Yu Liang1, Lidan Hou1, Linjing Li1, Lei Li1, Liming Zhu1, Yu Wang1, Xin Huang1, Yichao Hou1, Danxi Zhu1, Huimin Zou1, Yan Gu2, Xiaoling Weng3,4, Yingying Wang5, Yue Li6, Tianqi Wu3, Mengfei Yao3, Isabelle Gross7,8, Christian Gaiddon9,10, Meng Luo2, Jianhua Wang3, Xiangjun Meng1
1 Département de gastroentérologie, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Chine
2 Département de chirurgie générale, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Chine
3 Institut du cancer, Fudan University Shanghai Cancer Center, Fudan University, Shanghai, Chine
4 Ningbo Aitagene Technology Co. LTD, Shanghai, Chine
5 Département de biochimie et de biologie moléculaire et
Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, Chine
6 Pathology Center, Shanghai First People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, Chine
7 INSERM UMR_S1113, Strasbourg F-67200, France
8 FMTS, Universite de Strasbourg Strasbourg, Strasbourg F-67000, France
9Universitéde Strasbourg, Inserm IRFAC UMR_S1113, Laboratoire Stress Response and Innovative Therapy « Streinth », Strasbourg 67200, France
10 CLCC Paul Strauss, Strasbourg, France
Meng Luo [email protected]
Jianhua Wang [email protected]
Xiangjun Meng [email protected]
Ces auteurs ont contribué à parts égales : Yu Liang, Lidan Hou
Reçu : 16 mars 2019
Révisé : 17 septembre 2019
Accepté : 19 septembre 2019
Publié : 9 octobre 20199 octobre 2019
Résumé
Le développement de la chimiorésistance reste un défi majeur qui explique la létalité du cancer colorectal (CCR). Le dichloroacétate (DCA) était à l’origine utilisé comme régulateur métabolique dans le traitement des maladies métaboliques ; ici, le DCA a été testé pour identifier les mécanismes sous-jacents à la chimiorésistance du CCR. Nous avons constaté que le DCA augmentait considérablement la chimiosensibilité des cellules du CCR au fluorouracile (5-FU) et réduisait la formation de colonies en raison de niveaux élevés d’apoptose. En utilisant l’essai microarray, nous avons noté que miR-149-3p était impliqué dans la chimiorésistance du CRC, qui a été modulée par p53 de type sauvage après le traitement DCA. De plus, PDK2 a été identifié comme une cible directe de miR-149-3p. Les analyses mécanistiques ont montré que la surexpression de miR-149-3p augmentait l’apoptose induite par le 5-FU et réduisait le métabolisme du glucose, de manière similaire aux effets du knockdown de PDK2. De plus, la surexpression de PDK2 a partiellement inversé l’effet inhibiteur du miR-149-3p sur le métabolisme du glucose. Enfin, on a constaté que le traitement au DCA et la surexpression du miR-149-3p dans les cellules de CCR résistantes au 5-FU sensibilisent nettement l’effet chimiothérapeutique du 5-FU in vivo, et cet effet a également été validé dans une petite cohorte rétrospective de patients atteints de CCR. Dans l’ensemble, nous avons déterminé que la voie de signalisation p53/miR-149-3p/PDK2 peut potentiellement être ciblée par un traitement au DCA pour vaincre le CCR chimiorésistant.
Informations supplémentaires : La version en ligne de cet article (https:// doi.org/10.1038/s41388-019-1035-8) contient du matériel supplémentaire, qui est disponible pour les utilisateurs autorisés.
Introduction
Le cancer colorectal (CCR) est la quatrième cause de mortalité liée au cancer en Chine [1] et la deuxième cause de mortalité liée au cancer aux États-Unis [2]. Il est principalement dû aux métastases et à l’échec de la chimiothérapie en raison de la résistance aux médicaments, ce qui entraîne environ 50 000 décès par an [3].
Récemment, alors que l’étoile émergente PD1/PDL1 a suscité un grand intérêt, et que de plus en plus d’agents biothérapeutiques ont montré des résultats encourageants dans le traitement du cancer, le taux d’efficacité limité et les effets indésirables inévitables freinent son utilisation en clinique [4,5]. Actuellement, la chimiothérapie reste un choix majeur en clinique, en particulier pour les patients atteints de cancers non résécables à un stade avancé ou métastatique [6], mais le développement de la résistance aux médicaments reste la plus grande limite de la chimiothérapie [7]. Par conséquent, l’exploration des mécanismes de résistance aux médicaments et la recherche de nouvelles combinaisons de médicaments anticancéreux classiques afin d’optimiser leur efficacité pourraient être bénéfiques pour le traitement du CCR. Le fluorouracil (5-FU) étant le médicament chimiothérapeutique le plus couramment utilisé pour le CCR, des lignées cellulaires CCR résistantes au 5-FU ont été utilisées dans cette étude [8,9].
Une anomalie du métabolisme du glucose représente l’un des aspects majeurs des caractéristiques du cancer [10]. On sait que le développement d’une masse cellulaire incontrôlée entraîne une mauvaise vascularisation de la tumeur, ce qui réduit l’apport en oxygène. Par conséquent, les cellules cancéreuses s’adaptent aux altérations du microenvironnement en déplaçant leur métabolisme du métabolisme oxydatif vers le métabolisme glycolytique, qui est basé sur un approvisionnement en glucose et produit du lactate. Ce changement est appelé « effet Warburg » et est couramment observé dans diverses cellules cancéreuses comme l’une des caractéristiques remarquables [11- 13]. L’accumulation récente de données de recherche a conduit à la nécessité d’affiner la théorie de Warburg [14]; par exemple, il a été démontré que le changement de métabolisme est impliqué dans la chimiorésistance [15]; par conséquent, le ciblage des modèles de métabolisme des cellules cancéreuses pourrait être exploité pour surmonter la chimiorésistance [16,17].
Les miARN représentent une catégorie de petits ARN non codants endogènes qui régulent la traduction et la dégradation des ARNm [21] et sont impliqués dans de nombreux autres processus biologiques, notamment la prolifération, la migration, l’apoptose, l’auto-renouvellement, l’initiation, le développement du cancer et la chimiorésistance des cellules [22-24].
Les preuves indiquent que cibler le métabolisme anormal des cellules cancéreuses a été une voie de recherche intense visant à » asphyxier la tumeur « , dont les stratégies consistent à inhiber les enzymes clés impliquées dans le métabolisme glycolytique [15]. Dans ce manuscrit, le dichloroacétate (DCA) a été initialement utilisé pour traiter l’acidose lactique et la maladie mitochondriale héréditaire [25]. Le DCA inhibe l’activité enzymatique des kinases de la pyruvate déshydrogénase (PDK1-4), qui est nécessaire pour transformer le pyruvate en acétyl-CoA, reliant le métabolisme glycolytique au cycle de l’acide citrique [26,27]; le DCA a récemment été signalé comme ayant des effets anticancéreux [28-31]. Cependant, le mécanisme qui sous-tend l’effet du DCA sur le traitement du cancer colorectal reste insaisissable.
La présente étude s’est concentrée sur le mécanisme moléculaire impliqué dans la régulation du métabolisme du glucose et la résistance à la chimiothérapie dans le cancer colorectal. En utilisant le DCA dans les cellules du cancer colorectal, nous avons étudié les rôles des miARN associés et ainsi révélé une voie de signalisation qui explique la résistance au traitement par 5-FU.
Résultats
LeDCA restaure la chimiosensibilité des cellules de CCR résistantes au 5-FU
Il a été signalé que le DCA est un médicament antitumoral efficace qui agit en ciblant les voies liées à l’énergie dans certains cancers [32]; cependant, l’effet du DCA sur les cellules de CCR chimiorésistantes n’a pas été bien étudié. À l’aide du test CCK8, nous avons constaté que, par rapport aux lignées cellulaires parentales HCT-8 et HCT116, les cellules HCT-8/F et HCT116/F résistantes au 5-FU étaient insensibles au 5-FU (figure supplémentaire 1A) et que les concentrations inhibitrices semi-maximales (CI50) du DCA dans les cellules HCT-8/F et HCT116/F étaient d’environ 15 et 20 mM, respectivement, ce qui est conforme aux rapports précédents [33, 34] (figure supplémentaire 1B). Nous avons ensuite constaté que le DCA inhibait de manière significative la synthèse de l’ADN (figure supplémentaire 1C, panneau supérieur) et induisait la génération de ROS (figure supplémentaire 1C, panneau inférieur) dans les cellules CRC résistantes au 5-FU. Les marqueurs du métabolisme énergétique, notamment la consommation de glucose, la production de lactate et la glycolyse, étaient nettement plus élevés dans les cellules de cancer colorectal résistantes au 5-FU que dans les cellules de cancer colorectal sensibles au 5-FU, tandis que l’ajout de DCA réduisait nettement l’expression de ces marqueurs (Fig. 1a). Compte tenu d’un prétraitement sans sérum de 6 h dans la mesure de la glycolyse, un kit de dosage de la vitesse glycolytique Seahorse XF a été utilisé pour éliminer l’effet du prétraitement et pour mesurer la vitesse glycolytique en temps réel. Le taux d’efflux de protons glycolytiques (glycoPER) reflète le taux d’acidification extracellulaire de la glycolyse. L’ajout de DCA a réduit de manière significative la glycolyse induite par rapport aux contrôles respectifs (Fig. 1b et Fig. 1D supplémentaire).
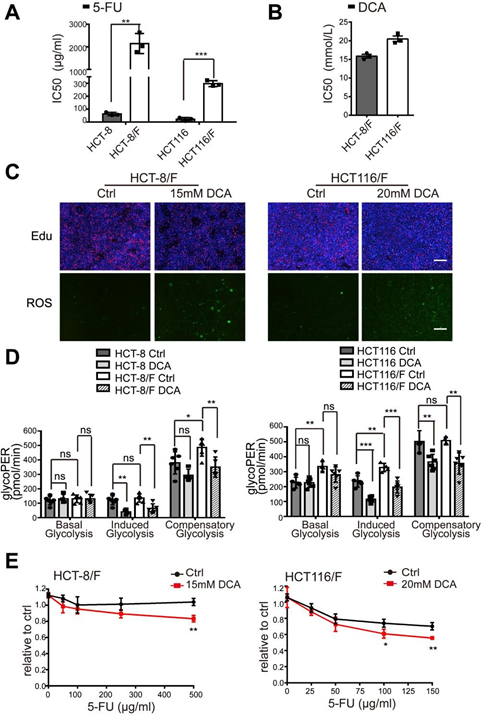
(A) Les cellules de CRC ont été traitées avec différentes concentrations de 5-FU pendant 24 heures. La CI50 du 5-FU dans chaque cellule a été calculée. (B) Les cellules HCT-8/F et HCT116/F ont été traitées avec différentes concentrations de DCA pendant 24 heures. La CI50 du DCA dans chaque cellule a été calculée. (C) Les cellules HCT-8/F et HCT116/F ont été traitées avec 15 mM et 20 mM de DCA, respectivement, pendant 24 heures. Images représentatives de la coloration par immunofluorescence d’Edu (panneau supérieur) et images représentatives de la coloration par immunofluorescence de ROS (panneau inférieur), barre d’échelle : 200 μm. (D) La détermination du taux de glycolyse, y compris le taux de glycolyse basal, le taux de glycolyse induit et le taux de glycolyse compensatoire, a été calculée par le Seahorse Glycolytic Rate Assay Report Generator. (E) La croissance cellulaire a été déterminée par un test CCK8 après traitement avec 5-FU et DCA. Les gammes de dosage du 5-FU étaient de 100 ug/ml à 500 ug/ml pour les cellules HCT-8/F et de 25 ug/ml à 150 ug/ml pour les cellules HCT116/F. Les résultats de trois expériences indépendantes sont présentés sous forme de moyenne ± SEM. Chaque expérience a été réalisée en 3 à 6 répétitions biologiques. *p < 0,05 ; **, P < 0,01 ; ***, P < 0,001.
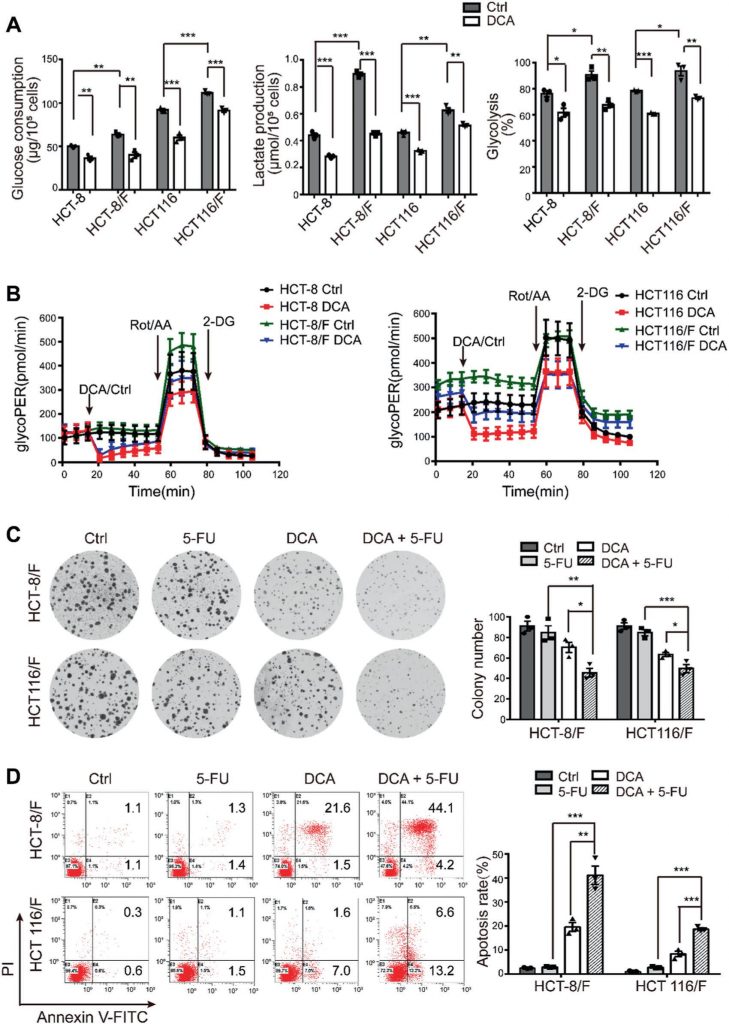
Le DCA a permis de surmonter de manière significative la résistance au 5-FU des cellules HCT-8/F et HCT116/F, comme le montrent les mesures de la croissance cellulaire (figure supplémentaire 1E). La capacité de formation de colonies a été considérablement inhibée (Fig. 1c) et l’apoptose a été induite de façon marquée dans un traitement combiné (Fig. 1d), tous ces effets ayant été quantifiés. Ces résultats suggèrent que le DCA peut restaurer la chimiosensibilité des cellules de CCR résistantes au 5-FU.
lemiR-149-3p joue un rôle crucial dans la chimiosensibilité des cellules du CCR
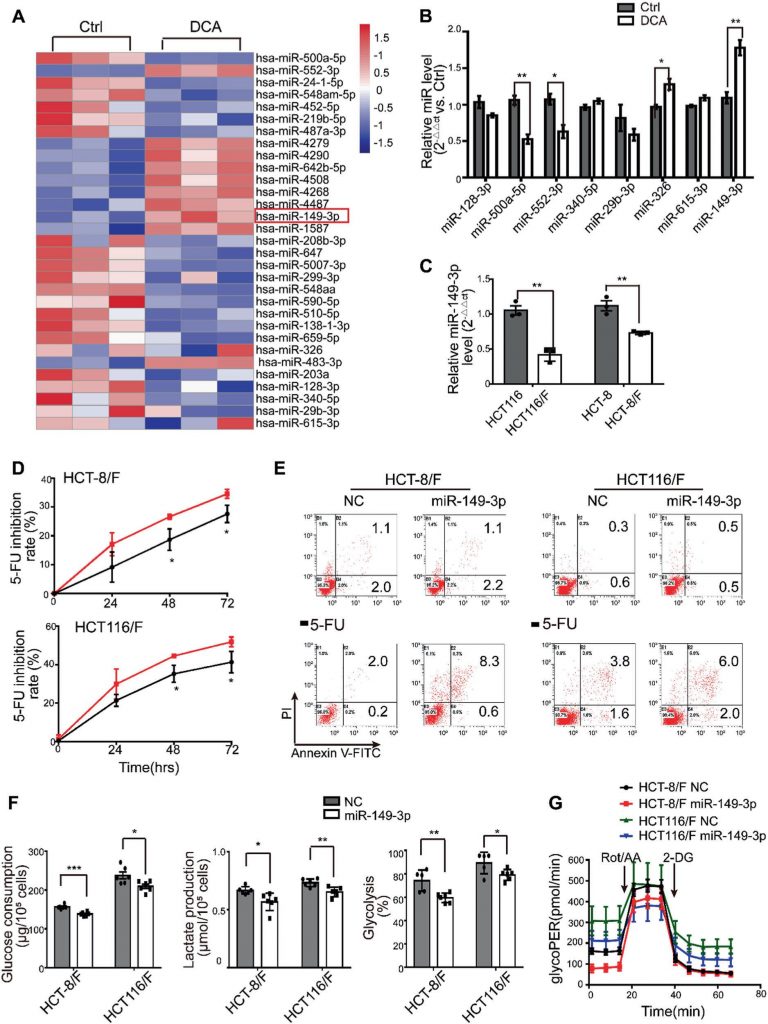
Les miRNA ont été considérés comme un outil thérapeutique prometteur pour leurs effets sur la suppression des tumeurs [35]. À cet égard, nous avons d’abord déterminé les profils d’expression des miRNA en utilisant la matrice de miRNA contenant 2059 miRNA humains. Un total de 119 miRNAs ont été différentiellement exprimés en réponse au DCA dans les cellules HCT116 (Fig. 2a et Fig. S2A supplémentaire). Parmi eux, les niveaux d’expression de huit miRNA ont été confirmés par PCR quantitative en temps réel (Fig. 2b), et miR-149-3p a finalement été trouvé pour être régulé par DCA d’une manière dose-dépendante (Supplementary Fig. S2B).
Ensuite, nous avons constaté que les cellules HCT116 ayant des niveaux basaux plus élevés de miR-149-3p conféraient une plus grande sensibilité au 5-FU et au L-OHP, comme le montre la figure supplémentaire 2C. De plus, la transfection anti-miR-149-3p des cellules HCT116 a remarquablement réduit l’effet chimiothérapeutique du 5-FU (Fig. 2D-E supplémentaires). Notamment, les niveaux de miR-149-3p dans les cellules de CRC chimiosensibles étaient significativement plus élevés que les niveaux dans les cellules de CRC chimiorésistantes (Fig. 2c). Par conséquent, la transfection de mimiques miR-149-3p a augmenté de manière significative le taux d’inhibition ; a favorisé l’apoptose cellulaire induite par le 5-FU ; a réduit la consommation de glucose, la production de lactate et la glycolyse dans les cellules HCT-8/F et HCT116/F (Fig. 2d-f). Un test de taux glycolytique Seahorse XF a montré que l’expression de miR-149-3p réduisait la glycolyse basale dans les cellules CRC résistantes au 5-FU, ce qui était cohérent avec les résultats ci-dessus (Fig. 2g et Fig. 2F supplémentaire). Ces résultats suggèrent que miR-149-3p est favorable pour surmonter la chimiorésistance dans les cellules CRC.
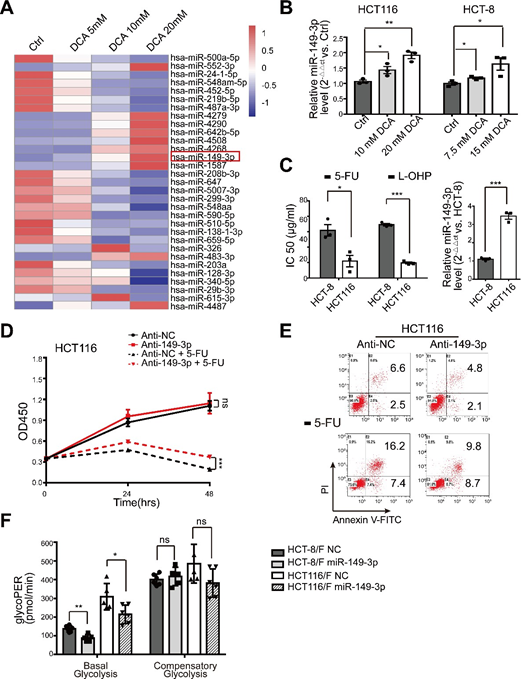
(A) La carte thermique du profil des microARN différentiellement exprimés dans les cellules HCT116 traitées avec le contrôle, 5 mM, 10 mM, 20 mM DCA pendant 24 heures. (B) L’ARN total a été préparé 24 heures après le traitement au DCA 10/7,5 mM et 20/15 mM des cellules HCT116 et HCT-8. Le niveau de miR-149-3p a été analysé par PCR quantitative en temps réel. (C) Les cellules HCT-8 et HCT116 ont été traitées avec différentes concentrations de 5-FU et d’oxaliplatine (L-OHP) respectivement pendant 24 heures et la concentration inhibitrice demi-maximale (IC50) a été calculée (panneau de gauche). Les niveaux basaux de miR-149-3p ont été déterminés par PCR quantitative en temps réel dans les cellules HCT-8 et HCT116 (panneau de droite). (D-E) Les cellules HCT116 ont été transfectées de manière transitoire avec l’Anti-NC ou l’inhibiteur de miR-149-3p. Après transfection, les cellules ont été traitées avec 25 μg/ml de 5-FU pendant 24 heures. La croissance cellulaire et l’apoptose ont été déterminées respectivement par CCK8 et cytométrie de flux. (F) La détermination du taux de glycolyse incluant le glycoPER basal et le glycoPER compensatoire dans les cellules CRC résistantes au 5-FU transfectées avec NC ou une mimique de miR-149-3p ont été calculées par Seahorse Glycolytic Rate Assay Report Generator. Les données de trois expériences indépendantes sont présentées sous forme de moyenne ± SEM. Chaque expérience a été réalisée dans 3 à 6 répétitions biologiques. *p < 0,05 ; **, P < 0,01 ; ***, P < 0,001 ; ns, non significatif.
Le DCA induit l’expression de miR-149-3p par le biais de p53 de type sauvage (wt)
Sachant que plusieurs études récentes ont révélé que miR-149-3p est régulé par plusieurs médicaments [36,37], nous avons ensuite déterminé comment miR-149-3p est régulé par le DCA. Nous avons constaté que le DCA augmente de manière significative l’expression de p53 wt et ses signaux en aval, y compris l’expression de p21, PUMA, et MDM2, dans les cellules CRC (Fig. 3a, b). De plus, nous avons noté que les altérations de l’expression de p53 wt étaient capables de moduler de manière significative l’expression de miR-149-3p, comme le montrent les Fig. 3c, d, indiquant que miR-149-3p était régulé positivement par p53 wt. Par conséquent, en utilisant la lignée cellulaire HCT116 p53-null (TP53-/-), nous avons constaté que miR-149-3p n’était pas régulé par le traitement DCA, mais l’expression ectopique de p53 a inversé cet effet. En outre, nous avons utilisé la nutlin-3, un inhibiteur puissant qui inhibe l’interaction MDM2-p53, conduisant à l’activation de p53 comme un contrôle positif, avec l’expression de miR-149-3p étant élevé dans la wt HCT116 lignée cellulaire (TP53+/+) (Fig. 3e-g). Mécaniquement, quatre sites de liaison putatifs de p53 à la région d’ADN génomique flanquante de miR-149 ont été prédits en utilisant le logiciel d’analyse bioinformatique (IGV). Des essais ChIP ont ensuite été réalisés dans les cellules en utilisant un anticorps contre p53 wt. L’ADN arraché a été amplifié par PCR ordinaire avec des amorces conçues sur la base de ces sites. Nos résultats ont montré que, par rapport à la région 4, la région 3 était nettement enrichie après traitement par le DCA dans la chromatine de HCT116 immunoprécipitée par la p53 wt (Fig. 3h), ce qui suggère que seule la région 3 contient un site de liaison spécifique activé par le DCA. Ces résultats indiquent que le DCA module le miR-149-3p par l’intermédiaire de wt p53.
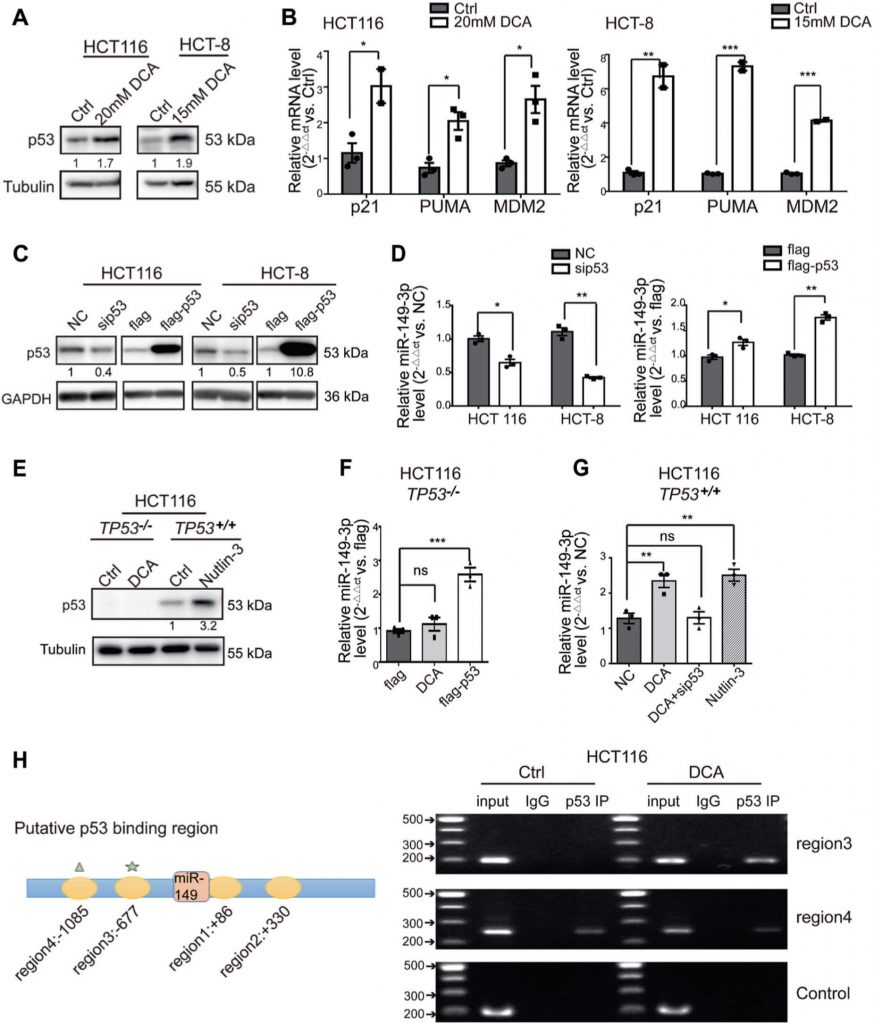
PDK2 est une cible directe du miR-149-3p
Pour élucider les mécanismes par lesquels le miR-149-3p régule la chimiosensibilité des cellules du CCR, nous avons analysé les gènes associés au métabolisme énergétique, qui sont régulés par le miR-149-3p en utilisant deux plateformes publiques (TargetScan et miRDB). Finalement, nous avons identifié que la pyruvate déshydrogénase kinase 2 (PDK2) et l’hexokinase 1 (HK1) sont des candidats potentiels (Fig. 4a). Pour confirmer ces résultats, la mimique ou l’inhibiteur du miR-149-3p ont été transfectés dans des cellules de CRC. Nous avons trouvé que les niveaux de mRNA de PDK2 ont été négativement réglés par miR-149-3p, mais pas par HK1 (Fig. 4b et Fig. 3A supplémentaire). Deux sites de liaison possibles du miR-149-3p dans le 3′-UTR de PDK2 ont été trouvés, et le test de rapporteur de double-luciferase a indiqué que le miR-149-3p se lie au site prédit (686-693) du 3′-UTR de PDK2 (Fig. 4c, d). Nous avons alors confirmé que les niveaux de protéine de PDK2 ont été négativement réglés par miR-149-3p (Fig. 4e).
PDK possède quatre isozymes nommés PDK1, 2, 3 et 4, qui ont tous été rapportés comme étant régulés par le DCA [26, 38, 39]. Nous avons ensuite analysé l’expression de l’ARNm de PDK1-4 dans les cellules HCT8 et HCT116 après traitement au DCA. Le DCA a inhibé de manière significative l’expression de l’ARNm de PDK2, mais pas celle des autres isozymes de PDK (figure supplémentaire S3B). En outre, la transfection d’anti-149-3p a partiellement inversé la réduction de PDK2 par le DCA (Fig. 4f). Ensuite, nous avons déterminé les niveaux de protéines de PDK2 et de sa sous-unité pyruvate déshydrogénase E1-alpha (PDHA1) en aval dans les cellules de CRC sensibles au 5-FU et résistantes au 5-FU. Les niveaux basaux de PDK2 étaient élevés dans les cellules de CRC chimiorésistantes, par rapport aux niveaux des cellules chimiosensibles. En accord avec cette élévation, la phosphorylation de PDHA1 était également élevée (Fig. 4g).
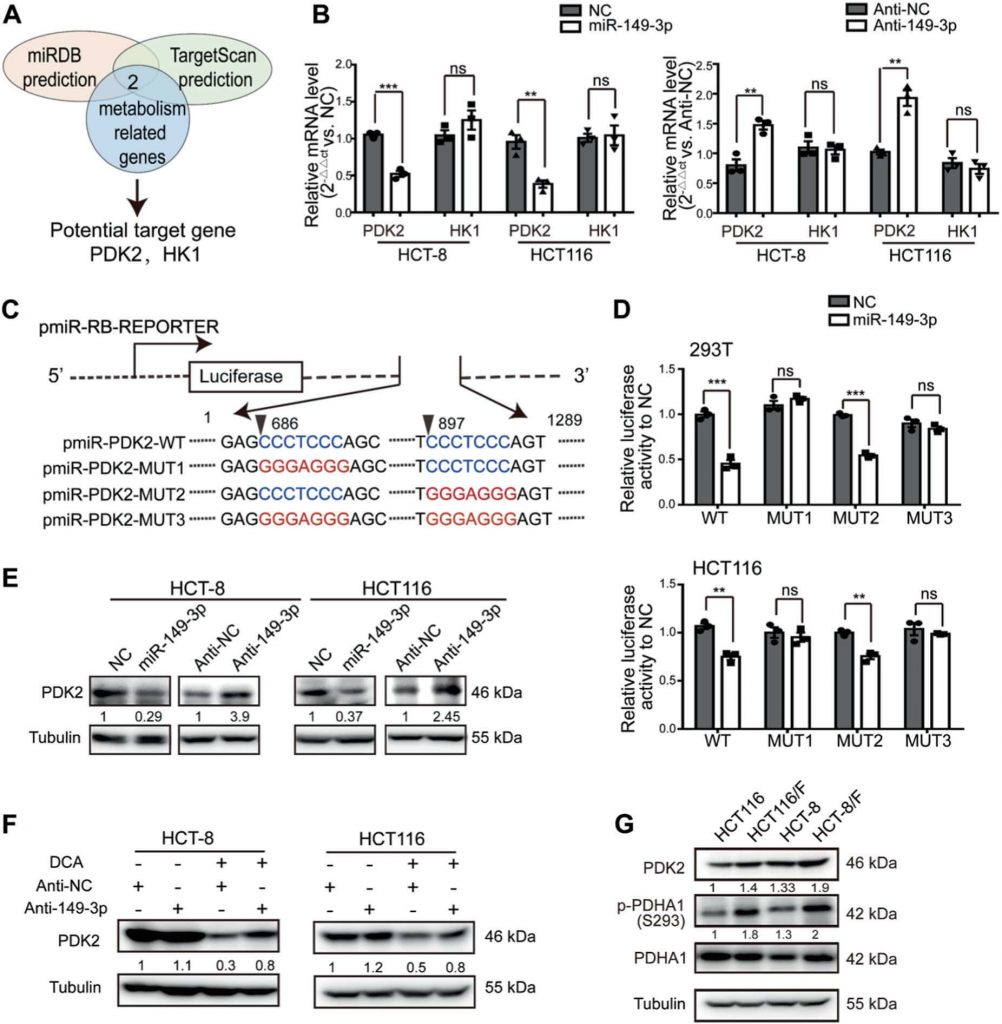
La voie miR-149-3p/PDK2 régule la chimiosensibilité
Pour comprendre comment la voie miR-149-3p/PDK2 régule la réponse des cellules du CCR au 5-FU, la lignée cellulaire HCT-8/F a été choisie comme lignée cellulaire représentative pour étudier si les niveaux d’expression de miR-149-3p et de PDK2 affectaient la réponse cellulaire au 5-FU. Le knockdown de PDK2 dans les cellules HCT-8/F a inhibé la phosphorylation de PDHA1 (Fig. 5a) et réduit les marqueurs du métabolisme énergétique, tels que la consommation de glucose, la production de lactate et la glycolyse (Fig. 5b). De plus, les réductions de PDK2 ont pu renforcer les effets du 5-FU sur l’augmentation des niveaux de PARP clivée (c-PARP) et de Bax, qui sont tous deux des biomarqueurs reconnus de l’apoptose cellulaire dans les cellules HCT-8/F (Fig. 5c). En outre, le knockdown de PDK2 a augmenté la chimiosensibilité au 5-FU des cellules HCT-8/F, comme le montrent les tests CCK8 et de formation de colonies (Fig. 5d, e). Le knockdown de PDK2 a favorisé l’apoptose induite par le 5-FU dans les cellules HCT-8/F et HCT116/F, comme le montre la figure supplémentaire S4A, tandis que la surexpression de PDK2 a favorisé la phosphorylation de PDHA1 et atténué l’apoptose cellulaire induite par le 5-FU dans les cellules HCT-8 et HCT116 (figures supplémentaires S4B-S4C).
En outre, la surexpression de PDK2 a inversé l’effet inhibiteur du miR-149-3p sur PDK2 (Fig. 5f) et a partiellement aboli l’effet inhibiteur du miR-149-3p sur la consommation de glucose, la production de lactate et la glycolyse (Fig. 5g). L’expression ectopique de PDK2 a nettement aboli l’effet inhibiteur de miR-149-3p sur la croissance cellulaire et la formation de colonies dans les cellules HCT-8/F traitées au 5-FU (Fig. 5h, i). L’ensemble de nos résultats indique que la voie miR-149-3p/PDK2 restaure la chimiosensibilité en ciblant, au moins partiellement, le métabolisme du glucose dans les cellules CRC chimiorésistantes.
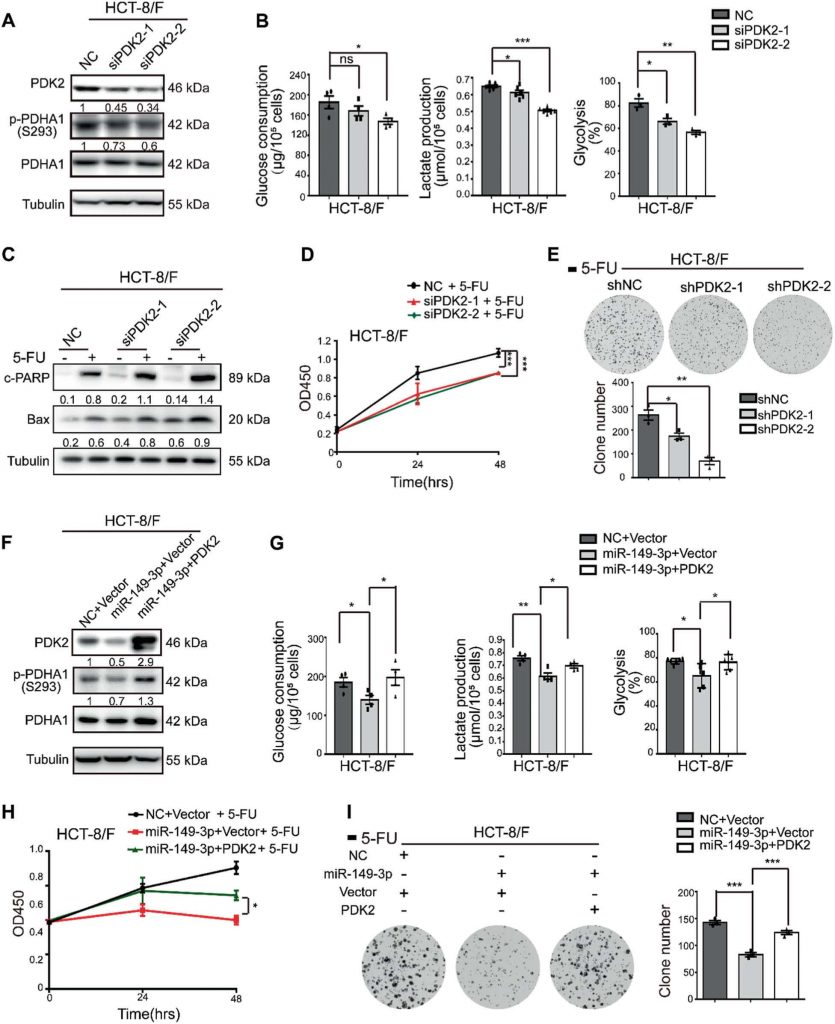
LeDCA améliore la chimiosensibilité du 5-FU in vivo
Ensuite, le 5-FU, le DCA ou une combinaison de 5-FU et de DCA ont été injectés par voie intrapéritonéale dans le modèle de xénogreffe sous-cutanée (appelé groupe intrapéritonéal). Compte tenu de l’insuffisance de l’apport sanguin dans la partie centrale des tumeurs sous-cutanées, nous avons également injecté par voie intratumorale du DCA ou du PBS plus une injection intrapéritonéale de 5-FU (groupe intratumoral). Les combinaisons de DCA et de 5-FU ont montré de meilleurs effets inhibiteurs sur la croissance tumorale que le DCA ou le 5-FU seuls après quatre semaines d’injection dans le groupe intrapéritonéal (Fig. 6a, b). L’expression de Ki-67 a été réduite après le traitement combiné dans le groupe intrapéritonéal (Fig. 6c). Le DCA a inhibé de manière significative la croissance tumorale dans le groupe intratumoral par rapport à celle du groupe PBS (Fig. 6d), et le miR-149-3p a été régulé à la hausse dans le groupe ayant reçu une injection intratumorale de DCA, ce qui correspond aux résultats obtenus in vitro (Fig. 6e). L’injection intratumorale de DCA a également favorisé l’apoptose tumorale (Fig. 6F).
Pour évaluer davantage l’effet du miR-149-3p sur la réponse des cellules du CCR au 5-FU in vivo, le modèle de xénogreffe sous-cutanée a été soumis à une injection intratumorale d’agomiR-149-3p ou d’agomiR-NC plus une injection intrapéritonéale de 5-FU. Le miR-149-3p a significativement inhibé la croissance tumorale dans le groupe intratumoral par rapport à celle du groupe témoin dans lequel le contrôle agomiR-négatif (NC) a été injecté par voie intratumorale (Fig. 6g, h). La surexpression de miR-149-3p a été validée par PCR quantitative en temps réel (Fig. 6i), et la surexpression de miR-149-3p a favorisé l’apoptose (Fig. 6j) et réduit l’expression de PDK2 et de p-PDHA1 (Fig. 6k, l). L’imagerie tumorale a été réalisée après 3 semaines de traitement par microtomographie par émission de positons (TEP) au 18F-fluorodésoxyglucose (FDG). la captation du 18F-FDG a été observée sur les sites d’implantation de la tumeur, et la captation maximale standardisée (SUVmax) ainsi que les volumes tumoraux métaboliques (MTV) ont été mesurés. Aucune différence dans le SUVmax n’a été observée entre les deux groupes, tandis que le MTV était significativement diminué dans le groupe miR-149-3p (Fig. 6m).
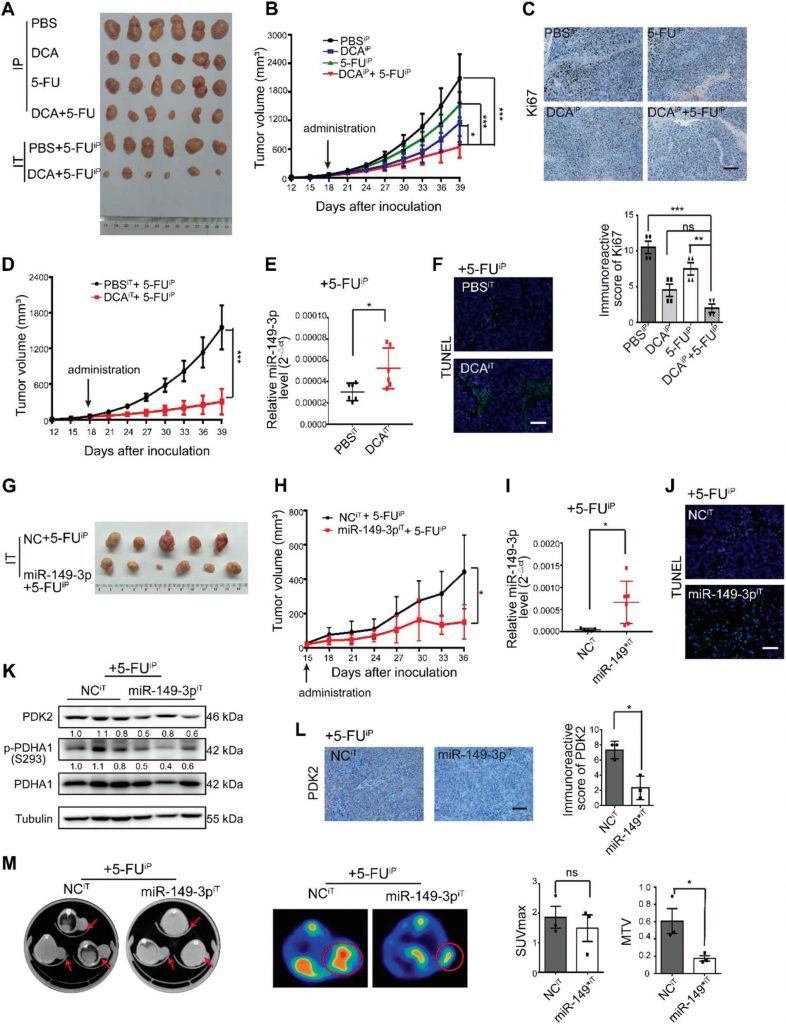
miR-149-3p est inversement corrélé avec PDK2 chez les patients atteints de CRC
Une corrélation inverse significative entre les niveaux d’ARNm de miR-149-3p et de PDK2 a été observée dans les tissus humains de CRC (Fig. 7a, b). Parmi eux, huit patients en condition/maladie stable (SD) dans les 3 ans après la chimiothérapie ont exprimé un niveau plus élevé de miR-149-3p que cinq patients avec une maladie progressive (PD) (Fig. S5A supplémentaire). Cinq paires de patients PD et SD avec la même pathologie et le même stade TNM ont été analysées, et la coloration de PDK2 a varié de manière significative dans les échantillons des patients PD et SD (Supplementary Fig. S5B). Les patients CRC de la base de données TCGA présentant une forte expression de PDK2 étaient également caractérisés par une survie globale (OS) plus faible (Fig. 7c). La base de données TCGA a également montré que l’expression de PDK2 dans le groupe p53 wt était réduite par rapport à celle du groupe p53 mutant (Fig. 7d). Ces résultats suggèrent que PDK2 est régulé négativement par miR-149-3p chez les patients atteints de CRC.
Discussion
Le CCR se caractérise par des anomalies tumorigènes et des voies métaboliques altérées. Il est l’une des principales causes de décès par cancer [2]. La résistance à la chimiothérapie est la principale cause d’échec du traitement [7]. Dans cette étude, nous avons découvert que le DCA pouvait augmenter l’effet chimiothérapeutique du 5-FU dans les cellules de CCR chimiorésistantes et que l’activation de la voie p53/miR-149-3p/PDK2 était capable d’augmenter la chimiosensibilité in vitro et in vivo.
De plus en plus de preuves indiquent que l’augmentation de la glycolyse est étroitement liée à la résistance à la chimiothérapie [15, 17, 40]. Ici, nous avons également constaté que, par rapport à leurs lignées cellulaires parentales, les cellules de CCR chimiorésistantes présentaient une consommation de glucose, une production de lactate et une glycolyse élevées, ce qui suggère que les anomalies métaboliques sont une caractéristique typique, mais que les mécanismes moléculaires restent encore flous dans les cellules de CCR.
On a observé que le DCA diminuait les niveaux de lactate sanguin in vivo chez les rongeurs à des doses de ~25-50 mg/kg/24 h [41] et que le DCA était utilisé avec une large gamme de doses allant de 1 à 50 mM [26]. Notamment, l’effet indésirable du DCA chez l’homme se limite généralement à une neuropathie périphérique sensorielle et motrice réversible, qui est influencée par l’âge et le génotype [42]. Récemment, le DCA a été identifié comme une nouvelle thérapie métabolique pour divers patients atteints de cancer [26, 29].
Il a été signalé que le DCA est capable d’inhiber l’activité de la PDK et de convertir le pyruvate en acétyl-CoA, ce qui entraîne un déplacement de la production d’énergie de la glycolyse vers la phosphorylation oxydative mitochondriale [43,44]. Il a également été démontré que le DCA atténue la résistance au 5-FU induite par l’hypoxie dans le cancer gastrique [45], qu’il surmonte la résistance au sorafénib dans le carcinome hépatocellulaire [46] et qu’il atténue la résistance au cisplatine dans le cancer de la tête et du cou [47]. Notre étude a révélé que le DCA était capable d’atténuer la chimiorésistance des cellules du CCR au 5-FU. De plus, nous avons démontré que le DCA réduisait la consommation de glucose et la production de lactate dans les cellules de CCR chimiorésistantes au niveau basal des cellules de CCR chimiosensibles. Étant donné que l’autophagie a le potentiel d’alimenter le métabolisme du cancer [48], nous avons déterminé l’effet du DCA et du miR-149-3p sur l’autophagie. Nous avons constaté que le DCA active l’autophagie et que le miR-149-3p n’a aucune influence sur l’autophagie. Cela suggère que l’autophagie n’est pas impliquée dans l’effet du DCA/miR-149-3p dans la régulation du métabolisme du glucose (figure supplémentaire S6).
Les PDK, en tant que régulateurs clés de la glycolyse du cancer, ont suscité de grandes inquiétudes en raison des résultats de nombreuses études [49]. Il existe quatre isoformes de PDK (PDK1-4), et chacune d’entre elles s’exprime d’une manière spécifique au tissu comme suit : La PDK1 est fortement exprimée dans le cœur, la PDK2 est exprimée de manière ubiquitaire, la PDK3 a une distribution tissulaire relativement limitée et la PDK4 est exprimée dans le cœur et les muscles squelettiques [27]. La PDK2 est exprimée à des niveaux plus élevés que les autres isoenzymes, ce qui suggère qu’elle pourrait être la principale isoforme responsable de la régulation de l’activité enzymatique du complexe pyruvate déshydrogénase (PDHC) [50]. En outre, les isoenzymes PDK diffèrent dans leur régulation aiguë par les métabolites [51]. Nous nous sommes concentrés ici sur les PDK2, les isoformes les plus sensibles au DCA [31]. Bien que les interactions moléculaires entre le DCA et les PDK aient été étudiées, le mécanisme potentiel de la régulation transcriptionnelle des PDK n’est toujours pas clair. Une étude récente a rapporté que miR-182 joue un rôle régulateur dans les voies métaboliques du cancer du poumon en ciblant PDK4 [52]. La présente étude a démontré que PDK2 était régulé par miR-149-3p dans le CRC et que les niveaux de PDK2 dans les tumeurs primaires des patients CRC étaient inversement corrélés à l’expression de miR-149-3p. Puisque PDK2 est largement présent dans la plupart des tissus, cibler PDK2 peut être un moyen plus important et plus efficace de tuer les cellules tumorales et de surmonter la chimiorésistance.
Il a été signalé que miR-149-3p joue un rôle vital dans divers cancers et est induit par certains médicaments antitumoraux [36, 37, 53]. Notamment, nous avons constaté que le traitement au DCA pouvait induire la liaison de p53 à la région en amont (-677 à -477) du miR-149 et que le miR-149-3p était régulé à la hausse par le traitement au DCA d’une manière p53-dépendante. TP53, un suppresseur de tumeur classique, est fréquemment inactivé dans les tumeurs [54] et a été récemment signalé comme régulant le métabolisme du glucose dans le cancer. Il a été démontré que la protéine p53 Wt est capable d’inhiber » l’effet Warburg » en contrôlant PDK2. Cependant, la fréquence des mutations de TP53 dans le cancer colorectal est de 40 à 50 % [55, 56], ce qui entraîne une perte de sa fonction suppressive. Il a été signalé que les patients atteints de CCR avec TP53 wt bénéficient d’un avantage de survie après une chimiothérapie à base de 5-FU, mais pas ceux avec TP53 mutant [57]. Nos résultats révèlent un nouveau mécanisme entre p53 et PDK2 qui est modulé par miR-149-3p. Ces résultats suggèrent que les patients présentant une mutation de TP53 pourraient bénéficier davantage d’une chimiothérapie d’adjonction avec miR-149-3p plutôt qu’avec DCA. Compte tenu de la fréquence élevée des mutations de TP53 dans le CCR, nous pensons que le miR-149-3p joue un rôle essentiel dans la surveillance et la modulation de la chimiosensibilité dans le CCR.
Les cellules cancéreuses consomment une grande quantité de glucose et présentent un état de glycolyse aérobie élevé. Par conséquent, la réduction de l’absorption de glucose est une stratégie prometteuse pour limiter la croissance du cancer [58]. Nous avons observé que l’élévation du miR-149-3p inhibait remarquablement la glycolyse dans les cellules chimiorésistantes du CCR ; cependant, par rapport au groupe témoin, le groupe de xénogreffes auquel on avait injecté la mimique du miR-149-3p par voie intratumorale présentait un SUVmax inchangé. Ce résultat contradictoire pourrait être dû au développement de la nécrose dans la partie centrale des tissus tumoraux sous-cutanés, ce qui mérite d’être étudié.
Dans l’ensemble, nous révélons que la voie de signalisation p53/miR-149-3p/PDK2 peut potentiellement être ciblée pour vaincre le CCR chimiorésistant après un traitement au DCA, ce qui constitue une stratégie potentielle pour le traitement du CCR en intervenant sur le métabolisme tumoral (Fig. 7e).
Matériel et méthodes
Tissu cancéreux
Vingt-huit patients atteints de CRC du Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine ont été inclus entre 2013 et 2016. Treize de ces patients ont reçu une chimiothérapie postopératoire à base de 5-FU et ont été suivis pendant au moins 3 ans. Tous les tissus ont été collectés après avoir obtenu un consentement éclairé, et toutes les procédures impliquant des patients humains ont été menées conformément aux règlements établis par le Comité d’éthique du Neuvième hôpital populaire affilié au Collège médical de l’Université Jiao Tong de Shanghai. Les informations cliniques des patients atteints de CCR sont présentées dans le tableau supplémentaire 1.
| Caractéristiques des patients atteints de CCR | |
| Caractéristique | Total(n=28) |
| Age-an | 65.4±10.5 |
| Sexe-no. (%) | |
| Homme | 17(60.7) |
| Femme | 11(39.3) |
| Localisation-no. (%) | |
| Rectum | 10(35.7) |
| Colon | 18(64.3) |
| Stade – nbre (%) | |
| T1 ou T2 | 6(21.4) |
| T3 ou T4 | 22(78.6) |
| Ganglions lymphatiques impliqués – nbre (%) | |
| N0 | 12(42.9) |
| N1 ou N2 | 16(57.1) |
| Métastases – nbre (%) | |
| M0 | 27(96.4) |
| M1 | 1(3.6) |
| Chimiothérapie postopératoire avec un régime à base de fluorouracil | |
| Oui | 21(75) |
| Suivi pendant 3 ans | |
| Oui | 13(61.9) |
| Non | 8(38.1) |
| Non | 7(25) |
Culture cellulaire
La lignée cellulaire HCT-8/F résistante au 5-FU et sa lignée cellulaire parentale HCT-8 ont été achetées auprès de iCell Bioscience, Inc. (Shanghai, Chine). La lignée cellulaire résistante au 5-FU HCT116/F et sa lignée cellulaire parentale HCT116 ont été aimablement fournies par le Dr Gu (Yanhong Gu, Nanjing Medical University, Jiangsu, Chine). Les cellules HCT116-/- ont été offertes par le Dr Lu (Hua Lu, Université Fudan, Shanghai, Chine). La lignée cellulaire 293T de rein embryonnaire humain a été obtenue auprès de l’American Type Culture Collection (ATCC, Manassas, VA, USA). Les lignées cellulaires 293T, HCT116 et HCT-8 ont été cultivées dans du milieu Dulbecco’s Modified Eagle’s (HyClone, Utah, États-Unis) ou du milieu RPMI-1640 (HyClone, Utah, États-Unis) contenant 10 % de sérum bovin fœtal (Gemini, Californie, États-Unis), 100 U/mL de pénicilline et 100 μg/mL de streptomycine (HyClone, Utah, États-Unis) dans un incubateur humidifié à 37 °C et 5 % deCO2. Les milieux de culture des lignées cellulaires HCT-8/F et HCT116/F ont été complétés par 15 μg/ml de 5-FU et 5 μg/ml de 5-FU, respectivement. Toutes les lignées cellulaires ont été authentifiées par séquençage des répétitions en tandem courtes par Genetic Testing Biotechnology Corporation (Suzhou, Jiangsu, Chine). Le DCA a été acheté auprès de Sigma-Aldrich Co. Ltd. (MO, ÉTATS-UNIS).
Coloration par immunofluorescence de Edu et ROS
Les cellulesHCT-8/Fet HCT116/F ont été ensemencées dans des plaques à 96 puits à raison de 15 000 cellules/puits. Après une incubation d’une nuit, les cellules ont été traitées avec 15 mM et 20 mM de DCA, respectivement, pendant 24 h. La coloration de l’Edu a été réalisée conformément aux instructions du fabricant (Ribobio, Guangzhou, Chine). Les niveaux de ROS ont été mesurés dans les cellules incubées avec 10 μM de 2′,7′-dichlorofluorescéine diacétate (DCF-DA) (Beyotime, Shanghai, Chine) pendant 30 min à 37 °C. Les plaques ont ensuite été lavées deux fois et les cellules ont été analysées à l’aide d’un microscope à fluorescence.
Croissance cellulaire
Les cellules ont été ensemencées dans des plaques à 96 puits à raison de 5 000 cellules/puits pendant une nuit, traitées avec des médicaments pendant 24 h, et chaque puits a ensuite été remplacé par des mélanges de 10 μl de CCK8 (Dojindo, Japon) et 90 μl de milieu de culture. L’absorbance a été mesurée à une valeur de DO de 450 nm à l’aide d’un lecteur de microplaques enzymatiques (BioTeck, Vermont, États-Unis) deux heures plus tard. Le rapport d’inhibition du médicament a été calculé à l’aide de la formule suivante : 1-ODdrug/ODctrl. La CI50 de chaque cellule a été calculée par GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Test d’apoptose cellulaire
Les cellulesHCT-8/Fet HCT116/F ont été ensemencées dans des plaques à six puits à une concentration de 2 ×105 cellules/puits. Les cellules ont été traitées avec DCA (15 mM) / 5-FU (50 μg/ml) et DCA (20 mM) /5-FU (25 μg/ml), respectivement, pendant 48 h. Les cellules ont ensuite été trypsinées, lavées et colorées avec des anticorps Annexin V-FITC/PI ou Annexin V-PE/7-AAD selon le protocole du fabricant (BD, CA, USA). L’apoptose a été mesurée par cytométrie de flux (BD, CA, USA).
Test de formation de colonies
Les cellulesHCT-8/Fet HCT116/F ont été traitées respectivement par DCA (15 mM)/5-FU (50 μg/ml) et DCA (20 mM)/5-FU (25 μg/ml) pendant 24 heures. Ensuite, les cellules ont été ensemencées dans des plaques à six puits à raison de 1000 cellules par puits et cultivées dans du milieu frais à 37 °C pendant 1 à 2 semaines, puis fixées avec du paraformaldéhyde à 4 % pendant 30 min ; les cellules ont ensuite été colorées avec du cristal violet à 1 %, et le nombre de colonies cellulaires a été compté par un compteur (Gelcount, Optronix, Oxford).
Transfection génétique transitoire
Les mimiques et inhibiteurs de miR-149-3p, siPDK2, sip53, et leurs séquences d’oligonucléotides NC correspondantes ont été synthétisés par GenePharma (Shanghai, Chine). Le plasmide Flag-p53 a été aimablement fourni par le Dr Lu (Hua Lu, Université Fudan, Shanghai, Chine). La transfection a été réalisée avec Lipofectamine 3000 (Invitrogen, CA, USA) à une concentration finale de 50 nmol/L (mimiques et siRNA) ou 100 nmol/L (inhibiteurs). Les cellules ont été récoltées pour les analyses 24 ou 48 heures après la transfection. Les séquences des siRNA, des mimiques et des inhibiteurs sont indiquées dans le tableau supplémentaire 2.
| séquence du siRNA, de la mimique et de l’inhibiteur | |
| siPDK2-1 | sens : 5′-GACCGAUGCUGUCAUCUAUU-3′ |
| antisens : 5′-AAUAGAUGACAGCAUCGGUC-3′ | |
| siPDK2-2 | sens : 5′-GACUCUUCAGCUACAUGUA-3 |
| anti-sens : 5′-UACAUGUAGCUGAAGAGAGUC-3 | |
| NC | sens : 5′-UUCCGAACGUGUCACGUTT-3′ |
| anti-sens : 5′-ACGUGACACGUUCGGAGAATT-3 | |
| mimic-149-3p | sens : 5′-AGGGAGGGACGGGGGCUGUGC-3′ ; – antisens : 5′-ACAGACGUUCGGAGAATT-3′ |
| antisens : 5′-ACAGCCCCCGUCCCCCUUUU-3 | |
| inhibiteur-NC | 5′-CAGUACUUUUUGUGUAGUACAA-3′ |
| inhibiteur-149-3p | 5′-GCACAGCCCCCGUCCCUCCCU-3′ |
| sip53 | sens : 5′-GUAAUCUACUGGGACGGAAtt-3′ ; et |
| anti-sens : 5′-UUCCGUCCCAGUAGAUUACca-3′ |
Transfection de gènes stables
LV-PDK2 et le virus NC correspondant ont été achetés chez GeneChem (Shanghai, Chine). shPDK2-1, shPDK2-2 et le plasmide de contrôle ont été achetés chez GeneChem (Shanghai, Chine). Le surnageant viral a été récolté à partir de cellules T 293. Par la suite, les cellules CRC ont été infectées avec le virus et criblées avec de la puromycine. L’efficacité de l’infection a été validée par cytométrie de flux et microscopie à fluorescence. L’expression de l’ARNm et de la protéine PDK2 a été analysée par PCR quantitative en temps réel et par Western blot.
essais de luciférase reporter 3′-UTR
Les séquences de liaison du miR-149-3p wt ou mutant dans le 3′UTR de PDK2 humain ont été clonées en aval de la luciférase pmiR-RB-Reporter (Ribobio, Guangzhou, Chine), se référant à WT, MUT1, MUT2, et MUT3 dans la Fig. 3c. les cellules T 293 et les cellules HCT116 ont été ensemencées dans des plaques à 24 puits, puis on a procédé à la cotransfection avec 500 ng de constructions rapporteurs et soit 50 nmol/L de mimique miR-149-3p, soit une NC, en utilisant la Lipofectamine 3000 (Thermo Fisher Scientific, Waltham MA). L’activité luciférase a été mesurée après 48 h d’incubation à l’aide du Dual-Luciferase Reporter Assay System (Promega, Madison, USA) selon le protocole du fabricant.
Consommation de glucose et production de lactate
Les cellulesHCT-8/Fet HCT116/F ont été ensemencées dans des plaques à 24 puits à raison de 1 ×105 cellules/puits pendant la nuit, puis traitées avec 15 mM et 20 mM de DCA, respectivement, pendant 24 h. Après le traitement, les cellules ont été cultivées dans un milieu sans rouge de phénol contenant 10 % de sérum bovin fœtal pendant 24 h. Le milieu de culture a été récolté, et la consommation de glucose et la production de lactate ont été mesurées. Le lactate a été mesuré à l’aide d’un kit de dosage du lactate (Njjcbio, Nanjin, Chine), et le glucose a été mesuré à l’aide d’un kit de dosage du glucose (Rsbio, Shanghai, Chine). Toutes les valeurs ont été normalisées en comptant un nombre égal de cellules. Pour évaluer l’état de la glycolyse, 100 ng/mL d’oligomycine (un inhibiteur de l’ATP synthase ; Sangon Biotech) ont été ajoutés aux cellules en culture pendant 6 h. Le rapport de la concentration de lactate en présence et en l’absence d’oligomycine a été mesuré et déterminé comme décrit précédemment [46].
Test de la vitesse glycolytique Seahorse XF-96
Les cellules ont été ensemencées dans une plaque de culture à 96 puits à une densité de 25 000 cellules/puits et ont été incubées pendant la nuit dans un milieu de croissance contenant 10 % de sérum bovin fœtal. La cartouche du capteur a été hydratée pendant la nuit. Le lendemain, le milieu cellulaire a été remplacé par un milieu d’essai faiblement tamponné sans bicarbonate, complété par du glucose, du pyruvate de sodium et de la glutamine. Après que les cellules aient été incubées pendant 1 h à 37 °C dans un incubateur sans CO2, le taux de consommation d’oxygène et le taux d’acidification extracellulaire ont été mesurés avant et après l’injection de DCA/ctrl, Roténone (Rot) + antimycine A (AA) et 2-désoxy-d-glucose (2-DG) en utilisant l’instrument Seahorse XF (Agilent, Santa Clara, CA) comme décrit précédemment [59, 60]. Les expériences ont été réalisées en temps réel dans cinq à six puits répétés. Les glycoPER comprennent le glycoPER basal, le glycoPER induit et le glycoPER compensatoire et ont été automatiquement calculés par le logiciel Wave (Agilent, Santa Clara, CA).
PCR quantitative en temps réel et microarray miRNA
L’ARN total a été extrait des tissus ou des cellules de CRC avec le réactif TRIzol (Life, CA, USA). L’ADNc a été synthétisé en utilisant le kit de réactif RT PrimeScript (TaKaRa, Tokyo, Japon). Le test de microréseau a été réalisé avec trois répliques de cellules HCT116 traitées avec 5 mM, 10 mM ou 20 mM de DCA pendant 12, 24 ou 48 h. Les données originales ont été téléchargées dans la base de données GEO (GSE125309). La PCR quantitative en temps réel a été réalisée à l’aide du prémélange Ex Taq 420 A (TaKaRa, Tokyo, Japon) sur la plateforme ABI-7500. L’actine et l’U6 ont été utilisées comme contrôles internes. Les séquences d’amorces sont présentées dans le tableau supplémentaire 3.
| séquences d’amorces pour la PCR quantitative en temps réel par transcription inverse | |
| Actine | F : 5′-CTCCATCCTGGCCTCGCTGT-3′ |
| R : 5′-GCTGTCACCTCACCGTTCC-3 | |
| PDK2 | F : 5′-CGCTGGCTGGCTTTTTGGTTATG-3′ |
| R : 5′-ACAGGGCCTTGAGATAGATG-3 | |
| PDK1 | F : 5′-GCTGTATGGCCTGCAAGATG-3′ -GCTGTATGGCCTGCAAGATG-3′ -GCTGTATGGCCTGCAAGATG-3 |
| R : 5′-GCTGTCCTGGTGATTTTGCA-3′ | |
| PDK3 | F : 5′-GGTTTGCCAATTTCCCGTCTG-3′ ; R : 5′-CATCGGGGTGATTTTGCA-3′ ; ET |
| R : 5′-CATCGGCTCAGGCGTGGTC-3 | |
| PDK4 | F : 5′-GAGAATTATTGACCGCCTCT-3′ |
| R : 5′-CGAGAAATTGGCAAGCCGTAA-3′ | |
| HK1 | F : 5′-CTTACTAAGGGATGCGATAAA-3′ |
| R : 5′-TCCCAACAATGAGTCCAACC-3 | |
| TP53 | F : 5′-CCCAAGCAATGGATGATTTGA-3′ |
| R : 5′-GGCATTCTGGAGCTTCATCT-3 | |
| p21 | F : 5′-CTGGACTGTTTTCTCTCGGCTC-3′ |
| R : 5′-TGTATATTCAGCATTGTGGGAGGA-3 | |
| MDM2 | F : 5′-ATGAATCCCCCCCTTCCAT-3′ |
| R : 5′-CAGGAAGCCAATTCTCACGAA-3′ | |
| PUMA | F : 5′-ACAGTACGAGCGGCGGAGACAA-3′ |
| R : 5′-GGCGGGTGCAGGCACCTAATT-3 | |
| miR-149-3p | RT : 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGG ATACGACGCACAG-3′ |
| F : 5′-ACAGGGAGGGACGGGGGGG-3 | |
| R : 5′-ATCCAGTGCAGGGTCCGAGG-3′ | |
| miR-128-3p | RT : 5′-TCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGA TACGACAAAGAG-3′ |
| F : 5′-CGCGTCACAGTGAACCGGT-3 | |
| R : 5′-AGTGCAGGGTCCGAGGTATT-3′ | |
| miR-500a-5p | RT : 5′-GTCGTATCCAGTGCGTCGGAGTCGGCAATTGCAC TGGATACGACTCTCACC-3′ |
Western blots
Trente microgrammes de lysats de protéines totales ont été chargés, et les anticorps primaires ont été appliqués : anti-PDK2 (sc-100534, Santa Cruz, Californie, US), anti-p-PDHA1 (S293) (ABS204, Merck, Darmstadt, Allemagne), anti-PDHA1 (ab168379, Abcam, Cambridge, UK), anti-p53 (sc-126, Santa Cruz, Californie, US), anti-c-PARP (D64E10, CST, Massachusetts, US), anti-Bax (D2E11, CST, Massachusetts, US), anti-LC3B (L7543, sigma, MO, USA), anti-GAPDH (Proteintech, Wuhan, Chine), et anti-α-tubuline (Proteintech, Wuhan, Chine). Les anticorps secondaires ont été achetés auprès de Sungene (Tianjin, Chine). Les essais de transfert ont été imagés par un système d’imagerie par chimioluminescence (Bioshine, Shanghai, Chine).
Immunoprécipitation de la chromatine (ChIP)
Les cellulesHCT116ensemencées dans des plaques de 10 cm ont été traitées avec ou sans 20 mM de DCA pendant 24 h, puis la fixation des cellules et la fragmentation des chromosomes ont été réalisées conformément aux instructions du fabricant (Pierce Agarose ChIP Kit, Thermo). La chromatine a été incubée avec des anticorps IgG et anti-p53 (Sigma, MO, USA) à 4°C pendant la nuit. Après incubation, 60 ul d’agarose protéine A/ADN de sperme de saumon ont été ajoutés. Ensuite, le complexe précipité a été lavé avec les tampons de lavage IP 1, 2, 3, et élué avec le tampon d’élution. La réticulation a été inversée par l’ajout de 6 μl de NaCl 5 M et de 2 ul de protéinase K à 65°C pendant 1,5 h. L’ADN immunoprécipité et l’ADN de l’extrait de cellule entière (entrée) ont été purifiés, puis utilisés pour des analyses PCR à l’aide des amorces correspondantes. Une amorce de contrôle a été utilisée pour le suivi de l’expérience. Les séquences d’amorces pour la PCR sont présentées dans le tableau supplémentaire 3.
Xénogreffe tumorale sous-cutanée chez les souris nude et imagerie par micro-TEP/CT
Tout d’abord, 1 ×107 cellules HCT-8/F diluées dans 100 μl de PBS ont été implantées par voie sous-cutanée chez des souris nude (mâles, 6 semaines). Les souris ont été divisées aléatoirement en six groupes (six par groupe) après 12 jours. Les souris du groupe I au groupe IV ont reçu une injection intrapéritonéale quotidienne de PBS, DCA (50 mg/kg)/PBS, 5-FU (10 mg/kg)/PBS et 5-FU (10 mg/kg)/DCA (50 mg/kg), respectivement. Les souris du groupe V et du groupe VI ont reçu une injection intratumorale de PBS ou de DCA (50 mg/kg), respectivement, tous les deux jours et une injection intrapéritonéale de 5-FU (10 mg/kg) tous les deux jours. Le volume tumoral a été mesuré en aveugle tous les 3 jours. Les souris ont été sacrifiées après 3 semaines de traitement, et les tumeurs ont été disséquées, pesées et congelées à -80 °C pour une étude ultérieure.
Pour évaluer si le miR-149-3p exerce un effet de chimiosensibilisation, deux autres groupes de modèles animaux ont été établis. En bref, des souris ont été implantées par voie sous-cutanée avec 6 ×106 cellules HCT-8/F. Après que la taille des tumeurs ait atteint ~50 mm3, les souris ont reçu une dose intrapéritonéale de 5-FU (10 mg/kg) tous les deux jours ainsi qu’une injection intratumorale de 5 nmol de mimiques miR-149-3p conjuguées à du cholestérol ou d’un NC tous les 3 jours pendant 3 semaines. Ensuite, trois souris de chaque groupe ont été mises à jeun pendant la nuit et ont reçu une injection intraveineuse de 0,15 mCi 18F-FDG. un balayage micro-PET-CT au 18F-FDG (Siemens, Berlin, Allemagne) a été effectué après 60 minutes. Les images d’acquisition TEP ont été représentées à l’aide d’une carte pseudo-colorée, la couleur rouge indiquant une forte captation de 18F-FDG. Le SUVmax et le MTV ont été utilisés pour déterminer l’activité du 18F-FDG-PET. Toutes les expériences et les soins aux animaux ont été approuvés par le comité d’éthique du Ninth People’s Hospital Affiliated to the Medical College of Shanghai Jiao Tong University.
Coloration immunohistochimique et immunofluorescence
En bref, les coupes de tissus ont été incubées avec les anticorps primaires contre Ki67 (Servicebio, Wuhan, Chine) et PDK2 (Proteintech, Wuhan, Chine) à 4 °C pendant une nuit, puis incubées avec l’anticorps secondaire. La réaction chromogène a été réalisée avec de la 3,3-diaminobenzidine et contre-colorée avec de l’hématoxyline. Le score d’immunoréactivité (IRS) a été calculé par deux investigateurs en aveugle de l’affectation des groupes. IRS = SI (intensité de la coloration) × PP (pourcentage de cellules positives). Le SI a été attribué comme suit : 0 × négatif ; 1 × faible ; 2 × modéré ; 3 × fort. Le PP est défini comme suit : 0 × 0 % ; 1 × 0-25 % ; 2 × 25-50 % ; 3 × 50-75 % ; 4 × 75-100 %. Des sections congelées de six millimètres ont été colorées à l’aide d’un kit de réaction TUNEL (Roche, Basel, Suisse) et contre-colorées avec du DAPI. Les images ont été capturées à l’aide d’un microscope à fluorescence équipé de filtres d’excitation et d’émission appropriés.
Analyse statistique
Les données ont été analysées par le logiciel GraphPad Prism 6.0. Les données sont présentées comme les moyennes ± SD/SEM de trois expériences indépendantes. Chaque expérience a été réalisée au moins trois fois. Le test t de Student bilatéral a été utilisé pour comparer les différences entre les deux groupes. L’ANOVA à sens unique suivie du test post-hoc de Bonferroni a été utilisée pour les comparaisons multiples. Les courbes de Kaplan-Meier pour les analyses de survie ont été déterminées à l’aide du test log-rank. La relation entre miR-149-3p et PDK2 a été évaluée en utilisant l’analyse du coefficient de corrélation de rang de Spearman. Une valeur P <0,05 a été considérée comme statistiquement significative.
Remerciements
Ce travail a été soutenu par la Fondation nationale des sciences naturelles de Chine (81272745, 81872419 et 81272404) et le programme pour les professeurs à nomination spéciale (Eastern Scholar to JW) des institutions d’enseignement supérieur de Shanghai. Nous remercions le Dr Yanhong Gu pour avoir fourni les lignées cellulaires HCT116 résistantes au 5-FU et le Dr Hua Lu pour avoir fourni le plasmide p53.
Respect des normes éthiques
Conflit d’intérêts
Les auteurs déclarent ne pas avoir de conflit d’intérêts.
Note de l’éditeur
Springer Nature reste neutre en ce qui concerne les revendications juridictionnelles dans les cartes publiées et les affiliations institutionnelles.
Accès libre
Cet article est placé sous licence Creative Commons Attribution 4.0 International License, qui autorise l’utilisation, le partage, l’adaptation, la distribution et la reproduction sur tout support ou dans tout format, à condition de citer de manière appropriée le ou les auteurs originaux et la source, de fournir un lien vers la licence Creative Commons et d’indiquer si des modifications ont été apportées. Les images ou tout autre matériel tiers figurant dans cet article sont inclus dans la licence Creative Commons de l’article, sauf indication contraire dans une ligne de crédit vers le matériel. Si le matériel n’est pas inclus dans la licence Creative Commons de l’article et que l’usage que vous souhaitez en faire n’est pas autorisé par la réglementation ou dépasse l’usage autorisé, vous devrez obtenir l’autorisation directement auprès du détenteur du droit d’auteur. Pour consulter une copie de cette licence, rendez-vous sur http://creativecommons. org/licenses/by/4.0/.
RÉFÉRENCES
1 Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, et al. Incidence du cancer et mortalité en Chine, 2014. Chin J Cancer Res. 2018;30:1-12.2 Siegel RL, Miller KD, Jemal A. Statistiques sur le cancer, 2018. CA : Cancer J Clin. 2018;68:7-30.
3 Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB 3rd, et al. Le dichloroacétate modifie le métabolisme de Warburg, inhibe la croissance cellulaire et augmente la sensibilité aux rayons X des cellules cancéreuses pulmonaires humaines A549 et H1299 NSC. Free Radic Biol Med. 2015;89:263-73.
4 Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. La signalisation de l’interféron tumoral régule un programme de résistance multigénique au blocage des points de contrôle immunitaires. Cell. 2016;167 : 1540-54 e12.
5 Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Rapport de cas d’un événement indésirable grave après l’administration de cellules T transduites avec un récepteur d’antigène chimérique reconnaissant ERBB2. Mol Ther : J Am Soc Gene Ther. 2010;18:843-51. Le dichloroacétate rétablit la chimiosensibilité du cancer colorectal par le biais de la médiation p53/miR-149-3p/PDK2. . . 483
6 Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Statistiques sur le traitement du cancer et la survie, 2016. CA : Cancer J Clin. 2016;66:271-89.
7 Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer : a review. Therapeutic Adv Med Oncol. 2016;8:57-84.
8 Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combiné au fluorouracile comparé au fluorouracile seul comme traitement de première ligne du cancer colorectal métastatique : un essai randomisé multicentrique. Lancet. 2000;355:1041-7.
9 Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil et leucovorin pour le cancer colorectal métastatique. Groupe d’étude sur l’irinotécan. New Engl J Med. 2000;343:905-14.
10 Hanahan D, Weinberg RA. Hallmarks of cancer : the next generation. Cell. 2011;144:646-74.
11 Matthew G, Vander Heiden LCC, Craig BT. Comprendre l’effet Warburg : les exigences métaboliques de la prolifération cellulaire. Science. 2009;324:1029-33.
12 Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
13 Shaw RJ. Le métabolisme du glucose et le cancer. Curr Opin Cell Biol. 2006;18:598-608.
14 Van Dang C, Pollak M. Pourquoi le cancer et le métabolisme ? Why now ? Cancer Metab. 2013;1:1.
15 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532.
16 Cairns RA, Harris IS, Mak TW. Régulation du métabolisme des cellules cancéreuses. Nat Rev Cancer. 2011;11:85-95.
17 Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition de la glycolyse dans les cellules cancéreuses : une nouvelle stratégie pour surmonter la résistance aux médicaments associée à un défaut respiratoire mitochondrial et à l’hypoxie. Cancer Res. 2005;65:613-21.
18 Guo WQZ, Wang Z, et al. MiR-199a-5p est négativement associé aux malignités et régule la glycolyse et la production de lactate en ciblant l’hexokinase 2 dans le cancer du foie. Hépatologie. 2015;62:1132-44.
19 Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 réduit la voie du pentose phosphate et la prolifération en ciblant les ARNm PRPS1 et RPIA dans les cellules cancéreuses colorectales humaines. Gastroenterology. 2015;149:1587-98 e11.
20 Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. Le microARN-129- 5p régule la glycolyse et la prolifération cellulaire en ciblant le transporteur de glucose SLC2A3 dans les cellules cancéreuses gastriques. Front Pharmacol. 2018;9:502.
21 Bartel DP. MicroARNs : génomique, biogénèse, mécanisme et fonction. Cell. 2004;116:281-97.
22 Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167-79.
23 Huang S, He X. microRNAs : tiny RNA molecules, huge driving forces to move the cell. Protein Cell. 2010;1:916-26.
24 Zhang Y, Wang J. Les microARN sont des régulateurs importants de la résistance aux médicaments dans le cancer colorectal. Biol Chem. 2017;398:929-38.
25 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacité du dichloroacétate comme médicament abaissant le taux de lactate. J Clin Pharmacol. 2003;43:683-91.
26 Kankotia S, Stacpoole PW. Le dichloroacétate et le cancer : un nouveau foyer pour un médicament orphelin ? Biochim Biophys Acta. 2014;1846:617-29.
27 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Preuve de l’existence d’une régulation spécifique aux tissus du complexe pyruvate déshydrogénase des mammifères. Biochemical J. 1998;329(Pt 1):191-6.
28 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell 2007;11:37-51.
29 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4.
30 Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33:603-10.
31 Papandreou I, Goliasova T, Denko NC. Les médicaments anticancéreux qui ciblent le métabolisme : le dichloroacétate est-il le nouveau paradigme ? Int J Cancer. 2011;128:1001-8.
32 Michelakis ED, Webster L, Mackey JR. Le dichloroacétate (DCA) comme thérapie potentielle ciblant le métabolisme pour le cancer. Br J Cancer. 2008;99:989-94.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Le dichloroacétate induit l’apoptose et l’arrêt du cycle cellulaire dans les cellules cancéreuses colorectales. Br J Cancer. 2010;102:1746-52.
34 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett. 2010;297:75-83.
35 Bertoli G, Cava C, Castiglioni I. MicroRNAs : nouveaux biomarqueurs pour le diagnostic, le pronostic, la prédiction thérapeutique et outils thérapeutiques pour le cancer du sein. Theranostics 2015;5:1122-43.
36 Cao D, Jia Z, You L, Wu Y, Hou Z, Suo Y, et al. L’acide 18bétaglycyrrhétinique supprime le cancer gastrique par l’activation de la signalisation miR149-3p-Wnt-1. Oncotarget. 2016;7:71960-73.
37 Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, et al. Effets puissants de la dioscine contre le cancer du pancréas via l’inhibition de la voie de signalisation Akt1 médiée par miR-149-3P. Br J Pharm. 2017;174:553-68.
38 Kato M, Li J, Chuang JL, Chuang DT. Mécanismes structurels distincts pour l’inhibition des isoformes de la pyruvate déshydrogénase kinase par l’AZD7545, le dichloroacétate et le radicicol. Structure. 2007;15:992-1004.
39 Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. 2005;272:3004-14.
40 Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, Beloueche-Babari M, Koh KX, et al. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br J Pharm. 2014;171:3255-67.
41 Stacpoole PW. La pharmacologie du dichloroacétate. Metab : Clin Exp. 1989;38:1124-44.
42 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate : possible relevance to toxicity. J Pharmacol Exp Therap. 2008;324:1163-71.
43 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Le ciblage combiné de PDK1 et EGFR déclenche la régression du glioblastome en inversant l’effet Warburg. Cancer Res. 2013;73:7277-89.
44 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. L’inactivation de l’axe de signalisation HIF-1alpha/ PDK3 conduit le mélanome vers un métabolisme oxydatif mitochondrial et potentialise l’activité thérapeutique des pro-oxydants. Cancer Res. 2012;72:5035-47.
45 Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, et al. Le dichloroacétate atténue la résistance au 5-fluorouracile induite par l’hypoxie dans le cancer gastrique par la régulation du métabolisme du glucose. Exp Cell Res. 2014;321:219-30.
46 Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, et al. L’activation de la phosphorylation oxydative par un inhibiteur de la pyruvate déshydrogénase kinase permet de surmonter la résistance au sorafénib du carcinome hépatocellulaire. Br J Cancer. 2013;108:72-81. 484 Y. Liang et al.
47 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. L’activation de l’oxydation mitochondriale par l’inhibition de PDK2 renverse la résistance au cisplatine dans le cancer de la tête et du cou. Cancer Lett. 2016;371:20-9.
48 Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab 2017;25:1037-43.
49 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. L’activation mitochondriale par l’inhibition de PDKII supprime la signalisation HIF1a et l’angiogenèse dans le cancer. Oncogene. 2013;32:1638-50.
50 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversité de la famille de gènes de la pyruvate déshydrogénase kinase chez l’homme. J Biol Chem. 1995;270:28989-94.
51 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003 ; 284 : E855-62.
52 Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182- PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98.
53 Bellazzo A, Di Minin G, Valentino E, Sicari D, Torre D, Marchionni L, et al. Cell-autonomous and cell non-autonomous downregulation of tumor suppressor DAB2IP by microRNA-149- 3p promotes aggressiveness of cancer cells. Cell Death Differ. 2018;25:1224-38.
54 Vazquez A, Bond EE, Levine AJ, Bond GL. La génétique de la voie p53, l’apoptose et la thérapie du cancer. Nat Rev Drug Discov. 2008;7:979-87.
55 Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. Le suppresseur de tumeur p53 dans le contrôle du métabolisme et de la ferroptose. Front Endocrinol. 2018;9:124.
56 Contractor T, Harris CR. p53 régule négativement la transcription de la pyruvate déshydrogénase kinase Pdk2. Cancer Res. 2012;72:560-7.
57 Iacopetta B. Mutation de TP53 dans le cancer colorectal. Hum Mutat. 2003;21:271-6.
58 Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927-30.
59 Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, Tempera I. La poly(ADP-ribose) polymérase 1 est nécessaire pour coactiver l’expression génique dépendante du facteur inductible d’hypoxie 1 par la protéine de membrane latente 1 du virus d’EpsteinBarr. PLoS Pathog. 2018;14 : e1007394.
60 Hlouschek J, Ritter V, Wirsdorfer F, Klein D, Jendrossek V, Matschke J. Le ciblage de SLC25A10 atténue l’amélioration de la capacité antioxydante et la radiorésistance associée des cellules cancéreuses induites par l’hypoxie à cycle chronique. Cancer Lett. 2018;439:24-38.
Contenu connexe :