Yu Liang1, Lidan Hou1, Linjing Li1, Lei Li1, Liming Zhu1, Yu Wang1, Xin Huang1, Yichao Hou1, Danxi Zhu1, Huimin Zou1, Yan Gu2, Xiaoling Weng3,4, Yingying Wang5, Yue Li6, Tianqi Wu3, Mengfei Yao3, Isabelle Gross7,8, Christian Gaiddon9,10, Meng Luo2, Jianhua Wang3, Xiangjun Meng1
1 Abteilung für Gastroenterologie, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China2
Abteilung für Allgemeine Chirurgie, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China3
Krebsinstitut, Fudan University Shanghai Cancer Center, Fudan University, Shanghai, China
4 Ningbo Aitagene Technology Co. LTD, Shanghai, China
5 Abteilung für Biochemie und Molekular- und Zellbiologie, Shanghai Jiao Tong University School of Medicine, Shanghai, China
6 Pathology Center, Shanghai First People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
7 INSERM UMR_S1113, Strasbourg F-67200, Frankreich
8 FMTS, Universite de Strasbourg Strasbourg, Strasbourg F-67000, Frankreich
9Universitede Strasbourg, Inserm IRFAC UMR_S1113, Laboratory Stress Response and Innovative Therapy „Streinth“, Strasbourg 67200, Frankreich
10 CLCC Paul Strauss, Strasbourg, FrankreichMeng
Luo
[email protected] Wang
[email protected] Meng
[email protected] Autoren haben gleichermaßen beigetragen: Yu Liang, Lidan HouReceived
: 16 March 2019Revised
: 17 September 2019Accepted
: 19. September 2019Veröffentlicht
: 9. Oktober 20199. Oktober 2019
Zusammenfassung
Die Entwicklung von Chemoresistenz ist nach wie vor eine große Herausforderung, die für die Letalität von Darmkrebs (CRC) verantwortlich ist. Dichloracetat (DCA) wurde ursprünglich als Stoffwechselregulator bei der Behandlung von Stoffwechselkrankheiten eingesetzt; hier wurde DCA untersucht, um die der Chemoresistenz von CRC zugrunde liegenden Mechanismen zu identifizieren. Wir fanden heraus, dass DCA die Chemosensitivität von CRC-Zellen gegenüber Fluorouracil (5-FU) deutlich erhöhte und die Koloniebildung aufgrund der hohen Apoptosewerte reduzierte. Mithilfe des Microarray-Assays stellten wir fest, dass miR-149-3p an der Chemoresistenz von CRC beteiligt war, die nach DCA-Behandlung durch Wildtyp-p53 moduliert wurde. Darüber hinaus wurde PDK2 als ein direktes Ziel von miR-149-3p identifiziert. Mechanistische Analysen zeigten, dass die Überexpression von miR-149-3p die 5-FU-induzierte Apoptose verstärkte und den Glukosestoffwechsel verringerte, ähnlich wie die Auswirkungen des Knockdowns von PDK2. Darüber hinaus hob die Überexpression von PDK2 die hemmende Wirkung von miR-149-3p auf den Glukosestoffwechsel teilweise auf. Schließlich wurde festgestellt, dass sowohl die DCA-Behandlung als auch die Überexpression von miR-149-3p in 5-FU-resistenten CRC-Zellen die chemotherapeutische Wirkung von 5-FU in vivo deutlich sensibilisiert, und dieser Effekt wurde auch in einer kleinen retrospektiven Kohorte von CRC-Patienten validiert. Zusammengenommen haben wir festgestellt, dass der p53/miR-149-3p/PDK2-Signalweg potenziell mit einer DCA-Behandlung angegangen werden kann, um chemoresistentes CRC zu überwinden.
Ergänzende Informationen: Die Online-Version dieses Artikels (https:// doi.org/10.1038/s41388-019-1035-8) enthält ergänzendes Material, das autorisierten Benutzern zur Verfügung steht.
Einleitung
Darmkrebs (CRC) ist die vierthäufigste krebsbedingte Todesursache in China [1] und die zweithäufigste krebsbedingte Todesursache in den Vereinigten Staaten [2], die hauptsächlich auf Metastasenbildung und Versagen der Chemotherapie aufgrund von Medikamentenresistenz zurückzuführen ist und jährlich zu etwa 50.000 Todesfällen
führt[3].
In jüngster Zeit hat der neue Star PD1/PDL1 zwar großes Interesse geweckt, und immer mehr Biotherapeutika zeigen vielversprechende Ergebnisse in der Krebsbehandlung, doch die begrenzte Wirksamkeit und die unvermeidlichen unerwünschten Wirkungen schränken ihren Einsatz in der Klinik ein [4,5]. Derzeit ist die Chemotherapie immer noch die erste Wahl in der Klinik, insbesondere für Patienten mit inoperablen Krebserkrankungen im Spätstadium und mit Metastasen [6], aber die Entwicklung von Arzneimittelresistenzen ist nach wie vor die größte Einschränkung bei der Chemotherapie [7]. Die Erforschung der Mechanismen der Arzneimittelresistenz und die Erforschung neuer Kombinationen klassischer Krebsmedikamente zur Optimierung der Wirksamkeit können daher für die Behandlung von Darmkrebs von Nutzen sein. Da Fluorouracil (5-FU) das am häufigsten verwendete Chemotherapeutikum bei CRC ist, wurden in dieser Studie 5-FU-resistente CRC-Zelllinien verwendet [8,9].
Eine Anomalie des Glukosestoffwechsels ist eines der wichtigsten Merkmale von Krebs [10]. Es ist bekannt, dass die Entwicklung einer unkontrollierten Zellmasse zu einer schlechten Vaskularisierung des Tumors und damit zu einer reduzierten Sauerstoffversorgung führt. Die Krebszellen passen sich daher an die veränderte Mikroumgebung an, indem sie ihren Stoffwechsel vom oxidativen Stoffwechsel auf den glykolytischen Stoffwechsel umstellen, der auf einer Glukoseversorgung beruht und Laktat produziert. Diese Verlagerung wird als „Warburg-Effekt“ bezeichnet und wird häufig bei verschiedenen Krebszellen als eines der bemerkenswerten Merkmale beobachtet [11- 13]. Die jüngste Anhäufung von Forschungsdaten hat dazu geführt, dass die Warburg-Theorie verfeinert werden muss [14]; so hat sich beispielsweise gezeigt, dass die Stoffwechselverschiebung an der Chemoresistenz beteiligt ist [15]; daher könnte ein gezielter Eingriff in die Stoffwechselmuster von Krebszellen potenziell zur Überwindung der Chemoresistenz genutzt werden [16,17].
Es wurde berichtet, dass mehrere Mechanismen die Stoffwechselverschiebung in Krebszellen steuern, darunter auch mikroRNAs (miRNAs) [18-20]. miRNAs stellen eine Klasse kleiner, endogener, nicht kodierender RNAs dar, die die Übersetzung und den Abbau von mRNAs regulieren [21] und an vielen weiteren biologischen Prozessen beteiligt sind, darunter Zellproliferation, Migration, Apoptose, Selbsterneuerung, Initiation, Krebsentwicklung und Chemoresistenz [22-24].
Es gibt Hinweise darauf, dass die gezielte Beeinflussung des abnormalen Stoffwechsels von Krebszellen ein intensiver Forschungszweig ist, der auf die „Erstickung des Tumors“ abzielt und dessen Strategien darin bestehen, Schlüsselenzyme zu hemmen, die am glykolytischen Stoffwechsel beteiligt sind [15]. In diesem Manuskript wurde Dichloracetat (DCA) ursprünglich zur Behandlung von Laktatazidose und erblicher mitochondrialer Erkrankungen eingesetzt [25]. DCA hemmt die enzymatische Aktivität der Pyruvat-Dehydrogenase-Kinasen (PDK1-4), die für die Umwandlung von Pyruvat in Acetyl-CoA erforderlich ist und den glykolytischen Stoffwechsel mit dem Zitronensäurezyklus verbindet [26,27]; in jüngster Zeit wurde DCA auch eine krebshemmende Wirkung zugeschrieben [28-31]. Der Mechanismus, der der Wirkung von DCA auf die Behandlung von Darmkrebs zugrunde liegt, ist jedoch nach wie vor nicht klar.
Die vorliegende Studie konzentrierte sich auf den molekularen Mechanismus, der an der Regulierung des Glukosestoffwechsels und der Chemotherapieresistenz bei CRC beteiligt ist. Unter Verwendung von DCA in CRC-Zellen untersuchten wir die Rolle verwandter miRNAs und deckten damit einen Signalweg auf, der für die 5-FU-Behandlungsresistenz verantwortlich ist.
Ergebnisse
DCA stellt die Chemosensitivität von 5-FU-resistenten CRC-Zellen wieder her
Es wurde berichtet, dass DCA ein wirksames Antitumormittel ist, das bei bestimmten Krebsarten auf energiebezogene Signalwege abzielt [32]die Wirkung von DCA in chemoresistenten CRC-Zellen ist jedoch noch nicht ausreichend untersucht worden. Mit Hilfe des CCK8-Assays stellten wir fest, dass die 5-FU-resistenten HCT-8/F- und HCT116/F-Zellen im Vergleich zu ihren Elternzellen HCT-8 und HCT116 unempfindlich gegenüber 5-FU waren (ergänzende Abb. 1A) und die halbmaximalen Hemmkonzentrationen (IC50) von DCA in HCT-8/F- und HCT116/F-Zellen ~15 bzw. 20 mM betrugen, was mit früheren Berichten [33, 34] übereinstimmt (ergänzende Abb. 1B). Als Nächstes stellten wir fest, dass DCA in 5-FU-resistenten CRC-Zellen die DNA-Synthese signifikant hemmte (ergänzende Abb. 1C, oberes Feld) und die ROS-Bildung induzierte (ergänzende Abb. 1C, unteres Feld). Die Marker des Energiestoffwechsels, einschließlich des Glukoseverbrauchs, der Laktatproduktion und der Glykolyse, waren in 5-FU-resistenten CRC-Zellen im Vergleich zu denen in 5-FU-sensitiven CRC-Zellen deutlich erhöht, während die Zugabe von DCA die Expression dieser Marker deutlich reduzierte (Abb. 1a). Unter Berücksichtigung einer 6-stündigen serumfreien Vorbehandlung bei der Messung der Glykolyse wurde ein Seahorse XF Glykolyse-Assay-Kit verwendet, um den Effekt der Vorbehandlung zu eliminieren und die Glykolyse-Rate in Echtzeit zu messen. Die glykolytische Protonen-Efflux-Rate (glycoPER) spiegelt die Rate der extrazellulären Ansäuerung durch Glykolyse wider. Die Zugabe von DCA verringerte die induzierte Glykolyse im Vergleich zu den entsprechenden Kontrollen erheblich (Abb. 1b und ergänzende Abb. 1D).
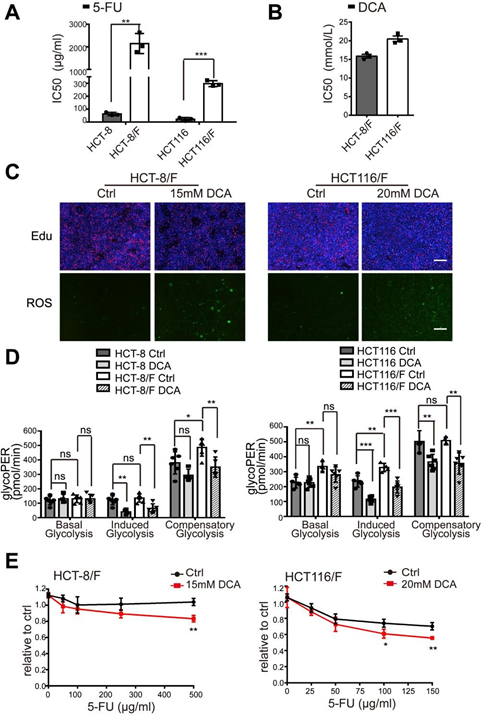
(A) CRC-Zellen wurden 24 Stunden lang mit verschiedenen Konzentrationen von 5-FU behandelt. Die IC50 von 5-FU in jeder Zelle wurde berechnet. (B) HCT-8/F- und HCT116/F-Zellen wurden 24 Stunden lang mit unterschiedlichen Konzentrationen von DCA behandelt. Die IC50 von DCA in jeder Zelle wurde berechnet. (C) HCT-8/F- und HCT116/F-Zellen wurden 24 Stunden lang mit 15 mM bzw. 20 mM DCA behandelt. Repräsentative Bilder der Edu-Immunfluoreszenzfärbung (obere Tafel) und repräsentative Bilder der ROS-Immunfluoreszenzfärbung (untere Tafel), Maßstabsleiste: 200 μm. (D) Die Bestimmung der Glykolyse-Rate einschließlich der Basal-Glykolyse, der induzierten Glykolyse und der kompensatorischen Glykolyse wurde mit dem Seahorse Glycolytic Rate Assay Report Generator berechnet. (E) Das Zellwachstum wurde mit einem CCK8-Assay nach Behandlung mit 5-FU und DCA bestimmt. Die Dosierungsbereiche von 5-FU waren 100 ug/ml bis 500 ug/ml für HCT-8/F Zellen und 25 ug/ml bis 150 ug/ml für HCT116/F Zellen. Die Ergebnisse von drei unabhängigen Experimenten sind als Mittelwert ± SEM angegeben. Jedes Experiment wurde mit 3-6 biologischen Replikaten durchgeführt. *p < 0,05; **, P < 0,01; ***, P < 0,001.
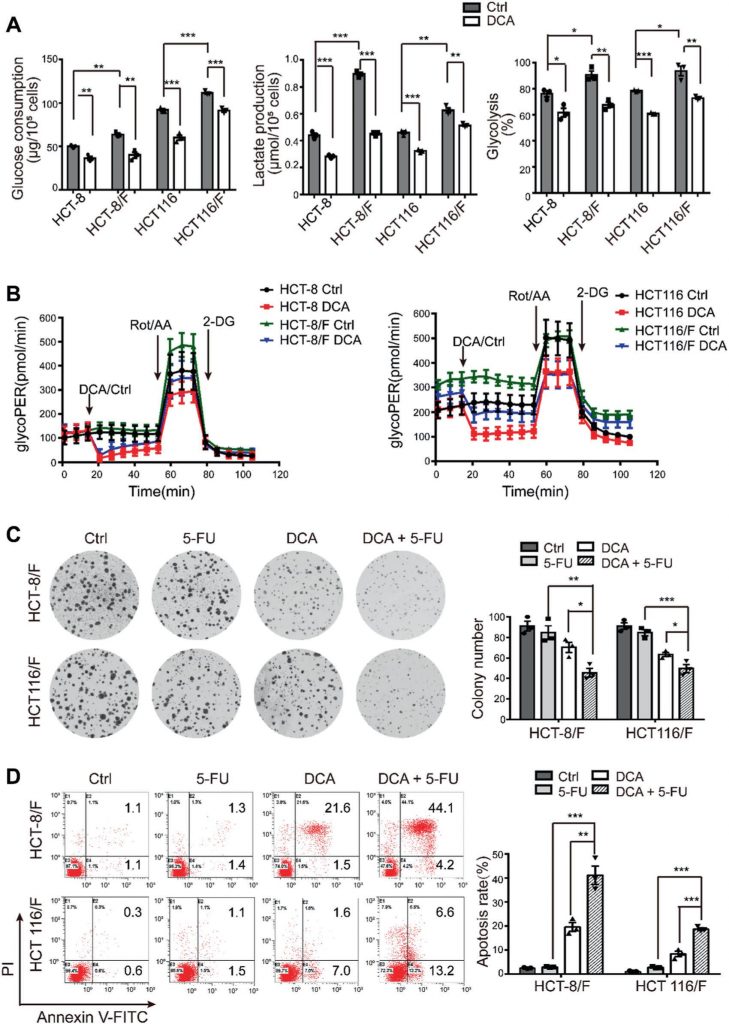
DCA überwand die 5-FU-Resistenz in HCT-8/F- und HCT116/F-Zellen deutlich, wie die Messungen des Zellwachstums zeigten (ergänzende Abb. 1E). Die Fähigkeit zur Koloniebildung wurde signifikant gehemmt (Abb. 1c), und die Apoptose wurde in einer Kombinationsbehandlung deutlich induziert (Abb. 1d), was alles weiter quantifiziert wurde. Diese Ergebnisse legen nahe, dass DCA die Chemosensitivität von 5-FU-resistenten CRC-Zellen wiederherstellen kann.
miR-149-3p spielt eine entscheidende Rolle bei der Chemosensitivität von CRC-Zellen
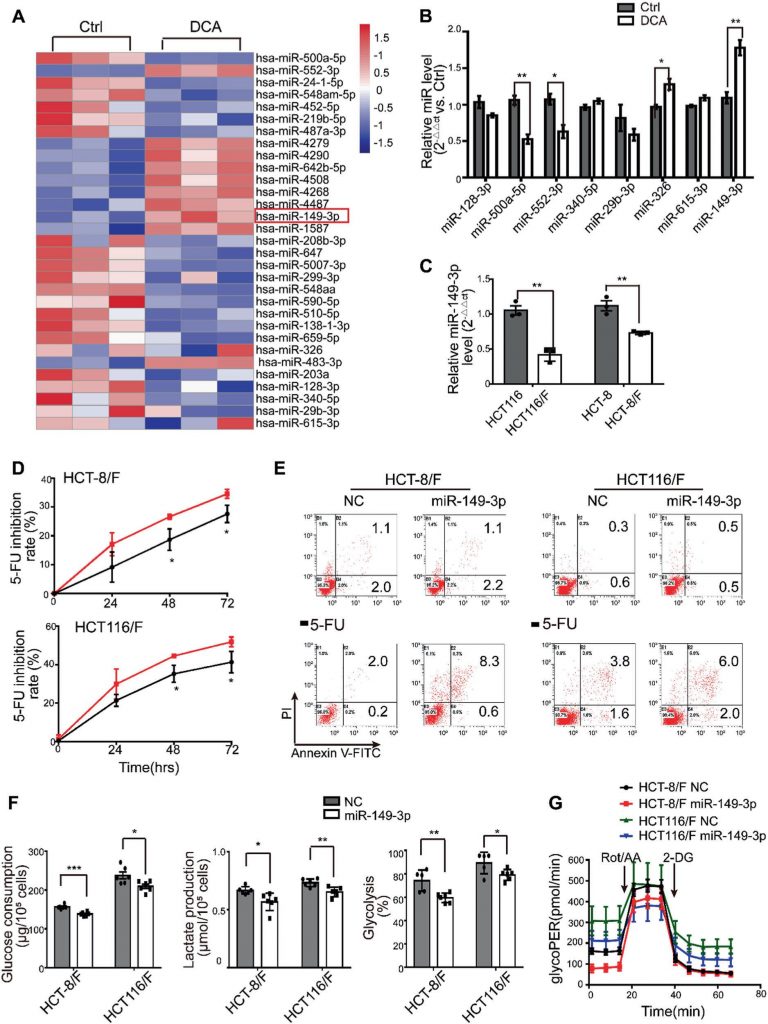
miRNAs werden aufgrund ihrer Wirkung auf die Tumorunterdrückung als vielversprechendes therapeutisches Mittel angesehen [35]. In diesem Zusammenhang haben wir zunächst miRNA-Expressionsprofile mithilfe des miRNA-Arrays bestimmt, der 2059 menschliche miRNAs enthält. Insgesamt 119 miRNAs wurden als Reaktion auf DCA in HCT116-Zellen differenziell exprimiert (Abb. 2a und ergänzende Abb. S2A). Unter ihnen wurden die Expressionsniveaus von acht miRNAs weiter durch quantitative Echtzeit-PCR (Abb. 2b) bestätigt, und miR-149-3p wurde schließlich durch DCA in einer dosisabhängigen Weise hochreguliert (ergänzende Abb. S2B).
Als nächstes stellten wir fest, dass HCT116-Zellen mit höheren Basalwerten von miR-149-3p empfindlicher auf 5-FU und L-OHP reagieren, wie in der ergänzenden Abb. 2C gezeigt. Darüber hinaus verringerte die Transfektion von HCT116-Zellen mit anti-miR-149-3p die chemotherapeutische Wirkung von 5-FU deutlich (siehe ergänzende Abb. 2D-E). Bemerkenswerterweise waren die Spiegel von miR-149-3p in chemosensitiven CRC-Zellen signifikant höher als die Spiegel in chemoresistenten CRC-Zellen (Abb. 2c). Daher erhöhte die Transfektion von miR-149-3p-Mimikern die Hemmungsrate signifikant, förderte die durch 5-FU induzierte Zellapoptose und reduzierte den Glukoseverbrauch, die Laktatproduktion und die Glykolyse in HCT-8/F und HCT116/F Zellen (Abb. 2d-f). Ein Seahorse XF Glykolyse-Assay zeigte, dass die Expression von miR-149-3p die basale Glykolyse in 5-FU-resistenten CRC-Zellen reduzierte, was mit den oben genannten Ergebnissen übereinstimmte (Abb. 2g und ergänzende Abb. 2F). Diese Ergebnisse deuten darauf hin, dass miR-149-3p für die Überwindung der Chemoresistenz in CRC-Zellen günstig ist.
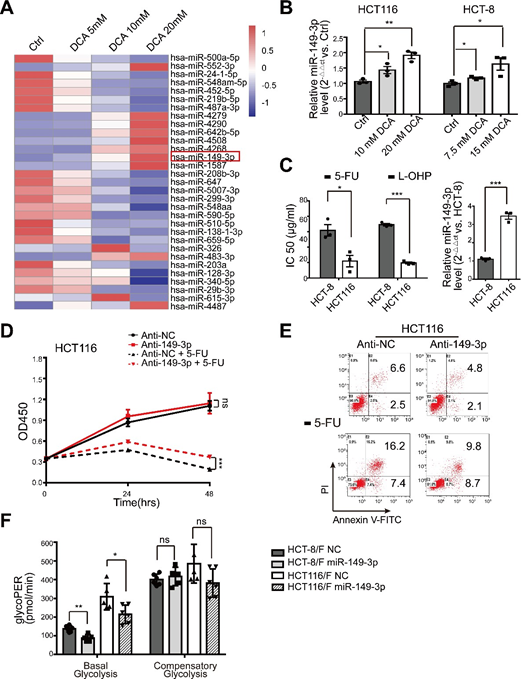
(A) Heatmap des Profils der differenziell exprimierten microRNA in HCT116-Zellen, die 24 Stunden lang mit Kontrolle, 5 mM, 10 mM und 20 mM DCA behandelt wurden. (B) Die Gesamt-RNA wurde 24 Stunden nach der Behandlung mit 10/7,5 mM und 20/15 mM DCA aus HCT116- und HCT-8-Zellen gewonnen. Der miR-149-3p-Spiegel wurde durch quantitative Echtzeit-PCR analysiert. (C) HCT-8- und HCT116-Zellen wurden 24 Stunden lang mit verschiedenen Konzentrationen von 5-FU bzw. Oxaliplatin (L-OHP) behandelt, und die halbmaximale Hemmkonzentration (IC50) wurde berechnet (linkes Feld). Die Basalwerte von miR-149-3p wurden mittels quantitativer Echtzeit-PCR in HCT-8-Zellen und HCT116-Zellen bestimmt (rechtes Feld). (D-E) HCT116-Zellen wurden transient mit Anti-NC oder miR-149-3p-Inhibitor transfiziert. Nach der Transfektion wurden die Zellen 24 Stunden lang mit 25 μg/ml 5-FU behandelt. Das Zellwachstum und die Apoptose wurden mittels CCK8 bzw. Durchflusszytometrie bestimmt. (F) Die Bestimmung der Glykolyse-Rate einschließlich der basalen Glykolyse und der kompensatorischen Glykolyse in 5-FU-resistenten CRC-Zellen, die mit NC oder einem miR-149-3p-Mimic transfiziert wurden, wurde mit dem Seahorse Glycolytic Rate Assay Report Generator berechnet. Die Daten von drei unabhängigen Experimenten sind als Mittelwert ± SEM angegeben. Jedes Experiment wurde mit 3-6 biologischen Replikaten durchgeführt. *p < 0,05; **, P < 0,01; ***, P < 0,001; ns, keine Bedeutung.
DCA induziert die Expression von miR-149-3p durch Wildtyp (wt) p53
In Anbetracht der Tatsache, dass mehrere neuere Studien gezeigt haben, dass miR-149-3p durch verschiedene Medikamente reguliert wird [36,37], haben wir als nächstes untersucht, wie miR-149-3p durch DCA reguliert wird. Wir fanden heraus, dass DCA die Expression von wt p53 und seiner nachgeschalteten Signale, einschließlich der Expression von p21, PUMA und MDM2, in CRC-Zellen signifikant erhöhte (Abb. 3a, b). Darüber hinaus stellten wir fest, dass Veränderungen in der wt p53-Expression die miR-149-3p-Expression signifikant modulieren konnten (Abb. 3c, d), was darauf hindeutet, dass miR-149-3p durch wt p53 positiv reguliert wird. Bei Verwendung der p53-null HCT116-Zelllinie (TP53-/-) stellten wir fest, dass miR-149-3p durch DCA-Behandlung nicht hochreguliert wurde, aber die ektopische Expression von p53 kehrte diesen Effekt um. Darüber hinaus verwendeten wir Nutlin-3, einen potenten Inhibitor, der die Interaktion zwischen MDM2 und p53 hemmt und zur Aktivierung von p53 führt, als Positivkontrolle, wobei die Expression von miR-149-3p in der wt HCT116-Zelllinie (TP53+/+) erhöht war (Abb. 3e-g). Mit Hilfe einer bioinformatischen Analysesoftware (IGV) wurden vier mutmaßliche p53-Bindungsstellen für die miR-149 flankierende genomische DNA-Region vorhergesagt. Anschließend wurden in den Zellen ChIP-Assays mit einem Antikörper gegen wt p53 durchgeführt. Die heruntergezogene DNA wurde durch gewöhnliche PCR mit Primern amplifiziert, die auf der Grundlage dieser Stellen entwickelt worden waren. Unsere Ergebnisse zeigten, dass im Vergleich zu Region 4 die Region 3 nach DCA-Behandlung in wt p53-immungefälltem HCT116-Chromatin deutlich angereichert war (Abb. 3h), was darauf hindeutet, dass nur Region 3 eine spezifische, durch DCA aktivierte Bindungsstelle enthält. Diese Ergebnisse zeigen, dass DCA miR-149-3p durch wt p53 moduliert.
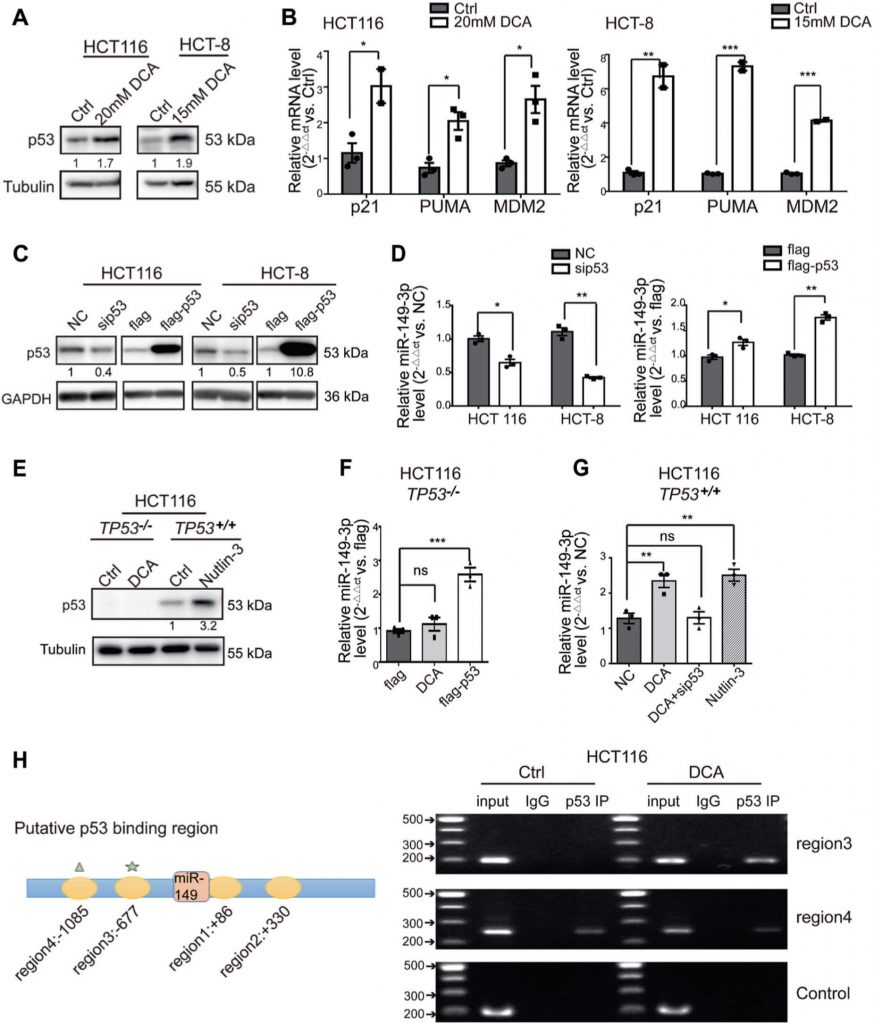
PDK2 ist ein direktes Ziel von miR-149-3p
Um die Mechanismen aufzuklären, durch die miR-149-3p die Chemosensitivität in CRC-Zellen reguliert, untersuchten wir Gene, die mit dem Energiestoffwechsel in Verbindung stehen und durch miR-149-3p reguliert werden, mithilfe zweier öffentlicher Plattformen (TargetScan und miRDB). Schließlich stellten wir fest, dass Pyruvat-Dehydrogenase-Kinase 2 (PDK2) und Hexokinase 1 (HK1) potenzielle Kandidaten sind (Abb. 4a). Um diese Ergebnisse zu bestätigen, wurde der miR-149-3p-Mimiker oder -Inhibitor in CRC-Zellen transfiziert. Wir fanden heraus, dass die mRNA-Spiegel von PDK2 durch miR-149-3p negativ reguliert wurden, nicht aber durch HK1 (Abb. 4b und ergänzende Abb. 3A). Es wurden zwei mögliche miR-149-3p-Bindungsstellen in der 3′-UTR von PDK2 gefunden, und der Dual-Luciferase-Reporter-Assay zeigte, dass miR-149-3p an die vorhergesagte Stelle (686-693) der 3′-UTR von PDK2 bindet (Abb. 4c, d). Anschließend bestätigten wir, dass der PDK2-Proteinspiegel durch miR-149-3p negativ reguliert wurde (Abb. 4e).
PDK hat vier Isoenzyme namens PDK1, 2, 3 und 4, von denen berichtet wurde, dass sie alle durch DCA reguliert werden [26, 38, 39]. Anschließend analysierten wir die mRNA-Expression von PDK1-4 sowohl in HCT8- als auch in HCT116-Zellen nach DCA-Behandlung. DCA hemmte signifikant die mRNA-Expression von PDK2, aber nicht die anderer PDK-Isoenzyme (ergänzende Abb. S3B). Darüber hinaus kehrte die Transfektion von anti-149-3p die Reduzierung von PDK2 durch DCA teilweise um (Abb. 4f). Als Nächstes bestimmten wir die PDK2 und ihre nachgeschaltete Pyruvat-Dehydrogenase E1-alpha-Untereinheit (PDHA1) in 5-FU-sensitiven und 5-FU-resistenten CRC-Zellen. Die Basalwerte von PDK2 waren in chemoresistenten CRC-Zellen im Vergleich zu den Werten in chemosensitiven Zellen erhöht. Im Einklang mit dieser Erhöhung war auch die Phosphorylierung von PDHA1 erhöht (Abb. 4g).
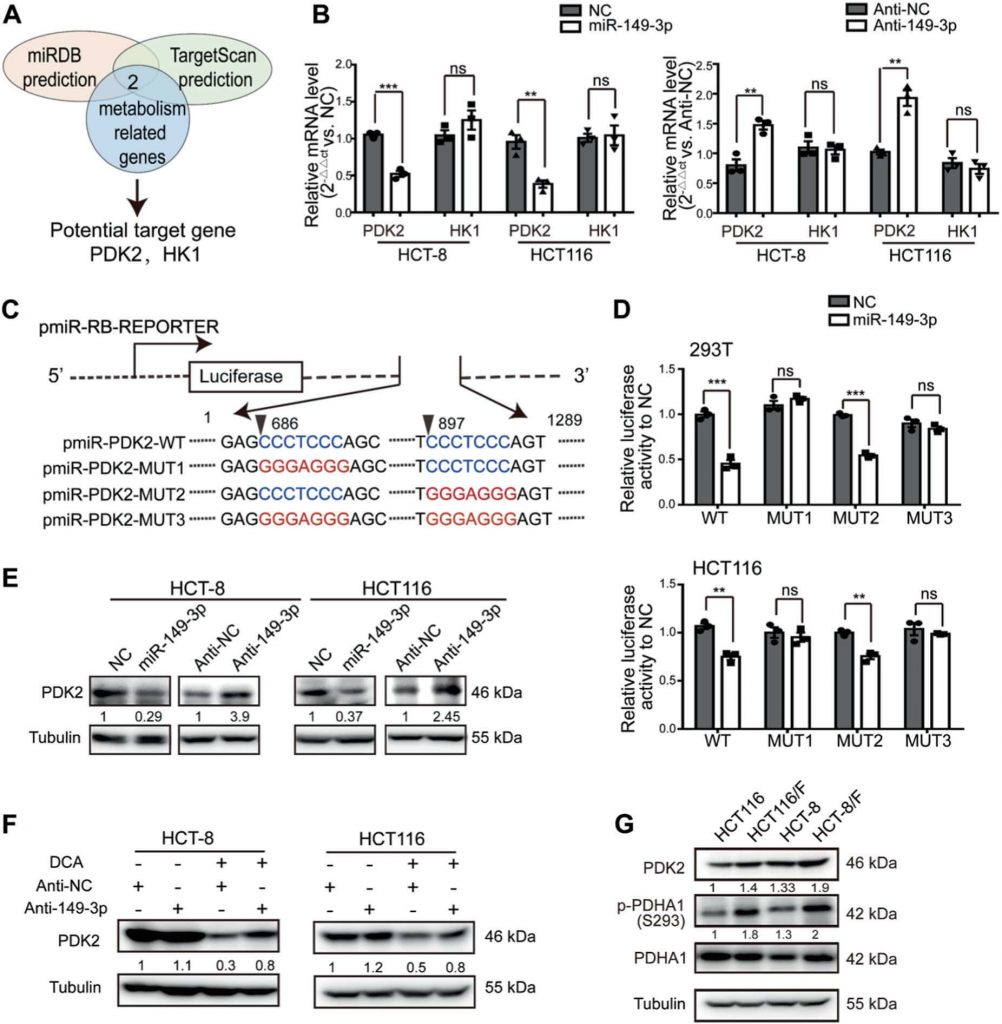
Der miR-149-3p/PDK2-Signalweg reguliert die Chemosensitivität
Um zu verstehen, wie der miR-149-3p/PDK2-Signalweg die Reaktion der CRC-Zellen auf 5-FU reguliert, wurde die HCT-8/F-Zelllinie als repräsentative Zelllinie ausgewählt, um zu untersuchen, ob die Expressionsniveaus von miR-149-3p und PDK2 die Reaktion der Zellen auf 5-FU beeinflussen. Der Knockdown von PDK2 in HCT-8/F-Zellen hemmte die Phosphorylierung von PDHA1 (Abb. 5a) und reduzierte Energiestoffwechselmarker wie Glukoseverbrauch, Laktatproduktion und Glykolyse (Abb. 5b). Darüber hinaus konnte eine Verringerung von PDK2 die Wirkung von 5-FU auf die Erhöhung der Spiegel von gespaltenem PARP (c-PARP) und Bax verstärken, die beide anerkannte Biomarker für die Zellapoptose in HCT-8/F-Zellen sind (Abb. 5c). Darüber hinaus erhöhte der Knockdown von PDK2 die Chemosensitivität von HCT-8/F-Zellen gegenüber 5-FU, wie mit CCK8- und Koloniebildungstests ermittelt wurde (Abb. 5d, e). Der Knockdown von PDK2 förderte die 5-FU-induzierte Apoptose in HCT-8/F- und HCT116/F-Zellen und ist in der ergänzenden Abb. S4A dargestellt, während die Überexpression von PDK2 die Phosphorylierung von PDHA1 förderte und die durch 5-FU induzierte Zellapoptose in HCT-8- und HCT116-Zellen abmilderte (ergänzende Abb. S4B-S4C).
Darüber hinaus kehrte die Überexpression von PDK2 die hemmende Wirkung von miR-149-3p auf PDK2 um (Abb. 5f) und hob die hemmende Wirkung von miR-149-3p auf Glukoseverbrauch, Laktatproduktion und Glykolyse teilweise auf (Abb. 5g). Die ektopische Expression von PDK2 hob die hemmende Wirkung von miR-149-3p auf das Zellwachstum und die Koloniebildung in mit 5-FU behandelten HCT-8/F-Zellen deutlich auf (Abb. 5h, i). Insgesamt deuten unsere Ergebnisse darauf hin, dass der miR-149-3p/PDK2-Signalweg die Chemosensitivität wiederherstellt, indem er zumindest teilweise auf den Glukosestoffwechsel in chemoresistenten CRC-Zellen abzielt.
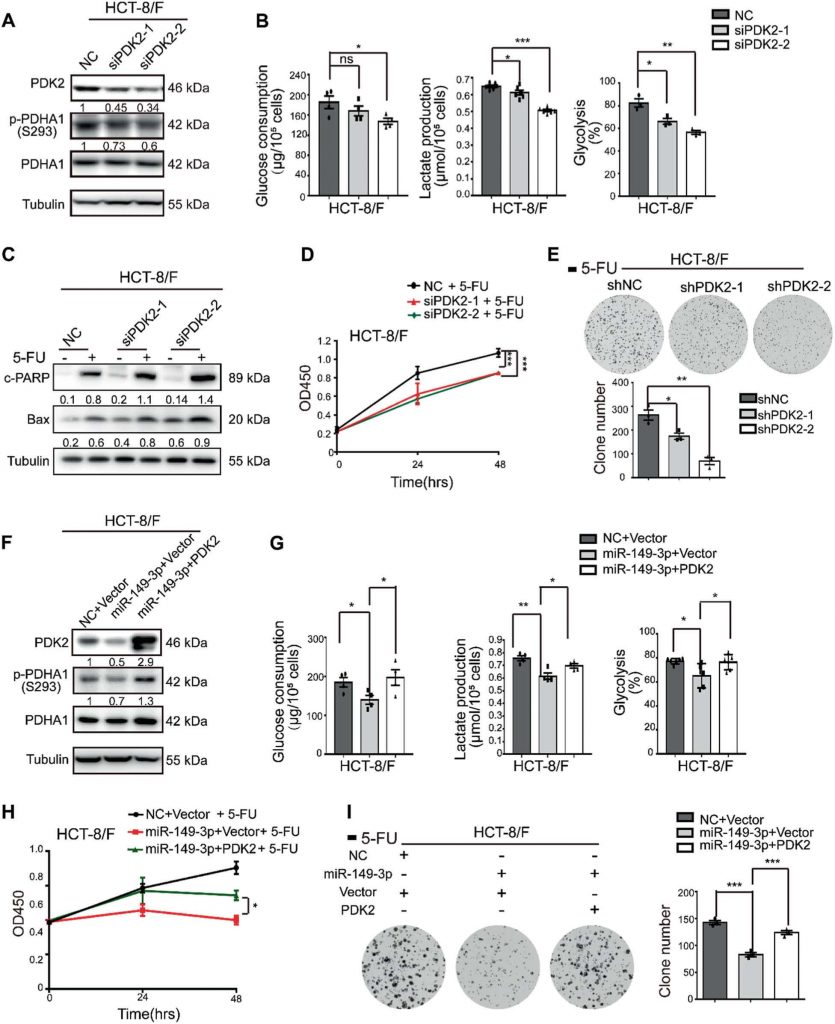
DCA erhöht die Chemosensitivität von 5-FU in vivo
Anschließend wurden 5-FU, DCA oder eine Kombination aus 5-FU und DCA intraperitoneal in das subkutane Xenotransplantatmodell injiziert (als intraperitoneale Gruppe bezeichnet). In Anbetracht der unzureichenden Blutversorgung im zentralen Teil der subkutanen Tumoren haben wir auch DCA oder PBS plus eine intraperitoneale Injektion von 5-FU intratumoral injiziert (als intratumorale Gruppe bezeichnet). Die Kombination von DCA mit 5-FU zeigte nach vierwöchiger Injektion in der intraperitonealen Gruppe eine bessere Hemmwirkung auf das Tumorwachstum als DCA oder 5-FU allein (Abb. 6a, b). Die Expression von Ki-67 war nach der Kombinationsbehandlung in der intraperitonealen Gruppe reduziert (Abb. 6c). DCA hemmte das Tumorwachstum in der intratumoralen Gruppe im Vergleich zur PBS-Gruppe signifikant (Abb. 6d), und miR-149-3p wurde in der intratumoralen DCA-Injektionsgruppe hochreguliert, was mit den Ergebnissen in vitro übereinstimmt (Abb. 6e). Die intratumorale Injektion von DCA förderte auch die Tumorapoptose (Abb. 6F).
Um die Wirkung von miR-149-3p auf die Reaktion von CRC-Zellen auf 5-FU in vivo weiter zu untersuchen, wurde das subkutane Xenotransplantatmodell einer intratumoralen Injektion von agomiR-149-3p oder agomiR-NC plus einer intraperitonealen Injektion von 5-FU unterzogen. miR-149-3p hemmte das Tumorwachstum in der intratumoralen Gruppe signifikant im Vergleich zu dem in der Kontrollgruppe, in der agomiR-negative Kontrolle (NC) intratumoral injiziert wurde (Abb. 6g, h). Die Überexpression von miR-149-3p wurde durch quantitative Echtzeit-PCR validiert (Abb. 6i), und die Überexpression von miR-149-3p förderte die Apoptose (Abb. 6j) und reduzierte die Expression von PDK2 und p-PDHA1 (Abb. 6k, l). Nach dreiwöchiger Behandlung wurde eine 18F-Fluordesoxyglucose (FDG)-Mikro-Positronen-Emissions-Tomographie (PET)-CT-Untersuchung des Tumors durchgeführt. die 18F-FDG-Aufnahme wurde an den Stellen der Tumorimplantation beobachtet, und die maximale standardisierte Aufnahme (SUVmax) sowie die metabolischen Tumorvolumina (MTVs) wurden gemessen. Zwischen den beiden Gruppen wurde kein Unterschied in der SUVmax festgestellt, während das MTV in der miR-149-3p-Gruppe signifikant verringert wurde (Abb. 6m).
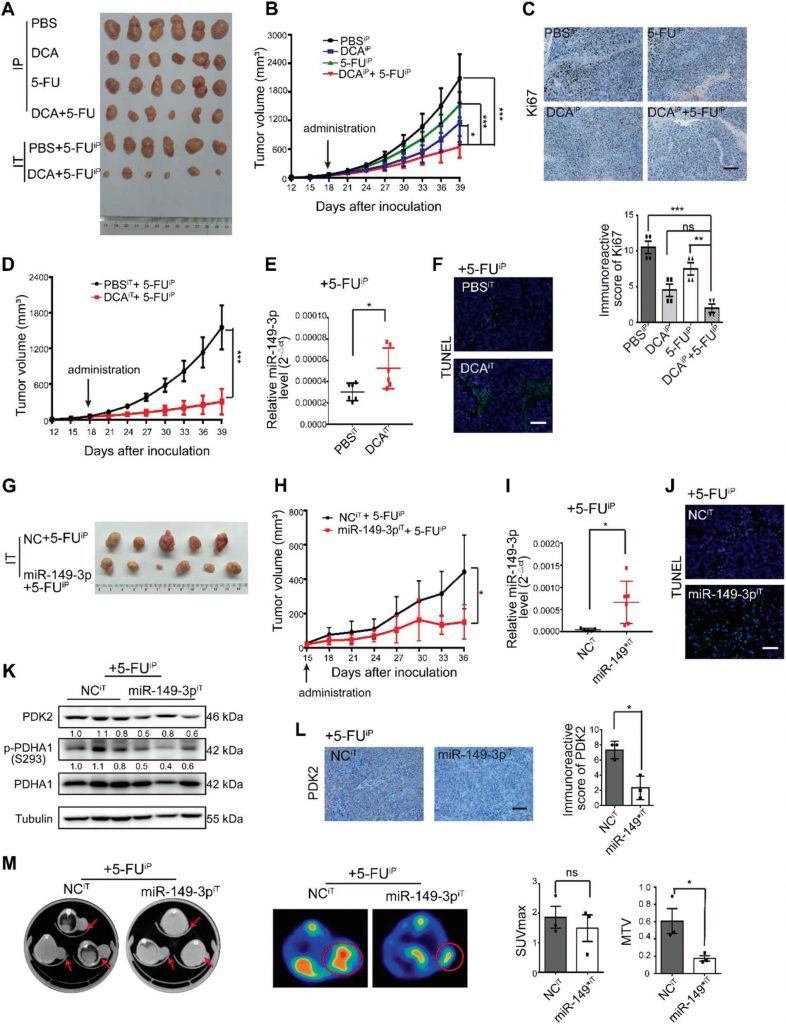
miR-149-3p steht in CRC-Patienten in umgekehrter Korrelation zu PDK2
Eine signifikante umgekehrte Korrelation zwischen den miR-149-3p- und PDK2-mRNA-Spiegeln wurde in menschlichem CRC-Gewebe beobachtet (Abb. 7a, b). Bei acht Patienten in stabilem Zustand (SD) innerhalb von drei Jahren nach der Chemotherapie war die miR-149-3p-Konzentration höher als bei fünf Patienten mit fortschreitender Erkrankung (PD) (ergänzende Abb. S5A). Fünf Paare von PD- und SD-Patienten mit der gleichen Pathologie und dem gleichen TNM-Stadium wurden analysiert, und die PDK2-Färbung variierte signifikant in den Proben der PD- und SD-Patienten (ergänzende Abb. S5B). CRC-Patienten aus der TCGA-Datenbank mit hoher PDK2-Expression zeichneten sich auch durch ein schlechteres Gesamtüberleben (OS) aus (Abb. 7c). Die TCGA-Datenbank zeigte auch, dass die Expression von PDK2 in der wt p53-Gruppe im Vergleich zu derjenigen in der Gruppe mit mutiertem p53 reduziert war (Abb. 7d). Diese Ergebnisse legen nahe, dass PDK2 bei CRC-Patienten durch miR-149-3p negativ reguliert wird.
Diskussion
Darmkrebs (CRC) zeichnet sich durch tumorigene Anomalien und veränderte Stoffwechselwege aus und ist eine der häufigsten Todesursachen bei Krebserkrankungen [2]. Resistenz gegen Chemotherapie ist die Hauptursache für das Scheitern der Behandlung [7]. In dieser Studie haben wir festgestellt, dass DCA die chemotherapeutische Wirkung von 5-FU in chemoresistenten CRC-Zellen verstärken kann und dass die Aktivierung des p53/miR-149-3p/PDK2-Stoffwechselwegs die Chemosensitivität in vitro und in vivo erhöhen kann.
Es gibt immer mehr Hinweise darauf, dass eine erhöhte Glykolyse in engem Zusammenhang mit der Resistenz gegenüber Chemotherapie steht [15, 17, 40]. In diesem Fall haben wir auch festgestellt, dass chemoresistente CRC-Zellen im Vergleich zu ihren Elternzelllinien einen erhöhten Glukoseverbrauch, eine erhöhte Laktatproduktion und eine erhöhte Glykolyse aufwiesen, was darauf hindeutet, dass Stoffwechselanomalien ein typisches Merkmal sind, aber die molekularen Mechanismen in CRC-Zellen noch unklar sind.
Es wurde beobachtet, dass DCA bei Nagetieren in vivo den Laktatspiegel im Blut bei Dosierungen von ~25-50 mg/kg/24 h senkt [41] und dass DCA in einem breiten Dosierungsbereich von 1 bis 50 mM eingesetzt wird [26]. Bemerkenswert ist, dass sich die schädliche Wirkung von DCA beim Menschen im Allgemeinen auf eine reversible periphere sensorische und motorische Neuropathie beschränkt, die durch Alter und Genotyp beeinflusst wird [42]. Kürzlich wurde DCA als neuartige Stoffwechseltherapie für verschiedene Krebspatienten identifiziert [26, 29].
Es wurde berichtet, dass DCA in der Lage ist, die PDK-Aktivität zu hemmen und Pyruvat in Acetyl-CoA umzuwandeln, was zu einer Verlagerung der Energiegewinnung von der Glykolyse zur mitochondrialen oxidativen Phosphorylierung führt [43,44]. Es wurde auch gezeigt, dass DCA die Hypoxie-induzierte Resistenz gegen 5-FU bei Magenkrebs abschwächt [45], die Sorafenib-Resistenz bei hepatozellulärem Karzinom überwindet [46] und die Cisplatin-Resistenz bei Kopf- und Halskrebs abschwächt [47]. Unsere Studie zeigte, dass DCA in der Lage war, die Chemoresistenz von CRC-Zellen gegenüber 5-FU zu vermindern. Darüber hinaus konnten wir zeigen, dass DCA den Glukoseverbrauch und die Laktatproduktion in chemoresistenten CRC-Zellen auf das Ausgangsniveau chemosensitiver CRC-Zellen reduziert. Da die Autophagie das Potenzial hat, den Krebsstoffwechsel anzukurbeln [48], wurde die Wirkung von DCA und miR-149-3p auf die Autophagie untersucht. Wir fanden heraus, dass DCA die Autophagie aktiviert und miR-149-3p keinen Einfluss auf die Autophagie hat. Dies deutet darauf hin, dass die Autophagie nicht an der Wirkung von DCA/miR-149-3p bei der Regulierung des Glukosestoffwechsels beteiligt ist (ergänzende Abbildung S6).
Die PDKs als Schlüsselregulatoren der Glykolyse bei Krebs haben aufgrund der Ergebnisse vieler Studien große Besorgnis ausgelöst [49]. Es gibt vier PDK-Isoformen (PDK1-4), und jede von ihnen tritt in einer gewebespezifischen Weise auf: PDK1 wird stark im Herzen exprimiert, PDK2 wird ubiquitär exprimiert, PDK3 hat eine relativ begrenzte Gewebeverteilung, und PDK4 wird im Herzen und in der Skelettmuskulatur exprimiert [27]. PDK2 wird im Vergleich zu anderen Isoenzymen in höherem Maße exprimiert, was darauf hindeutet, dass sie möglicherweise die Hauptisoform ist, die für die Regulierung der enzymatischen Aktivität des Pyruvatdehydrogenase-Komplexes (PDHC) verantwortlich ist [50]. Darüber hinaus unterscheiden sich die PDK-Isoenzyme in ihrer akuten Regulierung durch Metaboliten [51]. Hier konzentrierten wir uns auf PDK2, die Isoform, die am empfindlichsten auf DCA reagiert [31]. Obwohl die molekularen Wechselwirkungen zwischen DCA und PDKs bereits untersucht wurden, ist der potenzielle Mechanismus der Transkriptionsregulierung von PDK noch unklar. In einer kürzlich durchgeführten Studie wurde berichtet, dass miR-182 eine regulierende Rolle in den Stoffwechselwegen von Lungenkrebs spielt, indem es auf PDK4 abzielt [52]. In der vorliegenden Studie wurde gezeigt, dass PDK2 bei CRC durch miR-149-3p reguliert wird und dass die PDK2-Konzentrationen in den Primärtumoren von CRC-Patienten umgekehrt mit der miR-149-3p-Expression korreliert sind. Da PDK2 in den meisten Geweben weit verbreitet ist, könnte ein gezielter Einsatz von PDK2 ein wichtiger und effizienter Weg sein, um Tumorzellen abzutöten und Chemoresistenz zu überwinden.
Es wurde berichtet, dass miR-149-3p bei verschiedenen Krebsarten eine wichtige Rolle spielt und durch einige Antitumormittel induziert wird [36, 37, 53]. Wir stellten fest, dass eine DCA-Behandlung die Bindung von p53 an die Upstream-Region (-677 bis -477) von miR-149 induzieren kann und dass miR-149-3p durch eine DCA-Behandlung in einer p53-abhängigen Weise hochreguliert wird. TP53, ein klassischer Tumorsuppressor, ist in Tumoren häufig inaktiviert [54] und reguliert nach jüngsten Berichten den Glukosestoffwechsel bei Krebs. Es wurde gezeigt, dass wt p53 in der Lage ist, den „Warburg-Effekt“ durch Kontrolle von PDK2 zu hemmen. Die Häufigkeit von TP53-Mutationen bei CRC beträgt jedoch ca. 40-50 % [55, 56], was zum Verlust der suppressiven Funktion führt. Es wurde berichtet, dass CRC-Patienten mit wt TP53 einen Überlebensvorteil durch eine 5-FU-basierte Chemotherapie haben, diejenigen mit mutiertem TP53 jedoch nicht [57]. Unsere Ergebnisse zeigen einen neuen Mechanismus zwischen p53 und PDK2, der durch miR-149-3p moduliert wird. Diese Ergebnisse legen nahe, dass Patienten mit mutiertem TP53 eher von einer adjuvanten Chemotherapie mit miR-149-3p als mit DCA profitieren könnten. In Anbetracht der hohen Häufigkeit von TP53-Mutationen bei CRC glauben wir, dass miR-149-3p eine wichtige Rolle bei der Überwachung und Modulation der Chemosensitivität bei CRC spielt.
Krebszellen verbrauchen eine große Menge an Glukose und weisen einen hohen aeroben Glykolysezustand auf, daher ist die Verringerung der Glukoseaufnahme eine vielversprechende Strategie zur Begrenzung des Krebswachstums [58]. Wir beobachteten, dass die Erhöhung von miR-149-3p die Glykolyse in chemoresistenten CRC-Zellen deutlich hemmte; im Vergleich zur Kontrollgruppe hatte die Xenotransplantatgruppe, der das miR-149-3p-Mimik intratumoral injiziert wurde, jedoch eine unveränderte SUVmax. Vielleicht ist dieses widersprüchliche Ergebnis auf die Entwicklung von Nekrose im Kernbereich des subkutanen Tumorgewebes zurückzuführen, was eine Untersuchung rechtfertigt.
Insgesamt zeigen wir, dass der p53/miR-149-3p/PDK2-Signalweg bei der Überwindung von chemoresistentem CRC nach DCA-Behandlung möglicherweise gezielt eingesetzt werden kann, was eine potenzielle Strategie für die CRC-Behandlung unter dem Gesichtspunkt des Eingriffs in den Tumorstoffwechsel darstellt (Abb. 7e).
Materialien und Methoden
Krebsgewebe
Achtundzwanzig CRC-Patienten aus dem Neunten Volkskrankenhaus, das der Shanghai Jiao Tong University School of Medicine angegliedert ist, wurden zwischen 2013 und 2016 aufgenommen. Dreizehn dieser Patienten erhielten eine postoperative Chemotherapie auf 5-FU-Basis und wurden mindestens 3 Jahre lang nachbeobachtet. Alle Gewebe wurden nach Einholung einer informierten Zustimmung entnommen, und alle Verfahren, an denen menschliche Patienten beteiligt waren, wurden in Übereinstimmung mit den Vorschriften der Ethikkommission des Neunten Volkskrankenhauses der Medizinischen Hochschule der Jiao-Tong-Universität Shanghai durchgeführt. Die klinischen Daten der CRC-Patienten sind in der ergänzenden Tabelle 1 aufgeführt.
| Charakteristisch für CRC-Patienten | |
| Charakteristisch | Insgesamt (n=28) |
| Alter-yr | 65.4±10.5 |
| Geschlecht-no. (%) | |
| Männlich | 17(60.7) |
| Weiblich | 11(39.3) |
| Lokalisation – Nr. (%) | |
| Rektum | 10(35.7) |
| Dickdarm | 18(64.3) |
| Staging – Anzahl (%) | |
| T1 oder T2 | 6(21.4) |
| T3 oder T4 | 22(78.6) |
| Befallene Lymphknoten- Nr. (%) | |
| N0 | 12(42.9) |
| N1 oder N2 | 16(57.1) |
| Metastasierung- Nr. (%) | |
| M0 | 27(96.4) |
| M1 | 1(3.6) |
| Postoperative Chemotherapie mit einem Fluorouracil-basierten Regime | |
| Ja | 21(75) |
| Nachbeobachtung für 3 Jahre | |
| Ja | 13(61.9) |
| Nein | 8(38.1) |
| Nein | 7(25) |
Zellkultur
Die 5-FU-resistente Zelllinie HCT-8/F und die elterliche HCT-8-Zelllinie wurden von iCell Bioscience, Inc. (Shanghai, China) erworben. Die 5-FU-resistente Zelllinie HCT116/F und die elterliche HCT116-Zelllinie wurden freundlicherweise von Dr. Gu (Yanhong Gu, Nanjing Medical University, Jiangsu, China) zur Verfügung gestellt. HCT116-/–Zellen waren ein Geschenk von Dr. Lu (Hua Lu, Fudan-Universität, Shanghai, China). Die humane embryonale Nierenzelllinie 293T wurde von der American Type Culture Collection (ATCC, Manassas, VA, USA) bezogen. Die Zelllinien 293T, HCT116 und HCT-8 wurden in Dulbecco’s Modified Eagle’s Medium (HyClone, Utah, USA) oder RPMI-1640 Medium (HyClone, Utah, USA) mit 10 % fötalem Rinderserum (Gemini, Kalifornien, USA), 100 U/mL Penicillin und 100 μg/mL Streptomycin (HyClone, Utah, USA) in einem 37 °C, 5 %CO2 befeuchteten Inkubator kultiviert. Die Kulturmedien der Zelllinien HCT-8/F und HCT116/F wurden mit 15 μg/ml 5-FU bzw. 5 μg/ml 5-FU angereichert. Alle Zelllinien wurden von der Genetic Testing Biotechnology Corporation (Suzhou, Jiangsu, China) durch Short-Tandem-Repeats-Sequenzierung authentifiziert. DCA wurde von Sigma-Aldrich Co. Ltd. (MO, USA).
Immunfluoreszenzfärbung von Edu und ROS
HCT-8/F- und HCT116/F-Zellen wurden in 96-Well-Platten mit 15.000 Zellen/Well ausgesät. Nach einer Inkubation über Nacht wurden die Zellen 24 Stunden lang mit 15 mM bzw. 20 mM DCA behandelt. Die Edu-Färbung wurde gemäß den Anweisungen des Herstellers (Ribobio, Guangzhou, China) durchgeführt. Die ROS-Konzentrationen wurden in Zellen gemessen, die mit 10 μM 2′,7′-Dichlorfluoresceindiacetat (DCF-DA) (Beyotime, Shanghai, China) für 30 Minuten bei 37 °C inkubiert wurden. Die Platten wurden dann zweimal gewaschen und die Zellen wurden mit einem Fluoreszenzmikroskop analysiert.
Zellwachstum
Die Zellen wurden über Nacht in Platten mit 96 Vertiefungen (5000 Zellen/Vertiefung) ausgesät, 24 Stunden lang mit Arzneimitteln behandelt und anschließend wurde jede Vertiefung durch eine Mischung aus 10 μl CCK8 (Dojindo, Japan) und 90 μl Kulturmedium ersetzt. Die Absorption wurde zwei Stunden später bei einem OD-Wert von 450 nm mit einem Enzym-Mikroplattenlesegerät (BioTeck, Vermont, USA) gemessen. Das Hemmungsverhältnis des Arzneimittels wurde mit der folgenden Formel berechnet: 1-ODdrug/ODctrl. Die IC50 für jede Zelle wurde mit GraphPad Prism 6 (GraphPad Software, San Diego, CA) berechnet.
Zellapoptosetest
HCT-8/F- und HCT116/F-Zellen wurden in 6-Well-Platten in einer Konzentration von 2 ×105 Zellen/Well ausgesät. Die Zellen wurden 48 Stunden lang mit DCA (15 mM) / 5-FU (50 μg/ml) bzw. DCA (20 mM) /5-FU (25 μg/ml) behandelt. Anschließend wurden die Zellen trypsinisiert, gewaschen und mit Annexin V-FITC/PI- oder Annexin V-PE/7-AAD-Antikörpern gemäß dem Herstellerprotokoll (BD, CA, USA) gefärbt. Die Apoptose wurde mittels Durchflusszytometrie (BD, CA, USA) gemessen.
Koloniebildungstest
HCT-8/F- und HCT116/F-Zellen wurden 24 Stunden lang mit DCA (15 mM)/5-FU (50 μg/ml) bzw. DCA (20 mM)/5-FU (25 μg/ml) behandelt. Anschließend wurden die Zellen in 6-Well-Platten mit 1000 Zellen pro Vertiefung ausgesät und in frischem Medium bei 37 °C 1 bis 2 Wochen lang kultiviert. Anschließend wurden die Zellen 30 Minuten lang mit 4 % Paraformaldehyd fixiert, mit 1 % Kristallviolett angefärbt und die Anzahl der Zellkolonien mit einem Zähler (Gelcount, Optronix, Oxford) gezählt.
Transiente Gentransfektion
miR-149-3p Mimics, Inhibitoren, siPDK2, sip53 und ihre entsprechenden NC-Oligonukleotidsequenzen wurden von GenePharma (Shanghai, China) synthetisiert. Das Flag-p53-Plasmid wurde freundlicherweise von Dr. Lu (Hua Lu, Fudan University, Shanghai, China) zur Verfügung gestellt. Die Transfektion erfolgte mit Lipofectamine 3000 (Invitrogen, CA, USA) in einer Endkonzentration von 50 nmol/L (Mimics und siRNAs) oder 100 nmol/L (Inhibitoren). Die Zellen wurden 24 oder 48 Stunden nach der Transfektion für die Tests geerntet. Die siRNA-, Mimik- und Inhibitorsequenzen sind in der ergänzenden Tabelle 2 aufgeführt.
| siRNA-, Mimik- und Inhibitor-Sequenz | |
| siPDK2-1 | sinn: 5′-GACCGAUGCUGUCAUCUAUU-3′ |
| antisense: 5′-AAUAGAUGACAGCAUCGGUC-3′ | |
| siPDK2-2 | sinn: 5′-GACUCUUUCAGCUACAUGUA-3′ |
| antisense: 5′-UACAUGUAGCUGAAGAGUC-3′ | |
| NC | sinn: 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| antisinn: 5′-ACGUGACACGUUCGGAGAATT-3′ | |
| mimic-149-3p | sinn: 5′-AGGGAGGGGACGGGGGCUGUGC-3′ |
| antisense: 5′-ACAGCCCCCGUCCCUCCCUU-3′ | |
| hemmstoff-NC | 5′-CAGUACUUUUGUGUAGUACAA-3′ |
| hemmstoff-149-3p | 5′-GCACAGCCCCCGUCCCUCCCU-3′ |
| sip53 | sinn: 5′-GUAAUCUACUGGGACGGAAtt-3′ |
| antisense: 5′-UUCCGUCCCAGUAGAUUACca-3′ |
Stabile Gentransfektion
LV-PDK2 und das entsprechende NC-Virus wurden von GeneChem (Shanghai, China) erworben. shPDK2-1, shPDK2-2 und das Kontrollplasmid wurden von GeneChem (Shanghai, China) erworben. Der Virusüberstand wurde aus 293 T-Zellen gewonnen. Anschließend wurden die CRC-Zellen mit dem Virus infiziert und mit Puromycin getestet. Die Effizienz der Infektion wurde durch Durchflusszytometrie und Fluoreszenzmikroskopie überprüft. Die mRNA- und Proteinexpression von PDK2 wurde mittels quantitativer Echtzeit-PCR und Western Blot weiter analysiert.
3′-UTR-Reporter-Luciferase-Assays
Die wt- oder mutierten miR-149-3p-Bindungssequenzen in der humanen PDK2 3′UTR wurden in die stromabwärts gelegene Luciferase pmiR-RB-Reporter (Ribobio, Guangzhou, China) kloniert, siehe WT, MUT1, MUT2 und MUT3 in Abb. 3c. 293 T-Zellen und HCT116-Zellen wurden in 24-Well-Platten ausgesät, gefolgt von einer Cotransfektion mit 500 ng Reporterkonstrukten und entweder 50 nmol/L miR-149-3p-Mimik oder einem NC mit Lipofectamine 3000 (Thermo Fisher Scientific, Waltham MA). Die Luziferaseaktivität wurde nach 48 Stunden Inkubation mit dem Dual-Luciferase-Reporter-Assay-System (Promega, Madison, USA) gemäß dem Protokoll des Herstellers gemessen.
Glukoseverbrauch und Laktatproduktion
HCT-8/F- und HCT116/F-Zellen wurden über Nacht in 24-Well-Platten mit 1 ×105 Zellen/Vertiefung ausgesät und dann 24 Stunden lang mit 15 mM bzw. 20 mM DCA behandelt. Nach der Behandlung wurden die Zellen 24 Stunden lang in phenolrotfreiem Medium mit 10 % fötalem Rinderserum kultiviert. Das Kulturmedium wurde geerntet, und der Glukoseverbrauch und die Laktatproduktion wurden gemessen. Das Laktat wurde mit einem Laktat-Assay-Kit (Njjcbio, Nanjin, China) und die Glukose mit einem Glukose-Assay-Kit (Rsbio, Shanghai, China) gemessen. Alle Werte wurden durch Zählung einer gleichen Anzahl von Zellen standardisiert. Zur Bewertung des Glykolysezustands wurde den kultivierten Zellen 6 Stunden lang 100 ng/ml Oligomycin (ein ATP-Synthase-Inhibitor; Sangon Biotech) zugesetzt. Das Verhältnis der Laktatkonzentration in An- und Abwesenheit von Oligomycin wurde wie zuvor beschrieben gemessen und bestimmt [46].
Seahorse XF-96 glycolytic rate assay
Die Zellen wurden in einer 96-Well-Kulturplatte mit einer Dichte von 25.000 Zellen/Well ausgesät und über Nacht in Wachstumsmedium mit 10 % fötalem Rinderserum inkubiert. Die Sensorpatrone wurde über Nacht hydratisiert. Am nächsten Tag wurde das Zellmedium durch ein bikarbonatfreies, niedrig gepuffertes Assay-Medium ersetzt, das mit Glukose, Natriumpyruvat und Glutamin angereichert war. Nach der Inkubation der Zellen für 1 Stunde bei 37 °C in einem CO2-freien Inkubator wurden die Sauerstoffverbrauchsrate und die extrazelluläre Versauerungsrate vor und nach der Injektion von DCA/ctrl, Rotenon (Rot) + Antimycin A (AA) und 2-Desoxy-d-Glucose (2-DG) mit dem Seahorse XF Instrument (Agilent, Santa Clara, CA) gemessen, wie zuvor beschrieben [59, 60]. Die Experimente wurden in Echtzeit in fünf bis sechs Wiederholungsvertiefungen durchgeführt. Die Glykoeffizienten, einschließlich des Basal-Glykoeffizienten, des induzierten Glykoeffizienten und des kompensatorischen Glykoeffizienten, wurden automatisch von der Wave-Software (Agilent, Santa Clara, CA) berechnet.
Quantitative Echtzeit-PCR und miRNA-Microarray
Die gesamte RNA wurde aus CRC-Geweben oder -Zellen mit TRIzol-Reagenz (Life, CA, USA) extrahiert. cDNA wurde mit dem PrimeScript RT Reagent Kit (TaKaRa, Tokio, Japan) synthetisiert. Der Microarray-Assay wurde mit drei Wiederholungen von HCT116-Zellen durchgeführt, die 12, 24 oder 48 Stunden lang mit 5 mM, 10 mM oder 20 mM DCA behandelt wurden. Die Originaldaten wurden in die GEO-Datenbank (GSE125309) hochgeladen. Die quantitative Echtzeit-PCR wurde mit dem Premix Ex Taq 420 A (TaKaRa, Tokio, Japan) auf der ABI-7500-Plattform durchgeführt. Actin und U6 wurden als interne Kontrollen verwendet. Die Primer-Sequenzen sind in der ergänzenden Tabelle 3 aufgeführt.
| primer-Sequenzen für die quantitative Echtzeit-PCR in umgekehrter Transkription | |
| Aktin | F: 5′-CTCCATCCTGGCCTCGCTGT-3′ |
| R: 5′-GCTGTCACCTTCACCGTTCC-3′ | |
| PDK2 | F: 5′-CGCTGGCTGGCTTTGGTTATG-3′ |
| R: 5′-ACAGGGCCTTGAGATAGATG-3′ | |
| PDK1 | F: 5′-GCTGTATGGCCTGCAAGATG-3′ |
| R: 5′-GCTGTCCTGGTGATTTTGCA-3′ | |
| PDK3 | F: 5′-GGTTTGCCAATTTCCCGTCTG-3′ |
| R: 5′-CATCGGCTTCAGGCGTGGTC-3′ | |
| PDK4 | F: 5′-GAGAATTATTGACCGCCTCT-3′ |
| R: 5′-CGAGAAATTGGCAAGCCGTAA-3′ | |
| HK1 | F: 5′-CTTACTAAGGGATGCGATAAA-3′ |
| R: 5′-TCCCAACAATGAGTCCAACC-3′ | |
| TP53 | F: 5′-CCCAAGCAATGGATGATTTGA-3′ |
| R: 5′-GGCATTCTGGGAGCTTCATCT-3′ | |
| p21 | F: 5′-CTGGACTGTTTTCTCTCGGCTC-3′ |
| R: 5′-TGTATATTCAGCATTGTGGGAGGA-3′ | |
| MDM2 | F: 5′-ATGAATCCCCCCCCCTTCCAT-3′ |
| R: 5′-CAGGAAGCCAATTCTCACGAA-3′ | |
| PUMA | F: 5′-ACAGTACGAGCGGCGGAGACAA-3′ |
| R: 5′-GGCGGGTGCAGGCACCTAATT-3′ | |
| miR-149-3p | RT: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGG ATACGACGCACAG-3′ |
| F: 5′-ACAGGGAGGGACGGGGG-3′ | |
| R: 5′-ATCCAGTGCAGGGTCCGAGG-3′ | |
| miR-128-3p | RT: 5′-TCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGA TACGACAAAGAG-3′ |
| F: 5′-CGCGTCACAGTGAACCGGT-3′ | |
| R: 5′-AGTGCAGGGTCCGAGGTATT-3′ | |
| miR-500a-5p | RT: 5′-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCAC TGGATACGACTCTCACC-3′ |
Western Blots
Dreißig Mikrogramm Gesamtproteinlysate wurden geladen, und die primären Antikörper wurden aufgetragen: anti-PDK2 (sc-100534, Santa Cruz, California, US), anti-p-PDHA1 (S293) (ABS204, Merck, Darmstadt, Deutschland), anti-PDHA1 (ab168379, Abcam, Cambridge, UK), anti-p53 (sc-126, Santa Cruz, California, US), anti-c-PARP (D64E10, CST, Massachusetts, USA), anti-Bax (D2E11, CST, Massachusetts, USA), anti-LC3B (L7543, sigma, MO, USA), anti-GAPDH (Proteintech, Wuhan, China) und anti-α-Tubulin (Proteintech, Wuhan, China). Die sekundären Antikörper wurden von Sungene (Tianjin, China) bezogen. Die Blot-Assays wurden mit einem Chemilumineszenz-Imaging-System (Bioshine, Shanghai, China) abgebildet.
Chromatin-Immunpräzipitation (ChIP)
HCT116-Zellen, die in 10-cm-Platten ausgesät worden waren, wurden 24 Stunden lang mit oder ohne 20 mM DCA behandelt, dann wurden die Zellen fixiert und die Chromosomenfragmentierung gemäß den Anweisungen des Herstellers durchgeführt (Pierce Agarose ChIP Kit, Thermo). Das Chromatin wurde mit IgG- und anti-p53-Antikörpern (Sigma, MO, USA) bei 4°C über Nacht inkubiert. Nach der Inkubation wurden 60 ul Protein-A-Agarose/Lachssperma-DNA hinzugefügt. Anschließend wurde der ausgefällte Komplex mit den IP-Waschpuffern 1, 2 und 3 gewaschen und mit Elutionspuffer eluiert. Die Vernetzung wurde durch Zugabe von 6 μl 5 M NaCl und 2 ul Proteinase K bei 65°C für 1,5 h aufgehoben. Die immungefällte DNA und die DNA aus dem Gesamtzellextrakt (Input) wurden gereinigt und dann für PCR-Analysen mit den entsprechenden Primern verwendet. Ein Kontrollprimer wurde zur Überwachung des Experiments verwendet. Die Primer-Sequenzen für die PCR sind in der ergänzenden Tabelle 3 aufgeführt.
Subkutane Tumor-Xenotransplantation in Nacktmäusen und Mikro-PET/CT-Bildgebung
Zunächst wurden 1 ×107 HCT-8/F-Zellen, verdünnt in 100 μl PBS, subkutan in Nacktmäuse (männlich, 6 Wochen) implantiert. Nach 12 Tagen wurden die Mäuse nach dem Zufallsprinzip in sechs Gruppen (sechs pro Gruppe) aufgeteilt. Die Mäuse der Gruppen I bis IV erhielten täglich eine intraperitoneale Injektion von PBS, DCA (50 mg/kg)/PBS, 5-FU (10 mg/kg)/PBS bzw. 5-FU (10 mg/kg)/DCA (50 mg/kg). Die Mäuse der Gruppen V und VI erhielten jeden zweiten Tag eine intratumorale Injektion von PBS bzw. DCA (50 mg/kg) und jeden zweiten Tag eine intraperitoneale Injektion von 5-FU (10 mg/kg). Das Tumorvolumen wurde alle 3 Tage blind gemessen. Die Mäuse wurden nach dreiwöchiger Behandlung getötet, und die Tumore wurden seziert, gewogen und bei -80 °C für weitere Untersuchungen eingefroren.
Um zu untersuchen, ob miR-149-3p eine chemosensibilisierende Wirkung hat, wurden zwei weitere Gruppen von Tiermodellen erstellt. Dazu wurden Mäusen 6 ×106 HCT-8/F-Zellen subkutan implantiert. Nach Erreichen einer Tumorgröße von ~50 mm3 erhielten die Mäuse jeden zweiten Tag eine intraperitoneale Dosis von 5-FU (10 mg/kg) sowie alle drei Tage eine intratumorale Injektion von 5 nmol Cholesterin-konjugierten miR-149-3p-Mimikern oder einem NC für drei Wochen. Danach wurden drei Mäuse aus jeder Gruppe über Nacht gefastet und intravenös mit 0,15 mCi 18F-FDG injiziert. nach 60 Minuten wurde ein 18F-FDG-Mikro-PET-CT-Scan (Siemens, Berlin, Deutschland) durchgeführt. Die Bilder der PET-Akquisition wurden in einer Pseudofarbkarte dargestellt, wobei die rote Farbe eine hohe 18F-FDG-Aufnahme anzeigte. SUVmax und MTV wurden zur Bestimmung der 18F-FDG-PET-Aktivität verwendet. Alle Experimente und die Behandlung der Tiere wurden von der Ethikkommission des Neunten Volkskrankenhauses der Medizinischen Hochschule der Jiao-Tong-Universität Shanghai genehmigt.
Immunhistochemische Färbung und Immunfluoreszenz
Die Gewebeschnitte wurden mit den primären Antikörpern gegen Ki67 (Servicebio, Wuhan, China) und PDK2 (Proteintech, Wuhan, China) über Nacht bei 4 °C inkubiert und anschließend mit den sekundären Antikörpern inkubiert. Die chromogene Reaktion wurde mit 3,3-Diaminobenzidin durchgeführt und mit Hämatoxylin gegengefärbt. Der immunreaktive Score (IRS) wurde von zwei Untersuchern berechnet, die für die Gruppenzuordnung blind waren. IRS = SI (Färbeintensität) × PP (Prozentsatz der positiven Zellen). SI wurde wie folgt zugeordnet: 0 × negativ; 1 × schwach; 2 × mäßig; 3 × stark. PP ist definiert als 0 × 0%; 1 × 0-25%; 2 × 25-50%; 3 × 50-75%; 4 × 75-100%. Sechs-Millimeter-Gefrierschnitte wurden mit einem TUNEL-Reaktionskit (Roche, Basel, Schweiz) gefärbt und mit DAPI gegengefärbt. Die Bilder wurden mit einem Fluoreszenzmikroskop mit entsprechenden Anregungs- und Emissionsfiltern aufgenommen.
Statistische Analyse
Die Daten wurden mit der Software GraphPad Prism 6.0 ausgewertet. Die Daten sind als Mittelwerte ± SD/SEM von drei unabhängigen Experimenten dargestellt. Jedes Experiment wurde mit mindestens drei Wiederholungen durchgeführt. Zum Vergleich der Unterschiede zwischen den beiden Gruppen wurde ein zweiseitiger Student’s t-Test verwendet. Für Mehrfachvergleiche wurde eine einseitige ANOVA, gefolgt von einem Bonferroni-Post-hoc-Test, durchgeführt. Die Kaplan-Meier-Kurven für Überlebensanalysen wurden mit dem Log-Rank-Test ermittelt. Die Beziehung zwischen miR-149-3p und PDK2 wurde mithilfe der Spearman-Rangkorrelationskoeffizienten-Analyse bewertet. Ein P-Wert <0,05 wurde als statistisch signifikant angesehen.
Danksagungen
Diese Arbeit wurde von der National Natural Science Foundation of China (81272745, 81872419 und 81272404) und dem Program for Professor of Special Appointment (Eastern Scholar to JW) der Shanghai Institutions of Higher Learning unterstützt. Wir danken Dr. Yanhong Gu für die Bereitstellung der 5-FU-resistenten HCT116-Zelllinien und Dr. Hua Lu für die Bereitstellung des p53-Plasmids.
Einhaltung der ethischen Standards
Interessenkonflikt
Die Autoren erklären, dass sie sich in keinem Interessenkonflikt befinden.
Anmerkung des Herausgebers
Springer Nature bleibt neutral in Bezug auf Rechtsprechungsansprüche in veröffentlichten Karten und institutionelle Zugehörigkeiten.
Open Access
Dieser Artikel steht unter einer Creative Commons Attribution 4.0 International License, die die Nutzung, Weitergabe, Anpassung, Verbreitung und Vervielfältigung in jedem Medium oder Format erlaubt, solange Sie den/die Originalautor(en) und die Quelle in angemessener Weise nennen, einen Link zur Creative Commons Lizenz angeben und angeben, ob Änderungen vorgenommen wurden. Die Bilder oder anderes Material von Dritten in diesem Artikel sind in der Creative-Commons-Lizenz des Artikels enthalten, es sei denn, es wird in einer Kreditlinie zu dem Material anders angegeben. Wenn das Material nicht in der Creative-Commons-Lizenz des Artikels enthalten ist und die von Ihnen beabsichtigte Nutzung nicht durch gesetzliche Bestimmungen erlaubt ist oder über die erlaubte Nutzung hinausgeht, müssen Sie die Erlaubnis direkt beim Urheberrechtsinhaber einholen. Eine Kopie dieser Lizenz finden Sie unter http://creativecommons. org/licenses/by/4.0/.
REFERENZEN
1 Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, et al. Cancer incidence and mortality in China, 2014. Chin J Cancer Res. 2018;30:1-12.2 Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA: Cancer J Clin. 2018;68:7-30.
3 Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB 3rd, et al. Dichloracetat verändert den Warburg-Stoffwechsel, hemmt das Zellwachstum und erhöht die Röntgenempfindlichkeit von menschlichen A549- und H1299-NSC-Lungenkrebszellen. Free Radic Biol Med. 2015;89:263-73.
4 Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor-Interferon-Signalisierung reguliert ein multigenes Resistenzprogramm gegen Immun-Checkpoint-Blockade. Cell. 2016;167: 1540-54 e12.
5 Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Fallbericht über ein schwerwiegendes unerwünschtes Ereignis nach der Verabreichung von T-Zellen, die mit einem chimären Antigenrezeptor transduziert wurden, der ERBB2 erkennt. Mol Ther: J Am Soc Gene Ther. 2010;18:843-51. Dichloracetat stellt die Chemosensitivität von Dickdarmkrebs durch die p53/miR-149-3p/PDK2-vermittelte. . . 483
6 Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA: Cancer J Clin. 2016;66:271-89.
7 Hammond WA, Swaika A, Mody K. Pharmakologic resistance in colorectal cancer: a review. Therapeutic Adv Med Oncol. 2016;8:57-84.
8 Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041-7.
9 Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus Fluorouracil und Leucovorin bei metastasiertem Kolorektalkarzinom. Irinotecan-Studiengruppe. New Engl J Med. 2000;343:905-14.
10 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74.
11 Matthew G, Vander Heiden LCC, Craig BT. Den Warburg-Effekt verstehen: die metabolischen Anforderungen der Zellproliferation. Science. 2009;324:1029-33.
12 Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
13 Shaw RJ. Glukosestoffwechsel und Krebs. Curr Opin Cell Biol. 2006;18:598-608.
14 Van Dang C, Pollak M. Why cancer & metabolism? Warum jetzt? Cancer Metab. 2013;1:1.
15 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532.
16 Cairns RA, Harris IS, Mak TW. Regulierung des Stoffwechsels von Krebszellen. Nat Rev Cancer. 2011;11:85-95.
17 Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613-21.
18 Guo WQZ, Wang Z, et al. MiR-199a-5p ist negativ mit bösartigen Erkrankungen assoziiert und reguliert die Glykolyse und die Laktatproduktion, indem es auf Hexokinase 2 bei Leberkrebs abzielt. Hepatology. 2015;62:1132-44.
19 Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduziert den Pentosephosphatweg und die Proliferation durch Targeting von PRPS1 und RPIA mRNAs in menschlichen Darmkrebszellen. Gastroenterology. 2015;149:1587-98 e11.
20 Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. MicroRNA-129- 5p reguliert die Glykolyse und die Zellproliferation, indem sie auf den Glukosetransporter SLC2A3 in Magenkrebszellen zielt. Front Pharmacol. 2018;9:502.
21 Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97.
22 Garzon R, Calin GA, Croce CM. MicroRNAs bei Krebs. Annu Rev Med. 2009;60:167-79.
23 Huang S, He X. microRNAs: winzige RNA-Moleküle, riesige Antriebskräfte, um die Zelle zu bewegen. Protein Cell. 2010;1:916-26.
24 Zhang Y, Wang J. MicroRNAs sind wichtige Regulatoren der Arzneimittelresistenz bei kolorektalem Krebs. Biol Chem. 2017;398:929-38.
25 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43:683-91.
26 Kankotia S, Stacpoole PW. Dichloracetat und Krebs: ein neues Zuhause für ein Arzneimittel für seltene Krankheiten? Biochim Biophys Acta. 2014;1846:617-29.
27 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochemical J. 1998;329(Pt 1):191-6.
28 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 2007;11:37-51.
29 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4.
30 Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33:603-10.
31 Papandreou I, Goliasova T, Denko NC. Krebsmedikamente, die auf den Stoffwechsel abzielen: Ist Dichloracetat das neue Paradigma? Int J Cancer. 2011;128:1001-8.
32 Michelakis ED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielle stoffwechselbedingte Therapie für Krebs. Br J Cancer. 2008;99:989-94.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in Darmkrebszellen. Br J Cancer. 2010;102:1746-52.
34 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Natriumdichloracetat (DCA) reduziert die Apoptose bei Hypoxie in kolorektalen Tumoren. Cancer Lett. 2010;297:75-83.
35 Bertoli G, Cava C, Castiglioni I. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015;5:1122-43.
36 Cao D, Jia Z, You L, Wu Y, Hou Z, Suo Y, et al. 18betaglycyrrhetinic acid suppresses gastric cancer by activation of miR149-3p-Wnt-1 signaling. Oncotarget. 2016;7:71960-73.
37 Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, et al. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br J Pharm. 2017;174:553-68.
38 Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15:992-1004.
39 Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Divergierende Regulierung der Genexpression der Pyruvat-Dehydrogenase-Kinase-Isoform in kultivierten menschlichen Muskelzellen. FEBS J. 2005;272:3004-14.
40 Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, BelouecheBabari M, Koh KX, et al. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br J Pharm. 2014;171:3255-67.
41 Stacpoole PW. The pharmacology of dichloroacetate. Metab: Clin Exp. 1989;38:1124-44.
42 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Altersabhängige Kinetik und Metabolismus von Dichloracetat: mögliche Bedeutung für die Toxizität. J Pharmacol Exp Therap. 2008;324:1163-71.
43 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013;73:7277-89.
44 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. Inactivation of the HIF-1alpha/ PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035-47.
45 Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res. 2014;321:219-30.
46 Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72-81. 484 Y. Liang et al.
47 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016;371:20-9.
48 Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab 2017;25:1037-43.
49 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32:1638-50.
50 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. 1995;270:28989-94.
51 Sugden MC, Holness MJ. Jüngste Fortschritte bei den Mechanismen zur Regulierung der Glukoseoxidation auf der Ebene des Pyruvatdehydrogenase-Komplexes durch PDKs. Am J Physiol Endocrinol Metab. 2003; 284: E855-62.
52 Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182- PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98.
53 Bellazzo A, Di Minin G, Valentino E, Sicari D, Torre D, Marchionni L, et al. Cell-autonomous and cell non-autonomous downregulation of tumor suppressor DAB2IP by microRNA-149- 3p promotes aggressiveness of cancer cells. Cell Death Differ. 2018;25:1224-38.
54 Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979-87.
55 Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. Der Tumorsuppressor p53 bei der Kontrolle von Stoffwechsel und Ferroptose. Front Endocrinol. 2018;9:124.
56 Contractor T, Harris CR. p53 reguliert negativ die Transkription der Pyruvat-Dehydrogenase-Kinase Pdk2. Cancer Res. 2012;72:560-7.
57 Iacopetta B. TP53 mutation in colorectal cancer. Hum Mutat. 2003;21:271-6.
58 Kim JW, Dang CV. Die molekulare Vorliebe von Krebs und der Warburg-Effekt. Cancer Res. 2006;66:8927-30.
59 Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, Tempera I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by EpsteinBarr virus latent membrane protein 1. PLoS Pathog. 2018;14: e1007394.
60 Hlouschek J, Ritter V, Wirsdorfer F, Klein D, Jendrossek V, Matschke J. Targeting SLC25A10 mildert die durch chronisch-zyklische Hypoxie induzierte verbesserte antioxidative Kapazität und damit verbundene Radioresistenz von Krebszellen. Cancer Lett. 2018;439:24-38.
Related content: