Kasirajan Ayyanathan1,2,*,#, Shailaja Kesaraju1,#, Ken Dawson-Scully1,2, Herbert Weissbach1
1 Center for Molecular Biology and Biotechnology, Charles E. Schmidt College of Science, Florida Atlantic University, Jupiter, Florida, Vereinigte Staaten von Amerika
,2 Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, Florida, Vereinigte Staaten von Amerika*E-Mail
:
[email protected]#TheseAutoren haben gleichermaßen zu dieser Arbeit beigetragen.
Received: 12 August 2011Accepted
: 4 June 2012Published
: 1 July 2012
Zusammenfassung
Sulindac ist ein von der FDA zugelassenes nichtsteroidales entzündungshemmendes Medikament mit nachgewiesener krebshemmender Wirkung. Unsere jüngsten Studien haben gezeigt, dass Sulindac die Abtötung von Krebszellen, die Oxidationsmitteln ausgesetzt sind, durch die Produktion reaktiver Sauerstoffspezies (ROS), die zu einer mitochondrialen Dysfunktion führen, selektiv verstärkt. Diese Wirkung von Sulindac und oxidativem Stress auf Krebszellen könnte mit der Störung der Atmung in Krebszellen zusammenhängen, die erstmals vor 50 Jahren von Warburg beschrieben wurde und als Warburg-Effekt bekannt ist. Wir postulierten, dass Sulindac die selektive Abtötung von Krebszellen verstärken könnte, wenn es mit einer Verbindung kombiniert wird, die die mitochondriale Atmung verändert. Um diese Hypothese zu testen, haben wir Dichloracetat (DCA) verwendet, von dem bekannt ist, dass es den Pyruvat-Stoffwechsel von der Milchsäurebildung zur Atmung verlagert. Man könnte erwarten, dass DCA, da es den aeroben Stoffwechsel anregt, die mitochondriale Atmung in Krebszellen belasten könnte, was zu einer verstärkten Abtötung in Gegenwart von Sulindac führen würde. In dieser Studie haben wir gezeigt, dass die Kombination von Sulindac und DCA die selektive Abtötung von A549- und SCC25-Krebszellen unter den verwendeten Bedingungen verstärkt. Wie vorhergesagt, beinhaltet der Mechanismus der Abtötung die ROS-Produktion, die mitochondriale Dysfunktion, die JNK-Signalisierung und den Tod durch Apoptose. Unsere Ergebnisse deuten darauf hin, dass die Wirkstoffkombination Sulindac-DCA eine wirksame Krebstherapie darstellen könnte.
Zitat: Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H (2012) Combination of Sulindac and Dichloroacetate Kills Cancer Cells via OxidativeDamage
. PLoS ONE 7(7): e39949. doi:10.1371/journal.pone.0039949Editor
: Joseph Alan Bauer, Bauer Research Foundation, Vereinigte Staaten von Amerika
© 2012 Ayyanathan et al. Dies ist ein frei zugänglicher Artikel, der unter den Bedingungen der Creative Commons Attribution License verbreitet wird, die die uneingeschränkte
Nutzung, Verbreitung und Vervielfältigung in jedem Medium
erlaubt
, sofern der ursprüngliche Autor und die Quelle genannt werden.
Finanzierung: Wir danken den National Institutes of Health (KA, Zuschuss 5K01CA95620) und HW (Zuschuss R15 CA122001) sowie dem Bundesstaat Florida (SURECAG-Zuschuss R94007) für die finanzielle Unterstützung bei der Durchführung dieser Arbeit. Die Geldgeber hatten keinen Einfluss auf das Studiendesign, die Datenerfassung und -analyse, die Entscheidung zur Veröffentlichung oder die Erstellung des Manuskripts.
Konkurrierende Interessen: Die Autoren haben erklärt, dass keine konkurrierenden Interessen bestehen.
EINLEITUNG
Sulindac ist ein von der FDA zugelassenes nichtsteroidales Antirheumatikum (NSAID), das nachweislich auch krebshemmende Wirkung hat [1–6]. Jüngste Studien unseres Labors haben gezeigt, dass die Krebszelllinien RKO, A549 und SCC25 empfindlich auf eine Kombination aus Sulindac und einem Oxidationsmittel wie TBHP oderH2O2[7]. Die Daten deuten darauf hin, dass die Wirkung von Sulindac nicht mit seiner NSAID-Wirkung zusammenhängt, sondern dass Sulindac die Krebszellen empfindlicher gegenüber oxidativem Stress macht, was zu mitochondrialer Dysfunktion und Verlust der Lebensfähigkeit führt. Im Gegensatz dazu zeigten normale Zellen unter ähnlichen Bedingungen keine verstärkte Abtötung [7]. In den letzten 10 Jahren gab es vereinzelte Berichte über eine verstärkte Krebsabtötung durch Sulindac in Kombination mit einer Reihe von Verbindungen wie Arsentrioxid, Bortezomib, Difluormethylornithin (DFMO) und Suberoylanilidhydroxamsäure (SAHA) [8-14]. Obwohl diese Verbindungen unterschiedliche Wirkorte haben, könnte ein gemeinsamer Mechanismus für die verstärkte Abtötung durch die Kombination von Sulindac und Medikamenten mit oxidativen Schäden einhergehen, wie in unseren früheren Studien mit Sulindac und einem Oxidationsmittel eindeutig nachgewiesen wurde [7], [15]. In der Tat wurden ROS in den Studien, in denen Sulindac in Kombination mit Arsentrioxid, Bortezomib und SAHA [10], [12], [14].
Unsere früheren Ergebnisse deuten darauf hin, dass die verstärkte Abtötung von Krebszellen durch die Kombination von Sulindac und einem Oxidationsmittel auf einen Defekt der Atmung in Krebszellen zurückzuführen sein könnte, wie er vor mehr als 50 Jahren erstmals von Warburg beschrieben wurde [16]der feststellte, dass Krebszellen im Gegensatz zu normalen Zellen die Glykolyse und nicht die Atmung zur Energiegewinnung bevorzugen. Einige Krebszellen beziehen bis zu 50 % ihrer Energie aus der Glykolyse, während die Glykolyse in normalen Zellen weniger als 5 % des Energiebedarfs ausmacht [16]. Um weitere Beweise für die mögliche Rolle einer veränderten Atmung und von ROS bei der Abtötung von Krebszellen durch Sulindac und oxidativen Stress zu erhalten, haben wir Studien mit Natriumdichloressigsäure (DCA) begonnen. DCA ist ein idealer Kandidat, da es bekanntermaßen eine Kinase hemmt, die die Aktivität der Pyruvat-Dehydrogenase herunterreguliert, was zu einer Verschiebung des Pyruvat-Stoffwechsels weg von der Milchsäurebildung hin zur Atmung führt [17], [18]. DCA wurde klinisch zur Behandlung von Patienten mit Laktatazidose eingesetzt [19]und aufgrund seiner biochemischen Eigenschaften wurde DCA auch als Krebsmittel getestet. Bonnet et al. 2007 haben gezeigt, dass DCA den Warburg-Effekt in Krebszellen umkehrt, indem es den Stoffwechsel der Krebszellen von der Glykolyse zur oxidativen Phosphorylierung umleitet. In diesen früheren Studien wurde gezeigt, dass DCA den Gehalt an ROS aus Komplex I erhöht. Dies wiederum löst einen „Umbau“ des mitochondrialen Stoffwechsels (verringert ΔΨm, öffnet die mitochondriale Übergangspore) in Krebszellen aus und treibt sie in Richtung Apoptose. Darüber hinaus haben mehrere neuere Studien bestätigt, dass DCA den ROS-Gehalt in Krebszellen erhöhen und die Mitochondrienmembran in Lungen-, Endometrium- und Glioblastom-Zelllinien depolarisieren kann, was sowohl in vitro als auch in vivo zur Apoptose führt [18], [20–22]. Interessant war die Beobachtung, dass DCA unter den verwendeten Bedingungen den mitochondrialen Stoffwechsel oder die Lebensfähigkeit normaler Zellen nicht signifikant zu beeinträchtigen scheint [18], [23].
Auf der Grundlage unserer früheren Beobachtungen über die krebsabtötende Wirkung von Sulindac und einem Oxidationsmittel, das den mitochondrialen Stoffwechsel beeinflusst [7]postulierten wir, dass die Kombination von Sulindac und DCA die krebsabtötende Wirkung synergistisch verstärken und einen wichtigen therapeutischen Wert haben könnte. In der vorliegenden Studie haben wir die Wirkung von Sulindac in Kombination mit DCA auf die Lebensfähigkeit der Krebszelllinien A549 und SCC25 untersucht. Wir haben auch die Rolle der mitochondrialen Funktion und der Apoptose bei der mit dieser Wirkstoffkombination beobachteten Krebsabtötung untersucht.
Materialien und Methoden
Materialien
Sulindac, N-Acetylcystein und Tiron wurden von Sigma (St. Louis, MO) bezogen. DCA-Natriumsalz wurde von Acros Organics (Geel, Belgien) bezogen. H2DCFDA und JC-1 wurden von Molecular Probes (Eugene, OR) bezogen. MTS-Assay-Reagenz und Deadend Tunel Kit wurden von Promega (Madison, WI) bezogen. Das Cytosol/Mitochondrien-Fraktionierungskit und das CBA077 InnoCyte™ Flow Cytometric Cytochrome c Release Kit wurden von Calbiochem, Gibbstown, NJ, bezogen. Alle Zellkulturmedien, fötales Rinderserum und andere Zusätze wie Penicillin/Streptomycin, Glutamin usw. wurden von American Type Culture Collection (ATCC; Rockville, MD) bezogen.
Zellkultur
Einenicht kleinzellige Lungenkarzinom-Zelllinie (NSCLC), A549, die normale menschliche Lungenzelllinie, MRC-5, und eine aus der Zunge stammende Plattenepithelkarzinom-Linie, SCC25, wurden von ATCC (Rockville, MD) erworben und in F12-K-Medium, das mit 10 % fötalem Rinderserum, 2 mM Glutamin, 100 IE/ml Penicillin und 100 µg/ml Streptomycin ergänzt wurde, in einem befeuchteten Inkubator mit 5 %CO2 bei 37 °C gehalten. Normale humane epidermale Keratinozyten wurden von der Promocell GmbH (Heidelberg, Deutschland) bezogen und in dem empfohlenen Kulturmedium gehalten. Für die Experimente wurden nicht-immortalisierte normale Zellen der ersten Passage verwendet.
Zellviabilitätstest
Die A549-Krebszellen und die normalen Lungenzellen wurden mit 3×103 Zellen pro Vertiefung, die SCC25-Krebszellen und die normalen Keratinozyten mit 7,5×103 Zellen pro Vertiefung in eine 96-Well-Platte gegeben. Die Zellen wurden 18-20 Stunden lang gezüchtet, das Medium wurde unter aseptischen Bedingungen verworfen und durch frisches Kulturmedium ersetzt, das die angegebenen Arzneimittelkombinationen enthielt. Wo angegeben, wurden 500 µM Sulindac bei A549-Krebszellen und normalen Lungenzellen und 100 µM Sulindac bei SCC25-Krebszellen und normalen Keratinozyten verwendet. Die Platten wurden 48 Stunden lang bei 37°C in einem 5%igenCO2-Inkubator bebrütet. Das Kulturmedium wurde verworfen und die Zellen wurden gründlich in 1× PBS gespült. Die Lebensfähigkeit der Zellen wurde mit dem CellTiter 96 Aqueous One Cell Proliferation Assay (Promega) nach den Anweisungen des Herstellers bestimmt. Der Assay verwendet eine Tetrazoliumverbindung, die durch die Wirkung zellulärer Dehydrogenasen, die in den metabolisch aktiven Zellen vorhanden sind, in ein wasserlösliches Formazan umgewandelt wird [24]. Das Formazan wurde durch Messung der Absorption bei 490 nm mit einem kolorimetrischen Mikrotiterplatten-Lesegerät (SpectraMax Plus; Molecular Devices) quantifiziert. Die Hintergrundabsorption wurde von jeder Probe abgezogen.
Intrazelluläre Messung von ROS
Die Krebszelllinien A549 und SCC25 wurden wie oben beschrieben ausgeplattet. Nach der 48-stündigen Medikamentenbehandlung wurden die Zellen mit 50 µM Dichlordihydrofluoresceindiacetat (H2DCFDA, Molecular Probes) in indikatorfreiem Medium 30 Minuten lang bei 37 °C inkubiert. Die Zellen wurden mit PBS gespült, und die ROS-Konzentrationen wurden durch Fluoreszenzmikroskopie sichtbar gemacht. Die Bilder wurden mit der Qcapture-Software aufgenommen und in Adobe Photoshop bearbeitet. Die Bildanalyse wurde mit der Slidebook-Software durchgeführt. Die aus einem repräsentativen Experiment gewonnenen Daten wurden zur Quantifizierung der DCF-positiven Zellen verwendet, die anhand der grünen Fluoreszenz aufgrund von oxidiertem DCF gemessen wurden.
JC-1-Färbung zur Überwachung des mitochondrialen Membranpotenzials
Das mitochondriale Membranpotenzial wurde mit dem JC-1-Farbstoff (Molecular Probes) bestimmt. Die Krebszelllinien A549 und SCC25 wurden wie oben beschrieben ausgeplattet. Nach der 48-stündigen Medikamentenbehandlung wurden die Zellen 30 Minuten lang bei 37 °C mit 5 ng/ml JC-1-Farbstoff in indikatorfreiem Medium inkubiert. Die Zellen wurden mit PBS gespült und durch Fluoreszenzmikroskopie sichtbar gemacht. Normale Mitochondrien nehmen den JC-1-Farbstoff aktiv und potenzialabhängig auf und bilden J-Aggregate, die eine rote Fluoreszenz aufweisen. Die Unterbrechung und der anschließende Verlust des mitochondrialen Membranpotenzials führt zu einer erhöhten grünen Fluoreszenz im Zytosol, die auf monomeres JC-1 zurückzuführen ist. Diese wird durch Verfolgung des Auftretens der grünen Fluoreszenz mit einem FITC-Filter bestimmt (Zeiss-Inversmikroskop – Axiovert 40 CFL). Bildaufnahme, -verarbeitung und -analyse wurden wie oben beschrieben durchgeführt. Die aus einem repräsentativen Experiment gewonnenen Daten wurden zur Quantifizierung der JC-1-grün-positiven Zellen verwendet.
Wirkung von ROS-Fängern auf die Zellviabilität in Gegenwart von Sulindac und DCA
Die Krebszelllinien A549 und SCC25 wurden wie oben beschrieben ausgeplattet. Um ROS abzufangen, wurden entweder 2 mM N-Acetylcystein (NAC) oder 2 mM Tiron (4,5-Dihydroxy-1,3-benzoldisulfonsäure-Dinatriumsalz) zusammen mit Sulindac und DCA für 48 h bei 37°C zugegeben. Die Lebensfähigkeit der Zellen wurde mit dem MTS-Assay überwacht und die statistische Analyse wie oben beschrieben durchgeführt.
TUNEL-Färbung zur Überwachung der in Apoptose befindlichen Zellen
Der TUNEL-Assaywurde in 96-Well-Platten unter Verwendung des kolorimetrischen DeadEnd-TUNEL-Assay-Kits (Promega) nach dem Protokoll des Herstellers durchgeführt. Die Krebszelllinien A549 und SCC25 wurden wie oben beschrieben plattiert und 48 Stunden lang mit keinem Medikament, Sulindac, DCA oder einer Medikamentenkombination behandelt. Nach der Medikamentenbehandlung wurden die Zellen mit Formalin fixiert und mit 0,2 % Triton X-100 in PBS permeabilisiert. Die Zellen wurden mit rekombinanter terminaler Desoxynukleotidyltransferase (TdT) und biotinylierten Nukleotiden inkubiert. Endogene Peroxidasen wurden vor der Inkubation mit Meerrettichperoxidase-Streptavidin (HRP-Streptavidin), das an die biotinylierten Nukleotide bindet, die in die eingekerbten Enden von Zellen, die sich in der Apoptose befinden, eingebaut sind, mit 0,3 %H2O2blockiert. Mit HRP-Streptavidin markierte Zellen wurden mit Wasserstoffperoxid und Diaminobenzidin (DAB) nachgewiesen. Zellen, die eine dunkelbraune Kernfärbung aufweisen, sind ein Anzeichen für Apoptose.
Western-Blot-Analyse
Die Zellen wurden bis zu einer Konfluenz von 70 % gezüchtet, mit den angegebenen Medikamenten für die angegebene Dauer behandelt und die zytosolischen Fraktionen mit dem Zytosol/Mitochondrien-Fraktionierungskit (Calbiochem, Gibbstown, NJ) nach dem Protokoll des Herstellers isoliert. Kurz gesagt wurden die Zellen zu verschiedenen Zeitpunkten geerntet und dann bei 600×g für 5 Minuten bei 4°C zentrifugiert. Die pelletierten Zellen wurden in dem mitgelieferten Puffer suspendiert und 10 Minuten auf Eis inkubiert. Die Zellen wurden dann mit einem Glas-Douncer homogenisiert und das Homogenat bei 700×g für 10 min bei 4°C zentrifugiert, um Zellkerne und Zelltrümmer zu sedimentieren. Der Überstand wurde bei 10.000×g für 30 min bei 4°C geschleudert, um das mitochondriale Pellet zu erhalten, und der Überstand wurde als zytosolische Fraktion betrachtet. Die Proteinkonzentration wurde mit einem Standard-Bradford-Assay bestimmt.
Sechzig Mikrogramm des Gesamtproteins wurden auf ein 4-12%iges NuPage Bis-Tris-Gel (Invitrogen, Eugene, OR) geladen und auf eine PVDF-Membran übertragen, die mit den primären Antikörpern sondiert wurde. Die primären Antikörper, JNK, pJNK, Cytochrom c und PARP (Cell Signaling Technology, Danvers, MA), wurden in einer Verdünnung von 1∶1000 verwendet. β-Actin (Santa Cruz Biotechnologies, Santa Cruz, Kalifornien) wurde in einer Verdünnung von 1∶4000 verwendet. Mit Meerrettichperoxidase konjugierte sekundäre Antikörper wurden verwendet, und die Banden wurden mit einer verstärkten Chemilumineszenzmethode (GE Healthcare, Piscataway, NJ) sichtbar gemacht.
Ligationsvermittelter PCR-basierter DNA-Laddering-Assay zur Überwachung des Ausmaßes der Apoptose unterworfenen Zellen
Um das Ausmaß der Apoptose zu bestätigen, wurde ein ligationsvermittelter PCR-basierter nukleosomaler DNA-Laddering-Assay wie beschrieben durchgeführt [25]. Die A549- und SCC25-Krebszelllinien wurden mit 5×104 und 1×105 Zellen pro Vertiefung in 35-mm-Schalen plattiert. Die A549-Krebszellen wurden 48 Stunden lang mit a) keinem Medikament, b) 500 µM Sulindac, c) 20 mM DCA und d) 500 µM Sulindac plus 20 mM DCA behandelt. In ähnlicher Weise wurden SCC25-Krebszellen mit den oben genannten vier verschiedenen Wirkstoffkombinationen behandelt, mit der Ausnahme, dass Sulindac und DCA in Konzentrationen von 100 µm bzw. 10 mM verwendet wurden. Nach der Behandlung wurde die gesamte zelluläre DNA extrahiert und an den Adaptor aus 27-mer 5′-GACGTCGACGTCGTACACGTGTCGACT-3′ und 12-mer 5′- AGTCGACACGTGTAC-3′ ligiert. Nach der Ligation wurde die DNA erhitzt, um das 12-mer freizusetzen, mit Taq-Polymerase aufgefüllt, einer semiquantitativen PCR unterzogen und auf einem 1,2%igen Agarosegel zusammen mit Größenmarkern analysiert.
In-situ-Lokalisierung von Cytochrom c durch Immunfluoreszenz
Die intrazelluläre Lokalisierung von Cytochrom c wurde durch Immunfluoreszenz unter Verwendung des CBA077 InnoCyte™ Flow Cytometric Cytochrome c Release Kits gemäß den Anweisungen des Herstellers überwacht. Kurz gesagt wurden die SCC25-Zellen mit 3,5×105 Zellen pro 35-mm-Glasbodenschale ausgeplattet und 15 Stunden lang mit den angegebenen Medikamenten behandelt. Die Zellen wurden in 5 ml 1× PBS gespült und 10 Minuten lang auf Eis in 300 µl des mitgelieferten Puffers permeabilisiert. Die Zellen wurden bei RT für 20 Minuten in 500 µl 4% Paraformaldehyd fixiert. Nach dem Waschen und Blockieren wurden die Zellen mit 250 µl Anti-Cytochrom-c-Antikörper (1∶500-Verdünnung) für 1 Stunde bei RT inkubiert. Nach dem Waschen wurden die Zellen mit 300 µl FITC-IgG (1∶300-Verdünnung) 1 Stunde lang bei RT inkubiert. Schließlich wurden die Zellen mit 300 µl DAPI (1 mg/ml) für 10 Minuten bei RT angefärbt. Die Zellen wurden mit einem inversen Fluoreszenzmikroskop von Olympus sichtbar gemacht. Die Bilder wurden wie oben beschrieben aufgenommen und verarbeitet. Es wurden mehrere Felder analysiert und repräsentative mikroskopische Aufnahmen angefertigt, die die Lokalisierungsmuster von Cytochrom c unter jeder Behandlungsbedingung zeigen. Die quantitativen Werte sind im Text aufgeführt.
Statistische Analyse und Bestimmung von Kombinationsindizes
Die Daten werden als Mittelwert ± SEM für die Zelllebensfähigkeitstests angegeben. Für die statistische Analyse wurde die Statistiksoftware Minitab verwendet, um den Student’s t-Test durchzuführen, und Werte mit p<0,05 wurden als statistisch signifikant angesehen. Um die synergistische Wirkung von Sulindac und DCA auf die Krebszelllinien A549 und SCC25 zu ermitteln, wurde eine quantitative Analyse der Dosis-Wirkungs-Beziehung durchgeführt, um die Kombinationsindizes zu bestimmen [26]. Sowohl Sulindac als auch DCA wurden in den angegebenen Konzentrationen allein an A549- und SCC25-Zellen getestet. Bei A549-Zellen wurde ein Verhältnis von 1∶50 für die Sulindac:DCA-Wirkstoffkombinationen von 0,2 mM:10 mM bis zu 1 mM:50 mM beibehalten. Bei SCC25-Zellen wurde ein Verhältnis von 1∶100 für die Sulindac:DCA-Wirkstoffkombinationen von 0,05 mM:5 mM bis zu 0,3 mM:30 mM beibehalten. Unsere experimentellen Ergebnisse und die ermittelten Kombinationsindexwerte sind im Text enthalten.
Ergebnisse
Sulindac und DCA bewirken eine verstärkte Abtötung von A549- und SCC25-Krebszellen, aber nicht von normalen Zellen
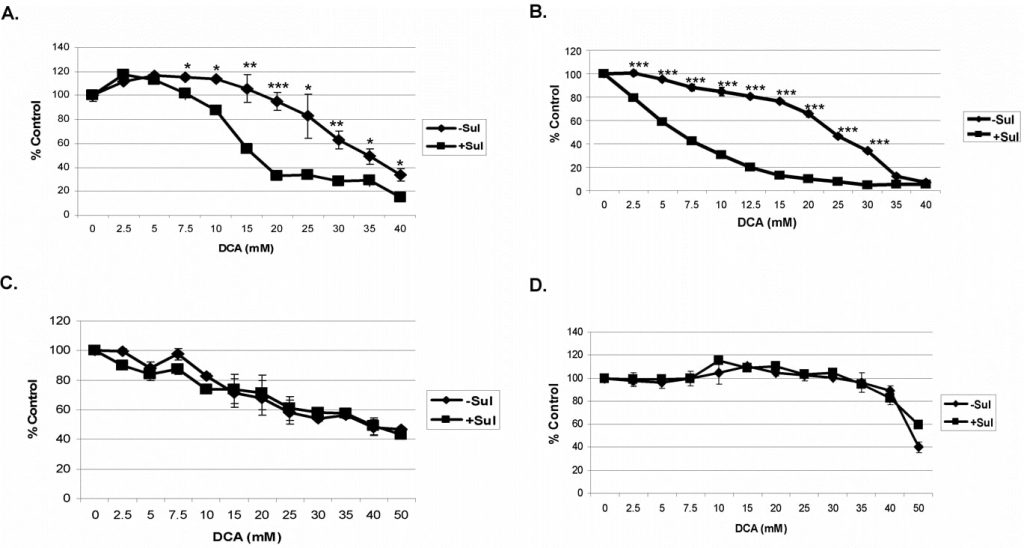
Für diese Studien haben wir die Kombination von Sulindac und DCA an A549- und SCC25-Krebszellen getestet. Die Zellen wurden 48 Stunden lang mit jeder Verbindung allein oder in Kombination inkubiert, bevor die Lebensfähigkeit getestet wurde (siehe Methoden). Eine Sulindac-Dosis-Wirkungs-Kurve unter diesen Bedingungen zeigte, dass A549- und SCC25-Krebszellen eine maximale Sulindac-Konzentration von 500 µM bzw. 100 µM tolerieren können, ohne signifikant abzutöten (Daten nicht gezeigt), und diese Konzentrationen wurden in allen Studien verwendet. DCA wurde in Konzentrationen von 0-40 mM verwendet, wie angegeben. Wir verwendeten diese Konzentrationen aufgrund früherer Berichte, die darauf hinwiesen, dass mehr als 5 mM erforderlich sind, um in In-vitro-Experimenten eine mitochondriale Dysfunktion zu verursachen [27]. Wie aus Abbildung 1A hervorgeht, ist DCA allein (ohne Sulindac) für A549-Krebszellen etwas toxisch, insbesondere bei Konzentrationen über 20 mM, aber in Gegenwart von Sulindac kommt es bei DCA-Konzentrationen über 5 mM zu einer verstärkten Abtötung dieser Zellen. Bei den SCC25-Krebszellen wurde selbst bei DCA-Konzentrationen unter 10 mM ein gewisser Verlust der Zelllebensfähigkeit mit DCA allein festgestellt (Abbildung 1B). In Anwesenheit von Sulindac kam es jedoch erneut zu einem deutlichen Anstieg des Zelltods, der zwischen DCA-Konzentrationen von 2-10 mM deutlich erkennbar war. Zuvor hatten wir gezeigt, dass die Kombination von Sulindac und einem Oxidationsmittel selektiv für Krebszellen ist und die Abtötung normaler Zellen nicht verstärkt [7]. Sulindac und DCA verstärkten unter den verwendeten Versuchsbedingungen auch nicht die Abtötung normaler Lungen- und Hautzellen, wie in den Abbildungen 1C und D dargestellt. Es ist zu beachten, dass die MRC-5-Zellen (normale Lungenzellen) besonders empfindlich auf DCA reagieren, wie bereits berichtet [28]aus nicht bekannten Gründen.
Um zu überprüfen, ob die Medikamentenkombination eine synergistische Wirkung hat, bestimmten wir die Kombinationsindizes, indem wir eine quantitative Analyse der Dosis-Wirkungs-Beziehung [26] an zwei verschiedenen Krebszelllinien (AbbildungS1). Die Kombinationsindizes betrugen 0,84 für die Krebszellen A549 und 0,73 für SCC25. Ein Wert von weniger als 1,00 deutet auf eine synergistische krebszerstörende Wirkung hin (Abbildung S2).
Der Sulindac-Effekt ist nicht auf seine NSAID-Aktivität zurückzuführen
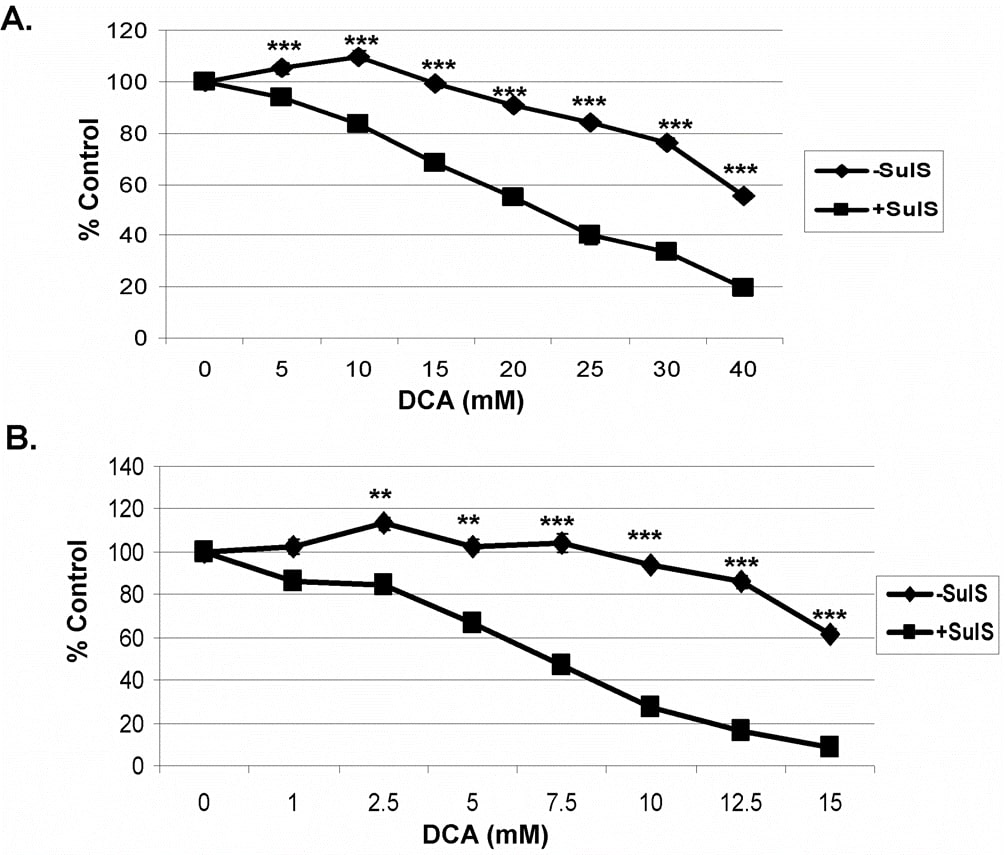
In früheren Studien mit Sulindac und einem Oxidationsmittel wurde gezeigt, dass die verstärkte und selektive Abtötung von Krebszellen durch Sulindac und ein Oxidationsmittel nicht mit der bekannten NSAID-Fähigkeit von Sulindac zusammenhing. Um die Rolle der COX-Hemmung zu bestimmen, kann ein Sulindac-Metabolit, Sulindacsulfon, verwendet werden, da es die COX 1 oder 2 nicht hemmt [7], [29]. Wie in Abbildung 2 dargestellt, zeigte die Kombination von Sulindacsulfon und DCA bei den Krebszellen A549 (A) und SCC25 (B) eine ähnliche abtötende Wirkung wie bei Sulindac. Diese Ergebnisse deuten darauf hin, dass die verstärkte krebsabtötende Wirkung von Sulindac in Gegenwart von DCA nicht mit seiner bekannten entzündungshemmenden Wirkung zusammenhängt.
Die Kombination von Sulindac und DCA erzeugt ROS
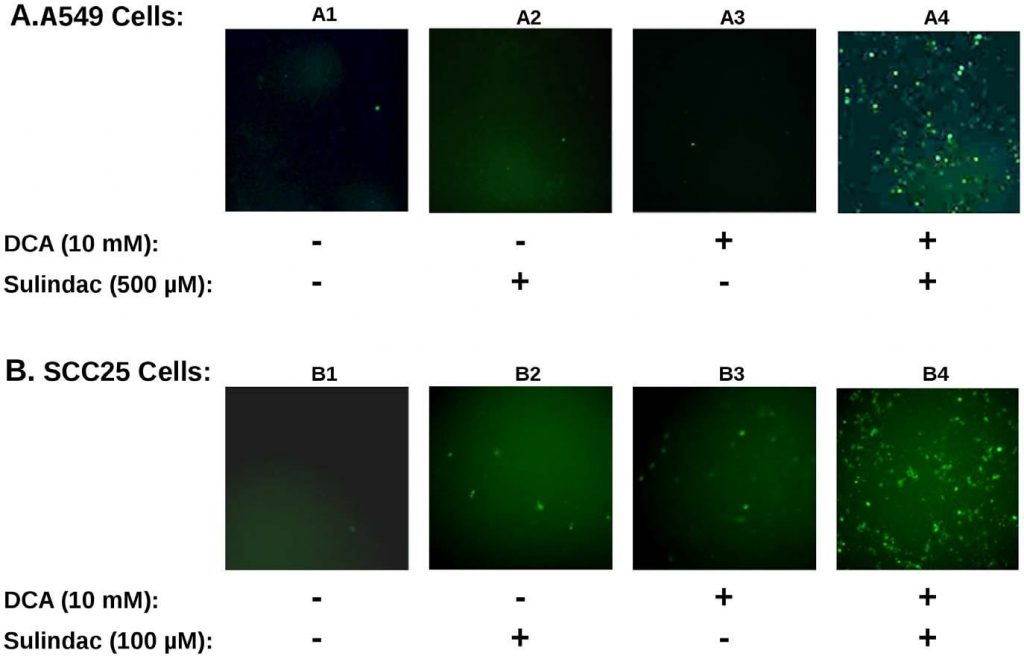
Die synergistische Wirkung auf die Lebensfähigkeit, die mit Sulindac und Dichloracetat sowohl bei A549- als auch bei SCC25-Krebszellen beobachtet wurde, ähnelt auffallend früheren Studien mit der Kombination von Sulindac und TBHP [7]. Um festzustellen, ob die ROS-Produktion an der in den vorliegenden Studien beobachteten selektiven Abtötung beteiligt war, wurde die ROS-Produktion mit dem IndikatorfarbstoffH2DCFDA(siehe Methoden) in den Krebszelllinien bestimmt, die Sulindac und DCA ausgesetzt waren. Die Ergebnisse sind in Abbildung 3 zusammengefasst. Abbildung 3A zeigt die Ergebnisse mit A549-Krebszellen. Aus den in Abbildung 3A dargestellten Ergebnissen ist ersichtlich, dass unbehandelte A549-Krebszellen (Feld A1) oder mit Sulindac allein (Feld A2) oder DCA allein (Feld A3) behandelte Zellen nur wenige positiv gefärbte Zellen aufweisen. Wurden die Zellen jedoch sowohl Sulindac als auch DCA ausgesetzt (Feld A4), ist ein starker Anstieg der positiv gefärbten Zellen für ROS (grüne Fluoreszenz) zu beobachten, was zeigt, dass die Anwesenheit von sowohl Sulindac als auch DCA zur Erzeugung signifikanter Mengen von ROS führt. Wie in Abbildung 3B dargestellt, sind ähnliche Ergebnisse bei den SCC25-Krebszellen zu sehen. Sulindac oder DCA allein führen zu einem geringen Anstieg der ROS-produzierenden Zellen (Tafeln B2 und B3), aber ein starker Anstieg der ROS-Produktion wird erneut beobachtet, wenn beide Medikamente hinzugefügt werden (Tafel B4). Die Quantifizierung unter Verwendung von SCC25-Zellen zeigt, dass die Anzahl der DCF-positiven Zellen (siehe Methoden) um das 9-10fache höher ist, wenn die Zellen mit Sulindac und DCA behandelt werden, als wenn jedes der beiden Medikamente allein verabreicht wird (siehe Abbildung S3A). Diese Ergebnisse und frühere Studien deuten darauf hin, dass die ROS-Produktion ein gemeinsames Merkmal bei der verstärkten Abtötung von Krebszellen sein könnte, wenn Sulindac in Kombination mit Verbindungen verwendet wird, die die mitochondriale Funktion beeinträchtigen.
Sulindac in Kombination mit DCA führt zu einem Verlust des mitochondrialen Membranpotenzials
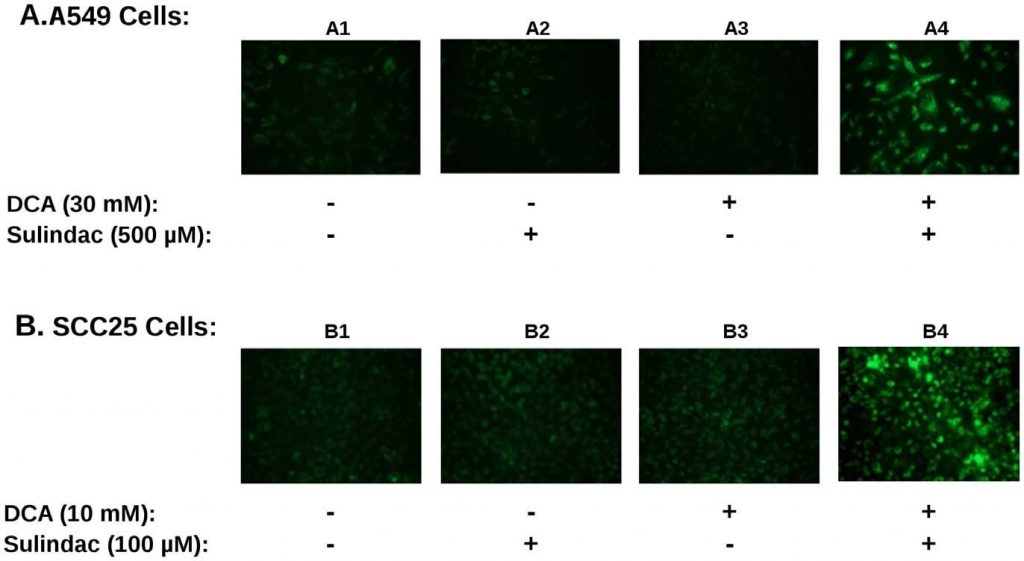
Wenn die ROS-Produktion an der verstärkten abtötenden Wirkung von Sulindac/DCA beteiligt ist, würde man erwarten, dass die Produktion von ROS durch die Medikamentenkombination die mitochondriale Funktion beeinträchtigt. Um dies festzustellen, wurde das mitochondriale Membranpotenzial mit Hilfe der JC-1-Färbung gemessen, wie in Methoden beschrieben. Ein Verlust des Membranpotenzials wird durch einen Anstieg der grünen Fluoreszenz angezeigt, wie in den Methoden beschrieben. Ein typisches Ergebnis ist in Abbildung 4 zusammengefasst. Sowohl A549- als auch SCC25-Krebszellen wurden 48 Stunden lang entweder allein oder in Kombination mit Sulindac und DCA behandelt und mit JC-1 angefärbt, um das mitochondriale Membranpotenzial zu überwachen. Abbildung 4A zeigt die Ergebnisse mit der Krebszelllinie A549. In Abwesenheit eines Medikaments scheinen die Mitochondrien intakt zu sein und ihr Membranpotenzial aufrechtzuerhalten, wie die geringe grüne Fluoreszenz zeigt (Tafel A1). In Gegenwart von Sulindac allein (Tafel A2) oder DCA allein (Tafel A3) ist ein geringer Anstieg der grünen Fluoreszenz zu beobachten, was auf einen gewissen Verlust des mitochondrialen Membranpotenzials hinweist. Wenn jedoch sowohl Sulindac als auch DCA vorhanden sind, kommt es zu einem auffälligen Verlust des mitochondrialen Membranpotenzials, der sich in einem starken Anstieg der grünen Fluoreszenz zeigt (Feld A4). Wir beobachteten dasselbe Muster, als wir mehrere unabhängige Felder mittels Fluoreszenzmikroskopie analysierten. Abbildung 4B zeigt ähnliche Ergebnisse mit den Krebszellen SCC25. Auch hier wurde ein signifikanter Verlust des mitochondrialen Membranpotenzials nur dann festgestellt, wenn die Zellen sowohl Sulindac als auch DCA ausgesetzt waren (Feld B4). Die Quantifizierung des Effekts ist in Abbildung S3B dargestellt. Es ist zu erkennen, dass der Prozentsatz der JC1-grün-positiven Zellen bei Verwendung der Wirkstoffkombination 3-4 mal so hoch ist wie bei Verwendung eines der beiden Wirkstoffe allein.
ROS sind an der Abtötung von Krebszellen durch die Kombination von Sulindac und DCA beteiligt
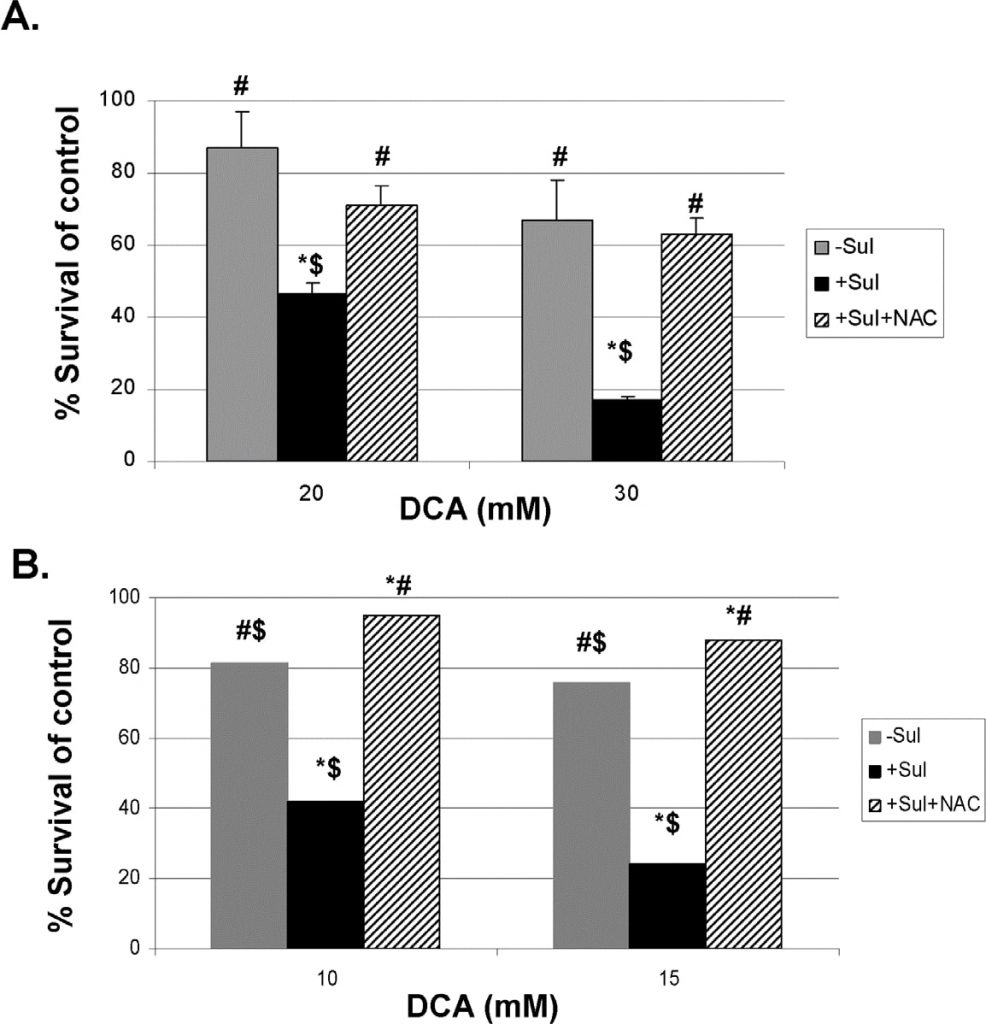
Um einen direkteren Nachweis zu erbringen, dass die erzeugten ROS an der verstärkten Abtötung der Krebszellen durch Sulindac und DCA beteiligt sind, haben wir zwei bekannte ROS-Fänger, N-Acetylcystein (NAC) und Tiron, verwendet (siehe Methoden). Die Ergebnisse mit NAC sind in Abbildung 5 dargestellt. Abbildung 5, Tafel A, zeigt, dass sowohl bei 20 als auch bei 30 mM DCA die verstärkte Abtötung von A549-Krebszellen, die in Gegenwart von Sulindac beobachtet wurde, weitgehend verhindert wird, wenn NAC (2 mM) während der 48-stündigen Inkubation vorhanden ist. Sehr ähnliche Ergebnisse wurden mit den SCC25-Krebszellen erzielt (siehe Abbildung 5, Tafel B). Vergleichbare Ergebnisse wurden erzielt, wenn Tiron anstelle von NAC verwendet wurde (Abbildung S4).
Sulindac und DCA töten Krebszellen unter Beteiligung des apoptotischen Todes
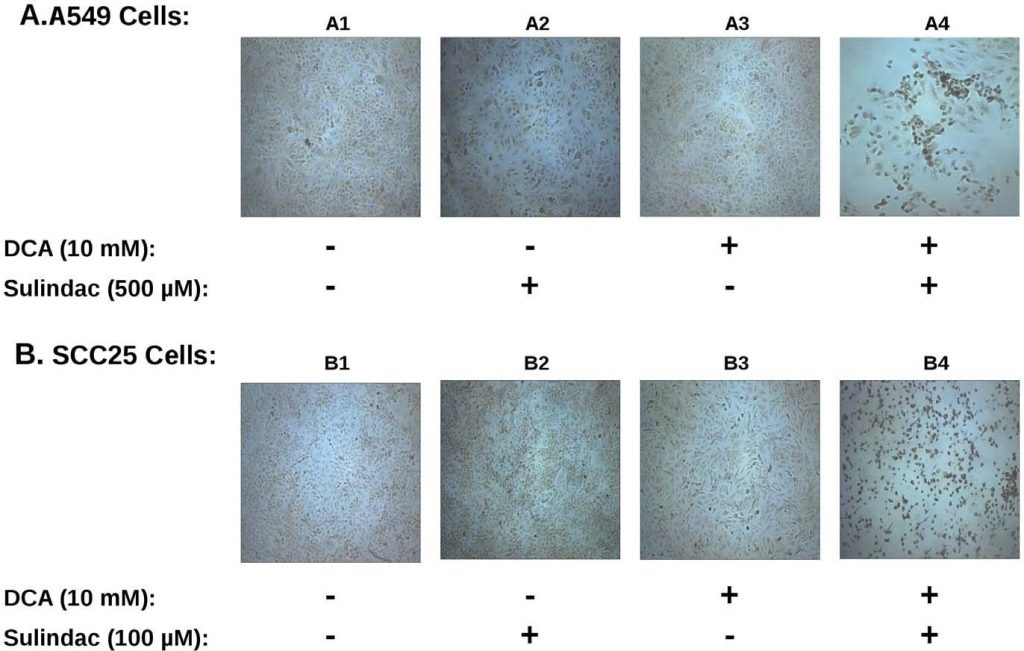
Die obigen Ergebnisse (Abbildungen 3, 4, 5) zeigen, dass die verstärkte Abtötung der Krebszelllinien mit einer mitochondrialen Dysfunktion einhergeht, was darauf schließen lässt, dass der beobachtete Zelltod durch Apoptose erfolgt. Frühere Studien haben gezeigt, dass Sulindac und seine Derivate proapoptotische Arzneimittel sind [5], [6]. Es gibt auch Berichte, dass DCA den Zelltod durch Apoptose verursachen kann [20], [23]. Um festzustellen, ob das durch ROS vermittelte Absterben der Krebszellen durch die Kombination dieser beiden Medikamente mit apoptotischem Tod einhergeht, führten wir TUNEL-Färbungen zur Messung der Apoptose durch (siehe Methoden). Für die TUNEL-Färbeversuche wurden mehrere Wiederholungen für Sulindac und DCA allein oder in Kombination getestet. Ein typisches Ergebnis ist in Abbildung 6 dargestellt, wobei die oberen Felder (Abbildung 6A, Felder A1-A4) die Ergebnisse mit den A549-Krebszellen und die unteren Felder (Abbildung 6B, Felder B1-B4) die Ergebnisse mit den SCC25-Krebszellen zeigen. Wenn die Zellen mit keinem Medikament, Sulindac allein oder DCA allein behandelt wurden (Abbildung 6, Felder A1-A3 und B1-B3), wurden nur wenige TUNEL-positive Zellen beobachtet. Wurden die Zellen jedoch sowohl mit Sulindac als auch mit DCA behandelt, kam es zu einem signifikanten Anstieg der TUNEL-positiven apoptotischen Zellen (Abbildung 6, Tafeln A4 und B4), was auf eine starke Induktion der Apoptose hindeutet. Zur Überprüfung der TUNEL-Ergebnisse wurde auch ein empfindlicherer ligationsvermittelter PCR-basierter DNA-Laddering-Assay verwendet, um die Apoptose zu überwachen [25]. Die Ergebnisse zeigten auch das Vorhandensein einer angereicherten starken nukleosomalen Leiter nur dann, wenn sowohl Sulindac als auch DCA in Kombination verwendet wurden (Abbildung S5; Spuren 4 und 8), was die TUNEL-Assay-Daten stark unterstützt.
Sulindac und DCA töten proapoptotische JNK-Signale
Von den bekannten mitogenaktivierten Proteinkinasen (MAP-Kinasen) ist die stressinduzierte Kinase c-Jun N-terminale Kinase (JNK/SAPK) direkt am apoptotischen Zelltod beteiligt [11]. Daher untersuchten wir die Rolle der JNK-Signalübertragung bei der durch Sulindac-DCA vermittelten Apoptose mit Hilfe von SP600125, einem JNK-spezifischen Inhibitor (JNKI), und die Ergebnisse sind in Abbildung 7A dargestellt. Wie oben gezeigt, starben die mit Sulindac behandelten SCC25-Zellen in Gegenwart steigender DCA-Konzentrationen verstärkt ab. Wenn diese Zellen jedoch mit Sulindac zusammen mit SP600125 inkubiert wurden, wurde der Sulindac-DCA-vermittelte Zelltod weitgehend verhindert. Diese Ergebnisse deuten darauf hin, dass die JNK-vermittelte proapoptotische Signalübertragung am Sulindac-DCA-vermittelten Zelltod beteiligt ist.
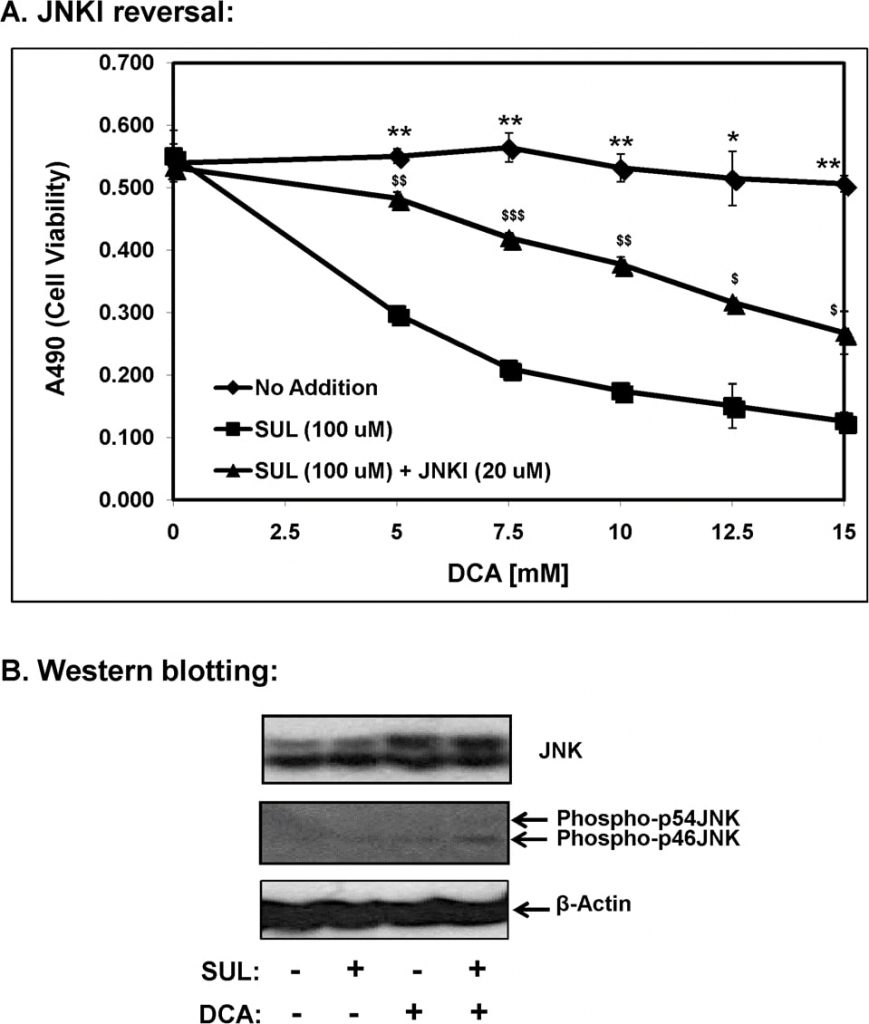
Durch Western-Blot-Analyse wurde außerdem festgestellt, dass die Kombination von Sulindac und DCA 12 Stunden nach der Exposition der Zellen gegenüber Sulindac und DCA die Phospho-JNK-Konzentration in den zytosolischen Fraktionen signifikant erhöhte (Abbildung 7B). Ein Anstieg der Gesamt-JNK-Konzentration (Proteinbanden bei 46 und 54 kDa) wurde sowohl bei der Behandlung der Zellen mit DCA allein als auch bei der Behandlung der Zellen mit der Kombination aus Sulindac und DCA festgestellt. Es ist anzumerken, dass sowohl die Phospho-p46JNK- als auch die Phospho-p54JNK-Isoformen durch die kombinierte Behandlung mit Sulindac und DCA induziert wurden, obwohl der Anstieg von Phospho-p46JNK signifikanter war (Abbildung 7B).
Es gibt zahlreiche Hinweise darauf, dass JNK die Freisetzung von apoptoseinduzierenden Faktoren aus den Mitochondrien, wie z. B. Cytochrom c, initiiert, die zur Spaltung von Caspasen und PARP (Poly(ADP-Ribose)-Polymerase) führen [30], [31]. Studien haben auch gezeigt, dass während der Apoptose das aus den Mitochondrien in das Zytoplasma freigesetzte Cytochrom c schließlich in den Zellkern gelangt [32]. Unsere Ergebnisse deuten darauf hin, dass die maximale Aktivierung von JNK etwa 12 Stunden nach der Exposition gegenüber Sulindac und DCA auftritt. Dies scheint zur Translokation von Cytochrom c in das Zytoplasma und zur Spaltung von PARP 18 Stunden nach der ersten Behandlung mit Sulindac und DCA zu führen (Abbildung S6A). Als positive Kontrolle für diese Experimente behandelten wir die Zellen mit 100 µM Etoposid, einem Apoptose-induzierenden Mittel. Unter der kombinierten Behandlung mit Sulindac und DCA kann eine erhöhte Kernfluoreszenz in der Mehrzahl der Zellen beobachtet werden, die sich aktiv in der Apoptose befinden (Abbildung S6B).
Eine detaillierte Analyse der experimentellen Daten der Ganzzell-Immunfluoreszenz ergab, dass ∼94% der Zellen, die weder mit Sulindac noch mit DCA behandelt wurden, eine punktförmige, mitochondriale Cytochrom-c-Fluoreszenz mit geringer diffuser Färbung im Zytoplasma oder in den Kernen aufwiesen. Im Gegensatz dazu zeigten nach einer Sulindac-Behandlung 81 % der Zellen eine diffuse, deutliche zytoplasmatische Fluoreszenz und nur sehr wenig Kernfluoreszenz. Nach DCA-Behandlung zeigten ∼83 % der Zellen eine diffuse, deutliche zytoplasmatische Fluoreszenz und <5 % der Zellen eine starke Kernfluoreszenz. Wurden die Zellen jedoch sowohl mit Sulindac als auch mit DCA behandelt, zeigten ∼72 % der Zellen sowohl Kern- als auch Zytoplasmafluoreszenz und ∼11 % der Zellen eine starke Kernfluoreszenz. Diese Ergebnisse deuten darauf hin, dass das aus den Mitochondrien freigesetzte Cytochrom c den intrinsischen apoptotischen Weg einleiten könnte, der bei der durch Sulindac und DCA vermittelten Krebsabtötung funktioniert.
Diskussion
Die vorliegende Studie stellt eine Erweiterung unserer früheren Arbeit dar, die gezeigt hat, dass Sulindac Krebszellen, nicht aber normale Zellen, empfindlicher gegenüber oxidativem Stress macht [7]. In diesen früheren Experimenten wurde Sulindac 24-48 Stunden lang mit den Zellen vorinkubiert und dann wurde das Sulindac entfernt, bevor die Zellen entweder TBHP oderH2O2für 2 Stunden ausgesetzt wurden. Aus den früheren Experimenten ging hervor, dass die Krebszellen durch die Vorbehandlung mit Sulindac sehr viel empfindlicher für das Oxidationsmittel wurden, was zu einem starken Anstieg der ROS und einem Verlust der Mitochondrienfunktion führte [7].
Auf der Grundlage dieser Ergebnisse lag es nahe, dass Sulindac in Kombination mit Verbindungen, die die mitochondriale Atmung beeinträchtigen, zu einer selektiven verstärkten Abtötung von Krebszellen, nicht aber von normalen Zellen führen würde. In der vorliegenden Studie mit den Krebszelllinien A549 und SCC25 verstärkte die Kombination von Sulindac und DCA die Abtötung dieser Krebszelllinien, nicht aber von normalen Lungen- oder Hautzellen. Unsere Ergebnisse zu den in ganzen Zellen benötigten DCA-Mengen stimmen mit früheren Berichten überein [28], [33], [34]. In unserem System liegt die IC50 für DCA bei SCC25-Zellen bei 23 mM und bei A549-Zellen bei 35 mM. Die IC50 für normale Keratinozyten liegt bei >50 mM und für normale Lungenzellen (MRC5) bei ∼40 mM. Die Ergebnisse deuten auch darauf hin, dass der beobachtete Krebszelltod mit der ROS-Produktion, der JNK-Aktivierung und der durch die Mitochondrien ausgelösten Apoptose einhergeht. Was die fehlende Wirkung auf normale Zellen betrifft, so hat sich gezeigt, dass Sulindac normale Lungenzellen vor oxidativer Schädigung infolge von TBHP-Exposition schützt [7] und wir haben kürzlich berichtet, dass Sulindac Herzzellen durch einen Präkonditionierungsmechanismus vor oxidativen Schäden infolge von Ischämie/Reperfusion schützen kann [35].
Nach unserem Kenntnisstand gibt es inzwischen mindestens acht Verbindungen, darunter unsere Studien mit TBHP,H2O2und DCA, die in Gegenwart von Sulindac eine verstärkte und selektive Krebsabtötung gezeigt haben [7]–[9], [12]–[15]. Obwohl ihre Stoffwechselziele innerhalb der Zelle bekannt und unterschiedlich sind, ist es sehr wahrscheinlich, dass sie alle direkt oder indirekt den Zelltod in Gegenwart von Sulindac durch einen Mechanismus verursachen, der eine Veränderung der mitochondrialen Atmung und der ROS-Produktion beinhaltet [10], [12], [14]. Wenn man ein Medikament findet, das in Kombination mit Sulindac selektiv Krebszellen, aber keine normalen Zellen abtötet, ist es wahrscheinlich, dass der Mechanismus der Abtötung oxidativen Stress beinhaltet, der zu mitochondrialer Dysfunktion führt. Eine veränderte Atmung könnte ein gemeinsamer Faktor bei diesen Experimenten mit Sulindac/Wirkstoff-Kombinationen sein, und die vorliegenden Ergebnisse mit DCA unterstützen diese Ansicht. Es ist durchaus möglich, dass die Sulindac-Wirkung mit den Beobachtungen zusammenhängt, die Warburg vor mehr als 50 Jahren gemacht hat, als er feststellte, dass normale Zellen zur Energiegewinnung die Atmung bevorzugen, während Krebszellen aufgrund eines Defekts in der Atmungskette die Glykolyse bevorzugen [16]. Dieser grundlegende Unterschied in der mitochondrialen Atmung zwischen normalen und Krebszellen kann Krebszellen empfindlicher gegenüber oxidativem Stress machen [36], [37]. Es scheint, dass Sulindac diesen grundlegenden Unterschied in der Biochemie von normalen Zellen und Krebszellen verstärken kann. Wie Sulindac Krebszellen für Medikamente sensibilisiert, die die mitochondriale Atmung beeinträchtigen, ist allerdings noch unklar, wird aber aktiv untersucht. Spitz und Mitarbeiter [38]kamen in Studien zum Glukoseentzug bei Krebszellen zu einer ähnlichen Schlussfolgerung hinsichtlich der Unterschiede im Stoffwechsel zwischen normalen und Krebszellen. In Übereinstimmung mit diesen Ergebnissen hat eine andere neuere Studie gezeigt, dass die pharmakologische Hemmung der Laktatdehydrogenase zu einer selektiven Krebsabtötung führen könnte [39]. In den letztgenannten Studien wurde gezeigt, dass die verstärkte selektive Abtötung von Krebszellen auch mit einer ROS-Produktion einhergeht, und die beobachtete Wirkung wurde auf einen veränderten Atmungsprozess in den Krebszellen zurückgeführt.
Es sei darauf hingewiesen, dass die Kombination von Sulindac mit einem Oxidationsmittel oder Medikamenten, die die mitochondriale Funktion beeinträchtigen können, bereits klinisch getestet wurde. Meyskens et al. 2008 zeigten, dass die Kombination von Sulindac mit DFMO in einer dreijährigen klinischen Studie eine signifikante Wirkung auf das Wiederauftreten von Dickdarmpolypen und das Auftreten von Dickdarmkrebs hatte [13]. Vor kurzem haben wir über die Verwendung von Sulindac mitH2O2in einer klinischen Studie zum Nachweis des Konzepts für die topische Behandlung von aktinischen Keratosen berichtet [15]. Einer der Nachteile dieser Kombination bestand darin, dass zwei topische Formulierungen benötigt wurden, da die Wirkstoffe nicht lange gelagert werden konnten, ohne dass das Sulindac durchH2O2zerstört wurde. Außerdem kann manH2O2nicht zur Behandlung von inneren Tumoren verwenden, da es nicht oral eingenommen werden kann. Die Kombination von Sulindac und DCA könnte jedoch in einer einzigen Formulierung für die topische Anwendung bereitgestellt werden, und die beiden Verbindungen können oral eingenommen werden. Tatsächlich wird DCA seit mehreren Jahren klinisch eingesetzt, um den Milchsäurespiegel bei Patienten zu senken, die an Laktatazidose leiden [40]–[42]. DCA wurde auch als Mittel gegen Krebs in vitro und in vivo bei verschiedenen Krebszelllinien eingesetzt, was darauf hindeutet, dass der mitochondriale Stoffwechsel in Krebszellen ein neues therapeutisches Ziel sein könnte [18], [20], [22]. Michelakis et al. (2010) haben gezeigt, dass die Behandlung mit DCA den mitochondrialen Stoffwechsel bei Glioblastom-Patienten mit reversiblen toxischen Wirkungen „umgestaltet“. Es sei darauf hingewiesen, dass sowohl Sulindac als auch DCA kostengünstig und relativ ungiftig sind und oral eingenommen werden können. Wenn sich die Kombination in vivo als erfolgreich erweist, wird sie eine neue Dimension in der Krebsbehandlung eröffnen, da beide Medikamente auf den mitochondrialen Stoffwechsel bei verschiedenen Krebsarten abzielen [22].
Zusammenfassend lässt sich sagen, dass unsere Studien mit der Kombination von Sulindac und DCA darauf hindeuten, dass Sulindac Krebszellen selektiv empfindlicher für Wirkstoffe macht, die die mitochondriale Atmung beeinträchtigen, was zu oxidativem Stress und mitochondrialer Dysfunktion führt. Diese Ergebnisse könnten mit dem Atmungsdefekt in Krebszellen zusammenhängen, der ursprünglich von Warburg beobachtet wurde [16]. Studien, die darauf abzielen, die grundlegenden Unterschiede zwischen der Reaktion von Krebszellen und normalen Zellen auf Sulindac und Wirkstoffe, die die mitochondriale Funktion beeinträchtigen, zu verstehen, werden derzeit untersucht.
Unterstützende Informationen
Abbildung S1 Zytotoxizität von Sulindac, DCA oder einer Wirkstoffkombination auf A549 und SCC25 Krebszellen
Die A549- und SCC25-Krebszellen wurden mit den angegebenen Konzentrationen von Sulindac (Felder 1 und 4) oder DCA (Felder 2 und 5) behandelt. In den Sulindac/DCA-Wirkstoffkombinationsexperimenten wurde für die A549-Zellen ein Verhältnis von 1:50 (Sulindac:DCA) beibehalten, wobei die Wirkstoffkombinationen von 0,2 mM:10 mM bis zu 1 mM:50 mM reichten (Tafel 3), und für die SCC25-Zellen wurde ein Verhältnis von 1:100 (Sulindac:DCA) beibehalten, wobei die Wirkstoffkombinationen von 0,05 mM:5 mM bis zu 0,3 mM:30 mM reichten (Tafel 6). (TIF) Abbildung S2 Bestimmung der Kombinationsindizes für die Krebszellen A549 und SCC25. Die Indizes für die Wirkstoffkombinationen wurden durch Einsetzen der oben ermittelten Werte für die Zellviabilität in die Gleichungen von Chou und Talalay [26] ermittelt. Siehe Text für weitere Details. (TIF) Abbildung S3 Quantifizierung der DCF- und JC-1-positiven grün fluoreszierenden Zellen. SCC25-Zellen wurden 48 Stunden lang mit Sulindac, DCA oder einer Wirkstoffkombination behandelt und mit den Farbstoffen H2DCFDA (Abb. S3A) oder JC-1 (Abb. S3B) gefärbt, wie in den Methoden beschrieben. Zur positiven Kontrolle wurden die Zellen 2 Stunden lang mit 200 mM TBHP behandelt und wie oben beschrieben gefärbt. Die Zellen wurden unter starker Vergrößerung mit dem Objektiv 1006 in einem inversen Fluoreszenzmikroskop von Olympus analysiert. Für jede Bedingung wurden mindestens 100 einzelne Zellen sichtbar gemacht, und die Prozentsätze der DCF-positiven und JC1-positiven grün fluoreszierenden Zellen sind in Tabellen und Grafiken dargestellt. (TIF) Abbildung S4 Der ROS-Fänger Tiron kehrt die Abtötung von Krebszellen durch die Kombination von Sulindac und DCA um. Die Krebszellen A549 und SCC25 wurden 48 Stunden lang mit den angegebenen Konzentrationen von DCA in Abwesenheit (grauer Balken) oder in Anwesenheit von Sulindac (schwarzer Balken) oder in Anwesenheit von Sulindac und Tiron (gestreifter Balken) behandelt. Die Lebensfähigkeit der Zellen wurde mit dem MTS-Test, wie in den Methoden beschrieben, überwacht. Die Zelllebensfähigkeit wird in % der Kontrolle (nicht mit Sulindac behandelte Zellen) angegeben. Die Fehlerbalken sind Standardfehler des Mittelwerts (SEM), ausgedrückt in % des Mittelwerts von Vierfachexemplaren aus einem repräsentativen Experiment. Die Hemmung des Krebszellwachstums erfolgte dosisabhängig während der Kombinationsbehandlung von DCA und Sulindac (schwarze Balken) sowohl bei A549-Krebszellen (A) als auch bei SCC25-Krebszellen (B). Diese verstärkte Abtötung wurde jedoch verhindert, wenn Tiron zusammen mit der Medikamentenkombination verabreicht wurde (gestreifte Balken in A und B). (TIF) Abbildung S5 Während der Abtötung von Krebszellen durch die Kombination von Sulindac und DCA kommt es zu einer ausgeprägten nukleosomalen DNA-Leiterbildung. Die Krebszellen A549 und SCC25 wurden 48 Stunden lang mit den angegebenen Medikamenten behandelt. Die nukleosomale DNA wurde extrahiert und einer ligationsvermittelten PCR unterzogen, wie in Methoden beschrieben, und auf einem 1,2%igen Agarosegel zusammen mit Größenmarkern analysiert. Spur ‚M‘ bezeichnet die Molekülgrößenmarker. Die Spuren 1-4 und 5-8 zeigen die Ergebnisse, die mit A549- bzw. SCC25-Krebszellen erzielt wurden. Die Ergebnisse sind in den Spuren 1 und 5 (ohne Wirkstoff), in den Spuren 2 und 6 (Sulindac allein), in den Spuren 3 und 7 (DCA allein) und in den Spuren 4 und 8 (Sulindac und DCA) dargestellt. Eine verstärkte nukleosomale DNA-Leiterbildung wurde nur bei der Kombinationsbehandlung mit Sulindac und DCA beobachtet (Spuren 4 und 8). (TIF)
Abbildung S6 Die Kombination von Sulindac und DCA führt zur Freisetzung von Cytochrom c aus den Mitochondrien und zur Spaltung von PARP
SCC25-Zellen wurden mit Sulindac, DCA, der Wirkstoffkombination oder Etoposid behandelt, um den intrazellulären Ort von Cytochrom c durch Western Blotting und Immunfluoreszenz zu bestimmen. A. Zytosolische Fraktionen wurden nach 18 Stunden isoliert, und das Vorhandensein von Cytochrom c im Zytoplasma und die Spaltung von PARP wurden durch Western Blotting bestimmt. Repräsentative Western Blots zeigen die Menge an Cytochrom c und gespaltenem PARP. b-Actin wurde als interne Kontrolle verwendet. B. Die Immunfluoreszenz wurde mit dem CBA077 InnoCyteTM Flow Cytometric Cytochrome c Release Kit gemäß den Anweisungen des Herstellers durchgeführt. Mehrere unabhängige Felder wurden analysiert, und die repräsentativen Mikroaufnahmen zeigen die Lokalisierungsmuster von Cytochrom c, wenn die Zellen Sulindac und/oder DCA ausgesetzt sind. Die quantitativen Werte sind im Text angegeben. (TIF)
Danksagungen
Die Autoren bedanken sich bei David Brunell für seine Hilfe bei der Bestimmung der Kombinationsindizes und bei Edna Gamliel für ihre Unterstützung bei der Zellkultur.
Beiträge der Autoren
Konzeption und Planung der Experimente: KA SK KDS HW. Durchführung der Experimente: KA SK. Analysierte die Daten: KA SK HW. Beigetragene Reagenzien/Materialien/Analysewerkzeuge: KDS. Schrieb die Arbeit: KA SK HW.
REFERENZEN
1 Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, et al. (1996) Cyclooxygenase-2-Überexpression und Tumorbildung werden durch Sulindac in einem Mausmodell der familiären adenomatösen Polyposis blockiert. Cancer Res 56: 2556-2560.
2 Taketo MM (1998) Cyclooxygenase-2-Inhibitoren in der Tumorentstehung (Teil II). J Natl
3 Taketo MM (1998) Cyclooxygenase-2-Hemmer in der Tumorentstehung (Teil I). J Natl Cancer Inst 90: 1529-1536.
4 Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, et al. (1995) Chemoprävention der Kolonkarzinogenese durch Sulindac, ein nichtsteroidales entzündungshemmendes Mittel. Cancer Res 55: 1464-1472.
5 Vogt T, McClelland M, Jung B, Popova S, Bogenrieder T, et al. (2001) Progression und NSAID-induzierte Apoptose in malignen Melanomen sind unabhängig von der Cyclooxygenase II. Melanoma Res 11: 587-599.
6 Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B (2001) Wachstumshemmung und Induktion von Apoptose in kolorektalen Tumorzellen durch Cyclooxygenase-Inhibitoren. Karzinogenese 22: 17-25.
7 Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, et al. (2009) Sulindac steigert die Abtötung von Krebszellen, die oxidativem Stress ausgesetzt sind. PLoS One 4: e5804.
8 Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, et al. (1999) Synergistische Effekte neuer chemopräventiver Wirkstoffe und konventioneller zytotoxischer Wirkstoffe gegen menschliche Lungenkrebszelllinien. Cancer Res 59: 6178-6184.
9 Jiang TT, Brown SL, Kim JH (2004) Kombinierte Wirkung von Arsentrioxid und Sulindacsulfid in A549 menschlichen Lungenkrebszellen in vitro. J Exp Clin Cancer Res 23: 259-262.
10 Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, et al. (2006) Synergistische Induktion der Apoptose durch Sulindac und Arsentrioxid in menschlichen Lungenkrebszellen A549 durch reaktive Sauerstoffspezies-abhängige Herunterregulierung von Survivin. Biochem Pharmacol 72: 1228-1236.
11 Jin HO, Seo SK, Woo SH, Lee HC, Kim ES, et al. (2008) A combination of sulindac and arsenic trioxide synergistically induces apoptosis in human lung cancer H1299 cells via c-Jun NH2-terminal kinase-dependent Bcl-xL phosphorylation. Lungenkrebs 61: 317-327.
12 Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, et al. (2005) Sulindac verstärkt den durch den Proteasom-Inhibitor Bortezomib vermittelten oxidativen Stress und die Antikrebsaktivität. Clin Cancer Res 11: 5248-5256.
13 Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, et al. (2008) Difluoromethylornithin plus Sulindac zur Vorbeugung von sporadischen kolorektalen Adenomen: eine randomisierte, placebokontrollierte Doppelblindstudie. Cancer Prev Res (Phila Pa) 1: 32-38.
14 Seo SK, Jin HO, Lee HC, Woo SH, Kim ES, et al. (2008) Combined effects of sulindac and suberoylanilide hydroxamic acid on apoptosis induction in human lung cancer cells. Mol Pharmacol 73: 1005-1012.
15 Resnick L, Rabinovitz H, Binninger D, Marchetti M, Weissbach H (2009) Topical sulindac combined with hydrogen peroxide in the treatment of actinic keratoses. J Drugs Dermatol 8: 29-32.
16 Warburg O (1956) On the origin of cancer cells. Science 123: 309-314.
17 Whitehouse S, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141: 761-774.
18 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 11: 37-51.
19 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60: 1478-1487.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol 109: 394-402.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2-Prostatakrebszellen in vitro für Strahlung. Prostate 68: 1223-1231.
<span id=“22″ class=“referencess blue-text „22 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, et al. (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2: 31ra34.
23 Michelakis ED, Webster L, Mackey JR (2008) Dichloracetat (DCA) als potenzielle metabolische Zieltherapie für Krebs. Br J Cancer 99: 989-994.
24 Cory AH, Owen TC, Barltrop JA, Cory JG (1991) Use of an a aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3: 207-212.
<span id=“25″ class=“referencess blue-text „25 Staley K, Blaschke AJ, Chun J (1997) Apoptotische DNA-Fragmentierung wird durch eine semi-quantitative ligationsvermittelte PCR von stumpfen DNA-Enden nachgewiesen. Zelltod Differ 4: 66-75.
26 Chou TC, Talalay P (1984) Quantitative Analyse von Dosis-Wirkungs-Beziehungen: die kombinierten Wirkungen von mehreren Medikamenten oder Enzyminhibitoren. Adv Enzyme Regul 22: 27-55.
<span id=“27″ class=“referencess blue-text „27 Papandreou I, Goliasova T, Denko NC (2010) Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer 128: 1001-1008.
28 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, et al. (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127: 2510-2519.
29 Babbar N, Ignatenko NA, Casero RA Jr, Gerner EW (2003) Cyclooxygenase-unabhängige Induktion der Apoptose durch Sulindacsulfon wird durch Polyamine bei Dickdarmkrebs vermittelt. J Biol Chem 278: 47762-47775.
30 Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haikel Y, et al. (2011) Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenesis 32: 1268-1278.
31 Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, et al. (2007) c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 14: 1001-1010.
<span id=“32″ class=“referencess blue-text „32 Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, et al. (2004) Nuclear translocation of cytochrome c during apoptosis. J Biol Chem 279: 24911-24914.
33 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, et al. (2011) Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 67: 647-655.
34 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in kolorektalen Krebszellen. Br J Cancer 102: 1746-1752.
35 Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac verleiht dem Herzen einen hochgradigen ischämischen Schutz durch späte Vorkonditionierungsmechanismen. Proc Natl Acad Sci U S A 106: 19611-19616.
<span id=“36″ class=“referencess blue-text „36 Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18: 54-61.
37 Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029-1033.
<span id=“38″ class=“referencess blue-text „38 Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, et al. (2005) Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280: 4254-4263.
<span id=“39″ class=“referencess blue-text „39 Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, et al. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107: 2037-2042.
<span id=“40″ class=“referencess blue-text „49 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI (1983) Treatment of lactic acidosis with dichloroacetate. N Engl J Med 309: 390-396.
41 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77: 535-541.
42 Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, et al. (2008) Evaluation der Langzeitbehandlung von Kindern mit kongenitaler Laktatazidose mit Dichloracetat. Pediatrics 121: e1223-1228.
Zugehöriger Inhalt: