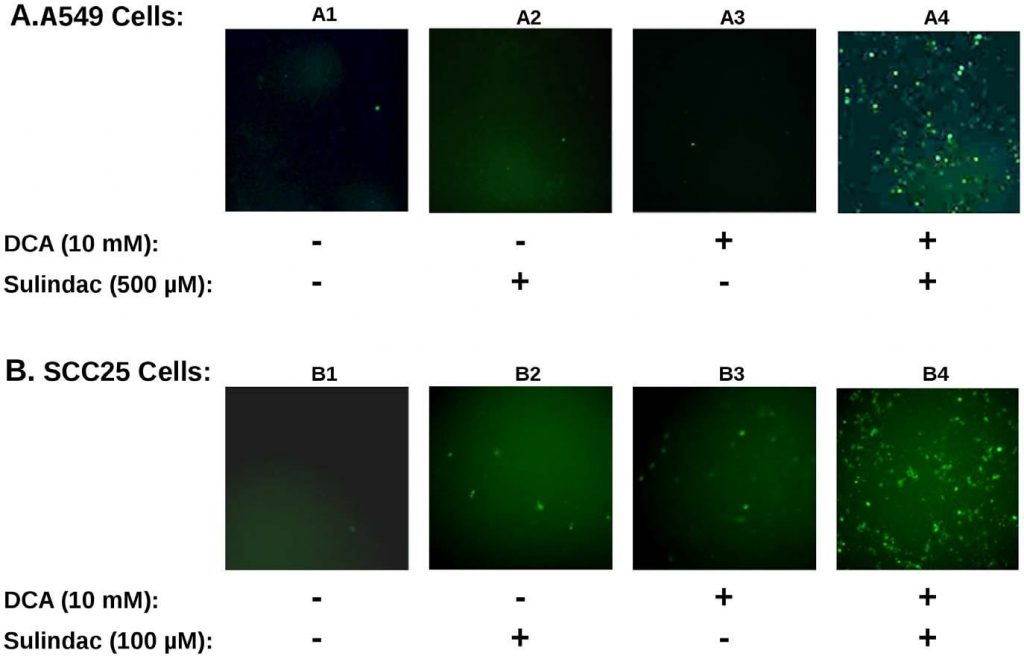
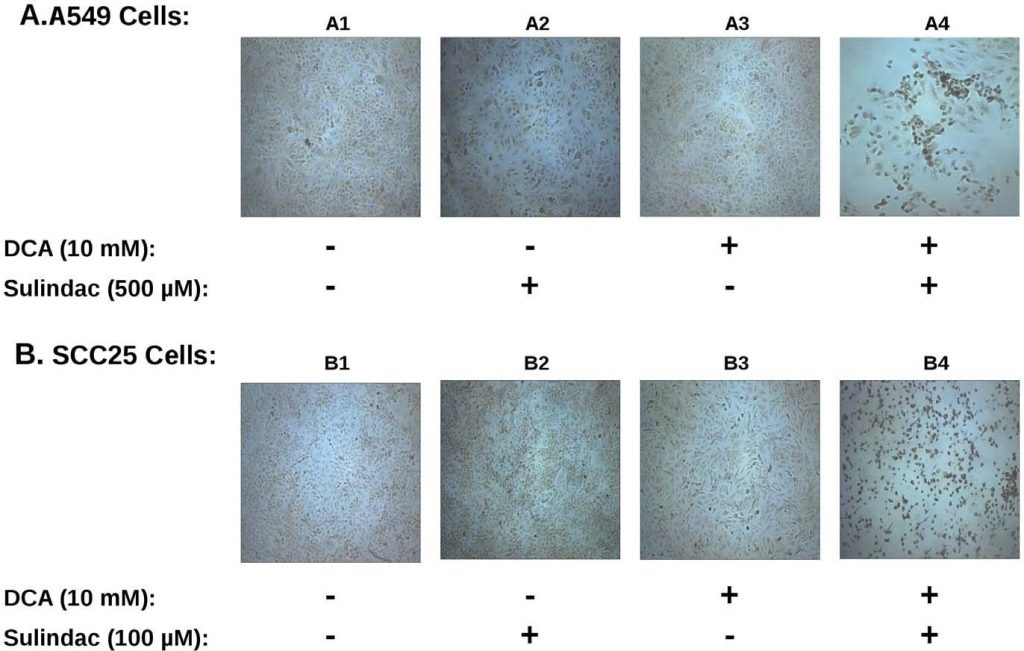
L’effet synergique sur la viabilité observé avec le sulindac et le dichloroacétate sur les cellules cancéreuses A549 et SCC25 est étonnamment similaire aux études précédentes utilisant la combinaison du sulindac et du TBHP [7]. Pour déterminer si la production de ROS était impliquée dans la destruction sélective observée dans les présentes études, la production de ROS, en utilisant le colorant indicateurH2DCFDA(voir Méthodes), a été déterminée dans les lignées de cellules cancéreuses exposées au sulindac et au DCA. Les résultats sont résumés dans la figure 3. La figure 3A montre les résultats obtenus avec les cellules cancéreuses A549. Il est évident, d’après les résultats décrits dans la figure 3A, que les cellules cancéreuses A549 non traitées (panneau A1), ou les cellules traitées avec le sulindac seul (panneau A2), ou le DCA seul (panneau A3), ne présentent que quelques cellules colorées positivement. Cependant, lorsque les cellules ont été exposées à la fois au sulindac et au DCA (panneau A4), une forte augmentation des cellules colorées positivement pour les ROS (fluorescence verte) est observée, montrant que la présence à la fois du sulindac et du DCA entraîne la génération de niveaux significatifs de ROS. Comme le montre la figure 3B, des résultats similaires sont observés avec les cellules cancéreuses SCC25. Le sulindac ou le DCA seuls entraînent une faible augmentation des cellules productrices de ROS (panneaux B2 et B3), mais une forte augmentation de la production de ROS est à nouveau observée lorsque les deux médicaments sont ajoutés (panneau B4). La quantification à l’aide de cellules SCC25 montre que le nombre de cellules positives au DCF (voir Méthodes) est 9-10× plus élevé lorsque les cellules sont traitées avec le sulindac et le DCA par rapport à chacun des médicaments seuls (voir Figure S3A). Il ressort de ces résultats et d’études antérieures que la production de ROS pourrait être une caractéristique commune de la destruction accrue des cellules cancéreuses lorsque le sulindac est utilisé en association avec des composés qui affectent la fonction mitochondriale.
L’association du sulindac et du DCA entraîne une perte du potentiel membranaire mitochondrial
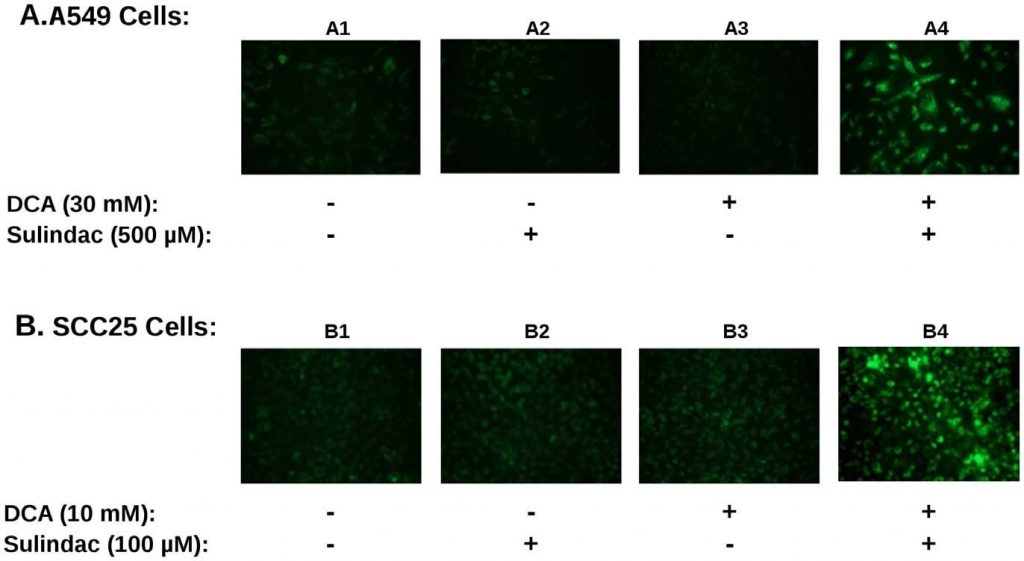
Si la production de ROS est impliquée dans l’effet meurtrier accru du sulindac/DCA, on pourrait s’attendre à ce que la production de ROS par l’association de médicaments affecte la fonction mitochondriale. Afin de le déterminer, le potentiel de la membrane mitochondriale a été mesuré en utilisant la coloration JC-1 comme décrit dans la section Méthodes. Une perte de potentiel membranaire est indiquée par une augmentation de la fluorescence verte, comme décrit dans les Méthodes. Un résultat typique est résumé dans la figure 4. Les cellules cancéreuses A549 et SCC25 ont été exposées au sulindac et au DCA, seuls ou en combinaison, pendant 48 heures et colorées avec JC-1 afin de surveiller le potentiel de la membrane mitochondriale. La figure 4A montre les résultats obtenus avec la lignée cellulaire cancéreuse A549. En l’absence de tout médicament, les mitochondries semblent intactes et maintiennent leur potentiel membranaire comme l’indique la faible fluorescence verte (panneau A1). En présence de sulindac seul (panneau A2) ou de DCA seul (panneau A3), il y a une petite augmentation de la fluorescence verte, indiquant une certaine perte du potentiel de la membrane mitochondriale. Cependant, lorsque le sulindac et le DCA sont tous deux présents, on observe une perte frappante du potentiel de la membrane mitochondriale, comme le montre une forte augmentation de la fluorescence verte (panneau A4). Nous avons observé le même schéma lorsque plusieurs champs indépendants ont été analysés par microscopie à fluorescence. La figure 4B montre des résultats similaires avec les cellules cancéreuses SCC25. Une fois de plus, une perte significative du potentiel de la membrane mitochondriale a été observée uniquement lorsque les cellules ont été exposées à la fois au sulindac et au DCA (panneau B4). La quantification de l’effet est présentée dans la figure S3B. On peut voir que le pourcentage de cellules positives au vert JC1 lorsque la combinaison de médicaments a été utilisée est 3-4× celui observé avec l’un ou l’autre des médicaments seuls.
Les ROS sont impliqués dans la destruction des cellules cancéreuses par l’association du sulindac et du DCA
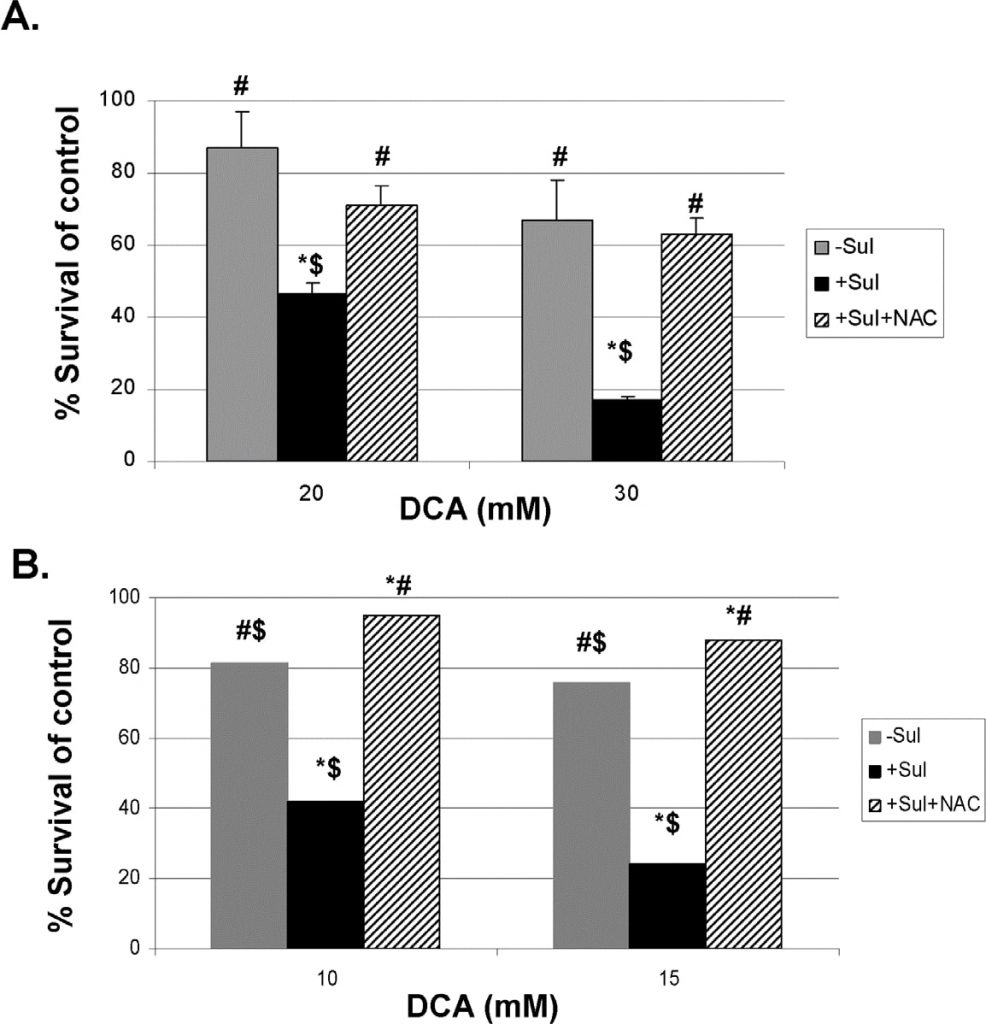
Pour fournir des preuves plus directes que les ROS produits sont impliqués dans la destruction accrue des cellules cancéreuses par le sulindac et le DCA, nous avons utilisé deux capteurs de ROS connus, la N-acétylcystéine (NAC) et le Tiron (voir Méthodes). Les résultats obtenus avec la NAC sont présentés dans la figure 5. La figure 5, panneau A, montre qu’à 20 et 30 mM de DCA, l’augmentation de la destruction des cellules cancéreuses A549 observée en présence de sulindac, est largement empêchée par la présence de NAC (2 mM) pendant les incubations de 48 heures. Des résultats très similaires sont observés avec les cellules cancéreuses SCC25, comme le montre la figure 5, panneau B. Des résultats comparables ont été obtenus lorsque le Tiron a été utilisé à la place de la NAC (figure S4).
La destruction des cellules cancéreuses par le sulindac et le DCA implique une mort apoptotique
Les résultats ci-dessus (figures 3, 4 et 5) montrent que la destruction accrue des lignées de cellules cancéreuses implique un dysfonctionnement mitochondrial, ce qui suggère que la mort cellulaire observée est due à l’apoptose. Des études antérieures ont indiqué que le sulindac et ses dérivés sont des médicaments proapoptotiques [5], [6]. Il existe également des rapports indiquant que le DCA peut provoquer la mort cellulaire par apoptose [20], [23]. Pour déterminer si la destruction des cellules cancéreuses par la combinaison de ces deux médicaments, médiée par les ROS, implique une mort apoptotique, nous avons effectué une coloration TUNEL pour mesurer l’apoptose (voir Méthodes). Des répétitions multiples ont été testées pour le sulindac et le DCA seuls, ou en combinaison, pour les expériences de coloration TUNEL. Un résultat typique est illustré à la figure 6, où les panneaux supérieurs (figure 6A, panneaux A1-A4) représentent les résultats obtenus avec les cellules cancéreuses A549 et les panneaux inférieurs (figure 6B, panneaux B1-B4) représentent les résultats obtenus avec les cellules cancéreuses SCC25. Lorsque les cellules ont été traitées sans médicament, avec le sulindac seul ou avec le DCA seul (figure 6, panneaux A1-A3 et B1-B3), seules quelques cellules TUNEL-positives ont été observées. Cependant, lorsque les cellules ont été exposées à la fois au sulindac et au DCA, on observe une augmentation significative des cellules apoptotiques TUNEL-positives (figure 6, panneaux A4 et B4), ce qui indique une induction importante de l’apoptose. Pour vérifier les résultats de la méthode TUNEL, un test plus sensible d’échelonnement de l’ADN basé sur la PCR avec ligature a également été utilisé pour surveiller l’apoptose [25]. Les résultats ont également montré la présence d’une échelle nucléosomique forte enrichie uniquement lorsque le sulindac et le DCA étaient utilisés en association (figure S5 ; voies 4 et 8), ce qui corrobore fortement les données du test TUNEL.
L’élimination du sulindac et du DCA implique la signalisation proapoptotique JNK
Parmi les protéines kinases activées par des mitogènes (MAP kinases) connues, la kinase induite par le stress, la c-Jun N-terminal kinase (JNK/SAPK) a été directement impliquée dans la mort cellulaire apoptotique [11]. Nous avons donc étudié le rôle de la signalisation JNK dans l’apoptose médiée par le sulindac-DCA en utilisant le SP600125, un inhibiteur spécifique de la JNK (JNKI), et les résultats sont présentés dans la Figure 7A. Comme indiqué ci-dessus, les cellules SCC25 traitées par le sulindac ont présenté une mort accrue en présence de concentrations croissantes de DCA. Cependant, lorsque ces cellules ont été incubées avec du sulindac et du SP600125, la mort cellulaire médiée par le sulindac-DCA a été largement évitée. Ces résultats indiquent la participation de la signalisation proapoptotique médiée par la JNK dans la mort cellulaire médiée par le sulindac-DCA.
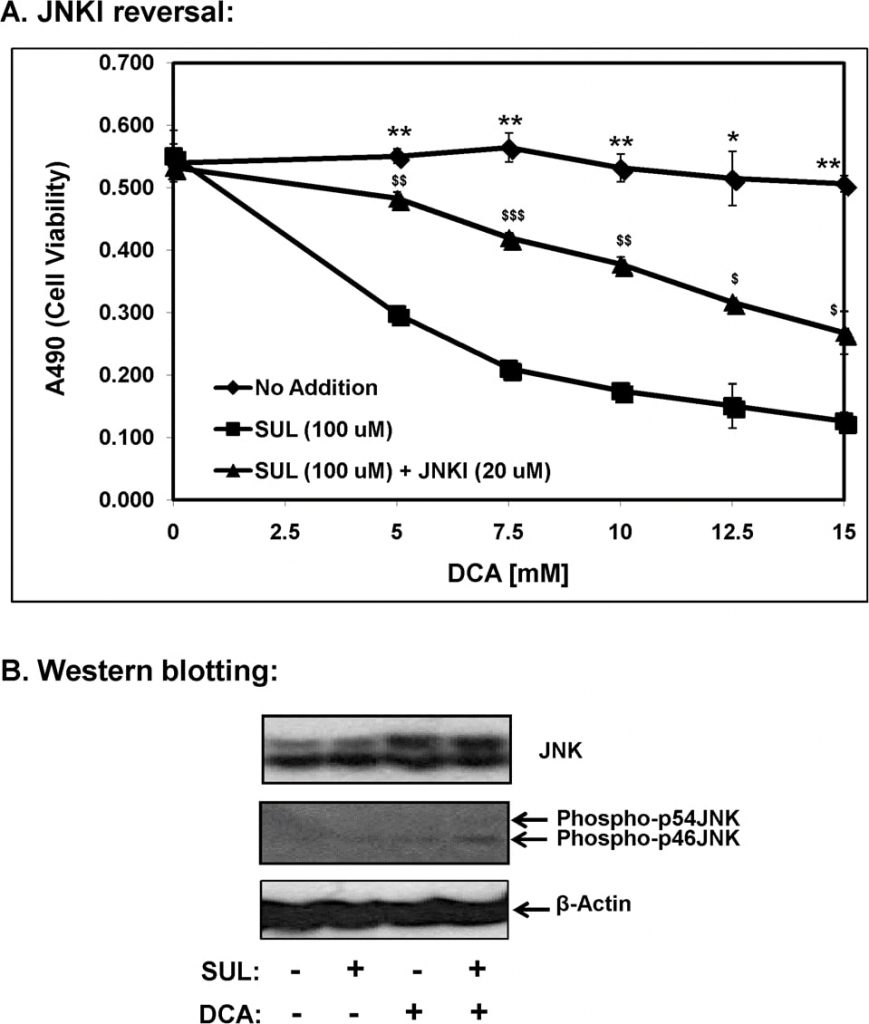
Par analyse western blot, nous avons également déterminé que la combinaison du sulindac et du DCA augmentait significativement les niveaux de phospho-JNK dans les fractions cytosoliques 12 h après l’exposition des cellules au sulindac et au DCA (Figure 7B). Une augmentation des niveaux de JNK total (bandes de protéines à 46 et 54 kDa) a été observée lorsque les cellules ont été traitées avec le DCA seul ainsi que lorsque les cellules ont été traitées avec la combinaison de sulindac et de DCA. Il convient de noter que les isoformes phospho-p46JNK et phospho-p54JNK ont toutes deux été induites par l’association du sulindac et du DCA, bien que l’augmentation de la phospho-p46JNK ait été plus significative (figure 7B).
Il existe un ensemble de preuves suggérant que JNK initie la libération de facteurs induisant l’apoptose à partir des mitochondries, tels que le cytochrome c, qui conduisent au clivage des caspases et de la PARP (poly(ADP-ribose) polymérase) [30], [31]. Des études ont également montré qu’au cours de l’apoptose, le cytochrome c libéré des mitochondries dans le cytoplasme pénètre finalement dans le noyau [32]. Nos résultats indiquent que l’activation maximale de JNK s’est produite environ 12 heures après l’exposition au sulindac et au DCA. Cela semble entraîner la translocation du cytochrome c dans le cytoplasme et le clivage de la PARP 18 heures après le traitement initial au sulindac et au DCA (figure S6A). Comme contrôle positif pour ces expériences, nous avons traité les cellules avec 100 µM d’étoposide, un agent induisant l’apoptose. Sous traitement combiné au sulindac et au DCA, une fluorescence nucléaire accrue peut être observée dans une majorité de cellules qui subissent activement l’apoptose (figure S6B).
L’analyse détaillée des données expérimentales d’immunofluorescence de cellules entières a révélé que ∼94 % des cellules non traitées par le sulindac ou le DCA présentaient une fluorescence ponctuelle du cytochrome c mitochondrial avec peu de coloration diffuse dans le cytoplasme ou dans les noyaux. En revanche, après le traitement au sulindac, 81 % des cellules présentaient une fluorescence cytoplasmique diffuse et distincte, et très peu de fluorescence nucléaire. Après le traitement au DCA, ∼83% des cellules ont montré une fluorescence cytoplasmique diffuse et distincte, et <5% des cellules ont montré une forte fluorescence nucléaire. Cependant, lorsque les cellules ont été traitées à la fois par le sulindac et le DCA, ∼72% des cellules ont montré une fluorescence à la fois nucléaire et cytoplasmique et ∼11% des cellules ont montré une forte fluorescence nucléaire. Ces résultats suggèrent que le cytochrome c libéré par les mitochondries peut initier la voie apoptotique intrinsèque fonctionnant dans la destruction du cancer médiée par le sulindac et le DCA.
Discussion
La présente étude est une extension de nos travaux précédents, qui ont démontré que le sulindac rendait les cellules cancéreuses, mais pas les cellules normales, plus sensibles au stress oxydatif [7]. Dans ces expériences précédentes, le sulindac a été pré-incubé avec les cellules pendant 24-48 heures, puis le sulindac a été retiré avant que les cellules ne soient exposées au TBHP ou au H2O2 pendant 2 heures. Il était évident, d’après les expériences précédentes, que le prétraitement au sulindac rendait les cellules cancéreuses beaucoup plus sensibles à l’agent oxydant, ce qui entraînait une forte augmentation des ROS et une perte de la fonction mitochondriale [7].
Il semblait raisonnable, sur la base de ces résultats, que le sulindac associé à des composés qui affectent la respiration mitochondriale entraîne une augmentation sélective de la destruction des cellules cancéreuses, mais pas des cellules normales. Dans la présente étude portant sur les lignées cellulaires cancéreuses A549 et SCC25, l’association du sulindac et du DCA a renforcé la destruction de ces lignées cellulaires cancéreuses, mais pas celle des cellules pulmonaires ou cutanées normales. Nos résultats sur les quantités de DCA nécessaires dans les cellules entières sont conformes à ce qui a été rapporté précédemment [28], [33], [34]. Dans notre système, la CI50 du DCA pour les cellules SCC25 est de 23 mM et pour les cellules A549 de 35 mM. La CI50 pour les kératinocytes normaux est >50 mM et pour les cellules pulmonaires normales (MRC5) est ∼40 mM. Les résultats indiquent également que la mort des cellules cancéreuses observée implique la production de ROS, l’activation de JNK et l’apoptose initiée par les mitochondries. En ce qui concerne l’absence d’effet sur les cellules normales, il a été montré que le sulindac protège les cellules pulmonaires normales contre les dommages oxydatifs résultant de l’exposition au TBHP [7] et nous avons également signalé récemment que le sulindac peut protéger les cellules cardiaques contre les dommages oxydatifs résultant de l’ischémie/reperfusion par un mécanisme de préconditionnement [35].
À notre connaissance, il existe maintenant au moins 8 composés, y compris nos études avec le TBHP, le H2O2 et le DCA, qui ont montré une destruction accrue et sélective du cancer en présence du sulindac [7]–[9], [12]–[15]. Bien que leurs cibles métaboliques au sein de la cellule soient connues et différentes, il est très probable qu’ils provoquent tous, directement ou indirectement, la mort cellulaire en présence de sulindac par un mécanisme qui implique une altération de la respiration mitochondriale et de la production de ROS [10], [12], [14]. Il semble probable que lorsqu’on trouve un médicament qui, associé au sulindac, tue sélectivement les cellules cancéreuses, mais pas les cellules normales, le mécanisme de destruction implique un stress oxydatif conduisant à un dysfonctionnement mitochondrial. L’altération de la respiration peut être un facteur commun dans ces expériences utilisant des combinaisons sulindac/médicament et les résultats actuels utilisant le DCA soutiennent ce point de vue. Il est tout à fait possible que l’effet du sulindac soit lié aux observations faites il y a plus de 50 ans par Warburg, qui a noté que les cellules normales préfèrent la respiration pour obtenir leur énergie, alors que les cellules cancéreuses préfèrent la glycolyse, en raison d’un défaut de la chaîne respiratoire [16]. Cette différence fondamentale dans la respiration mitochondriale entre les cellules normales et cancéreuses peut rendre les cellules cancéreuses plus sensibles au stress oxydatif [36], [37]. Il semble que le sulindac puisse amplifier cette différence fondamentale dans la biochimie des cellules normales et cancéreuses. Bien que la manière dont le sulindac sensibilise les cellules cancéreuses aux médicaments qui affectent la respiration mitochondriale ne soit pas encore claire, elle fait l’objet de recherches actives. Spitz et ses collègues [38], dans des études sur la privation de glucose des cellules cancéreuses, sont arrivés à une conclusion similaire concernant les différences de métabolisme entre les cellules normales et cancéreuses. En accord avec ces résultats, une autre étude récente a montré que l’inhibition pharmacologique de la lactate déshydrogénase pouvait entraîner une destruction sélective du cancer [39]. Dans ces dernières études, il a été démontré que l’amélioration de la destruction sélective des cellules cancéreuses impliquait également la production de ROS, et l’effet observé a été attribué à un processus respiratoire altéré dans les cellules cancéreuses.
Il convient de souligner que l’association du sulindac avec un agent oxydant ou des médicaments susceptibles d’affecter la fonction mitochondriale a déjà été testée en clinique. Meyskens et al. 2008 ont montré que l’association du sulindac avec le DFMO avait un effet significatif sur la récurrence des polypes du côlon et l’apparition du cancer du côlon dans une étude clinique de 3 ans [13]. Nous avons récemment rapporté l’utilisation du sulindac, avec H2O2, dans une étude clinique de preuve de concept pour le traitement topique des kératoses actiniques [15]. L’un des inconvénients de cette combinaison était la nécessité de disposer de deux formulations topiques, car les composés ne pouvaient pas être stockés pendant de longues périodes sans que le sulindac soit détruit par le H2O2. En outre, il n’est pas possible d’utiliser le H2O2 pour le traitement des tumeurs internes, car il ne peut être pris par voie orale. En revanche, l’association du sulindac et du DCA pourrait être délivrée sous la forme d’une formulation unique adaptée à un usage topique, et les deux composés peuvent être utilisés par voie orale. En fait, depuis plusieurs années, le DCA est utilisé en clinique pour réduire les niveaux d’acide lactique chez les patients souffrant d’acidose lactique [40]–[42]. Le DCA a également été utilisé comme agent anticancéreux in vitro et in vivo sur plusieurs lignées cellulaires cancéreuses différentes, ce qui indique que le métabolisme mitochondrial dans les cellules cancéreuses pourrait constituer une nouvelle cible thérapeutique [18], [20], [22]. Michelakis et al. (2010) ont montré que le traitement au DCA « remodèle » le métabolisme mitochondrial chez les patients atteints de glioblastome avec des effets toxiques réversibles. Il convient de noter que le sulindac et le DCA sont tous deux abordables, relativement non toxiques et peuvent être pris par voie orale. Si l’association s’avère efficace in vivo, elle ajoutera une nouvelle dimension au traitement du cancer, car les deux médicaments ciblent le métabolisme mitochondrial dans plusieurs cancers [22].
En résumé, nos études utilisant la combinaison du sulindac et du DCA suggèrent que le sulindac rend sélectivement les cellules cancéreuses plus sensibles aux agents qui affectent la respiration mitochondriale, ce qui entraîne un stress oxydatif et un dysfonctionnement mitochondrial. Ces résultats pourraient être liés au défaut de respiration des cellules cancéreuses, observé à l’origine par Warburg [16]. Des études visant à comprendre les différences fondamentales entre la façon dont les cellules cancéreuses et les cellules normales répondent au sulindac et aux agents qui affectent la fonction mitochondriale sont actuellement en cours.
Informations complémentaires
Figure S1 Cytotoxicité du sulindac, du DCA ou d’une combinaison de médicaments sur les cellules cancéreuses A549 et SCC25
Les cellules cancéreuses A549 et SCC25 ont été traitées avec les concentrations indiquées de sulindac (panneaux 1 et 4) ou de DCA (panneaux 2 et 5). Dans les expériences de combinaison de médicaments sulindac/DCA, un rapport de 1:50 (sulindac:DCA) a été maintenu pour les cellules A549, avec des combinaisons de médicaments allant de 0,2 mM:10 mM à 1 mM:50 mM (panneau 3) et un rapport de 1:100 (sulindac:DCA) a été maintenu pour les cellules SCC25 avec des combinaisons de médicaments allant de 0,05 mM:5 mM à 0,3 mM:30 mM (panneau 6). (TIF) Figure S2 Détermination des indices de combinaison pour les cellules cancéreuses A549 et SCC25. Les indices de combinaison des médicaments ont été déterminés en incorporant les valeurs de viabilité cellulaire obtenues ci-dessus dans les équations de Chou et Talalay [26]. Voir le texte pour plus de détails. (TIF) Figure S3 Quantification des cellules fluorescentes vertes positives DCF et JC-1. Les cellules SCC25 ont été traitées avec du sulindac, du DCA ou une combinaison de médicaments pendant 48 heures et colorées avec les colorants H2DCFDA (Fig. S3A) ou JC-1 (Fig. S3B) comme mentionné dans les Méthodes. Pour le contrôle positif, les cellules ont été traitées avec 200 mM de TBHP pendant 2 heures et la coloration a été effectuée comme ci-dessus. Les cellules ont été analysées sous fort grossissement en utilisant l’objectif 1006 dans un microscope inversé à fluorescence Olympus. Au moins 100 cellules individuelles ont été visualisées pour chaque condition et les pourcentages de cellules fluorescentes vertes DCF-positives et JC1-positives sont présentés sous forme de tableaux et de graphiques. (TIF) Figure S4 Le piégeur de ROS Tiron inverse la destruction des cellules cancéreuses par la combinaison du sulindac et du DCA. Les cellules cancéreuses A549 et SCC25 ont été traitées avec les concentrations indiquées de DCA en l’absence (barre grise) ou en présence de sulindac (barre noire) ou en présence de sulindac et de Tiron (barre rayée) pendant 48 heures. La viabilité cellulaire a été contrôlée par le test MTS comme mentionné dans les Méthodes. La viabilité cellulaire est exprimée en % du contrôle (cellules non traitées au sulindac). Les barres d’erreur représentent l’erreur standard de la moyenne (SEM) exprimée en % de la valeur moyenne des quadruplicats d’une expérience représentative. L’inhibition de la croissance des cellules cancéreuses s’est produite de manière dose-dépendante pendant le traitement combiné de DCA et de sulindac (barres noires) dans les cellules cancéreuses A549 (A) et SCC25 (B). Cependant, cette destruction accrue a été empêchée lorsque Tiron était présent en même temps que le traitement combiné (barres rayées en A et B). (TIF) Figure S5 Un échelonnement prononcé de l’ADN nucléosomal se produit pendant la destruction des cellules cancéreuses par l’association du sulindac et du DCA. Les cellules cancéreuses A549 et SCC25 ont été traitées avec les médicaments indiqués pendant 48 heures. L’ADN nucléosomique a été extrait et soumis à une PCR médiée par ligature comme décrit dans la section Méthodes et analysé sur un gel d’agarose à 1,2 % avec des marqueurs de taille. Le couloir « M » indique les marqueurs de taille moléculaire. Les voies 1-4 et 5-8 illustrent les résultats obtenus avec les cellules cancéreuses A549 et SCC25 respectivement. Les résultats sont illustrés dans les voies 1 et 5 (sans médicament), les voies 2 et 6 (sulindac seul), les voies 3 et 7 (DCA seul) et les voies 4 et 8 (sulindac et DCA). Une augmentation de l’échelonnement de l’ADN nucléosomal n’a été observée qu’avec le traitement associant le sulindac et le DCA (voies 4 et 8). (TIF)
Figure S6 La combinaison du sulindac et du DCA entraîne la libération du cytochrome c des mitochondries et le clivage de la PARP
Les cellulesSCC25ont été traitées avec du sulindac, du DCA, une combinaison de médicaments ou de l’étoposide afin de déterminer la localisation intracellulaire du cytochrome c par western blotting et immunofluorescence. A. Les fractions cytosoliques ont été isolées à 18 h et la présence de cytochrome c dans le cytoplasme et le clivage de PARP ont été déterminés par western blotting. Des western blots représentatifs montrent la quantité de cytochrome c et de PARP clivée. Les niveaux de b-actine ont été utilisés comme contrôle interne. B. L’immunofluorescence a été réalisée à l’aide du kit cytométrique de libération du cytochrome c CBA077 InnoCyteTM Flow Cytometric Kit selon les instructions du fabricant. Plusieurs champs indépendants ont été analysés et les micrographies représentatives montrent les schémas de localisation du cytochrome c lorsque les cellules sont exposées au sulindac et/ou au DCA. Les valeurs quantitatives sont présentées dans le texte. (TIF)
Remerciements
Les auteurs remercient David Brunell pour son aide dans la détermination des indices de combinaison et Edna Gamliel pour son assistance dans la culture cellulaire.
Contributions des auteurs
Conception et design des expériences : KA SK KDS HW. Réalisation des expériences : KA SK. Analyse des données : KA SK HW. A contribué aux réactifs/matériels/outils d’analyse : KDS. A rédigé l’article : KA SK HW.
RÉFÉRENCES
1 Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, et al. (1996) Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res 56 : 2556-2560.
2 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J Natl
3 Taketo MM (1998) Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst 90 : 1529-1536.
4 Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, et al. (1995) Chemoprevention of colon carcinogenesis by sulindac, a nonteroidal anti-inflammatory agent. Cancer Res 55 : 1464-1472.
5 Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, et al. (2001) Progression and NSAID-induced apoptosis in malignant melanomas are independent of cyclooxygenase II. Melanoma Res 11 : 587-599.
6 Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenèse 22 : 17-25.
7 Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, et al. (2009) Sulindac améliore l’élimination des cellules cancéreuses exposées au stress oxydatif. PLoS One 4 : e5804.
8 Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, et al. (1999) Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lines lung cancer cell. Cancer Res 59 : 6178-6184.
9 Jiang TT, Brown SL, Kim JH (2004) Combined effect of arsenic trioxide and sulindac sulfide in vitro human lung cancer cells. J Exp Clin Cancer Res 23 : 259-262.
10 Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, et al. (2006) Synergistic induction of apoptosis by sulindac and arsenic trioxide in human lung cancer A549 cells via reactive oxygen species-dependent down-regulation of survivin. Biochem Pharmacol 72 : 1228-1236.
11 Jin HO, Seo SK, Woo SH, Lee HC, Kim ES, et al. (2008) Une combinaison de sulindac et de trioxyde d’arsenic induit de manière synergique l’apoptose des cellules H1299 du cancer du poumon humain via la phosphorylation de Bcl-xL dépendante de la c-Jun NH2-terminal kinase. Cancer du poumon 61 : 317-327.
12 Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, et al. (2005) Le sulindac renforce le stress oxydatif et l’activité anticancéreuse de l’inhibiteur du protéasome bortezomib. Clin Cancer Res 11 : 5248-5256.
13 Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, et al. (2008) Difluoromethylornithine plus sulindac pour la prévention des adénomes colorectaux sporadiques : un essai randomisé en double aveugle contrôlé par placebo. Cancer Prev Res (Phila Pa) 1 : 32-38.
14 Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, et al. (2008) Combined effects of sulindac and suberoylanilide hydroxamic acid on apoptosis induction in human lung cancer cells. Mol Pharmacol 73 : 1005-1012.
15 Resnick L, Rabinovitz H, Binninger D, Marchetti M, Weissbach H (2009) Le sulindac topique combiné au peroxyde d’hydrogène dans le traitement des kératoses actiniques. J Drugs Dermatol 8 : 29-32.
16 Warburg O (1956) On the origin of cancer cells. Science 123 : 309-314.
17 Whitehouse S, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141 : 761-774.
18 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11 : 37-51.
19 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60 : 1478-1487.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induce apoptosis in endometrial cancer cells. Gynecol Oncol 109 : 394-402.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 68 : 1223-1231.
<span id= »22″ class= »referencess blue-text « 22 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, et al. (2010) Modulation métabolique du glioblastome avec le dichloroacétate. Sci Transl Med 2 : 31ra34.
23 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99 : 989-994.
24 Cory AH, Owen TC, Barltrop JA, Cory JG (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3 : 207-212.
<span id= »25″ class= »referencess blue-text « 25 Staley K, Blaschke AJ, Chun J (1997) Apoptotic DNA fragmentation is detected by a semi-quantitative ligation-mediated PCR of blunt DNA ends. Cell Death Differ 4 : 66-75.
26 Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships : the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22 : 27-55.
<span id= »27″ class= »referencess blue-text « 27 Papandreou I, Goliasova T, Denko NC (2010) Anticancer drugs that target metabolism : Is dichloroacetate the new paradigm ? Int J Cancer 128 : 1001-1008.
28 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, et al. (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127 : 2510-2519.
29 Babbar N, Ignatenko NA, Casero RA Jr, Gerner EW (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J Biol Chem 278 : 47762-47775.
30 Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haikel Y, et al. (2011) Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenèse 32 : 1268-1278.
31 Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, et al. (2007) c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 14 : 1001-1010.
<span id= »32″ class= »referencess blue-text « 32 Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, et al. (2004) Translocation nucléaire du cytochrome c pendant l’apoptose. J Biol Chem 279 : 24911-24914.
33 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, et al. (2011) La thérapie métabolique ciblée par le dichloroacétate déjoue la cytotoxicité des médicaments anticancéreux standard. Cancer Chemother Pharmacol 67 : 647-655.
34 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induit l’apoptose et l’arrêt du cycle cellulaire dans les cellules cancéreuses colorectales. Br J Cancer 102 : 1746-1752.
35 Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac confère une protection ischémique de haut niveau au cœur par des mécanismes de préconditionnement tardif. Proc Natl Acad Sci U S A 106 : 19611-19616.
<span id= »36″ class= »referencess blue-text « 36 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick : metabolism and tumor cell growth. Curr Opin Genet Dev 18 : 54-61.
37 Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect : the metabolic requirements of cell proliferation. Science 324 : 1029-1033.
<span id= »38″ class= »referencess blue-text « 38 Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, et al. (2005) Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280 : 4254-4263.
<span id= »39″ class= »referencess blue-text « 39 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. (2010) L’inhibition de la lactate déshydrogénase A induit un stress oxydatif et inhibe la progression tumorale. Proc Natl Acad Sci U S A 107 : 2037-2042.
<span id= »40″ class= »referencess blue-text « 49 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI (1983) Treatment of lactic acidosis with dichloroacetate. N Engl J Med 309 : 390-396.
41 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77 : 535-541.
42 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, et al. (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121 : e1223-1228.
Contenu connexe :