Yu Liang1, Lidan Hou1, Linjing Li1, Lei Li1, Liming Zhu1, Yu Wang1, Xin Huang1, Yichao Hou1, Danxi Zhu1, Huimin Zou1, Yan Gu2, Xiaoling Weng3,4, Yingying Wang5, Yue Li6, Tianqi Wu3, Mengfei Yao3, Isabelle Gross7,8, Christian Gaiddon9,10, Meng Luo2, Jianhua Wang3, Xiangjun Meng1
1 Dipartimento di Gastroenterologia, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Cina
2 Dipartimento di Sufergeria Generale, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Cina
3 Istituto di Cancro, Fudan University Shanghai Cancer Center, Fudan University, Shanghai, Cina
4 Ningbo Aitagene Technology Co. LTD, Shanghai, Cina
5 Dipartimento di Biochimica e Biologia Molecolare
Biologia cellulare, Shanghai Jiao Tong University School of Medicine, Shanghai, Cina
6 Centro di patologia, Shanghai First People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Cina
7 INSERM UMR_S1113, Strasburgo F-67200, Francia
8 FMTS, Universite de Strasbourg Strasbourg, Strasburgo F-67000, Francia
9Universitede Strasbourg, Inserm IRFAC UMR_S1113, Laboratory Stress Response and Innovative Therapy “Streinth”, Strasbourg 67200, Francia
10 CLCC Paul Strauss, Strasbourg, Francia
Meng Luo [email protected]
Jianhua Wang [email protected]
Xiangjun Meng [email protected]
Questi autori hanno contribuito in egual misura: Yu Liang, Lidan Hou
Ricevuto: 16 marzo 2019
Rivisto: 17 settembre 2019
Accettato: 19 settembre 2019
Pubblicato: 9 ottobre 20199 ottobre 2019
Abstract
Lo sviluppo della chemioresistenza rimane una sfida importante che spiega la letalità del cancro colorettale (CRC). Il dicloroacetato (DCA) è stato originariamente utilizzato come regolatore metabolico nel trattamento delle malattie metaboliche; in questo caso, il DCA è stato analizzato per identificare i meccanismi alla base della chemioresistenza del CRC. Abbiamo scoperto che il DCA aumenta notevolmente la chemiosensibilità delle cellule CRC al fluorouracile (5-FU) e riduce la formazione di colonie a causa di alti livelli di apoptosi. Utilizzando il saggio microarray, abbiamo notato che il miR-149-3p era coinvolto nella chemioresistenza del CRC, modulata da p53 wild-type dopo il trattamento con DCA. Inoltre, PDK2 è stato identificato come bersaglio diretto del miR-149-3p. L’analisi meccanica ha mostrato che la sovraespressione del miR-149-3p ha potenziato l’apoptosi indotta dal 5-FU e ha ridotto il metabolismo del glucosio, in modo simile agli effetti del knockdown di PDK2. Inoltre, la sovraespressione di PDK2 ha parzialmente invertito l’effetto inibitorio del miR-149-3p sul metabolismo del glucosio. Infine, sia il trattamento con DCA che la sovraespressione di miR-149-3p in cellule CRC resistenti al 5-FU sono risultati in grado di sensibilizzare notevolmente l’effetto chemioterapico del 5-FU in vivo e questo effetto è stato convalidato anche in una piccola coorte retrospettiva di pazienti CRC. Nel complesso, abbiamo stabilito che la via di segnalazione p53/miR-149-3p/PDK2 può essere potenzialmente bersagliata con il trattamento con DCA per superare il CRC chemioresistente.
Informazioni supplementari: La versione online di questo articolo (https:// doi.org/10.1038/s41388-019-1035-8) contiene materiale supplementare, disponibile per gli utenti autorizzati.
Introduzione
Il cancro del colon-retto (CRC) è la quarta causa di morte correlata al cancro in Cina [1] ed è la seconda causa di mortalità correlata al cancro negli Stati Uniti [2], principalmente attribuibile alle metastasi e al fallimento della chemioterapia a causa della resistenza ai farmaci, che porta a circa 50.000 morti ogni anno [3].
Recentemente, anche se l’emergente stella PD1/PDL1 ha suscitato grande interesse e un maggior numero di agenti bioterapeutici ha mostrato risultati incoraggianti nel trattamento del cancro, il limitato tasso di efficacia e gli inevitabili effetti avversi ne frenano l’uso in clinica [4,5]. Attualmente, la chemioterapia è ancora una scelta importante nella clinica, soprattutto per i pazienti non resecabili in fase avanzata e metastatica [6], ma lo sviluppo della resistenza ai farmaci rimane il limite maggiore della chemioterapia [7]. Pertanto, l’esplorazione dei meccanismi di resistenza ai farmaci e lo studio di nuove combinazioni di farmaci antitumorali classici per ottimizzarne l’efficacia possono fornire un vantaggio per il trattamento del CRC. Poiché il fluorouracile (5-FU) è il farmaco chemioterapico più comunemente usato nel CRC, in questo studio sono state utilizzate linee cellulari CRC resistenti al 5-FU [8,9].
Un’anomalia del metabolismo del glucosio rappresenta uno degli aspetti principali dei segni distintivi del cancro [10]. È noto che lo sviluppo di una massa cellulare incontrollata porta a una scarsa vascolarizzazione del tumore, causando un ridotto apporto di ossigeno. Di conseguenza, le cellule tumorali si adattano alle alterazioni del microambiente spostando il loro metabolismo dal metabolismo ossidativo al metabolismo glicolitico, che si basa sull’apporto di glucosio e produce lattato. Questo spostamento è chiamato “effetto Warburg” ed è comunemente osservato in varie cellule tumorali come una delle caratteristiche principali [11- 13]. Il recente accumulo di dati di ricerca ha portato all’esigenza di perfezionare la teoria di Warburg [14]; ad esempio, è stato dimostrato che il cambiamento metabolico è coinvolto nella chemioresistenza [15]; pertanto, i modelli di metabolismo delle cellule tumorali potrebbero essere sfruttati per superare la chemioresistenza [16,17].
In particolare, sono stati riportati molteplici meccanismi per controllare il cambiamento metabolico nelle cellule tumorali, tra cui i microRNA (miRNA) [18-20]. I miRNA rappresentano una classe di piccoli RNA endogeni non codificanti che regolano la traduzione e la degradazione degli mRNA [21] e sono coinvolti in molti altri processi biologici, tra cui la proliferazione cellulare, la migrazione, l’apoptosi, l’auto-rinnovamento, l’iniziazione, lo sviluppo del cancro e la chemioresistenza [22-24].
L’evidenza indica che il bersaglio del metabolismo anormale delle cellule tumorali è stato un’intensa attività di ricerca volta ad “asfissiare il tumore”, le cui strategie consistono nell’inibire gli enzimi chiave coinvolti nel metabolismo glicolitico [15]. In questo manoscritto, il dicloroacetato (DCA) è stato originariamente utilizzato per trattare l’acidosi lattica e la malattia mitocondriale ereditaria [25]. Il DCA inibisce l’attività enzimatica della piruvato deidrogenasi chinasi (PDK1-4), necessaria per trasformare il piruvato in acetil-CoA, collegando il metabolismo glicolitico al ciclo dell’acido citrico [26,27]; recentemente è stato riportato che il DCA ha effetti antitumorali [28-31]. Tuttavia, il meccanismo alla base dell’effetto del DCA sul trattamento del CRC rimane elusivo.
Il presente studio si è concentrato sul meccanismo molecolare coinvolto nella regolazione del metabolismo del glucosio e della resistenza alla chemioterapia nel CRC. Utilizzando il DCA nelle cellule di CRC, abbiamo analizzato il ruolo dei miRNA correlati, scoprendo così una via di segnalazione che spiega la resistenza al trattamento con 5-FU.
Risultati
IlDCA ripristina la chemiosensibilità delle cellule CRC resistenti al 5-FU
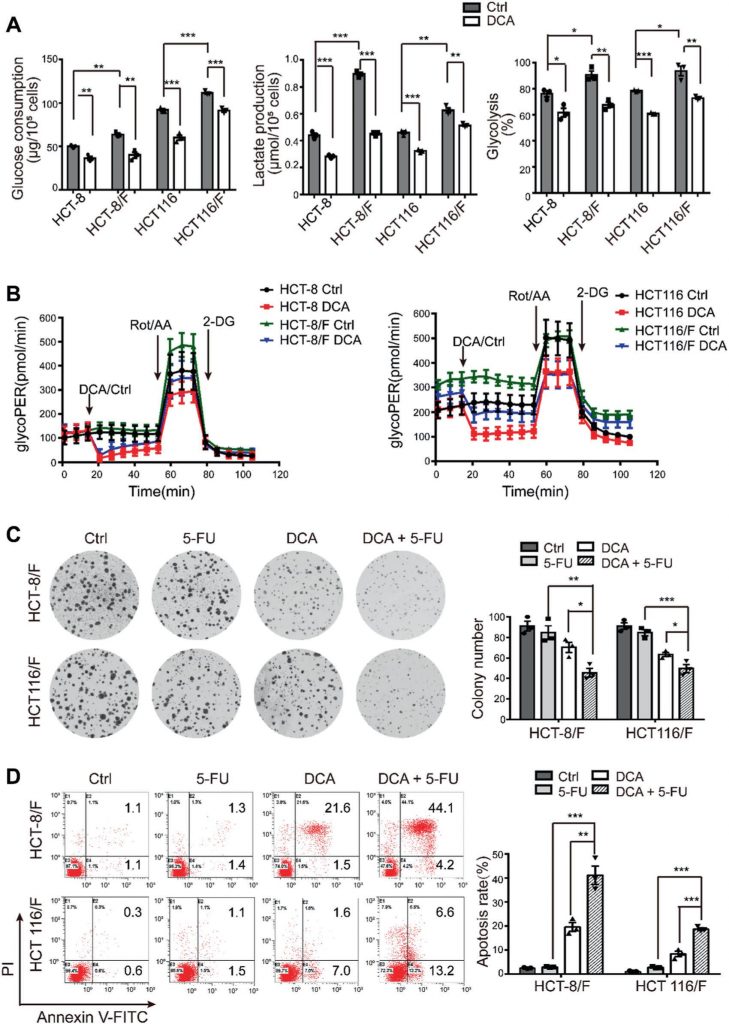
È stato riferito che il DCA è un efficace farmaco antitumorale che agisce colpendo le vie legate all’energia in alcuni tipi di cancro [32]; tuttavia, l’effetto del DCA nelle cellule CRC chemioresistenti non è stato ben studiato. Utilizzando il saggio CCK8, abbiamo scoperto che, rispetto alle linee cellulari parentali HCT-8 e HCT116, le cellule HCT-8/F e HCT116/F resistenti al 5-FU erano insensibili al 5-FU (Fig. 1A supplementare) e le concentrazioni inibitorie semimassimali (IC50) del DCA nelle cellule HCT-8/F e HCT116/F erano rispettivamente di ~15 e 20 mM, in accordo con quanto riportato in precedenza [33, 34] (Fig. 1B supplementare). Abbiamo poi notato che il DCA ha inibito significativamente la sintesi del DNA (Fig. 1C supplementare, pannello superiore) e ha indotto la generazione di ROS (Fig. 1C supplementare, pannello inferiore) nelle cellule CRC resistenti al 5-FU. I marcatori del metabolismo energetico, tra cui il consumo di glucosio, la produzione di lattato e la glicolisi, sono risultati notevolmente elevati nelle cellule CRC resistenti al 5-FU rispetto a quelle sensibili al 5-FU, mentre l’aggiunta di DCA ha ridotto notevolmente l’espressione di questi marcatori (Fig. 1a). Considerando un pretrattamento senza siero di 6 ore per la misurazione della glicolisi, è stato utilizzato un kit per il dosaggio del tasso glicolitico Seahorse XF per eliminare l’effetto del pretrattamento e misurare il tasso glicolitico in tempo reale. Il tasso di efflusso protonico glicolitico (glycoPER) riflette il tasso di acidificazione extracellulare della glicolisi. L’aggiunta di DCA ha ridotto significativamente la glicolisi indotta rispetto ai rispettivi controlli (Fig. 1b e Fig. 1D supplementare).
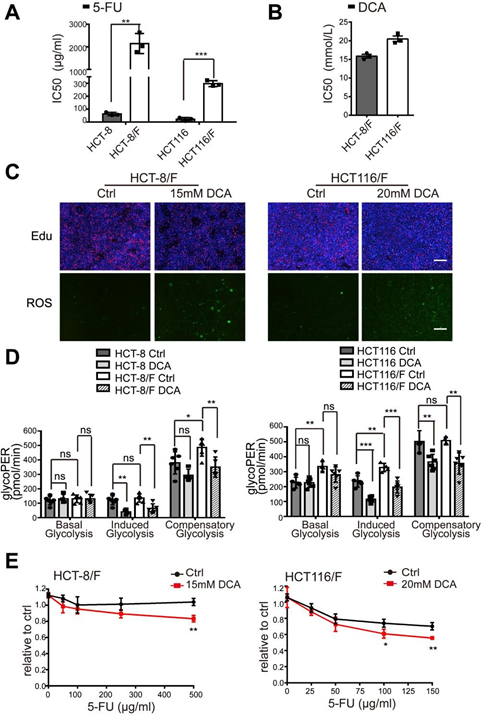
(A) Le cellule CRC sono state trattate con diverse concentrazioni di 5-FU per 24 ore. È stata calcolata l’IC50 del 5-FU in ciascuna cellula. (B) Le cellule HCT-8/F e HCT116/F sono state trattate con diverse concentrazioni di DCA per 24 ore. È stata calcolata l’IC50 del DCA in ogni cellula. (C) Le cellule HCT-8/F e HCT116/F sono state trattate con DCA 15 mM e 20 mM, rispettivamente, per 24 ore. Immagini rappresentative della colorazione di immunofluorescenza della Edu (pannello superiore) e immagini rappresentative della colorazione di immunofluorescenza dei ROS (pannello inferiore), barra di scala: 200 μm. (D) La determinazione del tasso di glicolisi, compreso il glicogeno basale, il glicogeno indotto e il glicogeno compensatorio, è stata calcolata con il Seahorse Glycolytic Rate Assay Report Generator. (E) La crescita cellulare è stata determinata mediante un saggio CCK8 dopo il trattamento con 5-FU e DCA. Gli intervalli di dosaggio del 5-FU erano da 100 ug/ml a 500 ug/ml per le cellule HCT-8/F e da 25 ug/ml a 150 ug/ml per le cellule HCT116/F. I risultati di tre esperimenti indipendenti sono mostrati come media ± SEM. Ogni esperimento è stato eseguito in 3-6 repliche biologiche. *p < 0,05; **, P < 0,01; ***, P < 0,001.
Il DCA ha superato in modo significativo la resistenza al 5-FU nelle cellule HCT-8/F e HCT116/F, come dimostrato dalla misurazione della crescita cellulare (Fig. 1E supplementare). La capacità di formazione di colonie è stata significativamente inibita (Fig. 1c) e l’apoptosi è stata marcatamente indotta in un trattamento combinato (Fig. 1d), tutti dati ulteriormente quantificati. Questi risultati suggeriscono che il DCA può ripristinare la chemiosensibilità nelle cellule CRC resistenti al 5-FU.
ilmiR-149-3p svolge un ruolo cruciale nella chemiosensibilità delle cellule CRC
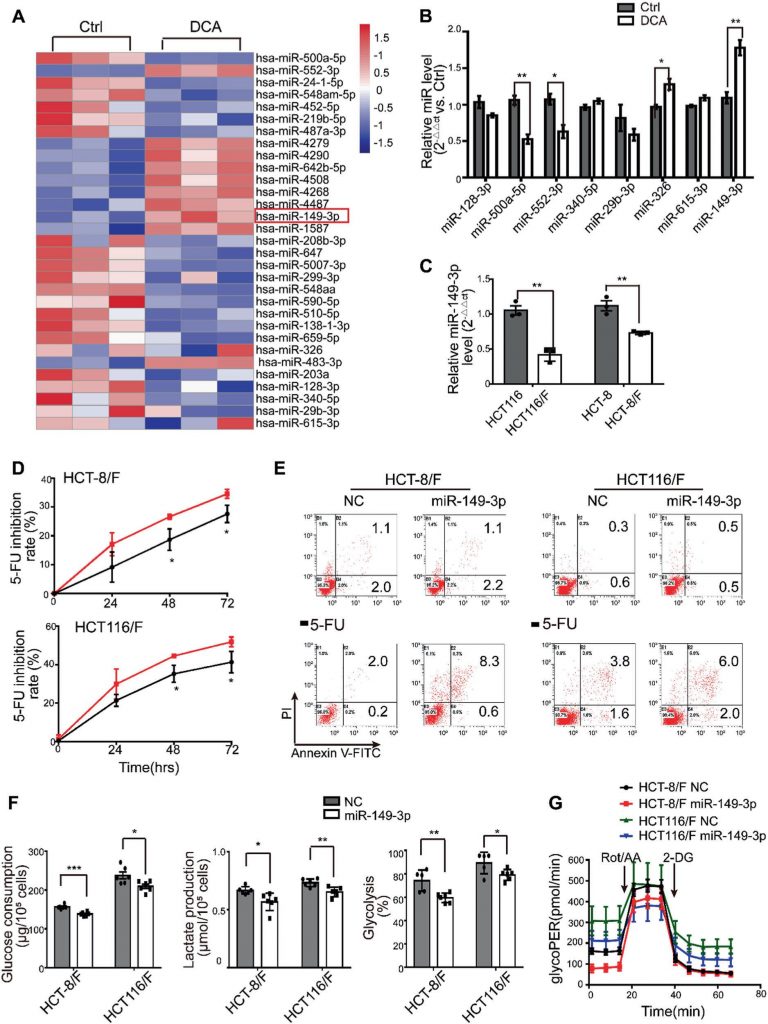
miRNA sono stati considerati un promettente strumento terapeutico per i loro effetti sulla soppressione del tumore [35]. A questo proposito, abbiamo innanzitutto determinato i profili di espressione dei miRNA utilizzando il miRNA array contenente 2059 miRNA umani. Un totale di 119 miRNA sono stati espressi in modo differenziato in risposta al DCA nelle cellule HCT116 (Fig. 2a e Fig. S2A supplementare). Tra questi, i livelli di espressione di otto miRNA sono stati ulteriormente confermati dalla PCR quantitativa in tempo reale (Fig. 2b) e il miR-149-3p è risultato essere upregolato dal DCA in modo dose-dipendente (Fig. S2B supplementare).
Abbiamo poi scoperto che le cellule HCT116 con livelli basali più elevati di miR-149-3p conferivano una maggiore sensibilità al 5-FU e all’L-OHP, come mostrato nella Fig. 2C supplementare. Inoltre, la trasfezione anti-miR-149-3p delle cellule HCT116 ha ridotto notevolmente l’effetto chemioterapico del 5-FU (Fig. 2D-E supplementare). In particolare, i livelli di miR-149-3p nelle cellule CRC chemioresistenti erano significativamente più alti di quelli delle cellule CRC chemioresistenti (Fig. 2c). Pertanto, la trasfezione dei mimici del miR-149-3p ha aumentato significativamente il tasso di inibizione; ha promosso l’apoptosi cellulare indotta dal 5-FU; ha ridotto il consumo di glucosio, la produzione di lattato e la glicolisi nelle cellule HCT-8/F e HCT116/F (Fig. 2d-f). Un saggio del tasso glicolitico Seahorse XF ha mostrato che l’espressione di miR-149-3p ha ridotto la glicolisi basale nelle cellule CRC resistenti al 5-FU, coerentemente con i risultati sopra riportati (Fig. 2g e Fig. 2F supplementare). Questi risultati suggeriscono che il miR-149-3p è favorevole al superamento della chemioresistenza nelle cellule CRC.
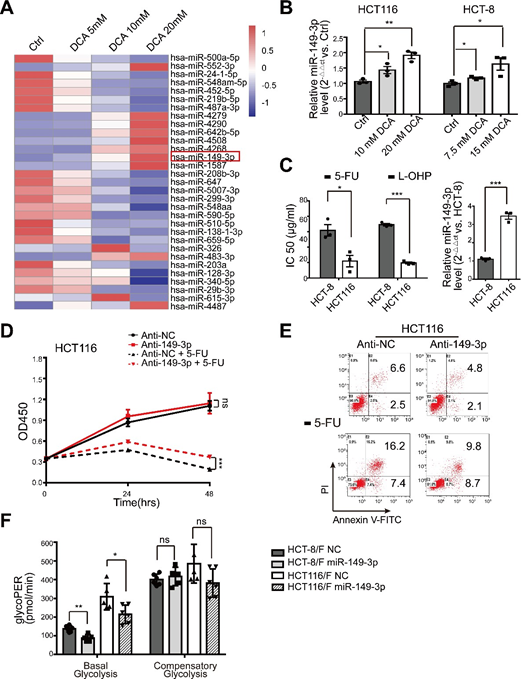
(A) La heatmap del profilo dei microRNA differenzialmente espressi nelle cellule HCT116 trattate con controllo, 5 mM, 10 mM, 20 mM DCA per 24 ore. (B) L’RNA totale è stato preparato a 24 ore dopo il trattamento con DCA 10/7,5 mM e 20/15 mM dalle cellule HCT116 e HCT-8. Il livello di miR-149-3p è stato analizzato mediante PCR quantitativa in tempo reale. (C) Le cellule HCT-8 e HCT116 sono state trattate con diverse concentrazioni di 5-FU e oxaliplatino (L-OHP) rispettivamente per 24 ore ed è stata calcolata la concentrazione inibitoria semimassimale (IC50) (pannello di sinistra). I livelli basali di miR-149-3p sono stati determinati mediante PCR quantitativa in tempo reale nelle cellule HCT-8 e HCT116 (pannello destro). (D-E) Le cellule HCT116 sono state trasfettate transitoriamente con Anti-NC o con l’inibitore del miR-149-3p. Dopo la trasfezione, le cellule sono state trattate con 25 μg/ml di 5-FU per 24 ore. La crescita cellulare e l’apoptosi sono state determinate rispettivamente mediante CCK8 e citometria a flusso. (F) La determinazione del tasso di glicolisi, compreso quello basale e quello compensatorio, in cellule CRC resistenti al 5-FU trasfettate con NC o con un miR-149-3p mimico è stata calcolata con il Seahorse Glycolytic Rate Assay Report Generator. I dati di tre esperimenti indipendenti sono mostrati come media ± SEM. Ogni esperimento è stato eseguito in 3-6 repliche biologiche. *p < 0,05; **, P < 0,01; ***, P < 0,001; ns, nessuna significatività.
IlDCA induce l’espressione del miR-149-3p attraverso la p53 wild-type (wt)
Considerando che diversi studi recenti hanno rivelato che il miR-149-3p è regolato da diversi farmaci [36,37], abbiamo poi determinato come il miR-149-3p sia regolato dal DCA. Abbiamo scoperto che il DCA aumenta significativamente l’espressione di p53 wt e dei suoi segnali a valle, compresa l’espressione di p21, PUMA e MDM2, nelle cellule CRC (Fig. 3a, b). Inoltre, abbiamo notato che le alterazioni dell’espressione di wt p53 erano in grado di modulare significativamente l’espressione di miR-149-3p, come mostrato in Fig. 3c, d, indicando che miR-149-3p era regolato positivamente da wt p53. Pertanto, utilizzando la linea cellulare HCT116 p53-null (TP53-/-), abbiamo riscontrato che il miR-149-3p non veniva upregolato dal trattamento con DCA, ma l’espressione ectopica di p53 invertiva questo effetto. Inoltre, abbiamo utilizzato nutlin-3, un potente inibitore che inibisce l’interazione MDM2-p53, portando all’attivazione di p53 come controllo positivo, mentre l’espressione di miR-149-3p è stata elevata nella linea cellulare HCT116 wt (TP53+/+) (Fig. 3e-g). Meccanicamente, sono stati previsti quattro siti di legame putativo di p53 alla regione del DNA genomico fiancheggiante il miR-149 utilizzando un software di analisi bioinformatica (IGV). Sono stati quindi eseguiti saggi ChIP nelle cellule utilizzando un anticorpo contro p53 wt. Il DNA pull-down è stato amplificato mediante PCR ordinaria con primer progettati in base a questi siti. I nostri risultati hanno mostrato che, rispetto alla regione 4, la regione 3 era marcatamente arricchita dopo il trattamento con DCA nella cromatina di HCT116 immunoprecipitata da p53 wt (Fig. 3h), suggerendo che solo la regione 3 contiene un sito di legame specifico attivato da DCA. Questi risultati indicano che il DCA modula il miR-149-3p attraverso wt p53.
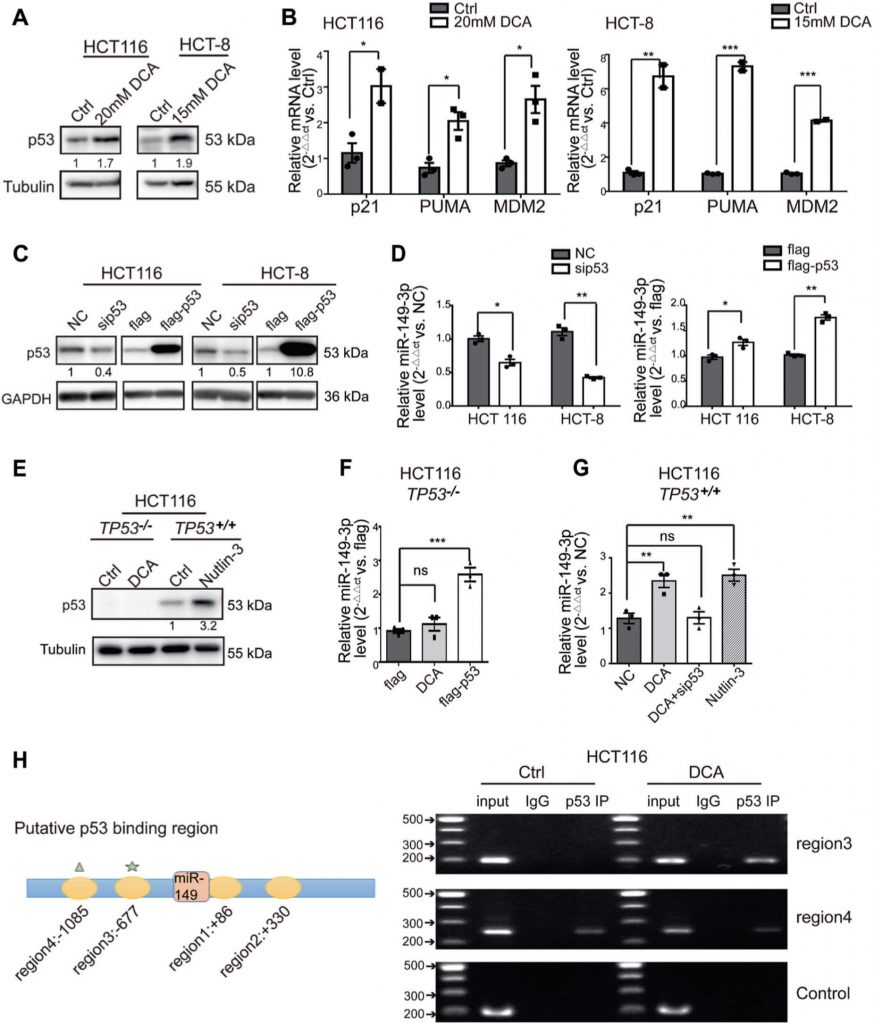
PDK2 è un bersaglio diretto del miR-149-3p
Per chiarire i meccanismi con cui il miR-149-3p regola la chemiosensibilità nelle cellule CRC, abbiamo analizzato i geni associati al metabolismo energetico, che sono regolati dal miR-149-3p, utilizzando due piattaforme pubbliche (TargetScan e miRDB). Infine, abbiamo identificato come potenziali candidati la piruvato deidrogenasi chinasi 2 (PDK2) e l’esochinasi 1 (HK1) (Fig. 4a). Per confermare questi risultati, il miR-149-3p mimico o inibitore è stato trasfettato in cellule CRC. Abbiamo scoperto che i livelli di mRNA di PDK2 erano regolati negativamente dal miR-149-3p, ma non da HK1 (Fig. 4b e Fig. 3A supplementare). Sono stati individuati due possibili siti di legame del miR-149-3p nel 3′-UTR di PDK2 e il saggio reporter a doppia luciferasi ha indicato che il miR-149-3p si lega al sito previsto (686-693) del 3′-UTR di PDK2 (Fig. 4c, d). Abbiamo poi confermato che i livelli della proteina PDK2 erano regolati negativamente dal miR-149-3p (Fig. 4e).
La PDK ha quattro isozimi denominati PDK1, 2, 3 e 4, tutti regolati dal DCA [26, 38, 39]. Abbiamo quindi analizzato l’espressione dell’mRNA di PDK1-4 nelle cellule HCT8 e HCT116 dopo il trattamento con DCA. Il DCA ha inibito significativamente l’espressione dell’mRNA di PDK2, ma non di altri isozimi PDK (Fig. S3B supplementare). Inoltre, la trasfezione di anti-149-3p ha parzialmente invertito la riduzione di PDK2 da parte di DCA (Fig. 4f). Successivamente, abbiamo determinato i livelli proteici di PDK2 e della sua subunità a valle piruvato deidrogenasi E1-alfa (PDHA1) in cellule CRC sensibili e resistenti al 5-FU. I livelli basali di PDK2 erano elevati nelle cellule CRC chemioresistenti, rispetto ai livelli delle cellule chemioresistenti. In linea con questo aumento, anche la fosforilazione di PDHA1 era elevata (Fig. 4g).
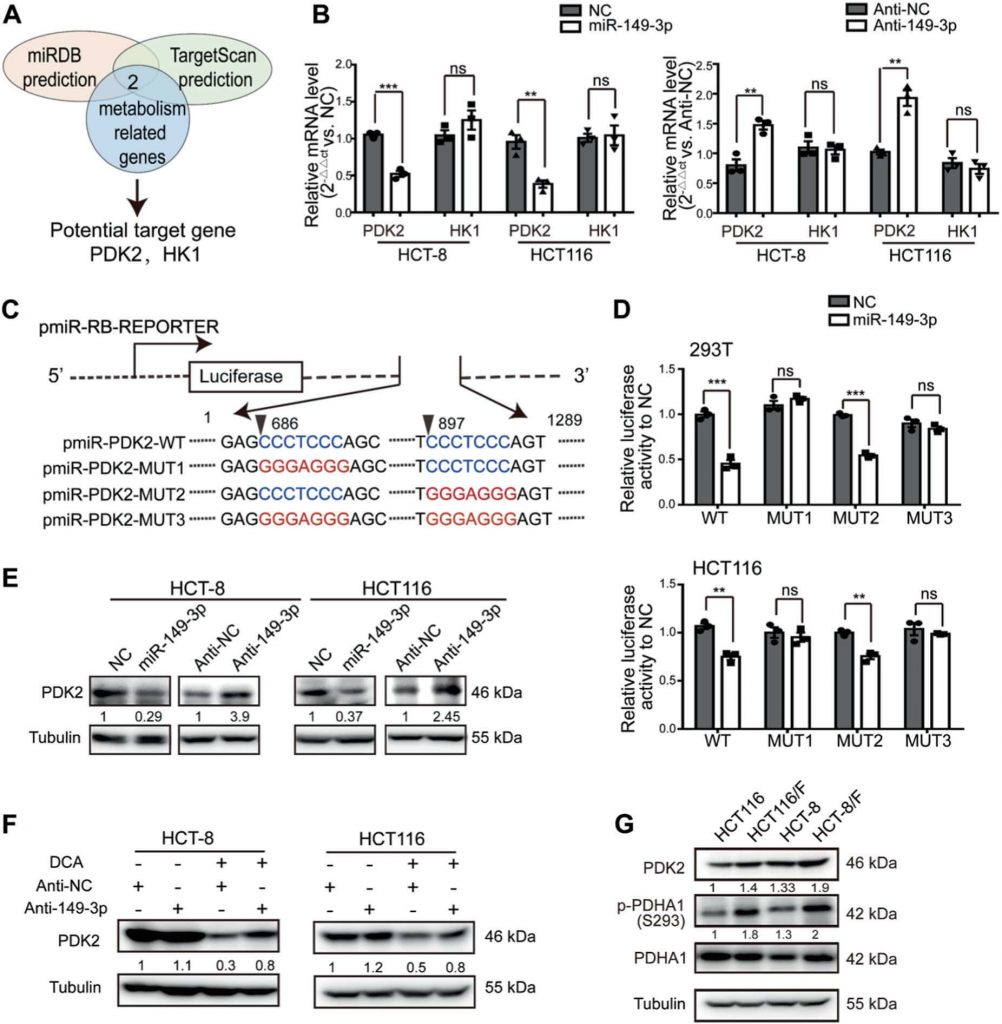
Il percorso miR-149-3p/PDK2 regola la chemiosensibilità
Per capire come il percorso miR-149-3p/PDK2 regoli la risposta delle cellule CRC al 5-FU, è stata selezionata la linea cellulare HCT-8/F come linea cellulare rappresentativa per studiare se i livelli di espressione di miR-149-3p e PDK2 influenzassero la risposta delle cellule al 5-FU. Il blocco di PDK2 nelle cellule HCT-8/F ha inibito la fosforilazione di PDHA1 (Fig. 5a) e ridotto i marcatori del metabolismo energetico, come il consumo di glucosio, la produzione di lattato e la glicolisi (Fig. 5b). Inoltre, le riduzioni di PDK2 sono state in grado di potenziare gli effetti del 5-FU sull’aumento dei livelli di PARP cleaved (c-PARP) e Bax, entrambi biomarcatori riconosciuti per l’apoptosi cellulare nelle cellule HCT-8/F (Fig. 5c). Inoltre, l’abbattimento di PDK2 ha aumentato la chemiosensibilità al 5-FU nelle cellule HCT-8/F, determinata mediante CCK8 e saggi di formazione di colonie (Fig. 5d, e). Il knockdown di PDK2 ha favorito l’apoptosi indotta dal 5-FU nelle cellule HCT-8/F e HCT116/F ed è mostrato nella Fig. S4A supplementare, mentre la sovraespressione di PDK2 ha favorito la fosforilazione di PDHA1 e mitigato l’apoptosi cellulare indotta dal 5-FU nelle cellule HCT-8 e HCT116 (Fig. S4B-S4C supplementare).
Inoltre, la sovraespressione di PDK2 ha invertito l’effetto inibitorio del miR-149-3p su PDK2 (Fig. 5f) e ha parzialmente abolito l’effetto inibitorio del miR-149-3p sul consumo di glucosio, sulla produzione di lattato e sulla glicolisi (Fig. 5g). L’espressione ectopica di PDK2 ha nettamente abolito l’effetto inibitorio del miR-149-3p sulla crescita cellulare e sulla formazione di colonie nelle cellule HCT-8/F trattate con 5-FU (Fig. 5h, i). Nel complesso, i nostri risultati indicano che il percorso miR-149-3p/PDK2 ripristina la chemiosensibilità indirizzando, almeno parzialmente, il metabolismo del glucosio nelle cellule CRC chemioresistenti.
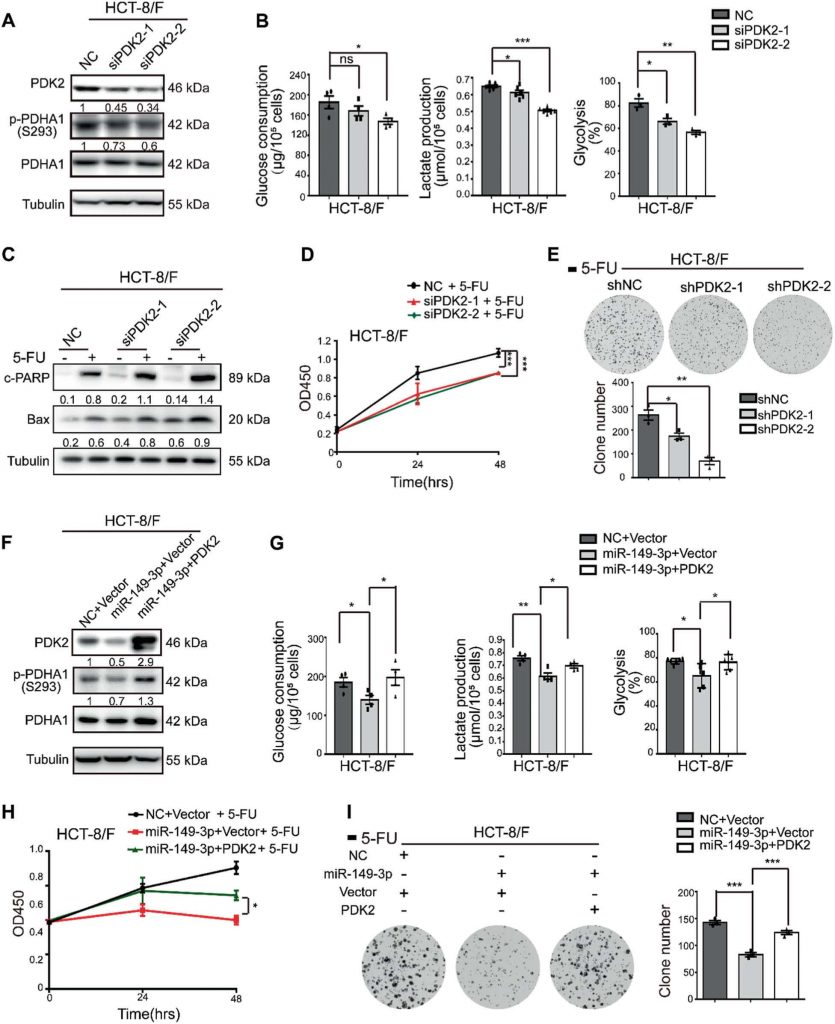
IlDCA aumenta la chemiosensibilità del 5-FU in vivo
Successivamente, 5-FU, DCA o una combinazione di 5-FU e DCA sono stati iniettati per via intraperitoneale nel modello di xenotrapianto sottocutaneo (indicato come gruppo intraperitoneale). Considerando l’insufficiente apporto di sangue nella parte centrale dei tumori sottocutanei, abbiamo anche iniettato per via intratumorale DCA o PBS più un’iniezione intraperitoneale di 5-FU (indicato come gruppo intratumorale). Le combinazioni di DCA e 5-FU hanno mostrato migliori effetti inibitori sulla crescita tumorale rispetto al solo DCA o 5-FU dopo quattro settimane di iniezione nel gruppo intraperitoneale (Fig. 6a, b). L’espressione di Ki-67 si è ridotta dopo il trattamento combinato nel gruppo intraperitoneale (Fig. 6c). Il DCA ha inibito significativamente la crescita tumorale nel gruppo intratumorale rispetto a quella del gruppo PBS (Fig. 6d) e il miR-149-3p è stato upregolato nel gruppo di iniezione intratumorale di DCA, il che è coerente con i risultati in vitro (Fig. 6e). L’iniezione intratumorale di DCA ha anche promosso l’apoptosi tumorale (Fig. 6F).
Per valutare ulteriormente l’effetto del miR-149-3p sulla risposta delle cellule CRC al 5-FU in vivo, il modello di xenotrapianto sottocutaneo è stato sottoposto a iniezione intratumorale di agomiR-149-3p o agomiR-NC più un’iniezione intraperitoneale di 5-FU. Il miR-149-3p ha inibito significativamente la crescita tumorale nel gruppo intratumorale rispetto a quello del gruppo di controllo in cui è stato iniettato per via intratumorale il controllo agomiR-negativo (NC) (Fig. 6g, h). La sovraespressione del miR-149-3p è stata convalidata mediante PCR quantitativa in tempo reale (Fig. 6i) e la sovraespressione del miR-149-3p ha promosso l’apoptosi (Fig. 6j) e ridotto l’espressione di PDK2 e p-PDHA1 (Fig. 6k, l). La tomografia a emissione di micropositroni (PET) con 18F-fluorodesiglucosio (FDG) è stata eseguita dopo 3 settimane di trattamento. la captazione di 18F-FDG è stata osservata nei siti di impianto del tumore e sono stati misurati la captazione massima standardizzata (SUVmax) e i volumi tumorali metabolici (MTV). Non è stata osservata alcuna differenza nel SUVmax tra i due gruppi, mentre il MTV è risultato significativamente ridotto nel gruppo miR-149-3p (Fig. 6m).
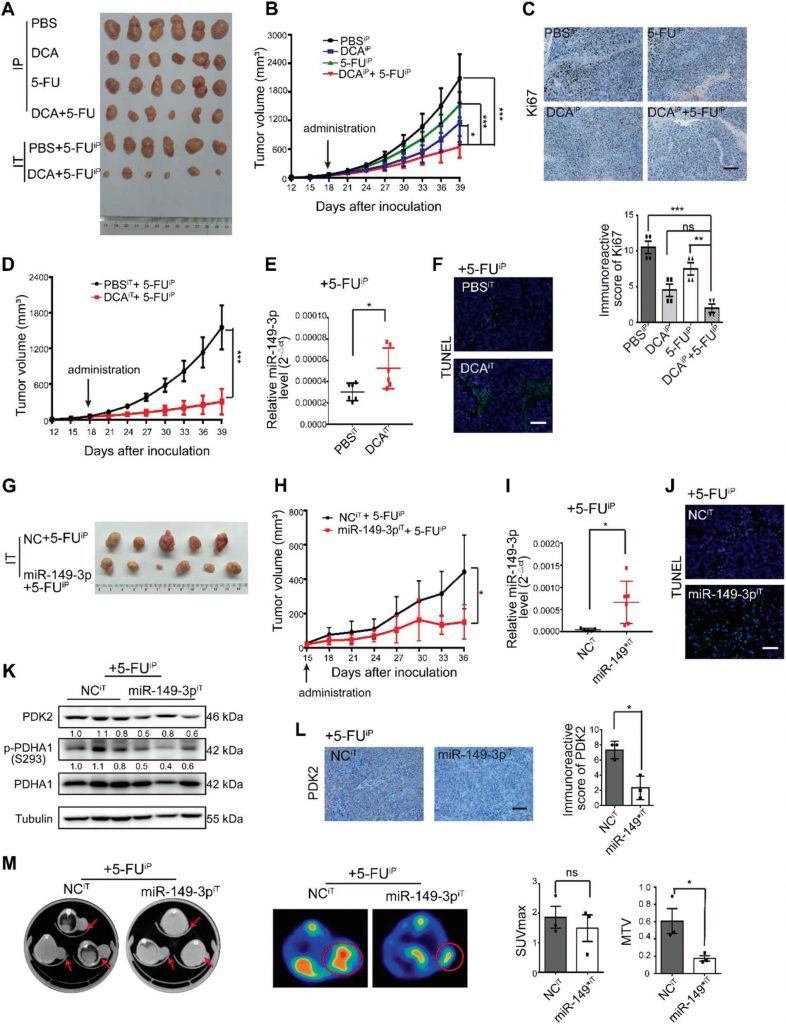
ilmiR-149-3p è inversamente correlato con PDK2 nei pazienti affetti da CRC
Nei tessuti umani di CRC è stata osservata una significativa correlazione inversa tra i livelli di mRNA del miR-149-3p e di PDK2 (Fig. 7a, b). Tra questi, otto pazienti in condizioni di malattia stabile (SD) entro 3 anni dalla chemioterapia hanno espresso un livello più elevato di miR-149-3p rispetto a cinque pazienti con malattia progressiva (PD) (Fig. S5A supplementare). Sono state analizzate cinque coppie di pazienti con PD e SD con la stessa patologia e lo stesso stadio TNM e la colorazione di PDK2 variava significativamente nei campioni dei pazienti con PD e SD (Fig. S5B supplementare). I pazienti CRC del database TCGA con elevata espressione di PDK2 erano anche caratterizzati da una peggiore sopravvivenza globale (OS) (Fig. 7c). Il database TCGA ha anche mostrato che l’espressione di PDK2 nel gruppo wt p53 era ridotta rispetto a quella del gruppo mutante p53 (Fig. 7d). Questi risultati suggeriscono che PDK2 è regolato negativamente dal miR-149-3p nei pazienti con CRC.
Discussione
Il CRC è caratterizzato da anomalie tumorali e alterazioni delle vie metaboliche ed è una delle principali cause di morte per cancro [2]. La resistenza alla chemioterapia è la causa principale del fallimento del trattamento [7]. In questo studio abbiamo scoperto che il DCA può aumentare l’effetto chemioterapico del 5-FU nelle cellule CRC chemioresistenti e che l’attivazione della via p53/miR-149-3p/PDK2 è in grado di aumentare la chemiosensibilità in vitro e in vivo.
Sempre più evidenze indicano che l’aumento della glicolisi è strettamente correlato alla resistenza alla chemioterapia [15, 17, 40]. In questo caso, abbiamo anche scoperto che, rispetto alle linee cellulari parentali, le cellule CRC chemioresistenti hanno mostrato un elevato consumo di glucosio, produzione di lattato e glicolisi, suggerendo che le anomalie metaboliche sono una caratteristica tipica, ma i meccanismi molecolari rimangono ancora poco chiari nelle cellule CRC.
È stato osservato che il DCA diminuisce i livelli di lattato nel sangue in vivo nei roditori a dosaggi di ~25-50 mg/kg/24 h [41] e che il DCA viene impiegato con un ampio intervallo di dosaggio da 1 a 50 mM [26]. In particolare, gli effetti avversi del DCA nell’uomo sono generalmente limitati alla neuropatia periferica sensoriale e motoria reversibile, che è influenzata dall’età e dal genotipo [42]. Recentemente, il DCA è stato identificato come una nuova terapia metabolica per diversi pazienti oncologici [26, 29].
È stato riportato che il DCA è in grado di inibire l’attività della PDK e di convertire il piruvato in acetil-CoA, determinando uno spostamento della produzione di energia dalla glicolisi alla fosforilazione ossidativa mitocondriale [43,44]. È stato inoltre dimostrato che il DCA attenua la resistenza al 5-FU indotta dall’ipossia nel carcinoma gastrico [45], supera la resistenza al sorafenib nel carcinoma epatocellulare [46] e attenua la resistenza al cisplatino nel carcinoma della testa e del collo [47]. Il nostro studio ha rivelato che il DCA è in grado di attenuare la chemioresistenza delle cellule CRC al 5-FU. Inoltre, abbiamo dimostrato che il DCA ha ridotto il consumo di glucosio e la produzione di lattato nelle cellule CRC chemioresistenti al livello basale delle cellule CRC chemioresistenti. Poiché l’autofagia ha il potenziale di alimentare il metabolismo del cancro [48], è stato determinato l’effetto del DCA e del miR-149-3p sull’autofagia. Abbiamo scoperto che il DCA attiva l’autofagia e il miR-149-3p non ha alcuna influenza sull’autofagia. Ciò suggerisce che l’autofagia non è coinvolta nell’effetto di DCA/miR-149-3p nella regolazione del metabolismo del glucosio (Figura S6 supplementare).
Le PDK, in quanto regolatori chiave della glicolisi del cancro, hanno suscitato grande preoccupazione a causa dei risultati di molti studi [49]. Esistono quattro isoforme di PDK (PDK1-4), ognuna delle quali si manifesta in modo specifico per ogni tessuto: PDK1 è altamente espressa nel cuore, PDK2 è espressa in modo ubiquitario, PDK3 ha una distribuzione tissutale relativamente limitata e PDK4 è espressa nel cuore e nel muscolo scheletrico [27]. La PDK2 è espressa a livelli più elevati rispetto agli altri isoenzimi, il che suggerisce che potrebbe essere la principale isoforma responsabile della regolazione dell’attività enzimatica del complesso piruvato deidrogenasi (PDHC) [50]. Inoltre, gli isoenzimi PDK differiscono nella loro regolazione acuta da parte dei metaboliti [51]. Qui ci siamo concentrati sulla PDK2, l’isoforma più sensibile al DCA [31]. Sebbene siano state messe a fuoco le interazioni molecolari tra DCA e PDK, il potenziale meccanismo della regolazione trascrizionale di PDK rimane poco chiaro. Uno studio recente ha riportato che il miR-182 svolge un ruolo regolatorio nelle vie metaboliche del cancro al polmone prendendo di mira la PDK4 [52]. Il presente studio ha dimostrato che PDK2 è regolata dal miR-149-3p nel CRC e che i livelli di PDK2 nei tumori primari dei pazienti CRC erano inversamente correlati all’espressione del miR-149-3p. Dal momento che PDK2 è ampiamente presente nella maggior parte dei tessuti, puntare su PDK2 potrebbe essere un modo più importante ed efficiente per uccidere le cellule tumorali e superare la chemioresistenza.
È stato riportato che il miR-149-3p svolge un ruolo vitale in vari tipi di cancro ed è indotto da alcuni farmaci antitumorali [36, 37, 53]. In particolare, abbiamo scoperto che il trattamento con DCA può indurre il legame di p53 alla regione a monte (da -677 a -477) del miR-149 e che il miR-149-3p è stato upregolato dal trattamento con DCA in modo p53-dipendente. TP53, un classico soppressore tumorale, è spesso inattivato nei tumori [54] ed è stato recentemente segnalato per regolare il metabolismo del glucosio nel cancro. È stato dimostrato che la p53 Wt è in grado di inibire l'”effetto Warburg” controllando PDK2. Tuttavia, la frequenza di mutazioni di TP53 nel CRC è di circa il 40-50% [55, 56], con conseguente perdita della sua funzione soppressiva. È stato riportato che i pazienti CRC con TP53 wt ottengono un beneficio in termini di sopravvivenza dalla chemioterapia a base di 5-FU, ma quelli con TP53 mutante no [57]. I nostri risultati rivelano un nuovo meccanismo tra p53 e PDK2, modulato dal miR-149-3p. Questi risultati suggeriscono che i pazienti con TP53 mutante possono trarre maggiore beneficio dalla chemioterapia in aggiunta con miR-149-3p piuttosto che con DCA. Considerando l’alta frequenza di mutazioni di TP53 nel CRC, riteniamo che il miR-149-3p svolga un ruolo fondamentale nel monitoraggio e nella modulazione della chemiosensibilità nel CRC.
Le cellule tumorali consumano una grande quantità di glucosio e mostrano un elevato stato di glicolisi aerobica, pertanto la riduzione dell’assorbimento di glucosio è una strategia promettente per limitare la crescita del cancro [58]. Abbiamo osservato che l’aumento del miR-149-3p ha notevolmente inibito la glicolisi nelle cellule CRC chemioresistenti; tuttavia, rispetto al gruppo di controllo, il gruppo di xenotrapianti a cui è stato iniettato per via intratumorale il miR-149-3p mimic ha presentato una SUVmax invariata. Forse questo risultato contraddittorio potrebbe essere dovuto allo sviluppo di necrosi nella parte centrale dei tessuti tumorali sottocutanei, che merita di essere indagato.
Nel complesso, abbiamo scoperto che la via di segnalazione p53/miR-149-3p/PDK2 può essere potenzialmente bersagliata per superare il CRC chemioresistente al trattamento con DCA, fornendo una potenziale strategia per il trattamento del CRC dal punto di vista dell’intervento sul metabolismo tumorale (Fig. 7e).
Materiali e metodi
Tessuto tumorale
Tra il 2013 e il 2016 sono stati inclusi 28 pazienti affetti da CRC provenienti dal Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. Tredici di questi pazienti hanno ricevuto una chemioterapia post-operatoria a base di 5-FU e sono stati seguiti per almeno 3 anni. Tutti i tessuti sono stati raccolti dopo aver ottenuto il consenso informato e tutte le procedure che coinvolgevano pazienti umani sono state condotte in conformità con le norme stabilite dal Comitato Etico del Nono Ospedale del Popolo Affiliato al Medical College della Shanghai Jiao Tong University. Le informazioni cliniche dei pazienti affetti da CRC sono riportate nella Tabella 1 supplementare.
| Caratteristiche dei pazienti con CRC | |
| Caratteristiche | Totale (n=28) |
| Età-anno | 65.4±10.5 |
| Sesso-no. (%) | |
| Maschio | 17(60.7) |
| Femmina | 11(39.3) |
| Localizzazione-no. (%) | |
| Retto | 10(35.7) |
| Colon | 18(64.3) |
| Staging – n. (%) | |
| T1 o T2 | 6(21.4) |
| T3 o T4 | 22(78.6) |
| Linfonodi coinvolti – n. (%) | |
| N0 | 12(42.9) |
| N1 o N2 | 16(57.1) |
| Metastasi – no (%) | |
| M0 | 27(96.4) |
| M1 | 1(3.6) |
| Chemioterapia postoperatoria con un regime a base di fluorouracile | |
| Sì | 21(75) |
| Follow-up per 3 anni | |
| Sì | 13(61.9) |
| No | 8(38.1) |
| No | 7(25) |
Coltura cellulare
La linea cellulare HCT-8/F resistente al 5-FU e la sua linea cellulare parentale HCT-8 sono state acquistate da iCell Bioscience, Inc. (Shanghai, Cina). La linea cellulare HCT116/F resistente al 5-FU e la sua linea cellulare parentale HCT116 sono state gentilmente fornite dal dottor Gu (Yanhong Gu, Nanjing Medical University, Jiangsu, Cina). Le cellule HCT116-/- sono state regalate dal Dr. Lu (Hua Lu, Fudan University, Shanghai, Cina). La linea cellulare 293T di rene embrionale umano è stata ottenuta dall’American Type Culture Collection (ATCC, Manassas, VA, USA). Le linee cellulari 293T, HCT116 e HCT-8 sono state coltivate in terreno Dulbecco’s Modified Eagle’s medium (HyClone, Utah, USA) o in terreno RPMI-1640 (HyClone, Utah, USA) contenente 10% di siero fetale bovino (Gemini, California, USA), 100 U/mL di penicillina e 100 μg/mL di streptomicina (HyClone, Utah, USA) in un incubatore umidificato a 37 °C e 5% diCO2. I terreni di coltura delle linee cellulari HCT-8/F e HCT116/F sono stati integrati rispettivamente con 15 μg/ml di 5-FU e 5 μg/ml di 5-FU. Tutte le linee cellulari sono state autenticate mediante sequenziamento di short tandem repeats da Genetic Testing Biotechnology Corporation (Suzhou, Jiangsu, Cina). Il DCA è stato acquistato da Sigma-Aldrich Co. Ltd. lTD. (MO, USA).
Colorazione in immunofluorescenza di Edu e ROS
Le cellule HCT-8/F e HCT116/F sono state seminate in piastre da 96 pozzetti a 15.000 cellule/pozzetto. Dopo l’incubazione notturna, le cellule sono state trattate con DCA 15 mM e 20 mM, rispettivamente, per 24 ore. La colorazione di Edu è stata eseguita secondo le istruzioni del produttore (Ribobio, Guangzhou, Cina). I livelli di ROS sono stati misurati in cellule incubate con 10 μM di 2′,7′-diclorofluoresceina diacetato (DCF-DA) (Beyotime, Shanghai, Cina) per 30 min a 37 °C. Le piastre sono state poi lavate due volte e le cellule sono state analizzate con un microscopio a fluorescenza.
Crescita cellulare
Le cellule sono state seminate in piastre da 96 pozzetti a 5000 cellule/pozzetto per una notte, trattate con i farmaci per 24 ore e successivamente ogni pozzetto è stato sostituito con miscele di 10 μl di CCK8 (Dojindo, Giappone) e 90 μl di terreno di coltura. L’assorbanza è stata misurata a un valore OD di 450 nm utilizzando un lettore di micropiastre per enzimi (BioTeck, Vermont, USA) due ore dopo. Il rapporto di inibizione del farmaco è stato calcolato con la seguente formula: 1-ODdrug/ODctrl. L’IC50 di ciascuna cellula è stato calcolato con GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Saggio di apoptosi cellulare
Le cellule HCT-8/F e HCT116/F sono state seminate in piastre a sei pozzetti a una concentrazione di 2 ×105 cellule/pozzetto. Le cellule sono state trattate con DCA (15 mM) / 5-FU (50 μg/ml) e DCA (20 mM) / 5-FU (25 μg/ml), rispettivamente, per 48 ore. Le cellule sono state poi tripsinizzate, lavate e colorate con anticorpi Annexin V-FITC/PI o Annexin V-PE/7-AAD secondo il protocollo del produttore (BD, CA, USA). L’apoptosi è stata misurata mediante citometria a flusso (BD, CA, USA).
Saggio di formazione di colonie
Le cellule HCT-8/F e HCT116/F sono state trattate rispettivamente con DCA (15 mM)/5-FU (50 μg/ml) e DCA (20 mM)/5-FU (25 μg/ml) per 24 ore. Quindi, le cellule sono state seminate in piastre a sei pozzetti a 1000 cellule per pozzetto e coltivate in terreno fresco a 37 °C per 1 o 2 settimane, seguite da fissazione con paraformaldeide al 4% per 30 minuti; le cellule sono state quindi colorate con cristalvioletto all’1% e il numero di colonie cellulari è stato contato con un contatore (Gelcount, Optronix, Oxford).
Trasfezione genica transitoria
I miR-149-3p mimici, gli inibitori, siPDK2, sip53 e le corrispondenti sequenze di oligonucleotidi NC sono stati sintetizzati da GenePharma (Shanghai, Cina). Il plasmide flag-p53 è stato gentilmente fornito dal Dr. Lu (Hua Lu, Fudan University, Shanghai, Cina). La trasfezione è stata eseguita con Lipofectamina 3000 (Invitrogen, CA, USA) a una concentrazione finale di 50 nmol/L (mimici e siRNA) o 100 nmol/L (inibitori). Le cellule sono state raccolte per i saggi 24 o 48 ore dopo la trasfezione. Le sequenze di siRNA, mimici e inibitori sono riportate nella Tabella 2 supplementare.
| sequenza di siRNA, mimici e inibitori | |
| siPDK2-1 | senso: 5′-GACCGAUGCUGUCAUCUAU-3′ |
| antisenso: 5′-AAUAGAUGACAGCAUCGGUC-3′ | |
| siPDK2-2 | senso: 5′-GACUCUAGCUACAUGUA-3′ |
| antisenso: 5′-UACAUGUAGCUGAAGAGUC-3′ | |
| NC | senso: 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| antisenso: 5′-ACGUGACGUUCGGAGAATT-3′ | |
| mimico-149-3p | senso: 5′-AGGGAGGGACGGGGCUGUGC-3′ |
| antisenso: 5′-ACAGCCCCCGUCCCUCCCUUU-3′ | |
| inibitore-NC | 5′-CAGUACUUUGUGUAGUACAA-3′ |
| inibitore-149-3p | 5′-GCACAGCCCCCGUCCCUCCCU-3′ |
| sip53 | senso: 5′-GUAAUCUACUGGACGGAAtt-3′ |
| antisenso: 5′-UUCCGUCCCAGUAGAUUACca-3′ |
Trasfezione genica stabile
LV-PDK2 e il corrispondente virus NC sono stati acquistati da GeneChem (Shanghai, Cina). shPDK2-1, shPDK2-2 e il plasmide di controllo sono stati acquistati da GeneChem (Shanghai, Cina). Il surnatante del virus è stato raccolto da cellule T 293. Successivamente, le cellule CRC sono state infettate con il virus e sottoposte a screening con puromicina. L’efficienza dell’infezione è stata convalidata mediante citometria a flusso e microscopia a fluorescenza. L’espressione mRNA e proteica di PDK2 è stata ulteriormente analizzata mediante PCR quantitativa in tempo reale e Western blot.
saggi di luciferasi reporter 3′UTR
Le sequenze di legame del miR-149-3p wt o mutante nel 3′UTR di PDK2 umana sono state clonate a valle della luciferasi pmiR-RB-Reporter (Ribobio, Guangzhou, Cina), riferite a WT, MUT1, MUT2 e MUT3 nella Fig. 3c. le cellule T 293 e le cellule HCT116 sono state seminate in piastre da 24 pozzetti, seguite dalla cotrasfezione con 500 ng di costrutti reporter e 50 nmol/L di miR-149-3p mimico o un NC utilizzando Lipofectamina 3000 (Thermo Fisher Scientific, Waltham MA). L’attività della luciferasi è stata misurata dopo 48 ore di incubazione utilizzando il Dual-Luciferase Reporter Assay System (Promega, Madison, USA) secondo il protocollo del produttore.
Consumo di glucosio e produzione di lattato
Le cellule HCT-8/F e HCT116/F sono state seminate in piastre da 24 pozzetti a 1 ×105 cellule/pozzetto per una notte e poi trattate con DCA 15 mM e 20 mM, rispettivamente, per 24 ore. Dopo il trattamento, le cellule sono state coltivate in terreno privo di rosso fenolo contenente il 10% di siero fetale bovino per 24 ore. Il terreno di coltura è stato raccolto e sono stati misurati il consumo di glucosio e la produzione di lattato. Il lattato è stato misurato con un Lactate Assay Kit (Njjcbio, Nanjin, Cina) e il glucosio con un Glucose Assay Kit (Rsbio, Shanghai, Cina). Tutti i valori sono stati standardizzati contando un numero uguale di cellule. Per valutare lo stato di glicolisi, 100 ng/mL di oligomicina (un inibitore dell’ATP sintasi; Sangon Biotech) sono stati aggiunti alle cellule coltivate per 6 ore. Il rapporto tra la concentrazione di lattato in presenza e in assenza di oligomicina è stato misurato e determinato come descritto in precedenza [46].
Saggio del tasso glicolitico Seahorse XF-96
Le cellule sono state seminate in una piastra di coltura a 96 pozzetti a una densità di 25.000 cellule/pozzetto e sono state incubate per una notte in terreno di crescita contenente il 10% di siero fetale bovino. La cartuccia del sensore è stata idratata per una notte. Il giorno successivo, il terreno di coltura è stato cambiato con un terreno di saggio a basso contenuto di bicarbonato, integrato con glucosio, piruvato di sodio e glutammina. Dopo l’incubazione delle cellule per 1 ora a 37 °C in un incubatore privo di CO2, il tasso di consumo di ossigeno e il tasso di acidificazione extracellulare sono stati misurati prima e dopo l’iniezione di DCA/ctrl, Rotenone (Rot) + antimicina A (AA) e 2-deossi-d-glucosio (2-DG) utilizzando lo strumento Seahorse XF (Agilent, Santa Clara, CA) come precedentemente descritto [59, 60]. Gli esperimenti sono stati eseguiti in tempo reale in cinque o sei pozzetti replicati. I glicogrammi, tra cui il glicogrammo basale, il glicogrammo indotto e il glicogrammo compensatorio, sono stati calcolati automaticamente dal software Wave (Agilent, Santa Clara, CA).
PCR quantitativa in tempo reale e microarray di miRNA
L’RNA totale è stato estratto da tessuti o cellule CRC con il reagente TRIzol (Life, CA, USA). Il cDNA è stato sintetizzato con il kit PrimeScript RT Reagent (TaKaRa, Tokyo, Giappone). Il saggio microarray è stato eseguito con tre repliche di cellule HCT116 trattate con DCA 5 mM, 10 mM o 20 mM per 12, 24 o 48 ore. I dati originali sono stati caricati nel database GEO (GSE125309). La PCR quantitativa in tempo reale è stata eseguita utilizzando la premiscela Ex Taq 420 A (TaKaRa, Tokyo, Giappone) sulla piattaforma ABI-7500. Actina e U6 sono stati utilizzati come controlli interni. Le sequenze dei primer sono riportate nella Tabella 3 supplementare.
| sequenze dei primer per la PCR quantitativa in tempo reale a trascrizione inversa | |
| Actina | F: 5′-CTCCATCCTGGCCTCGCTGT-3′ |
| R: 5′-GCTGTCACTTCACCGTTCC-3′ | |
| PDK2 | F: 5′-CGCTGGCTCTTTGGTTATG-3′ |
| R: 5′-ACAGGGCCTTGAGATAGATG-3′ | |
| PDK1 | F: 5′-GCTGTATGGCCTGCAAGATG-3′ |
| R: 5′-GCTGTCCTGGTGATTTTGCA-3′ | |
| PDK3 | F: 5′-GGTTTGCCAATTTCCCGTCTG-3′ |
| R: 5′-CATCGGCTTCAGGCGTGGTC-3′ | |
| PDK4 | F: 5′-GAGAATTGACCGCCTCT-3′ |
| R: 5′-CGAGAAATTGGCAAGCCGTAA-3′ | |
| HK1 | F: 5′-CTTACTAAGGGATGCGATAAA-3′ |
| R: 5′-TCCCAACAATGAGTCCAACC-3′ | |
| TP53 | F: 5′-CCCAAGCAATGGATGATTTGA-3′ |
| R: 5′-GGCATTCTGGGAGCTTCATCT-3′ | |
| p21 | F: 5′-CTGGACTGTTCTCTCGGCTC-3′ |
| R: 5′-TGTATATTCAGCATTGTGGGAGGA-3′ | |
| MDM2 | F: 5′-ATGAATCCCCCTTCCAT-3′ |
| R: 5′-CAGGAAGCCAATTCTCACGAA-3′ | |
| PUMA | F: 5′-ACAGTACGAGCGGCGGAGACAA-3′ |
| R: 5′-GGCGGGTGCAGGCACCTAATT-3′ | |
| miR-149-3p | RT: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGG ATACGACGCACAG-3′ |
| F: 5′-ACAGGGAGGGACGGGGG-3′ | |
| R: 5′-ATCCAGTGCAGGGTCCGAGG-3′ | |
| miR-128-3p | RT: 5′-TCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGA TACGACAAAGAG-3′ |
| F: 5′-CGCGTCACAGTGAACCGGT-3′ | |
| R: 5′-AGTGCAGGGTCCGAGGTATT-3′ | |
| miR-500a-5p | RT: 5′-GTCGTATCCAGTGCGTCGTGGAGTCGGCAATTGCAC TGGATACGACTCTCACC-3′ |
Western blots
Sono stati caricati trenta microgrammi di lisati proteici totali e sono stati applicati gli anticorpi primari: anti-PDK2 (sc-100534, Santa Cruz, California, USA), anti-p-PDHA1 (S293) (ABS204, Merck, Darmstadt, Germania), anti-PDHA1 (ab168379, Abcam, Cambridge, UK), anti-p53 (sc-126, Santa Cruz, California, USA), anti-c-PARP (D64E10, CST, Massachusetts, USA), anti-Bax (D2E11, CST, Massachusetts, USA), anti-LC3B (L7543, sigma, MO, USA), anti-GAPDH (Proteintech, Wuhan, Cina) e anti-α-tubulina (Proteintech, Wuhan, Cina). Gli anticorpi secondari sono stati acquistati da Sungene (Tianjin, Cina). I blot sono stati analizzati con un sistema di imaging a chemiluminescenza (Bioshine, Shanghai, Cina).
Immunoprecipitazione della cromatina (ChIP)
Le cellule HCT116 seminate in piastre da 10 cm sono state trattate con o senza 20 mM DCA per 24 ore, quindi sono state fissate e frammentate secondo le istruzioni del produttore (Pierce Agarose ChIP Kit, Thermo). La cromatina è stata incubata con anticorpi IgG e anti-p53 (Sigma, MO, USA) a 4°Covernight. Dopo l’incubazione, sono stati aggiunti 60 ul di agarosio proteico A/spermatozoi di salmone. Quindi, il complesso precipitato è stato lavato con i tamponi di lavaggio IP 1, 2, 3 ed eluito con il tampone di eluizione. La reticolazione è stata invertita aggiungendo 6 μl di NaCl 5 M e 2 ul di proteinasi K a 65°C per 1,5 h. Il DNA immunoprecipitato e il DNA dell’estratto di cellule intere (input) sono stati purificati e quindi utilizzati per le analisi PCR utilizzando i primer corrispondenti. Per il monitoraggio dell’esperimento è stato utilizzato un primer di controllo. Le sequenze dei primer per la PCR sono presentate nella Tabella 3 supplementare.
Xenotrapianto tumorale sottocutaneo in topi nudi e imaging micro-PET/CT
In primo luogo, 1 ×107 cellule HCT-8/F diluite in 100 μl di PBS sono state impiantate sottocute in topi nudi (maschi, 6 settimane). I topi sono stati divisi a caso in sei gruppi (sei per gruppo) dopo 12 giorni. I topi dal gruppo I al gruppo IV hanno ricevuto un’iniezione intraperitoneale giornaliera di PBS, DCA (50 mg/kg)/PBS, 5-FU (10 mg/kg)/PBS e 5-FU (10 mg/kg)/DCA (50 mg/kg), rispettivamente. I topi del gruppo V e del gruppo VI sono stati sottoposti a iniezioni intratumorali di PBS o DCA (50 mg/kg), rispettivamente, a giorni alterni e a un’iniezione intraperitoneale di 5-FU (10 mg/kg) a giorni alterni. Il volume del tumore è stato misurato in cieco ogni 3 giorni. I topi sono stati sacrificati dopo 3 settimane di trattamento e i tumori sono stati sezionati, pesati e congelati a -80 °C per ulteriori studi.
Per valutare se il miR-149-3p esercita un effetto di chemio-sensibilizzazione, sono stati creati altri due gruppi di modelli animali. In breve, i topi sono stati impiantati per via sottocutanea con 6 ×106 cellule HCT-8/F. Dopo che le dimensioni del tumore erano di circa 50 mm3, i topi hanno ricevuto una dose intraperitoneale di 5-FU (10 mg/kg) a giorni alterni e un’iniezione intratumorale di 5 nmol di miR-149-3p mimici coniugati con colesterolo o di un NC ogni 3 giorni per 3 settimane. Successivamente, tre topi di ciascun gruppo sono stati digiunati per una notte e sono stati iniettati per via endovenosa con 0,15 mCi di 18F-FDG. la scansione 18F-FDG micro-PET-CT (Siemens, Berlino, Germania) è stata eseguita dopo 60 minuti. Le immagini dell’acquisizione PET sono state mostrate utilizzando una mappa pseudocolore con il colore rosso che indica un’elevata captazione di 18F-FDG. Per determinare l’attività della 18F-FDG-PET sono stati utilizzati SUVmax e MTV. Tutti gli esperimenti e la cura degli animali sono stati approvati dal Comitato etico del Ninth People’s Hospital Affiliated to the Medical College of Shanghai Jiao Tong University.
Colorazione immunoistochimica e in immunofluorescenza
In breve, le sezioni di tessuto sono state incubate con gli anticorpi primari Ki67 (Servicebio, Wuhan, Cina) e PDK2 (Proteintech, Wuhan, Cina) a 4 °C per una notte e poi incubate con l’anticorpo secondario. La reazione cromogenica è stata eseguita con 3,3-diaminobenzidina e controcolorata con ematossilina. Il punteggio immunoreattivo (IRS) è stato calcolato da due ricercatori in cieco rispetto all’assegnazione del gruppo. IRS = SI (intensità di colorazione) × PP (percentuale di cellule positive). Il punteggio è stato assegnato come segue: 0 × negativo; 1 × debole; 2 × moderato; 3 × forte. PP è stato definito come 0 × 0%; 1 × 0-25%; 2 × 25-50%; 3 × 50-75%; 4 × 75-100%. Le sezioni congelate di sei millimetri sono state colorate con un kit di reazione TUNEL (Roche, Basilea, Svizzera) e controcolorate con DAPI. Le immagini sono state acquisite utilizzando un microscopio a fluorescenza con filtri di eccitazione ed emissione appropriati.
Analisi statistica
I dati sono stati analizzati mediante il software GraphPad Prism 6.0. I dati sono presentati come medie ± SD/SEM di tre esperimenti indipendenti. Ogni esperimento è stato eseguito in almeno tre repliche. Per confrontare le differenze tra i due gruppi è stato utilizzato il test t di Student a due code. Per i confronti multipli è stata utilizzata l’ANOVA a una via seguita dal test post-hoc di Bonferroni. Le curve di Kaplan-Meier per le analisi di sopravvivenza sono state determinate utilizzando il test log-rank. La relazione tra miR-149-3p e PDK2 è stata valutata utilizzando l’analisi del coefficiente di correlazione di rango di Spearman. Un valore P <0,05 è stato considerato statisticamente significativo.
Ringraziamenti
Questo lavoro è stato sostenuto dalla National Natural Science Foundation of China (81272745, 81872419 e 81272404) e dal Program for Professor of Special Appointment (Eastern Scholar to JW) delle Shanghai Institutions of Higher Learning. Si ringrazia il dottor Yanhong Gu per aver fornito le linee cellulari HCT116 resistenti al 5-FU e il dottor Hua Lu per aver fornito il plasmide p53.
Conformità alle norme etiche
Conflitto di interessi
Gli autori dichiarano di non avere alcun conflitto di interessi.
Nota dell’editore
Springer Nature rimane neutrale rispetto alle rivendicazioni giurisdizionali nelle mappe pubblicate e alle affiliazioni istituzionali.
Open Access
Questo articolo è concesso in licenza Creative Commons Attribution 4.0 International License, che ne permette l’uso, la condivisione, l’adattamento, la distribuzione e la riproduzione in qualsiasi mezzo o formato, a condizione che si dia adeguato credito all’autore o agli autori originali e alla fonte, si fornisca un link alla licenza Creative Commons e si indichi se sono state apportate modifiche. Le immagini o altro materiale di terze parti presenti in questo articolo sono incluse nella licenza Creative Commons dell’articolo, a meno che non sia indicato diversamente in una riga di credito del materiale. Se il materiale non è incluso nella licenza Creative Commons dell’articolo e l’uso che se ne intende fare non è consentito dalle norme di legge o eccede l’uso consentito, è necessario ottenere l’autorizzazione direttamente dal titolare del copyright. Per visualizzare una copia di questa licenza, visitare http://creativecommons. org/licenses/by/4.0/.
RIFERIMENTI
1 Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, et al. Cancer incidence and mortality in China, 2014. Chin J Cancer Res. 2018;30:1-12.2 Siegel RL, Miller KD, Jemal A. Statistiche sul cancro, 2018. CA: Cancer J Clin. 2018;68:7-30.
3 Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB 3rd, et al. Il dicloroacetato altera il metabolismo di Warburg, inibisce la crescita cellulare e aumenta la sensibilità ai raggi X delle cellule di cancro al polmone umano A549 e H1299 NSC. Free Radic Biol Med. 2015;89:263-73.
4 Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. La segnalazione dell’interferone tumorale regola un programma di resistenza multigenica al blocco dei checkpoint immunitari. Cell. 2016;167: 1540-54 e12.
5 Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Caso di un grave evento avverso in seguito alla somministrazione di cellule T trasdotte con un recettore chimerico dell’antigene che riconosce ERBB2. Mol Ther: J Am Soc Gene Ther. 2010;18:843-51. Il dicloroacetato ripristina la chemiosensibilità del cancro del colon-retto attraverso il meccanismo p53/miR-149-3p/PDK2. . . 483
6 Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Statistiche sul trattamento del cancro e sulla sopravvivenza, 2016. CA: Cancer J Clin. 2016;66:271-89.
7 Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Therapeutic Adv Med Oncol. 2016;8:57-84.
8 Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combinato con fluorouracile rispetto al solo fluorouracile come trattamento di prima linea per il carcinoma colorettale metastatico: uno studio multicentrico randomizzato. Lancet. 2000;355:1041-7.
9 Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan più fluorouracile e leucovorin per il cancro colorettale metastatico. Gruppo di studio sull’irinotecan. New Engl J Med. 2000;343:905-14.
10 Hanahan D, Weinberg RA. I segni distintivi del cancro: la prossima generazione. Cell. 2011;144:646-74.
11 Matthew G, Vander Heiden LCC, Craig BT. Capire l’effetto Warburg: i requisiti metabolici della proliferazione cellulare. Science. 2009;324:1029-33.
12 Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
13 Shaw RJ. Metabolismo del glucosio e cancro. Curr Opin Cell Biol. 2006;18:598-608.
14 Van Dang C, Pollak M. Perché cancro e metabolismo? Perché ora? Cancer Metab. 2013;1:1.
15 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532.
16 Cairns RA, Harris IS, Mak TW. Regolazione del metabolismo delle cellule tumorali. Nat Rev Cancer. 2011;11:85-95.
17 Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inibizione della glicolisi nelle cellule tumorali: una nuova strategia per superare la resistenza ai farmaci associata al difetto respiratorio mitocondriale e all’ipossia. Cancer Res. 2005;65:613-21.
18 Guo WQZ, Wang Z, et al. Il MiR-199a-5p è associato negativamente alle neoplasie e regola la glicolisi e la produzione di lattato prendendo di mira l’esochinasi 2 nel cancro del fegato. Epatologia. 2015;62:1132-44.
19 Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. Il microRNA-124 riduce la via del pentoso fosfato e la proliferazione prendendo di mira gli mRNA PRPS1 e RPIA in cellule di cancro colorettale umano. Gastroenterologia. 2015;149:1587-98 e11.
20 Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. Il microRNA-129-5p regola la glicolisi e la proliferazione cellulare prendendo di mira il trasportatore di glucosio SLC2A3 nelle cellule di cancro gastrico. Front Pharmacol. 2018;9:502.
21 Bartel DP. MicroRNA: genomica, biogenesi, meccanismo e funzione. Cell. 2004;116:281-97.
22 Garzon R, Calin GA, Croce CM. I microRNA nel cancro. Annu Rev Med. 2009;60:167-79.
23 Huang S, He X. I microRNA: minuscole molecole di RNA, enormi forze motrici per muovere la cellula. Protein Cell. 2010;1:916-26.
24 Zhang Y, Wang J. I microRNA sono importanti regolatori della resistenza ai farmaci nel cancro del colon-retto. Biol Chem. 2017;398:929-38.
25 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacia del dicloroacetato come farmaco per la riduzione del lattato. J Clin Pharmacol. 2003;43:683-91.
26 Kankotia S, Stacpoole PW. Dicloroacetato e cancro: una nuova casa per un farmaco orfano? Biochim Biophys Acta. 2014;1846:617-29.
27 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Prove dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochemical J. 1998;329(Pt 1):191-6.
28 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. Un asse mitocondriale-K + canale è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer cell 2007;11:37-51.
29 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci Transl Med. 2010;2:31ra4.
30 Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. Uno studio di fase I in aperto, a braccio singolo, di dose-escalation di dicloroacetato (DCA) in pazienti con tumori solidi avanzati. Invest New Drugs. 2015;33:603-10.
31 Papandreou I, Goliasova T, Denko NC. Farmaci antitumorali che colpiscono il metabolismo: il dicloroacetato è il nuovo paradigma? Int J Cancer. 2011;128:1001-8.
32 Michelakis ED, Webster L, Mackey JR. Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer. 2008;99:989-94.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Il dicloroacetato induce apoptosi e arresto del ciclo cellulare nelle cellule di cancro del colon-retto. Br J Cancer. 2010;102:1746-52.
34 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Il dicloroacetato di sodio (DCA) riduce l’apoptosi nell’ipossia del tumore colorettale. Cancer Lett. 2010;297:75-83.
35 Bertoli G, Cava C, Castiglioni I. I microRNA: nuovi biomarcatori per la diagnosi, la prognosi, la predizione della terapia e strumenti terapeutici per il cancro al seno. Theranostics 2015;5:1122-43.
36 Cao D, Jia Z, You L, Wu Y, Hou Z, Suo Y, et al. L’acido 18betaglicirretinico sopprime il tumore gastrico attraverso l’attivazione della segnalazione miR149-3p-Wnt-1. Oncotarget. 2016;7:71960-73.
37 Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, et al. Potenti effetti della dioscina contro il cancro al pancreas attraverso l’inibizione mediata dal miR-149-3P della via di segnalazione Akt1. Br J Pharm. 2017;174:553-68.
38 Kato M, Li J, Chuang JL, Chuang DT. Meccanismi strutturali distinti per l’inibizione delle isoforme della piruvato deidrogenasi chinasi da parte di AZD7545, dicloroacetato e radicicolo. Structure. 2007;15:992-1004.
39 Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Regolazione divergente dell’espressione genica delle isoforme della piruvato deidrogenasi chinasi in cellule muscolari umane in coltura. FEBS J. 2005;272:3004-14.
40 Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, BelouecheBabari M, Koh KX, et al. L’aumento della resistenza ai farmaci è associato a ridotti livelli di glucosio e a un fenotipo di glicolisi potenziata. Br J Pharm. 2014;171:3255-67.
41 Stacpoole PW. La farmacologia del dicloroacetato. Metab: Clin Exp. 1989;38:1124-44.
42 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Cinetica e metabolismo del dicloroacetato in funzione dell’età: possibile rilevanza per la tossicità. J Pharmacol Exp Therap. 2008;324:1163-71.
43 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. La combinazione di PDK1 ed EGFR innesca la regressione del glioblastoma invertendo l’effetto Warburg. Cancer Res. 2013;73:7277-89.
44 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. L’inattivazione dell’asse di segnalazione HIF-1alpha/ PDK3 spinge il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res. 2012;72:5035-47.
45 Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, et al. Il dicloroacetato attenua la resistenza al 5-fluorouracile indotta dall’ipossia nel cancro gastrico attraverso la regolazione del metabolismo del glucosio. Exp Cell Res. 2014;321:219-30.
46 Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, et al. L’attivazione della fosforilazione ossidativa mediante un inibitore della piruvato deidrogenasi chinasi supera la resistenza al sorafenib del carcinoma epatocellulare. Br J Cancer. 2013;108:72-81. 484 Y. Liang et al.
47 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. L’attivazione dell’ossidazione mitocondriale mediante l’inibizione di PDK2 inverte la resistenza al cisplatino nel cancro della testa e del collo. Cancer Lett. 2016;371:20-9.
48 Kimmelman AC, White E. Autofagia e metabolismo tumorale. Cell Metab 2017;25:1037-43.
49 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. L’attivazione mitocondriale tramite l’inibizione di PDKII sopprime la segnalazione di HIF1a e l’angiogenesi nel cancro. Oncogene. 2013;32:1638-50.
50 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversità della famiglia di geni della piruvato deidrogenasi chinasi nell’uomo. J Biol Chem. 1995;270:28989-94.
51 Sugden MC, Holness MJ. Recenti progressi nei meccanismi di regolazione dell’ossidazione del glucosio a livello del complesso piruvato deidrogenasi da parte delle PDK. Am J Physiol Endocrinol Metab. 2003; 284: E855-62.
52 Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182- PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98.
53 Bellazzo A, Di Minin G, Valentino E, Sicari D, Torre D, Marchionni L, et al. Cell-autonomous and cell non-autonomous downregulation of tumor suppressor DAB2IP by microRNA-149- 3p promotes aggressiveness of cancer cells. Cell Death Differ. 2018;25:1224-38.
54 Vazquez A, Bond EE, Levine AJ, Bond GL. La genetica della via p53, l’apoptosi e la terapia del cancro. Nat Rev Drug Discov. 2008;7:979-87.
55 Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. Il soppressore tumorale p53 nel controllo del metabolismo e della ferroptosi. Front Endocrinol. 2018;9:124.
56 Contractor T, Harris CR. p53 regola negativamente la trascrizione della piruvato deidrogenasi chinasi Pdk2. Cancer Res. 2012;72:560-7.
57 Iacopetta B. Mutazione di TP53 nel cancro colorettale. Hum Mutat. 2003;21:271-6.
58 Kim JW, Dang CV. La dolcezza molecolare del cancro e l’effetto Warburg. Cancer Res. 2006;66:8927-30.
59 Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, Tempera I. La poli(ADP-ribosio) polimerasi 1 è necessaria per coattivare l’espressione genica dipendente dall’hypoxia-inducible factor-1 da parte della proteina di membrana latente 1 del virus EpsteinBarr. PLoS Pathog. 2018;14: e1007394.
60 Hlouschek J, Ritter V, Wirsdorfer F, Klein D, Jendrossek V, Matschke J. Il targeting di SLC25A10 allevia il miglioramento della capacità antiossidante e la radioresistenza associata delle cellule tumorali indotte dall’ipossia cronica-ciclica. Cancer Lett. 2018;439:24-38.
Contenuti correlati: