Yu Liang1, Lidan Hou1, Linjing Li1, Lei Li1, Liming Zhu1, Yu Wang1, Xin Huang1, Yichao Hou1, Danxi Zhu1, Huimin Zou1, Yan Gu2, Xiaoling Weng3,4, Yingying Wang5, Yue Li6, Tianqi Wu3, Mengfei Yao3, Isabelle Gross7,8, Christian Gaiddon9,10, Meng Luo2, Jianhua Wang3, Xiangjun Meng1
1 Departamento de Gastroenterología, Noveno Hospital Popular de Shanghái, Facultad de Medicina de la Universidad Jiao Tong de Shanghái, China
2 Departamento de Cirugía General, Noveno Hospital Popular de Shanghái, Facultad de Medicina de la Universidad Jiao Tong de Shanghái, China
3 Instituto del Cáncer, Centro Oncológico de la Universidad Fudan de Shanghái, Universidad Fudan, Shanghái, China
4 Ningbo Aitagene Technology Co. LTD, Shanghai, China
5 Departamento de Bioquímica y Biología Molecular & Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China
6 Pathology Center, Shanghai First People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
7 INSERM UMR_S1113, Strasbourg F-67200, France
8 FMTS, Universite de Strasbourg Strasbourg, Strasbourg F-67000, Francia
9Universitede Strasbourg, Inserm IRFAC UMR_S1113, Laboratory Stress Response and Innovative Therapy «Streinth», Estrasburgo 67200, Francia
10 CLCC Paul Strauss, Estrasburgo, Francia
Meng Luo [email protected]
Jianhua Wang [email protected]
Xiangjun Meng [email protected]
Estos autores contribuyeron a partes iguales: Yu Liang, Lidan Hou
Recibido: 16 de marzo de 2019
Revisado: 17 de septiembre de 2019
Aceptado: 19 de septiembre de 2019
Publicado: 9 de octubre de 20199 Octubre de 2019
Resumen
El desarrollo de quimiorresistencia sigue siendo un reto importante que explica la letalidad del cáncer colorrectal (CCR). El dicloroacetato (DCA) se utilizó originalmente como regulador metabólico en el tratamiento de enfermedades metabólicas; aquí, el DCA se ensayó para identificar los mecanismos subyacentes a la quimiorresistencia del CCR. Descubrimos que el DCA aumentaba notablemente la quimiosensibilidad de las células del CCR al fluorouracilo (5-FU) y reducía la formación de colonias debido a los altos niveles de apoptosis. Utilizando el ensayo de microarrays, observamos que miR-149-3p estaba implicado en la quimiorresistencia del CCR, que fue modulada por p53 de tipo salvaje tras el tratamiento con DCA. Además, se identificó PDK2 como diana directa de miR-149-3p. Los análisis mecanísticos mostraron que la sobreexpresión de miR-149-3p potenciaba la apoptosis inducida por 5-FU y reducía el metabolismo de la glucosa, de forma similar a los efectos del knockdown de PDK2. Además, la sobreexpresión de PDK2 invirtió parcialmente el efecto inhibidor de miR-149-3p sobre el metabolismo de la glucosa. Por último, se observó que tanto el tratamiento con DCA como la sobreexpresión de miR-149-3p en células de CCR resistentes al 5-FU sensibilizaban notablemente el efecto quimioterapéutico del 5-FU in vivo, y este efecto también se validó en una pequeña cohorte retrospectiva de pacientes de CCR. En conjunto, determinamos que la vía de señalización p53/miR-149-3p/PDK2 puede ser potencialmente atacada con el tratamiento DCA para superar el CCR quimiorresistente.
Información suplementaria: La versión online de este artículo (https:// doi.org/10.1038/s41388-019-1035-8) contiene material suplementario, disponible para usuarios autorizados.
Introducción
El cáncer colorrectal (CCR) es la cuarta causa de muerte relacionada con el cáncer en China [1] y es la segunda causa de mortalidad relacionada con el cáncer en los Estados Unidos [2], que atribuye principalmente a la metástasis y el fracaso de la quimioterapia debido a la resistencia a los medicamentos, lo que lleva a ~ 50.000 muertes al año [3].
Recientemente, aunque la estrella emergente PD1/PDL1 suscitó un gran interés, y cada vez más agentes bioterapéuticos mostraban resultados alentadores en el tratamiento del cáncer, la limitada tasa de eficacia y los inevitables efectos adversos frenan su uso en la clínica [4,5]. En la actualidad, la quimioterapia sigue siendo una de las principales opciones en la clínica, especialmente para los cánceres metastásicos y en fase avanzada no resecables [6], pero el desarrollo de resistencia a los fármacos sigue siendo la mayor limitación de la quimioterapia [7]. Por lo tanto, explorar los mecanismos de resistencia a los fármacos y buscar nuevas combinaciones de fármacos anticancerosos clásicos para optimizar la eficacia puede ser beneficioso para el tratamiento del CCR. Dado que el fluorouracilo (5-FU) es el fármaco quimioterapéutico más utilizado en el CCR, en este estudio se utilizaron líneas celulares de CCR resistentes al 5-FU [8,9].
Una anomalía metabólica de la glucosa representa uno de los principales aspectos de las características distintivas del cáncer [10]. Se sabe que el desarrollo de una masa celular descontrolada conduce a una vascularización deficiente del tumor, lo que provoca una reducción del suministro de oxígeno. Por lo tanto, las células cancerosas se adaptan a las alteraciones del microenvío cambiando su metabolismo oxidativo por un metabolismo glucolítico, que se basa en el suministro de glucosa y produce lactato. Este cambio se denomina «efecto Warburg» y suele observarse en varias células cancerosas como una de sus características más notables [ 11-13]. La reciente acumulación de datos de investigación ha llevado a la necesidad de refinar la teoría de Warburg [14]; por ejemplo, se ha demostrado que el cambio metabólico está implicado en la quimiorresistencia [15]; por lo tanto, se podrían explotar los patrones metabólicos de las células cancerosas para superar la quimiorresistencia [16,17].
En particular, se han descrito múltiples mecanismos para controlar el cambio metabólico en las células cancerosas, incluidos los microARN (miARN) [18-20
]. Los miARN representan una clase de pequeños ARN endógenos no codificantes que regulan la traducción y degradación de los ARNm [21] y están implicados en muchos más procesos biológicos, como la proliferación celular, la migración, la apoptosis, la autorrenovación, la iniciación, el desarrollo del cáncer y la quimiorresistencia [22-24].
La evidencia indica que dirigirse al metabolismo anormal de las células cancerosas ha sido una intensa vía de investigación cuyo objetivo es «asfixiar el tumor», cuyas estrategias consisten en inhibir enzimas clave implicadas en el metabolismo glucolítico [15]. En este manuscrito, el dicloroacetato (DCA) se utilizó originalmente para tratar la acidosis láctica y la enfermedad mitocondrial hereditaria [25]. El DCA inhibe la actividad enzimática de la piruvato deshidrogenasa quinasa (PDK1-4), que es necesaria para transformar el piruvato en acetil-CoA, enlazando el metabolismo glucolítico con el ciclo del ácido cítrico [26,27]; recientemente se ha informado de que el DCA tiene efectos anticancerígenos [28-31]. Sin embargo, el mecanismo que subyace al efecto del DCA en el tratamiento del CCR sigue siendo incierto.
El presente estudio se centra en el mecanismo molecular implicado en la regulación del metabolismo de la glucosa y la resistencia a la quimioterapia en el CCR. Utilizando DCA en células de CCR, investigamos las funciones de los miARN relacionados y, de este modo, revelamos una vía de señalización que explica la resistencia al tratamiento con 5-FU.
Resultados
El DCArestaura la quimiosensibilidad de las células de CCR resistentes al 5-FU
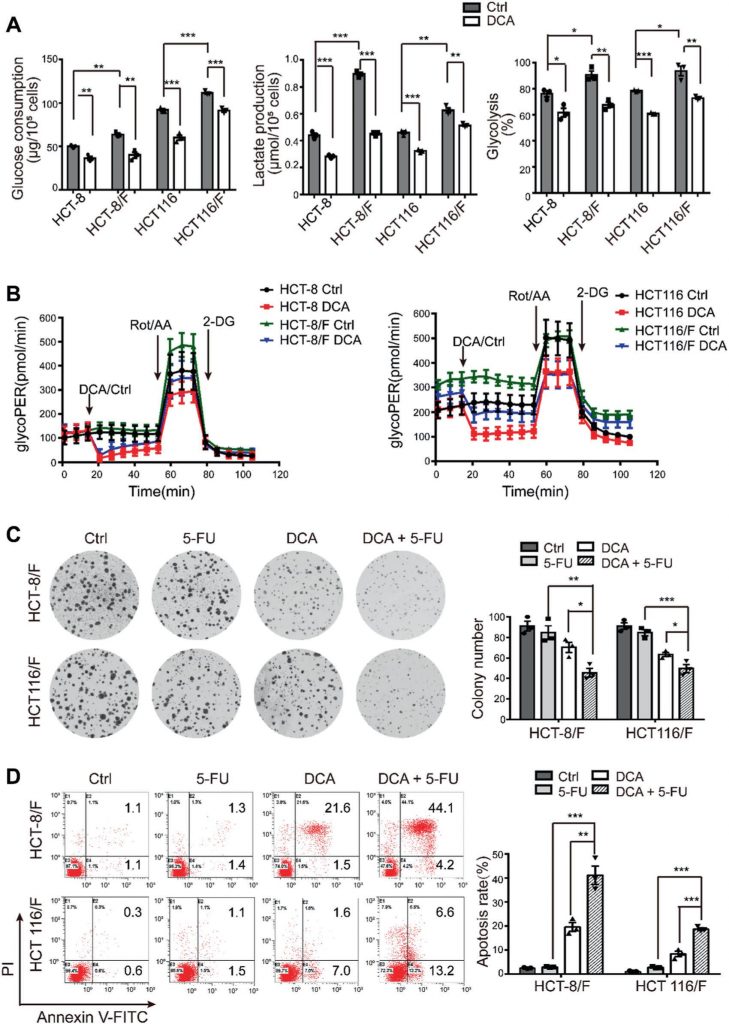
Se ha descrito que el DCA es un fármaco antitumoral eficaz que actúa dirigiéndose a las vías relacionadas con la energía en ciertos tipos de cáncer [32]; sin embargo, el efecto del DCA en las células de CCR quimiorresistentes no ha sido bien estudiado. Utilizando el ensayo CCK8, descubrimos que, en comparación con sus líneas celulares parentales HCT-8 y HCT116, las células HCT-8/F y HCT116/F resistentes al 5-FU eran insensibles al 5-FU (Fig. suplementaria 1A) y las concentraciones inhibitorias semimáximas (IC50) del DCA en las células HCT-8/F y HCT116/F eran de ~15 y 20 mM, respectivamente, lo que concuerda con informes anteriores [33, 34] (Fig. suplementaria 1B). A continuación observamos que el DCA inhibía significativamente la síntesis de ADN (Fig. suplementaria 1C, panel superior) e inducía la generación de ROS (Fig. suplementaria 1C, panel inferior) en células de CCR resistentes al 5-FU. Los marcadores del metabolismo energético, incluidos el consumo de glucosa, la producción de lactato y la glucólisis, se elevaron notablemente en las células de CCR resistentes al 5-FU en comparación con las células de CCR sensibles al 5-FU, mientras que la adición de DCA redujo notablemente la expresión de dichos marcadores (Fig. 1a). Considerando 6 h de pretratamiento sin suero en la medición de la glucólisis, se utilizó un kit de ensayo de la tasa glucolítica Seahorse XF para eliminar el efecto del pretratamiento y medir la tasa glucolítica en tiempo real. La tasa de eflujo de protones glucolítica (glycoPER) refleja la tasa de acidificación extracelular de la glucólisis. La adición de DCA redujo significativamente la glucólisis inducida en comparación con los controles respectivos (Fig. 1b y Fig. suplementaria 1D).
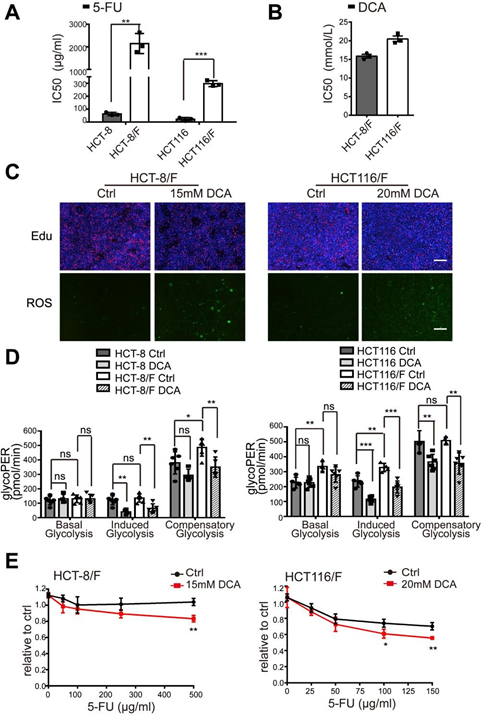
(A) Las células CRC fueron tratadas con diferentes concentraciones de 5-FU durante 24 horas. Se calculó el IC50 de 5-FU en cada célula. (B) Las células HCT-8/F y HCT116/F se trataron con diferentes concentraciones de DCA durante 24 horas. Se calculó el IC50 de DCA en cada célula. (C) Las células HCT-8/F y HCT116/F se trataron con 15 mM y 20 mM de DCA, respectivamente, durante 24 horas. Imágenes representativas de la tinción de inmunofluorescencia de Edu (panel superior) e imágenes representativas de la tinción de inmunofluorescencia de ROS (panel inferior), barra de escala: 200 μm. (D) La determinación de la tasa de glucólisis, incluidas la glucopresión basal, la glucopresión inducida y la glucopresión compensatoria, se calculó mediante el Seahorse Glycolytic Rate Assay Report Generator. (E) El crecimiento celular se determinó mediante un ensayo CCK8 tras el tratamiento con 5-FU y DCA. Los intervalos de dosificación de 5-FU fueron de 100 ug/ml a 500 ug/ml para las células HCT-8/F y de 25 ug/ml a 150 ug/ml para las células HCT116/F. Los resultados de tres experimentos independientes se muestran como media ± SEM. Cada experimento se realizó en 3-6 réplicas biológicas. *p < 0,05; **, P < 0,01; ***, P < 0,001.
El DCA superó significativamente la resistencia al 5-FU en las células HCT-8/F y HCT116/F, como se manifestó en las mediciones del crecimiento celular (Fig. suplementaria 1E). La capacidad de formación de colonias se inhibió significativamente (Fig. 1c), y la apoptosis se indujo notablemente en un tratamiento combinado (Fig. 1d), todo lo cual se cuantificó con más detalle. Estos resultados sugieren que el DCA puede restaurar la quimiosensibilidad en células de CCR resistentes al 5-FU.
miR-149-3p desempeña un papel crucial en la quimiosensibilidad de las células del CCR
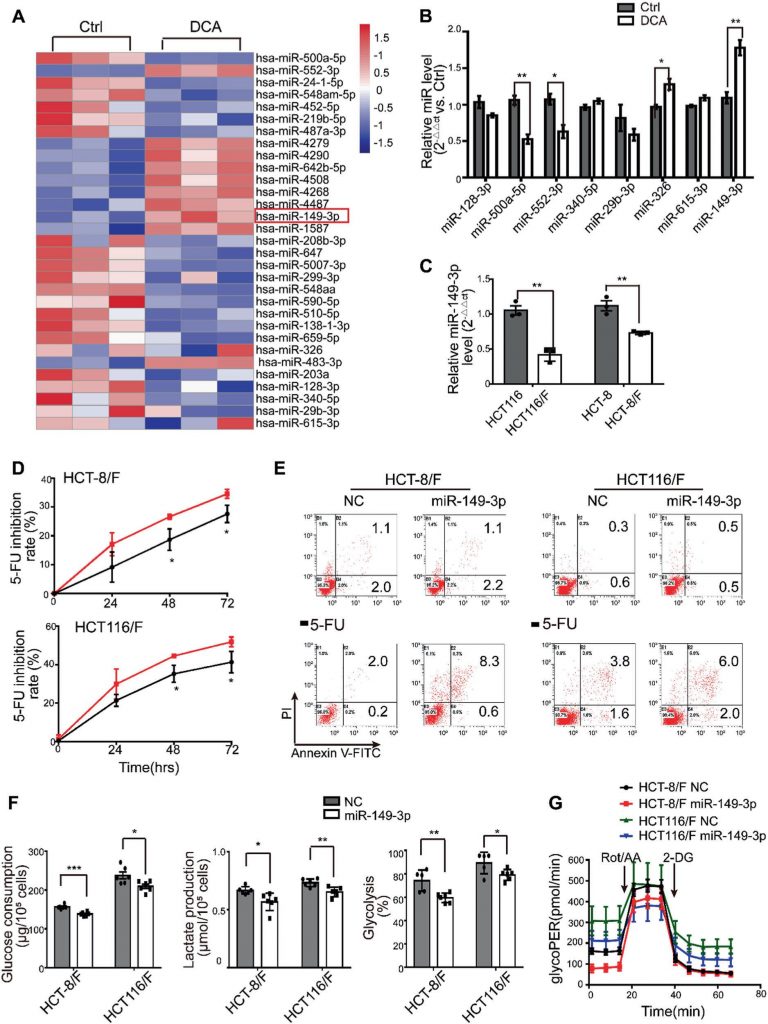
los miRNAs se han considerado una herramienta terapéutica prometedora por sus efectos en la supresión tumoral [35]. En este sentido, primero determinamos los perfiles de expresión de miARN utilizando el array de miARN que contiene 2059 miARN humanos. Un total de 119 miRNAs se expresaron diferencialmente en respuesta al DCA en células HCT116 (Fig. 2a y Fig. Suplementaria S2A). Entre ellos, los niveles de expresión de ocho miRNAs fueron confirmados por PCR cuantitativa en tiempo real (Fig. 2b), y miR-149-3p fue finalmente regulado por el DCA de forma dependiente de la dosis (Fig. Suplementaria S2B).
A continuación, descubrimos que las células HCT116 con niveles basales más altos de miR-149-3p conferían una mayor sensibilidad a 5-FU y L-OHP, como se muestra en la Fig. 2C suplementaria. Además, la transfección anti-miR-149-3p de células HCT116 redujo notablemente el efecto quimioterapéutico del 5-FU (Fig. suplementaria 2D-E). En particular, los niveles de miR-149-3p en células de CCR quimiosensibles fueron significativamente superiores a los niveles en células de CCR quimioresistentes (Fig. 2c). Por lo tanto, la transfección de imitadores de miR-149-3p aumentó significativamente la tasa de inhibición; promovió la apoptosis celular inducida por 5-FU; redujo el consumo de glucosa, la producción de lactato y la glucólisis en las células HCT-8/F y HCT116/F (Fig. 2d-f). Un ensayo de tasa glucolítica Seahorse XF mostró que la expresión de miR-149-3p reducía la glucoproteína basal en células de CCR resistentes a 5-FU, lo que concordaba con los resultados anteriores (Fig. 2g y Fig. 2F suplementaria). Estos resultados sugieren que miR-149-3p es favorable para superar la quimiorresistencia en células de CCR.
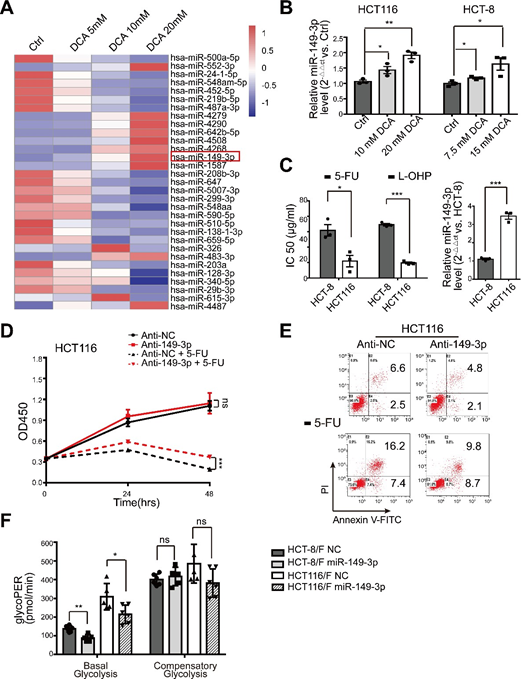
(A) El mapa de calor del perfil de microARN expresado diferencialmente en células HCT116 tratadas con control, 5 mM, 10 mM, 20 mM DCA durante 24 horas. (B) Se preparó ARN total de células HCT116 y HCT-8 a las 24 horas del tratamiento con 10/7,5 mM y 20/15 mM de DCA. El nivel de miR-149-3p se analizó mediante PCR cuantitativa en tiempo real. (C) Las células HCT-8 y HCT116 se trataron con diferentes concentraciones de 5-FU y oxaliplatino (L-OHP) respectivamente durante 24 horas y se calculó la concentración inhibitoria media máxima (IC50) (panel izquierdo). Los niveles basales de miR-149-3p se determinaron mediante PCR cuantitativa en tiempo real en células HCT-8 y HCT116 (panel derecho). (D-E) Las células HCT116 se transfectaron transitoriamente con Anti-NC o con un inhibidor de miR-149-3p. Tras la transfección, las células se trataron con 25 μg/ml de 5-FU durante 24 horas. El crecimiento celular y la apoptosis se determinaron mediante CCK8 y citometría de flujo respectivamente. (F) La determinación de la tasa de glicólisis, incluyendo la glicolisis basal y la glicolisis compensatoria en células de CCR resistentes a 5-FU transfectadas con NC o un imitador de miR-149-3p, se calculó mediante el Seahorse Glycolytic Rate Assay Report Generator. Los datos de tres experimentos independientes se muestran como media ± SEM. Cada experimento se realizó en 3-6 réplicas biológicas. *p < 0,05; **, P < 0,01; ***, P < 0,001; ns, sin significación.
El DCAinduce la expresión de miR-149-3p a través de p53 de tipo salvaje (wt)
Dado que varios estudios recientes han revelado que miR-149-3p está regulado por varios fármacos [36,37], a continuación determinamos cómo miR-149-3p está regulado por el DCA. Descubrimos que el DCA aumentaba significativamente la expresión de p53 wt y sus señales descendentes, incluyendo la expresión de p21, PUMA y MDM2, en células de CCR (Fig. 3a, b). Además, observamos que las alteraciones en la expresión de wt p53 eran capaces de modular significativamente la expresión de miR-149-3p, como se muestra en la Fig. 3c, d, lo que indica que miR-149-3p estaba regulado positivamente por wt p53. Por lo tanto, utilizando la línea celular HCT116 sin p53 (TP53-/-), observamos que miR-149-3p no se regulaba al alza por el tratamiento con DCA, pero la expresión ectópica de p53 invertía este efecto. Además, utilizamos nutlin-3, un potente inhibidor que inhibe la interacción MDM2-p53, provocando la activación de p53 como control positivo, junto con la expresión de miR-149-3p elevada en la línea celular HCT116 wt (TP53+/+) (Fig. 3e-g). Mecánicamente, se predijeron cuatro sitios putativos de unión de p53 a la región del ADN genómico que flanquea a miR-149 utilizando software de análisis bioinformático (IGV). A continuación, se realizaron ensayos ChIP en las células utilizando un anticuerpo contra p53 wt. El ADN extraído se amplificó mediante PCR ordinaria con cebadores diseñados a partir de estos sitios. Nuestros resultados mostraron que, en comparación con la región 4, la región 3 estaba marcadamente enriquecida tras el tratamiento con DCA en la cromatina del HCT116 inmunoprecipitada con p53 wt (Fig. 3h), lo que sugiere que sólo la región 3 contiene un sitio de unión específico activado por el DCA. Estos resultados indican que el DCA modula miR-149-3p a través de p53 wt.
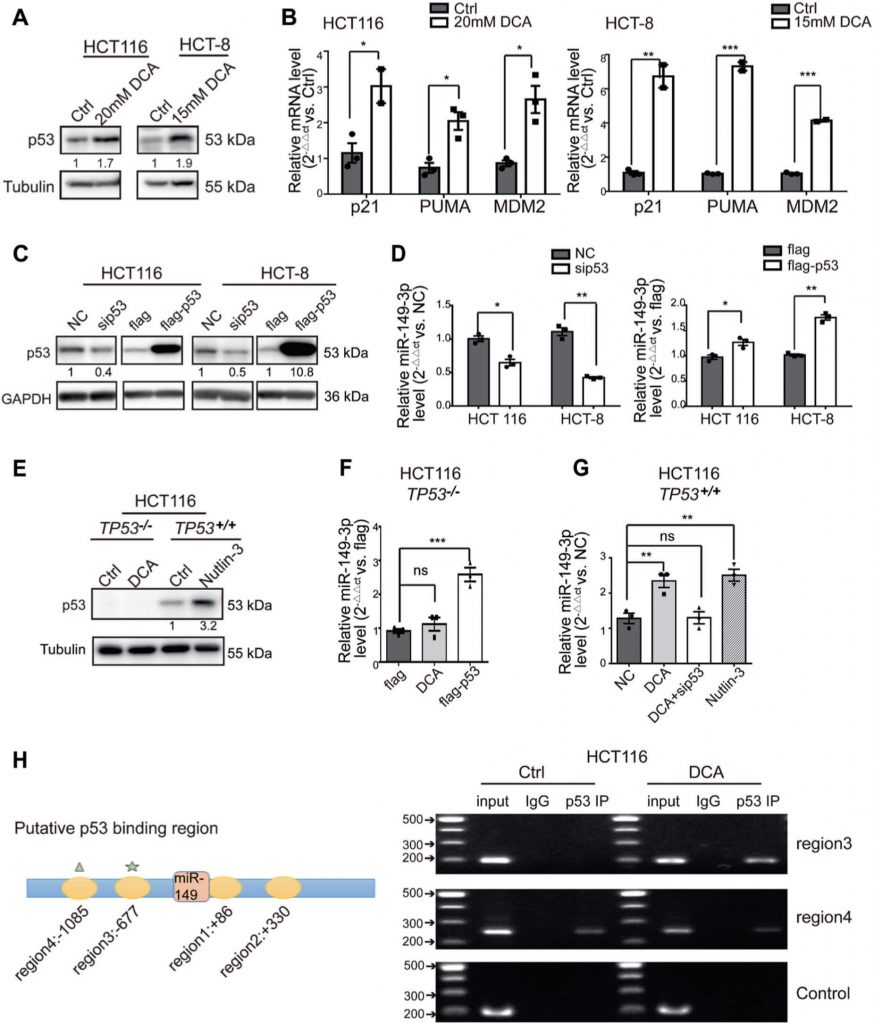
PDK2 es una diana directa de miR-149-3p
Para dilucidar los mecanismos por los que miR-149-3p regula la quimiosensibilidad en células de CCR, analizamos genes asociados con el metabolismo energético, que están regulados por miR-149-3p utilizando dos plataformas públicas (TargetScan y miRDB). Finalmente, identificamos que la piruvato deshidrogenasa quinasa 2 (PDK2) y la hexoquinasa 1 (HK1) son candidatos potenciales (Fig. 4a). Para confirmar estos hallazgos, se transfectó el imitador o inhibidor de miR-149-3p en células de CCR. Descubrimos que los niveles de ARNm de PDK2 estaban regulados negativamente por miR-149-3p, pero no por HK1 (Fig. 4b y Fig. suplementaria 3A). Se encontraron dos posibles sitios de unión de miR-149-3p en el 3′-UTR de PDK2, y el ensayo reportero de doble luciferasa indicó que miR-149-3p se une al sitio predicho (686-693) del 3′-UTR de PDK2 (Fig. 4c, d). A continuación, confirmamos que los niveles de proteína PDK2 estaban regulados negativamente por miR-149-3p (Fig. 4e).
La PDK tiene cuatro isozimas denominadas PDK1, 2, 3 y 4, todas las cuales han sido reguladas por el DCA [26, 38, 39]. A continuación, analizamos la expresión de ARNm de PDK1-4 en células HCT8 y HCT116 tras el tratamiento con DCA. El DCA inhibió significativamente la expresión de ARNm de PDK2, pero no de otras isozimas PDK (Fig. suplementaria S3B). Además, la transfección de anti-149-3p revirtió parcialmente la reducción de PDK2 por DCA (Fig. 4f). A continuación, determinamos los niveles proteicos de PDK2 y de su subunidad E1-alfa de la piruvato deshidrogenasa (PDHA1) en células de CCR sensibles al 5-FU y resistentes al 5-FU. Los niveles basales de PDK2 eran elevados en las células de CCR quimiorresistentes, en comparación con los niveles de las células quimiosensibles. En consonancia con esta elevación, la fosforilación de PDHA1 también fue elevada (Fig. 4g).
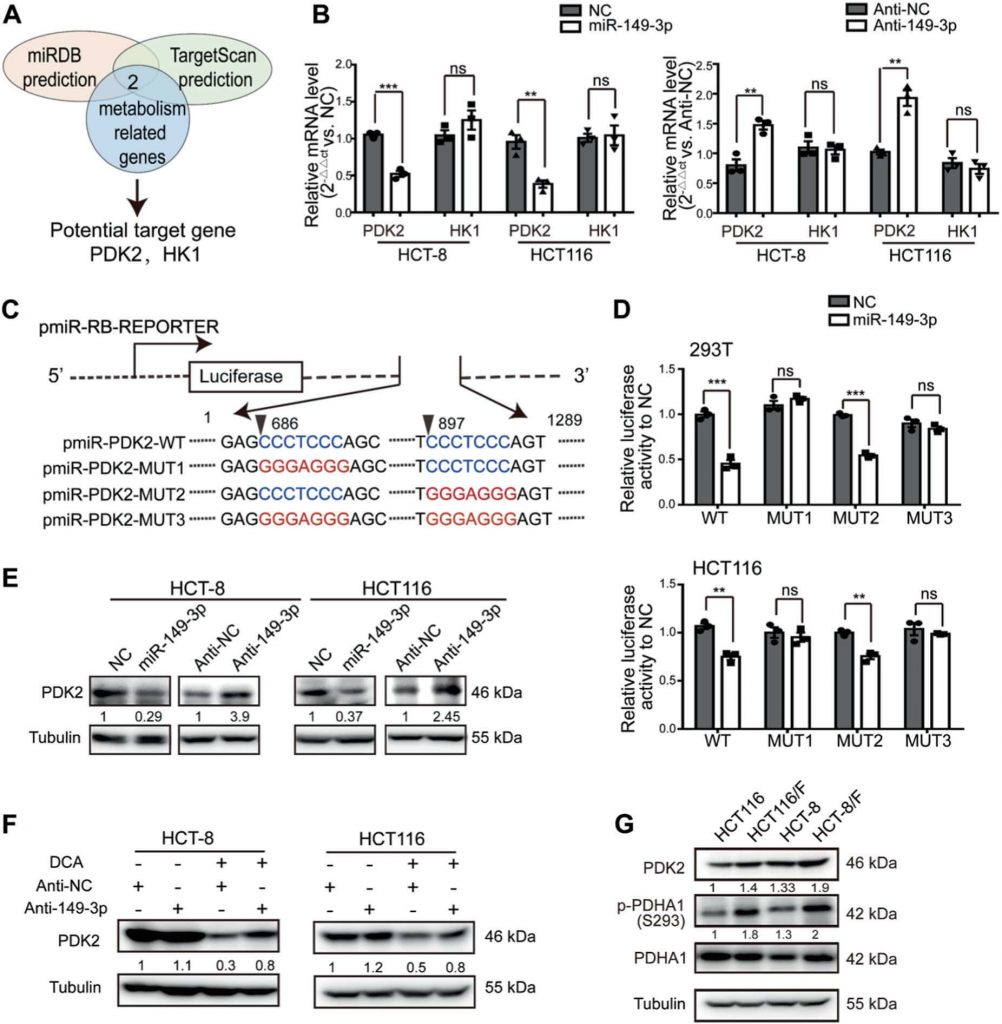
La vía miR-149-3p/PDK2 regula la quimiosensibilidad
Para entender cómo la vía miR-149-3p/PDK2 regula la respuesta celular del CCR al 5-FU, se seleccionó la línea celular HCT-8/F como línea celular representativa para investigar si los niveles de expresión de miR-149-3p y PDK2 afectaban a la respuesta celular al 5-FU. El knockdown de PDK2 en las células HCT-8/F inhibió la fosforilación de PDHA1 (Fig. 5a) y redujo los marcadores del metabolismo energético, como el consumo de glucosa, la producción de lactato y la glucólisis (Fig. 5b). Además, las reducciones de PDK2 fueron capaces de potenciar los efectos del 5-FU en el aumento de los niveles de PARP escindido (c-PARP) y Bax, ambos biomarcadores reconocidos de la apoptosis celular en células HCT-8/F (Fig. 5c). Además, el knockdown de PDK2 incrementó la quimiosensibilidad a 5-FU en células HCT-8/F, tal y como se determinó mediante CCK8 y ensayos de formación de colonias (Fig. 5d, e). El knockdown de PDK2 promovió la apoptosis inducida por 5-FU en células HCT-8/F y HCT116/F y se muestra en la Fig. Suplementaria S4A, mientras que la sobreexpresión de PDK2 promovió la fosforilación de PDHA1 y mitigó la apoptosis celular inducida por 5-FU en células HCT-8 y HCT116 (Fig. Suplementaria S4B-S4C).
Además, la sobreexpresión de PDK2 invirtió el efecto inhibidor de miR-149-3p sobre PDK2 (Fig. 5f) y abolió parcialmente el efecto inhibidor de miR-149-3p sobre el consumo de glucosa, la producción de lactato y la glucólisis (Fig. 5g. La expresión ectópica de PDK2 abolió marcadamente el efecto inhibidor de miR-149-3p sobre el crecimiento celular y la formación de colonias en células HCT-8/F tratadas con 5-FU (Fig. 5h, i). En conjunto, nuestros resultados indican que la vía miR-149-3p/PDK2 restablece la quimiosensibilidad, al menos parcialmente, mediante el metabolismo de la glucosa en células de CCR quimiorresistentes.
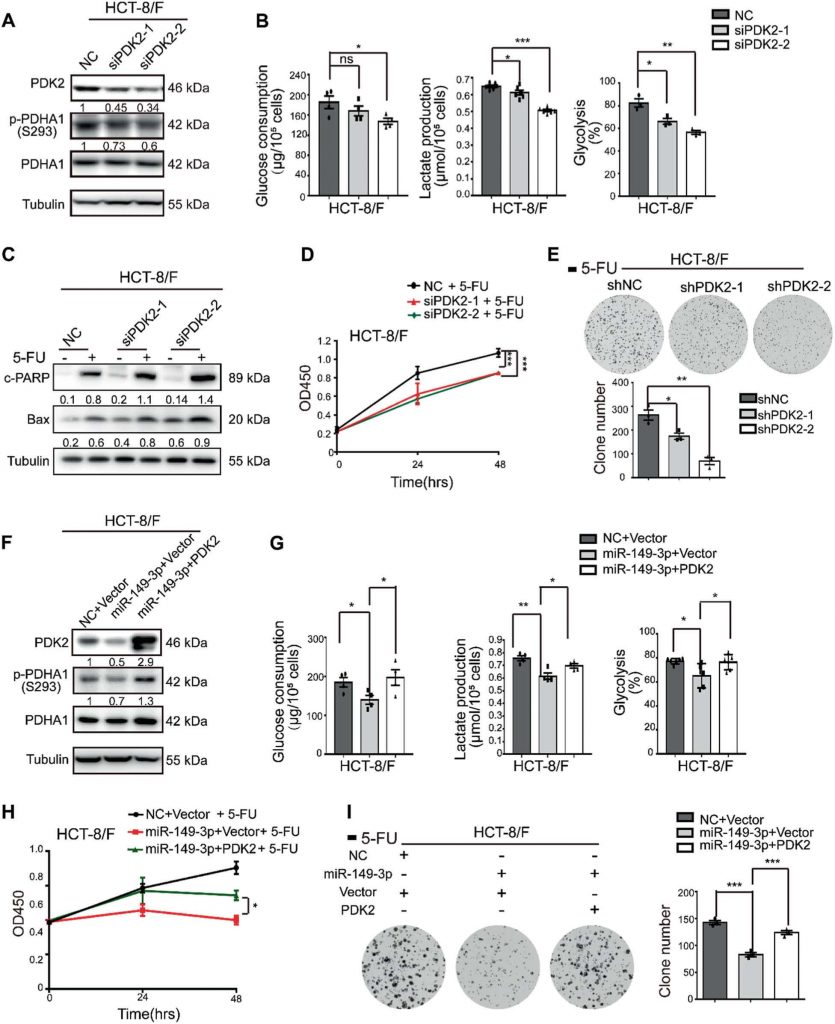
El DCA aumenta la quimiosensibilidad del 5-FU in vivo
A continuación, se inyectaron por vía intraperitoneal 5-FU, DCA o una combinación de 5-FU con DCA en el modelo de xenoinjerto subcutáneo (denominado grupo intraperitoneal). Teniendo en cuenta la insuficiente irrigación sanguínea en la parte central de los tumores subcutáneos, también inyectamos intratumoralmente DCA o PBS más una inyección intraperitoneal de 5-FU (grupo intratumoral). Las combinaciones de DCA con 5-FU mostraron mejores efectos inhibidores del crecimiento tumoral que el DCA o el 5-FU solos tras cuatro semanas de inyección en el grupo intraperitoneal (Fig. 6a, b). La expresión de Ki-67 se redujo tras el tratamiento combinado en el grupo intraperitoneal (Fig. 6c). El DCA inhibió significativamente el crecimiento tumoral en el grupo intratumoral en comparación con el grupo PBS (Fig. 6d), y miR-149-3p se incrementó en el grupo de inyección intratumoral de DCA, lo que concuerda con los resultados in vitro (Fig. 6e). La inyección intratumoral de DCA también promovió la apoptosis tumoral (Fig. 6F).
Para evaluar con más detalle el efecto de miR-149-3p en la respuesta de las células del CCR al 5-FU in vivo, el modelo de xenoinjerto subcutáneo fue sometido a la inyección intratumoral de agomiR-149-3p o agomiR-NC más una inyección intraperitoneal de 5-FU. miR-149-3p inhibió significativamente el crecimiento tumoral en el grupo intratumoral en comparación con el grupo control en el que se inyectó intratumoralmente agomiR-negativo (NC) (Fig. 6g, h). La sobreexpresión de miR-149-3p se validó mediante PCR cuantitativa en tiempo real (Fig. 6i), y la sobreexpresión de miR-149-3p promovió la apoptosis (Fig. 6j) y redujo la expresión de PDK2 y p-PDHA1 (Fig. 6k, l). Tras 3 semanas de tratamiento, se realizaron imágenes tumorales mediante tomografía por emisión de micropositrones (PET) con 18F-fluorodeoxiglucosa (FDG). se observó la captación de 18F-FDG en los lugares de implantación del tumor y se midieron la captación estandarizada máxima (SUVmáx) y los volúmenes tumorales metabólicos (MTV). No se observaron diferencias en el SUVmax entre los dos grupos, mientras que el MTV disminuyó significativamente en el grupo miR-149-3p (Fig. 6m).
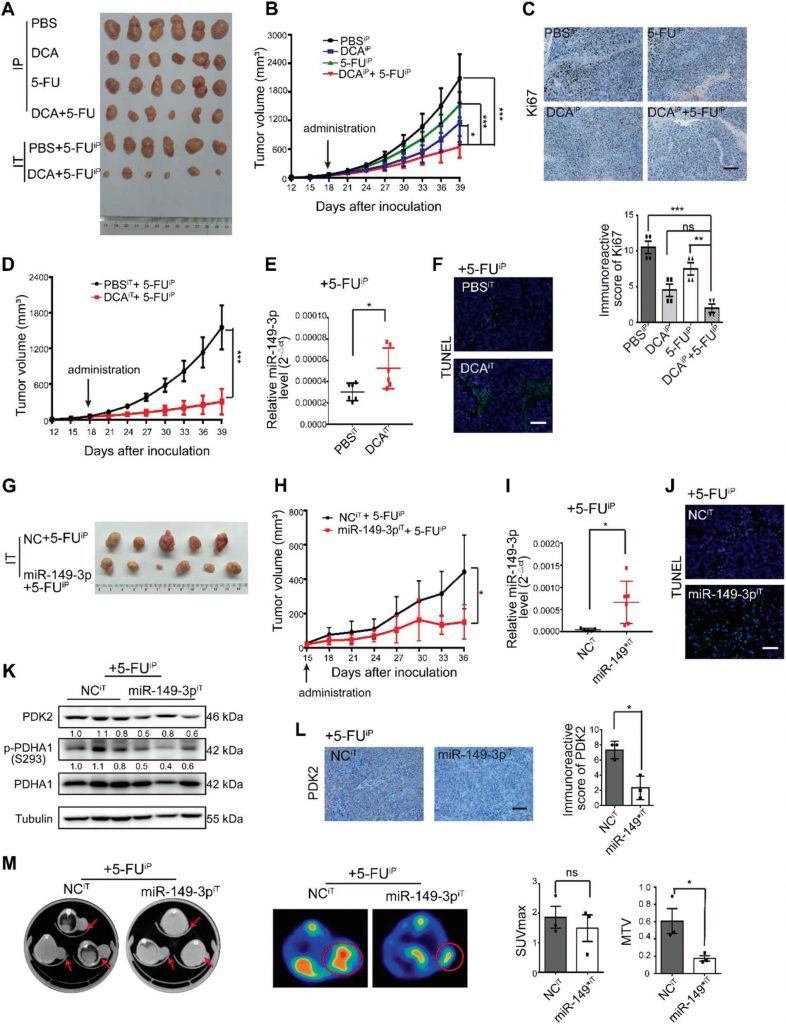
miR-149-3p está inversamente correlacionado con PDK2 en pacientes con CCR
Se observó una correlación inversa significativa entre los niveles de miR-149-3p y de ARNm de PDK2 en tejido de CCR humano (Fig. 7a, b). Entre ellos, ocho pacientes en condición/enfermedad estable (SD) dentro de los 3 años posteriores a la quimioterapia expresaron un mayor nivel de miR-149-3p que cinco pacientes con enfermedad progresiva (PD) (Fig. suplementaria S5A). Se analizaron cinco pares de pacientes con PD y SD con la misma patología y estadio TNM, y la tinción de PDK2 varió significativamente en las muestras de los pacientes con PD y SD (Fig. suplementaria S5B). Los pacientes de CCR de la base de datos TCGA con alta expresión de PDK2 también se caracterizaron por una peor supervivencia global (SG) (Fig. 7c). La base de datos TCGA también mostró que la expresión de PDK2 en el grupo wt p53 se redujo en comparación con la del grupo p53 mutante (Fig. 7d). Estos resultados sugieren que PDK2 está regulada negativamente por miR-149-3p en pacientes con CCR.
Discusión
El CCR se caracteriza por anomalías tumorigénicas y vías metabólicas alteradas y es una de las principales causas de muerte por cáncer [2]. La resistencia a la quimioterapia es la principal causa de fracaso del tratamiento [7]. En este estudio, descubrimos que el DCA podía aumentar el efecto quimioterapéutico del 5-FU en células de CCR quimiorresistentes y que la activación de la vía p53/miR-149-3p/PDK2 era capaz de aumentar la quimiosensibilidad in vitro e in vivo.
Cada vez hay más pruebas de que el aumento de la glucólisis está estrechamente relacionado con la resistencia a la quimioterapia [15, 17, 40]. En este caso, también descubrimos que, en comparación con sus líneas celulares parentales, las células de CCR quimiorresistentes mostraban un elevado consumo de glucosa, producción de lactato y glucólisis, lo que sugiere que las anomalías metabólicas son una característica típica, pero los mecanismos moleculares siguen sin estar claros en las células de CCR.
Se ha observado que el DCA disminuye los niveles de lactato en sangre in vivo en roedores a dosis de ~25-50 mg/kg/24 h [41] y que emplea DCA con un amplio rango de dosis de 1 a 50 mM [26]. En particular, el efecto adverso del DCA en humanos se limita generalmente a una neuropatía periférica sensitiva y motora reversible, que está influida por la edad y el genotipo [42]. Recientemente, el DCA ha sido identificado como una nueva terapia metabólica para varios pacientes con cáncer [ 26 , 29].
Se ha informado de que el DCA es capaz de inhibir la actividad PDK y convertir el piruvato en acetil-CoA, lo que conduce a un cambio en la generación de energía de la glucólisis a la fosforilación oxidativa mitocondrial [43,44]. También se ha demostrado que el DCA atenúa la resistencia inducida por la hipoxia al 5-FU en el cáncer gástrico [45], supera la resistencia al sorafenib en el carcinoma hepatocelular [46] y atenúa la resistencia al cisplatino en el cáncer de cabeza y cuello [47]. Nuestro estudio reveló que el DCA era capaz de aliviar la quimiorresistencia de las células del CCR al 5-FU. Además, demostramos que el DCA redujo el consumo de glucosa y la producción de lactato en células de CCR quimiorresistentes al nivel basal de las células de CCR quimiosensibles. Dado que la autofagia tiene el potencial de impulsar el metabolismo del cáncer [48], se determinó el efecto del DCA y miR-149-3p sobre la autofagia. Descubrimos que el DCA activa la autofagia y que miR-149-3p no influye en la autofagia. Esto sugería que la autofagia no estaba implicada en el efecto del DCA/miR-149-3p en la regulación del metabolismo de la glucosa (Figura suplementaria S6).
Las PDKs, como reguladores clave en la glucólisis del cáncer, han causado gran preocupación debido a los resultados de muchos estudios [49]. Existen cuatro isoformas de PDK (PDK1-4), y cada una de ellas se produce de forma específica en un tejido, como se indica a continuación: La PDK1 se expresa en gran medida en el corazón, la PDK2 se expresa de forma ubicua, la PDK3 tiene una distribución tisular relativamente limitada y la PDK4 se expresa en el corazón y el músculo esquelético [27]. La PDK2 se expresa a niveles más altos en comparación con otras isoenzimas, lo que sugiere que puede ser la principal isoforma responsable de la regulación de la actividad enzimática del complejo piruvato deshidrogenasa (PDHC) [50]. Además, las isoenzimas PDK difieren en su regulación aguda por metabolitos [51]. Aquí, nos centramos en la PDK2, la isoforma más sensible al DCA [31]. Aunque las interacciones moleculares entre DCA y PDKs han sido enfocadas, el mecanismo potencial de la regulación transcripcional de PDK permanece poco claro. Un estudio reciente informó de que miR-182 desempeña funciones reguladoras en las vías metabólicas del cáncer de pulmón dirigiéndose a PDK4 [52]. El presente estudio demostró que PDK2 estaba regulada por miR-149-3p en el CCR y que los niveles de PDK2 en los tumores primarios de pacientes con CCR estaban inversamente correlacionados con la expresión de miR-149-3p. Dado que PDK2 está ampliamente presente en la mayoría de los tejidos, dirigirse a PDK2 puede ser una forma más importante y eficiente de eliminar las células tumorales y superar la quimiorresistencia.
Se ha informado de que miR-149-3p desempeña un papel vital en varios tipos de cáncer y es inducido por algunos fármacos antitumorales [36, 37, 53]. En particular, descubrimos que el tratamiento con DCA podía inducir la unión de p53 a la región upstream (-677 a -477) de miR-149 y que miR-149-3p era regulado al alza por el tratamiento con DCA de forma dependiente de p53. TP53, un supresor tumoral clásico, se inactiva con frecuencia en los tumores [54] y recientemente se ha informado de que regula el metabolismo de la glucosa en el cáncer. Se ha demostrado que el p53 Wt es capaz de inhibir el «efecto Warburg» mediante el control de PDK2. Sin embargo, la frecuencia de mutaciones de TP53 en el CCR es de ~40-50% [55, 56], lo que provoca la pérdida de su función supresora. Se ha informado de que los pacientes con CCR con TP53 wt obtienen un beneficio de supervivencia de la quimioterapia basada en 5-FU, pero aquellos con TP53 mutante no [57]. Nuestros resultados revelan un nuevo mecanismo entre p53 y PDK2 que está modulado por miR-149-3p. Estos hallazgos sugieren que los pacientes con TP53 mutante pueden beneficiarse más de la quimioterapia adjunta con miR-149-3p que con DCA. Teniendo en cuenta la alta frecuencia de mutaciones TP53 en el CCR, creemos que miR-149-3p desempeña un papel vital en la monitorización y modulación de la quimiosensibilidad en el CCR.
Las células cancerosas consumen una gran cantidad de glucosa y muestran un elevado estado de glucólisis aeróbica, por lo que reducir la captación de glucosa es una estrategia prometedora para restringir el crecimiento del cáncer [58]. Observamos que la elevación de miR-149-3p inhibía notablemente la glucólisis en células de CCR quimiorresistentes; sin embargo, en comparación con el grupo de control, en el grupo de xenoinjerto al que se inyectó intratumoralmente el imitador de miR-149-3p el SUVmáx permaneció inalterado. Tal vez este resultado contradictorio podría deberse al desarrollo de necrosis en la parte central de los tejidos tumorales subcutáneos, lo que justifica la investigación.
En conjunto, revelamos que la vía de señalización p53/miR-149-3p/PDK2 puede ser un objetivo potencial en la superación del CCR quimiorresistente al tratamiento con DCA, proporcionando una estrategia potencial para el tratamiento del CCR desde el ángulo de la intervención en el metabolismo tumoral (Fig. 7e).
Materiales y métodos
Tejido canceroso
Se incluyeron 28 pacientes con CCR del Noveno Hospital Popular Afiliado a la Facultad de Medicina de la Universidad Jiao Tong de Shanghái entre 2013 y 2016. Trece de estos pacientes recibieron quimioterapia postoperatoria basada en 5-FU y fueron seguidos durante al menos 3 años. Todos los tejidos se recogieron tras obtener el consentimiento informado, y todos los procedimientos con pacientes humanos se llevaron a cabo de acuerdo con las normas establecidas por el Comité Ético del Noveno Hospital Popular Afiliado a la Facultad de Medicina de la Universidad Jiao Tong de Shanghái. La información clínica de los pacientes con CCR se presenta en la Tabla suplementaria 1.
| Características de los pacientes con CCR | |
| Características | Total (n=28) |
| Edad-año | 65.4±10.5 |
| Sexo-no. (%) | |
| Varón | 17(60.7) |
| Mujer | 11(39.3) |
| Localización-no. (%) | |
| Recto | 10(35.7) |
| Colon | 18(64.3) |
| Estadiaje- nº (%) | |
| T1 o T2 | 6(21.4) |
| T3 o T4 | 22(78.6) |
| Ganglios linfáticos afectados- nº (%) | |
| N0 | 12(42.9) |
| N1 o N2 | 16(57.1) |
| Metástasis- no. (%) | |
| M0 | 27(96.4) |
| M1 | 1(3.6) |
| Quimioterapia postoperatoria con un régimen basado en fluorouracilo | |
| Sí | 21(75) |
| Seguimiento durante 3 años | |
| Sí | 13(61.9) |
| No | 8(38.1) |
| No | 7(25) |
Cultivo celular
La línea celular HCT-8/F resistente al 5-FU y su línea celular parental HCT-8 se adquirieron a iCell Bioscience, Inc. (Shanghai, China). La línea celular resistente al 5-FU HCT116/F y su línea celular parental HCT116 fueron proporcionadas amablemente por el Dr. Gu (Yanhong Gu, Universidad Médica de Nanjing, Jiangsu, China). Las células HCT116-/- fueron un regalo del Dr. Lu (Hua Lu, Universidad de Fudan, Shanghai, China). La línea celular 293T de riñón embrionario humano se obtuvo de la American Type Culture Collection (ATCC, Manassas, VA, EE.UU.). Las líneas celulares 293T, HCT116 y HCT-8 se cultivaron en medio Eagle modificado de Dulbecco (HyClone, Utah, EE.UU.) o medio RPMI-1640 (HyClone, Utah, EE.UU.) con un 10% de suero bovino fetal (Gemini, California, EE.UU.), 100 U/mL de penicilina y 100 μg/mL de estreptomicina (HyClone, Utah, EE.UU.) en una incubadora humidificada a 37 °C y 5% deCO2. Los medios de cultivo de las líneas celulares HCT-8/F y HCT116/F se suplementaron con 15 μg/ml de 5-FU y 5 μg/ml de 5-FU, respectivamente. Todas las líneas celulares fueron autentificadas mediante secuenciación de repeticiones cortas en tándem por Genetic Testing Biotechnology Corporation (Suzhou, Jiangsu, China). El DCA se adquirió a Sigma-Aldrich Co. Ltd. (MO, EE.UU.). lTD. (MO, EE.UU.).
Tinción de inmunofluorescencia de Edu y ROS
Las célulasHCT-8/Fy HCT116/F se sembraron en placas de 96 pocillos a razón de 15.000 células/pocillo. Tras una incubación de una noche, las células se trataron con 15 mM y 20 mM de DCA, respectivamente, durante 24 h. La tinción Edu se realizó siguiendo las instrucciones del fabricante (Ribobio, Guangzhou, China). Los niveles de ROS se midieron en células incubadas con 10 μM de diacetato de 2′,7′-diclorofluoresceína (DCF-DA) (Beyotime, Shanghái, China) durante 30 min a 37 °C. A continuación, las placas se lavaron dos veces y las células se analizaron con un microscopio de fluorescencia.
Crecimiento celular
Las célulasse sembraron en placas de 96 pocillos a 5000 células/pocillo durante toda la noche, se trataron con fármacos durante 24 h y, posteriormente, cada pocillo se sustituyó por mezclas de 10 μl de CCK8 (Dojindo, Japón) y 90 μl de medio de cultivo. La absorbancia se midió a un valor DO de 450 nm utilizando un lector enzimático de microplacas (BioTeck, Vermont, EE.UU.) dos horas después. La relación de inhibición del fármaco se calculó con la siguiente fórmula: 1-ODdroga/ODctrl. El IC50 de cada célula se calculó mediante GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Ensayo de apoptosis celular
Las célulasHCT-8/Fy HCT116/F se sembraron en placas de seis pocillos a una concentración de 2 ×105 células/pocillo. Las células se trataron con DCA (15 mM)/5-FU (50 μg/ml) y DCA (20 mM)/5-FU (25 μg/ml), respectivamente, durante 48 h. A continuación, se tripsinizaron las células, se lavaron y se tiñeron con anticuerpos Annexin V-FITC/PI o Annexin V-PE/7-AAD según el protocolo del fabricante (BD, CA, EE.UU.). La apoptosis se midió mediante citometría de flujo (BD, CA, EE.UU.).
Ensayo de formación de colonias
Las célulasHCT-8/Fy HCT116/F se trataron con DCA (15 mM)/5-FU (50 μg/ml) y DCA (20 mM)/5-FU (25 μg/ml) durante 24 h, respectivamente. A continuación, las células se sembraron en placas de seis pocillos a 1000 células por pocillo y se cultivaron en medio fresco a 37 °C durante 1 a 2 semanas, tras lo cual se fijaron con paraformaldehído al 4% durante 30 min; a continuación, las células se tiñeron con violeta cristal al 1%, y el número de colonias celulares se contó con un contador (Gelcount, Optronix, Oxford).
Transfección génica transitoria
Los imitadores e inhibidores de miR-149-3p, siPDK2, sip53 y sus correspondientes secuencias de oligonucleótidos NC fueron sintetizados por GenePharma (Shanghai, China). El plásmido Flag-p53 fue proporcionado amablemente por el Dr. Lu (Hua Lu, Universidad de Fudan, Shanghai, China). La transfección se realizó con Lipofectamine 3000 (Invitrogen, CA, EE.UU.) a una concentración final de 50 nmol/L (imitadores y siRNAs) o 100 nmol/L (inhibidores). Las células se cosecharon para los ensayos 24 o 48 h después de la transfección. Las secuencias de los siRNAs, imitadores e inhibidores se muestran en la Tabla Suplementaria 2.
| secuencia de siARN, imitador e inhibidor | |
| siPDK2-1 | sentido: 5′-GACCGAUGCUGUCAUCUAUU-3′ |
| antisentido: 5′-AAUAGAUGACAGCAUCGGUC-3′ | |
| siPDK2-2 | sentido: 5′-GACUCUUCAGCUACAUGUA-3′ |
| antisentido: 5′-UACAUGUAGCUGAAGAGUC-3′ | |
| NC | sentido: 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| antisentido: 5′-ACGUGACACGUUCGGAGAATT-3′ | |
| mimic-149-3p | sentido: 5′-AGGGAGGGACGGGGGCUGUGC-3′ |
| antisentido: 5′-ACAGCCCCCGUCCCUCCCUUU-3′ | |
| inhibidor-NC | 5′-CAGUACUUUUGUGUAGUACAA-3′ |
| inhibidor-149-3p | 5′-GCACAGCCCCCGUCCCUCCCU-3′ |
| sip53 | sentido: 5′-GUAAUCUACUGGGACGGAAtt-3′ |
| antisentido: 5′-UUCCGUCCCAGUAGAUUACca-3′ |
Transfección génica estable
LV-PDK2 y el correspondiente virus NC se adquirieron en GeneChem (Shanghai, China). shPDK2-1, shPDK2-2 y el plásmido de control se adquirieron en GeneChem (Shanghai, China). El sobrenadante del virus se cosechó a partir de células 293 T. Posteriormente, las células CRC se infectaron con el virus y se analizaron con puromicina. La eficacia de la infección se validó mediante citometría de flujo y microscopía de fluorescencia. El ARNm y la expresión proteica de PDK2 se analizaron mediante PCR cuantitativa en tiempo real y Western blot.
ensayos de luciferasareportera 3′-UTR
Las secuencias de unión a miR-149-3p wt o mutantes en la 3′UTR de PDK2 humana se clonaron en la secuencia descendente de la luciferasa pmiR-RB-Reporter (Ribobio, Guangzhou, China), referidas a WT, MUT1, MUT2 y MUT3 en la Fig. 3c. las células 293 T y las células HCT116 se sembraron en placas de 24 pocillos seguidas de cotransfección con 500 ng de construcciones reporteras y 50 nmol/L de miR-149-3p mímico o un NC utilizando Lipofectamine 3000 (Thermo Fisher Scientific, Waltham MA). La actividad luciferasa se midió tras 48 h de incubación utilizando el Dual-Luciferase Reporter Assay System (Promega, Madison, EE.UU.) de acuerdo con el protocolo del fabricante.
Consumode glucosa yproducción de lactato
Las célulasHCT-8/Fy HCT116/F se sembraron en placas de 24 pocillos a razón de 1 ×105 células/pocillo durante la noche y, a continuación, se trataron con DCA 15 mM y 20 mM, respectivamente, durante 24 h. Tras el tratamiento, las células se cultivaron en medio libre de rojo de fenol que contenía un 10% de suero bovino fetal durante 24 h. Se cosechó el medio de cultivo y se midieron el consumo de glucosa y la producción de lactato. El lactato se midió con un kit de ensayo de lactato (Njjcbio, Nanjin, China), y la glucosa se midió con un kit de ensayo de glucosa (Rsbio, Shanghai, China). Todos los valores se estandarizaron contando un número igual de células. Para evaluar el estado de la glucólisis, se añadieron 100 ng/mL de oligomicina (un inhibidor de la ATP sintasa; Sangon Biotech) a las células cultivadas durante 6 h. Se midió y determinó la relación de la concentración de lactato en presencia y ausencia de oligomicina, tal como se ha descrito previamente [46].
Ensayo de tasa glucolítica Seahorse XF-96
Lascélulas se sembraron en una placa de cultivo de 96 pocillos a una densidad de 25.000 células/pocillo y se incubaron durante la noche en medio de crecimiento que contenía un 10% de suero bovino fetal. El cartucho sensor se hidrató durante la noche. Al día siguiente, el medio de las células se cambió a un medio de ensayo de tampón bajo sin bicarbonato suplementado con glucosa, piruvato sódico y glutamina. Después de incubar las células durante 1 h a 37 °C en una incubadora sin CO2, se midieron la tasa de consumo de oxígeno y la tasa de acidificación extracelular antes y después de la inyección de DCA/ctrl, Rotenona (Rot) + antimicina A (AA) y 2-deoxi-d-glucosa (2-DG) utilizando el instrumento Seahorse XF (Agilent, Santa Clara, CA) como se ha descrito previamente [59, 60]. Los experimentos se realizaron en tiempo real en cinco a seis pocillos replicados. El software Wave (Agilent, Santa Clara, CA) calculó automáticamente los GlycoPER, incluyendo el glycoPER basal, el glycoPER inducido y el glycoPER compensatorio.
PCR cuantitativa en tiempo real y microarrays de miARN
El ARN total se extrajo de tejidos o células de CCR con el reactivo TRIzol (Life, CA, EE.UU.). El ADNc se sintetizó utilizando el kit de reactivos PrimeScript RT (TaKaRa, Tokio, Japón). El ensayo de microarrays se realizó con tres réplicas de células HCT116 tratadas con 5 mM, 10 mM o 20 mM de DCA durante 12, 24 o 48 h. Los datos originales se subieron a la base de datos GEO (GSE125309). La PCR cuantitativa en tiempo real se realizó utilizando la premezcla Ex Taq 420 A (TaKaRa, Tokio, Japón) en la plataforma ABI-7500. Actina y U6 se utilizaron como controles internos. Las secuencias de los cebadores se presentan en la Tabla suplementaria 3.
| secuencias de cebadores para la PCR cuantitativa en tiempo real de transcripción inversa | |
| Actina | F: 5′-CTCCATCCTGGCCTCGCTGT-3′ |
| R: 5′-GCTGTCACCTTCACCGTTCC-3′ | |
| PDK2 | F: 5′-CGCTGGCTGGCTTTGGTTTTATG-3′ |
| R: 5′-ACAGGGCCTTGAGATAGATG-3′ | |
| PDK1 | F: 5′-GCTGTATGGCCTGCAAGATG-3′ |
| R: 5′-GCTGTCCTGGTGATTTTGCA-3′ | |
| PDK3 | F: 5′-GGTTTGCCAATTTCCCGTCTG-3′ |
| R: 5′-CATCGGCTTCAGGCGTGGTC-3′ | |
| PDK4 | F: 5′-GAGAATTATTGACCGCCTCT-3′ |
| R: 5′-CGAGAAATTGGCAAGCCGTAA-3′ | |
| HK1 | F: 5′-CTTACTAAGGGATGCGATAAA-3′ |
| R: 5′-TCCCAACAATGAGTCCAACC-3′ | |
| TP53 | F: 5′-CCCAAGCAATGGATGATTTGA-3′ |
| R: 5′-GGCATTCTGGGAGCTTCATCT-3′ | |
| p21 | F: 5′-CTGGACTGTTTTCTCTCGGCTC-3′ |
| R: 5′-TGTATATTCAGCATTGTGGGAGGA-3′ | |
| MDM2 | F: 5′-ATGAATCCCCCCCTTCCAT-3′ |
| R: 5′-CAGGAAGCCAATTCTCACGAA-3′ | |
| PUMA | F: 5′-ACAGTACGAGCGGCGGAGACAA-3′ |
| R: 5′-GGCGGGTGCAGGCACCTAATT-3′ | |
| miR-149-3p | RT: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGG ATACGACGCACAG-3′ |
| F: 5′-ACAGGGAGGGACGGGGGGG-3′ | |
| R: 5′-ATCCAGTGCAGGGTCCGAGG-3′ | |
| miR-128-3p | RT: 5′-TCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGA TACGACAAAGAG-3′ |
| F: 5′-CGCGTCACAGTGAACCGGT-3′ | |
| R: 5′-AGTGCAGGGTCCGAGGTATT-3′ | |
| miR-500a-5p | RT: 5′-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCAC TGGATACGACTCTCACC-3′ |
Western blots
Se cargaron 30 microgramos de lisados de proteína total y se aplicaron los anticuerpos primarios anti-PDK2 (sc-100534, Santa Cruz, California, EE.UU.), anti-p-PDHA1 (S293) (ABS204, Merck, Darmstadt, Alemania), anti-PDHA1 (ab168379, Abcam, Cambridge, Reino Unido), anti-p53 (sc-126, Santa Cruz, California, EE.UU.), anti-c-PARP (D64E10, CST, Massachusetts, EE.UU.), anti-Bax (D2E11, CST, Massachusetts, EE.UU.), anti-LC3B (L7543, sigma, MO, EE.UU.), anti-GAPDH (Proteintech, Wuhan, China) y anti-α-tubulina (Proteintech, Wuhan, China). Los anticuerpos secundarios se adquirieron a Sungene (Tianjin, China). Los ensayos blot se visualizaron mediante un sistema de imagen de quimioluminiscencia (Bioshine, Shanghai, China).
Inmunoprecipitación de la cromatina (ChIP)
Las célulasHCT116sembradas en placas de 10 cm se trataron con o sin 20 mM de DCA durante 24 h y, a continuación, se procedió a la fijación celular y a la fragmentación cromosómica siguiendo las instrucciones del fabricante (Pierce Agarose ChIP Kit, Thermo). La cromatina se incubó con anticuerpos IgG y anti-p53 (Sigma, MO, EE.UU.) a 4 °C durante la noche. Tras la incubación, se añadieron 60 ul de proteína A agarosa/ADN de esperma de salmón. A continuación, el complejo precipitado se lavó con los tampones de lavado IP 1, 2 y 3, y se eluyó con el tampón de elución. La reticulación se invirtió añadiendo 6 μl de NaCl 5 M y 2 ul de proteinasa K a 65 °C durante 1,5 h. El ADN inmunoprecipitado y el ADN de extracto de células enteras (entrada) se purificaron y luego se utilizaron para análisis PCR utilizando los cebadores pertinentes. Se utilizó un cebador de control para controlar el experimento. Las secuencias de los cebadores para la PCR se presentan en la Tabla suplementaria 3.
Xenoinjerto tumoral subcutáneo en ratones desnudos e imágenes micro-PET/CT
En primer lugar, se implantaron por vía subcutánea 1 ×107 células HCT-8/F diluidas en 100 μl de PBS en ratones desnudos (macho, 6 semanas). Los ratones se dividieron aleatoriamente en seis grupos (seis por grupo) al cabo de 12 días. Los ratones de los grupos I a IV recibieron una inyección intraperitoneal diaria de PBS, DCA (50 mg/kg)/PBS, 5-FU (10 mg/kg)/PBS y 5-FU (10 mg/kg)/DCA (50 mg/kg), respectivamente. A los ratones de los grupos V y VI se les inyectó intratumoralmente PBS o DCA (50 mg/kg), respectivamente, en días alternos y una inyección intraperitoneal de 5-FU (10 mg/kg) en días alternos. El volumen tumoral se midió a ciegas cada 3 días. Los ratones fueron sacrificados tras 3 semanas de tratamiento, y los tumores fueron disecados, pesados y congelados a -80 °C para su posterior estudio.
Para evaluar si miR-149-3p ejerce un efecto de quimiosensibilización, se establecieron otros dos grupos de modelos animales. Brevemente, se implantaron por vía subcutánea 6 ×106 células HCT-8/F en ratones. Una vez que el tamaño de los tumores fue de ~50 mm3, los ratones recibieron una dosis intraperitoneal de 5-FU (10 mg/kg) en días alternos, así como una inyección intratumoral de 5 nmol de imitadores de miR-149-3p conjugados con colesterol o un NC cada 3 días durante 3 semanas. Posteriormente, tres ratones de cada grupo fueron ayunados durante la noche y se les inyectó por vía intravenosa 0,15 mCi de 18F-FDG. se realizó una micro-PET-CT con 18F-FDG (Siemens, Berlín, Alemania) al cabo de 60 min. Las imágenes de adquisición de PET se mostraron mediante un mapa de pseudocolores en el que el color rojo indicaba una elevada captación de 18F-FDG. Se utilizaron SUVmáx y MTV para determinar la actividad de 18F-FDG-PET. Todos los experimentos y el cuidado de los animales fueron aprobados por el Comité Ético del Noveno Hospital Popular Afiliado a la Facultad de Medicina de la Universidad Jiao Tong de Shanghai.
Tinción inmunohistoquímica y de inmunofluorescencia
Brevemente, las secciones de tejido se incubaron con los anticuerpos primarios de nuevo Ki67 (Servicebio, Wuhan, China) y PDK2 (Proteintech, Wuhan, China) a 4 °C durante la noche, y después se incubaron con el anticuerpo secundario. La reacción cromogénica se realizó con 3,3-diaminobencidina y se contrateñó con hematoxilina. La puntuación inmunorreactiva (IRS) fue calculada por dos investigadores ciegos a la asignación de grupos. IRS = SI (intensidad de la tinción) × PP (porcentaje de células positivas). La SI se asignó de la siguiente manera: 0 × negativo; 1 × débil; 2 × moderado; 3 × fuerte. El PP se define como 0 × 0%; 1 × 0-25%; 2 × 25-50%; 3 × 50-75%; 4 × 75-100%. Las secciones congeladas de seis milímetros se tiñeron con un kit de reacción TUNEL (Roche, Basilea, Suiza) y se contrateñieron con DAPI. Las imágenes se capturaron utilizando un microscopio de fluorescencia con los filtros de excitación y emisión adecuados.
Análisis estadístico
Los datos se analizaron con el programa GraphPad Prism 6.0.
Los datos se presentan como la media ± DE/SEM de tres experimentos independientes. Cada experimento se realizó al menos tres veces. Se utilizó la prueba t de Student de dos colas para comparar las diferencias entre los dos grupos. Para las comparaciones múltiples se utilizó el ANOVA de una vía seguido de la prueba post hoc de Bonferroni. Las curvas de Kaplan-Meier para los análisis de supervivencia se determinaron mediante la prueba de rangos logarítmicos. La relación entre miR-149-3p y PDK2 se evaluó mediante el análisis del coeficiente de correlación de rangos de Spearman. Un valor P <0,05 se consideró estadísticamente significativo.
Agradecimientos
Este trabajo ha sido financiado por la Fundación Nacional de Ciencias Naturales de China (81272745, 81872419 y 81272404) y el Programa para Profesores de Nombramiento Especial (Eastern Scholar to JW) de las Instituciones de Enseñanza Superior de Shanghai. Damos las gracias al Dr. Yanhong Gu por facilitarnos las líneas celulares HCT116 resistentes al 5-FU y al Dr. Hua Lu por facilitarnos el plásmido p53.
Cumplimiento de las normas éticas
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Nota del editor
Springer Nature se mantiene neutral con respecto a las reclamaciones jurisdiccionales en los mapas publicados y las afiliaciones institucionales.
Acceso abierto
Este artículo está bajo una Licencia Creative Commons Atribución 4.0 Internacional, que permite su uso, compartición, adaptación, distribución y reproducción en cualquier medio o formato, siempre que se cite debidamente al autor o autores originales y la fuente, se proporcione un enlace a la licencia Creative Commons y se indique si se han realizado cambios. Las imágenes u otro material de terceros en este artículo están incluidos en la licencia Creative Commons del artículo, a menos que se indique lo contrario en una línea de crédito al material. Si el material no está incluido en la licencia Creative Commons del artículo y su uso previsto no está permitido por la normativa legal o excede el uso permitido, deberá obtener permiso directamente del titular de los derechos de autor. Para ver una copia de esta licencia, visite http://creativecommons. org/licenses/by/4.0/.
REFERENCIAS
1 Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, et al. Incidencia y mortalidad por cáncer en China, 2014. Chin J Cancer Res. 2018;30:1-12.2 Siegel RL, Miller KD, Jemal A. Estadísticas sobre el cáncer, 2018. CA: Cancer J Clin. 2018;68:7-30.
3 Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB 3rd, et al. El dicloroacetato altera el metabolismo de Warburg, inhibe el crecimiento celular y aumenta la sensibilidad a los rayos X de las células humanas de cáncer de pulmón A549 y H1299 NSC. Free Radic Biol Med. 2015;89:263-73.
4 Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167: 1540-54 e12.
5 Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Informe de caso de un acontecimiento adverso grave tras la administración de células T transducidas con un receptor de antígeno quimérico que reconoce ERBB2. Mol Ther: J Am Soc Gene Ther. 2010;18:843-51. El dicloroacetato restaura la quimiosensibilidad del cáncer colorrectal a través del mecanismo mediado por p53/miR-149-3p/PDK2. . . 483
6 Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Tratamiento del cáncer y estadísticas de supervivencia, 2016. CA: Cancer J Clin. 2016;66:271-89.
7 Hammond WA, Swaika A, Mody K. Resistencia farmacológica en el cáncer colorrectal: una revisión. Therapeutic Adv Med Oncol. 2016;8:57-84.
8 Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecán combinado con fluorouracilo en comparación con fluorouracilo solo como tratamiento de primera línea para el cáncer colorrectal metastásico: un ensayo aleatorizado multicéntrico. Lancet. 2000;355:1041-7.
9 Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. New Engl J Med. 2000;343:905-14.
10 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74.
11 Matthew G, Vander Heiden LCC, Craig BT. Comprender el efecto Warburg: los requisitos metabólicos de la proliferación celular. Science. 2009;324:1029-33.
12 Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
13 Shaw RJ. Glucose metabolism and cancer (Metabolismo de la glucosa y cáncer). Curr Opin Cell Biol. 2006;18:598-608.
14 Van Dang C, Pollak M. ¿Por qué cáncer y metabolismo? Why now? Cancer Metab. 2013;1:1.
15 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532.
16 Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism (Regulación del metabolismo de las células cancerosas). Nat Rev Cancer. 2011;11:85-95.
17 Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613-21.
18 Guo WQZ, Wang Z, et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatology. 2015;62:1132-44.
19 Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology. 2015;149:1587-98 e11.
20 Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. MicroRNA-129- 5p regulates glycolysis and cell proliferation by targeting the glucose transporter SLC2A3 in gastric cancer cells. Front Pharmacol. 2018;9:502.
21 Bartel DP. MicroRNAs: genómica, biogénesis, mecanismo y función. Cell. 2004;116:281-97.
22 Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167-79.
23 Huang S, He X. microRNAs: tiny RNA molecules, huge driving forces to move the cell. Protein Cell. 2010;1:916-26.
24 Zhang Y, Wang J. MicroRNAs are important regulators of drug resistance in colorectal cancer. Biol Chem. 2017;398:929-38.
25 Stacpoole PW, Nagaraja NV, Hutson AD. Eficacia del dicloroacetato como fármaco reductor del lactato. J Clin Pharmacol. 2003;43:683-91.
26 Kankotia S, Stacpoole PW. Dicloroacetato y cáncer: ¿nuevo hogar para un medicamento huérfano? Biochim Biophys Acta. 2014;1846:617-29.
27 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochemical J. 1998;329(Pt 1):191-6.
28 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell 2007;11:37-51.
29 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4.
30 Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33:603-10.
31 Papandreou I, Goliasova T, Denko NC. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer. 2011;128:1001-8.
32 Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989-94.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 2010;102:1746-52.
34 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett. 2010;297:75-83.
35 Bertoli G, Cava C, Castiglioni I. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015;5:1122-43.
36 Cao D, Jia Z, You L, Wu Y, Hou Z, Suo Y, et al. 18betaglycyrrhetinic acid suppresses gastric cancer by activation of miR149-3p-Wnt-1 signaling. Oncotarget. 2016;7:71960-73.
37 Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, et al. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br J Pharm. 2017;174:553-68.
38 Kato M, Li J, Chuang JL, Chuang DT. Mecanismos estructurales distintos para la inhibición de isoformas de piruvato deshidrogenasa quinasa por AZD7545, dicloroacetato y radicicol. Structure. 2007;15:992-1004.
39 Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. 2005;272:3004-14.
40 Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, BelouecheBabari M, Koh KX, et al. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br J Pharm. 2014;171:3255-67.
41 Stacpoole PW. La farmacología de dicloroacetato. Metab: Clin Exp. 1989;38:1124-44.
42 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Therap. 2008;324:1163-71.
43 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013;73:7277-89.
44 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. Inactivation of the HIF-1alpha/ PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035-47.
45 Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5- fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res. 2014;321:219-30.
46 Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72-81. 484 Y. Liang et al.
47 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016;371:20-9.
48 Kimmelman AC, White E. Autofagia y metabolismo tumoral. Cell Metab 2017;25:1037-43.
49 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. La activación mitocondrial mediante la inhibición de PDKII suprime la señalización de HIF1a y la angiogénesis en el cáncer. Oncogene. 2013;32:1638-50.
50 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversidad de la familia de genes de la piruvato deshidrogenasa quinasa en humanos. J Biol Chem. 1995;270:28989-94.
51 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003; 284: E855-62.
52 Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182- PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98.
53 Bellazzo A, Di Minin G, Valentino E, Sicari D, Torre D, Marchionni L, et al. Cell-autonomous and cell non-autonomous downregulation of tumor suppressor DAB2IP by microRNA-149- 3p promotes aggressiveness of cancer cells. Diferencia de muerte celular. 2018;25:1224-38.
54 Vázquez A, Bond EE, Levine AJ, Bond GL. La genética de la vía p53, la apoptosis y la terapia del cáncer. Nat Rev Drug Discov. 2008;7:979-87.
55 Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. El supresor tumoral p53 en el control del metabolismo y la ferroptosis. Front Endocrinol. 2018;9:124.
56 Contractor T, Harris CR. p53 regula negativamente la transcripción de la piruvato deshidrogenasa quinasa Pdk2. Cancer Res. 2012;72:560-7.
57 Iacopetta B. Mutación de TP53 en el cáncer colorrectal. Hum Mutat. 2003;21:271-6.
58 Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927-30.
59 Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, Tempera I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by EpsteinBarr virus latent membrane protein 1. PLoS Pathog. 2018;14: e1007394.
60 Hlouschek J, Ritter V, Wirsdorfer F, Klein D, Jendrossek V, Matschke J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018;439:24-38.
Contenido relacionado: