Yu Liang1, Lidan Hou1, Linjing Li1, Lei Li1, Liming Zhu1, Yu Wang1, Xin Huang1, Yichao Hou1, Danxi Zhu1, Huimin Zou1, Yan Gu2, Xiaoling Weng3,4, Yingying Wang5, Yue Li6, Tianqi Wu3, Mengfei Yao3, Isabelle Gross7,8, Christian Gaiddon9,10, Meng Luo2, Jianhua Wang3, Xiangjun Meng1
1 Отделение гастроэнтерологии, Девятая народная больница Шанхая, Медицинская школа Шанхайского университета Цзяо Тун, Шанхай, Китай
2 Отделение общей хирургии, Девятая народная больница Шанхая, Медицинская школа Шанхайского университета Цзяо Тун, Шанхай, Китай
Онкологический институт, Шанхайский онкологический центр Университета Фудань, Университет Фудань, Шанхай, Китай
Ningbo Aitagene Technology Co. LTD, Шанхай, Китай
5 Кафедра биохимии и молекулярной и Клеточная биология, Медицинская школа Шанхайского университета Цзяо Тун, Шанхай, Китай
6 Центр патологии, Первая народная больница Шанхая, Медицинская школа Шанхайского университета Цзяо Тун, Шанхай, Китай
INSERM UMR_S1113, Страсбург F-67200, Франция
FMTS, Страсбургский университет, Страсбург F-67000, Франция
9Universite de Strasbourg, Inserm IRFAC UMR_S1113, Laboratory Stress Response and Innovative Therapy «Streinth», Strasbourg 67200, France
10 CLCC Paul Strauss, Strasbourg, France
Meng Luo [email protected]
Jianhua Wang [email protected]
Xiangjun Meng [email protected]
Эти авторы внесли равный вклад: Yu Liang, Lidan Hou
Received: 16 марта 2019 г.
Пересмотрено: 17 сентября 2019 г.
Принято: 19 сентября 2019 г.
Опубликовано: 9 октября 20199 октября 2019 г
Аннотация
Развитие химиорезистентности остается одной из основных проблем, объясняющих летальность при колоректальном раке (КРР). Дихлорацетат (ДХА) первоначально использовался как регулятор метаболизма при лечении метаболических заболеваний; здесь ДХА был исследован для выявления механизмов, лежащих в основе химиорезистентности CRC. Мы обнаружили, что DCA заметно повышает химиочувствительность клеток CRC к флуороурацилу (5-FU) и снижает образование колоний из-за высокого уровня апоптоза. Используя анализ микрочипов, мы отметили, что miR-149-3p участвует в химиорезистентности CRC, которая была модулирована p53 дикого типа после лечения DCA. Кроме того, PDK2 был идентифицирован как прямая мишень miR-149-3p. Механический анализ показал, что сверхэкспрессия miR-149-3p усиливала 5-FU-индуцированный апоптоз и снижала метаболизм глюкозы, аналогично эффектам нокдауна PDK2. Кроме того, сверхэкспрессия PDK2 частично отменяла ингибирующий эффект miR-149-3p на метаболизм глюкозы. Наконец, было установлено, что лечение DCA и сверхэкспрессия miR-149-3p в клетках CRC, устойчивых к 5-FU, заметно повышают чувствительность к химиотерапевтическому эффекту 5-FU in vivo, и этот эффект также был подтвержден в небольшой ретроспективной когорте пациентов CRC. В целом, мы определили, что сигнальный путь p53/miR-149-3p/PDK2 потенциально может быть направлен на лечение DCA для преодоления химиорезистентности CRC.
Дополнительная информация: Онлайн-версия этой статьи (https:// doi.org/10.1038/s41388-019-1035-8) содержит дополнительные материалы, которые доступны авторизованным пользователям.
Введение
Колоректальный рак (КРР) является четвертой ведущей причиной смертности от рака в Китае [1] и второй ведущей причиной смертности от рака в США [2], что в основном объясняется метастазированием и неудачей химиотерапии из-за устойчивости к лекарствам, что приводит к ~50 000 смертей ежегодно [3].
В последнее время, хотя появившаяся звезда PD1/PDL1 привлекла большой интерес, и все больше биотерапевтических агентов показывают обнадеживающие результаты в лечении рака, ограниченный уровень эффективности и неизбежные побочные эффекты сдерживают их использование в клинике [4,5]. В настоящее время химиотерапия по-прежнему является основным методом выбора в клинике, особенно для пациентов с неоперабельными поздними стадиями и метастатическим раком [6], но развитие лекарственной устойчивости остается самым большим ограничением в химиотерапии [7]. Следовательно, изучение механизмов лекарственной устойчивости и поиск новых комбинаций классических противораковых препаратов для оптимизации эффективности может принести пользу в лечении КРР. Поскольку фторурацил (5-FU) является наиболее часто используемым химиотерапевтическим препаратом при РПК, в данном исследовании использовались 5-FUрезистентные клеточные линии РПК [8,9].
Нарушение метаболизма глюкозы представляет собой один из основных аспектов отличительных признаков рака [10]. Известно, что развитие неконтролируемой клеточной массы приводит к плохой васкуляризации опухоли, что вызывает снижение поступления кислорода. Следовательно, раковые клетки адаптируются к изменениям в микроэнвироменте, переключая свой метаболизм с окислительного обмена на гликолитический, который основан на снабжении глюкозой и производит лактат. Этот сдвиг называется «эффектом Варбурга» и обычно наблюдается в различных раковых клетках как одна из замечательных отличительных черт [11- 13]. Накопление данных последних исследований привело к необходимости уточнения теории Варбурга [14]; например, было показано, что метаболический сдвиг вовлечен в химиорезистентность [15]; следовательно, для преодоления химиорезистентности потенциально может быть использовано воздействие на метаболизм раковых клеток [16,17].
В частности, сообщалось о многочисленных механизмах контроля метаболического сдвига в раковых клетках, включая микроРНК (миРНК) [18-20]. МиРНК представляют собой класс малых эндогенных некодирующих РНК, которые регулируют трансляцию и деградацию мРНК [21] и вовлечены во многие другие биологические процессы, включая клеточную пролиферацию, миграцию, апоптоз, самообновление, инициацию, развитие рака и химиорезистентность [22-24].
Полученные данные свидетельствуют о том, что воздействие на аномальный метаболизм раковых клеток стало интенсивным направлением исследований, нацеленных на «удушение опухоли», стратегии которых заключаются в ингибировании ключевых ферментов, участвующих в гликолитическом метаболизме [15]. В данной рукописи дихлорацетат (ДХА) первоначально использовался для лечения молочнокислого ацидоза и наследственной митохондриальной болезни [25]. ДХА ингибирует ферментативную активность киназ пируватдегидрогеназы (PDK1-4), которая необходима для превращения пирувата в ацетил-КоА, связывая гликолитический метаболизм с циклом лимонной кислоты [26,27]; недавно сообщалось, что ДХА обладает противораковым действием [28-31]. Однако механизм, лежащий в основе влияния ДКА на лечение CRC, остается неустановленным.
Настоящее исследование было посвящено изучению молекулярного механизма, участвующего в регуляции метаболизма глюкозы и устойчивости к химиотерапии в CRC. Используя DCA в клетках CRC, мы изучили роль соответствующих миРНК и тем самым раскрыли сигнальный путь, объясняющий устойчивость к лечению 5-ФУ.
Результаты
DCA восстанавливает химиочувствительность клеток CRC, устойчивых к 5-FU
Сообщалось, что DCA является эффективным противоопухолевым препаратом, который действует на энергетические пути в некоторых видах рака [32]; однако влияние DCA на химиорезистентные клетки CRC не было изучено. Используя анализ CCK8, мы обнаружили, что по сравнению с родительскими клеточными линиями HCT-8 и HCT116, устойчивые к 5-ФУ клетки HCT-8/F и HCT116/F были нечувствительны к 5-ФУ (Дополнительный рис. 1A), а полумаксимальные ингибирующие концентрации (IC50) DCA в клетках HCT-8/F и HCT116/F составляли ~15 и 20 мМ, соответственно, что согласуется с предыдущими сообщениями [33, 34] (Дополнительный рис. 1B). Далее мы отметили, что DCA значительно подавлял синтез ДНК (дополнительная рис. 1С, верхняя панель) и индуцировал генерацию ROS (дополнительная рис. 1С, нижняя панель) в устойчивых к 5-FU клетках CRC. Маркеры энергетического метаболизма, включая потребление глюкозы, производство лактата и гликолиз, были заметно повышены в клетках CRC, устойчивых к 5-FU, по сравнению с клетками CRC, чувствительными к 5-FU, в то время как добавление DCA заметно снижало экспрессию этих маркеров (рис. 1a). Учитывая 6 ч предварительной обработки без сыворотки при измерении гликолиза, использовали набор для определения гликолитической скорости Seahorse XF, чтобы устранить эффект предварительной обработки и измерить гликолитическую скорость в реальном времени. Скорость гликолитического оттока протонов (glycoPER) отражает скорость внеклеточного закисления в результате гликолиза. Добавление DCA значительно снижало индуцированный гликолиз по сравнению с соответствующим контролем (рис. 1b и дополнительный рис. 1D).
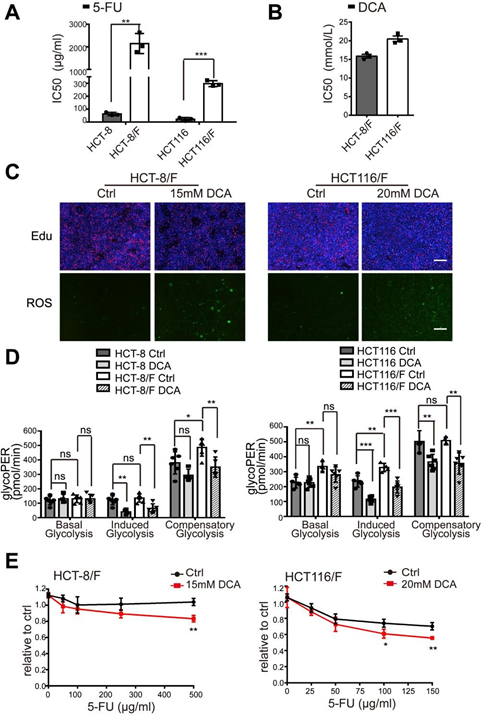
(A) Клетки CRC обрабатывали различными концентрациями 5-ФУ в течение 24 часов. Рассчитывали IC50 5-ФУ в каждой клетке. (B) Клетки HCT-8/F и HCT116/F обрабатывали различными концентрациями DCA в течение 24 часов. Рассчитывали IC50 DCA в каждой клетке. (C) Клетки HCT-8/F и HCT116/F обрабатывали 15 мМ и 20 мМ DCA, соответственно, в течение 24 часов. Репрезентативные изображения иммунофлуоресцентного окрашивания Edu (верхняя панель) и репрезентативные изображения иммунофлуоресцентного окрашивания ROS (нижняя панель), масштабная линейка: 200 мкм. (D) Определение скорости гликолиза, включая базальный гликопер, индуцированный гликопер и компенсаторный гликопер, рассчитывали с помощью Seahorse Glycolytic Rate Assay Report Generator. (E) Рост клеток определяли с помощью анализа CCK8 после обработки 5-FU и DCA. Диапазон доз 5-ФУ составлял от 100 мкг/мл до 500 мкг/мл для клеток HCT-8/F и от 25 мкг/мл до 150 мкг/мл для клеток HCT116/F. Результаты трех независимых экспериментов представлены как среднее ± SEM. Каждый эксперимент проводился в 3-6 биологических репликах. *, P < 0,05; **, P < 0,01; ***, P < 0,001.
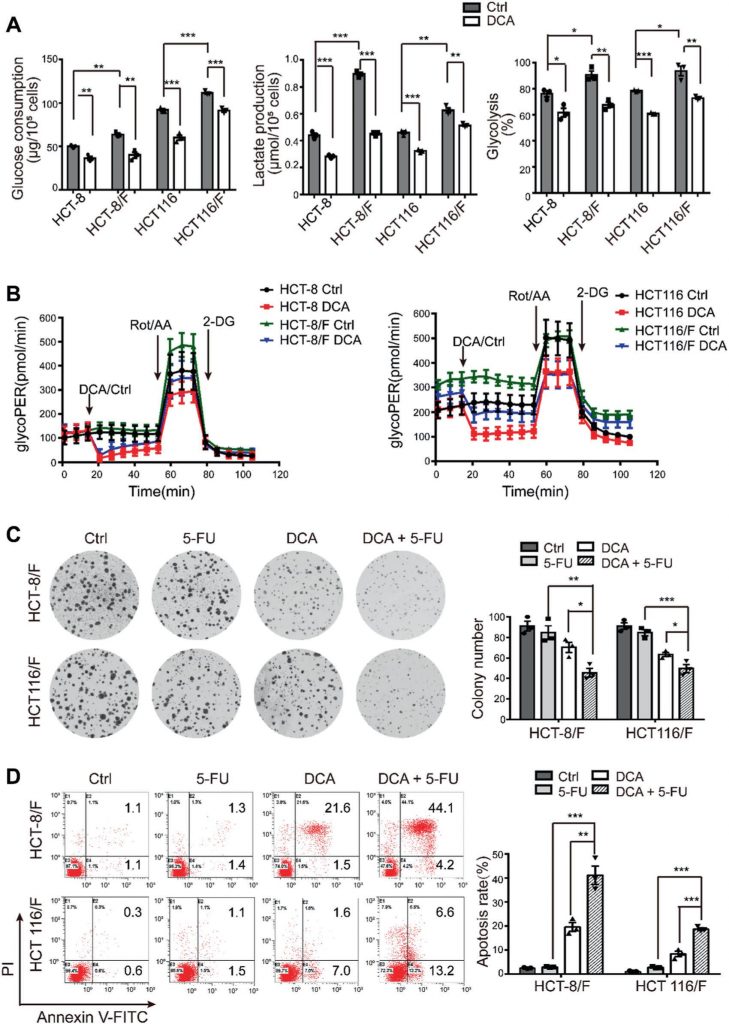
DCA значительно преодолевал резистентность к 5-ФУ в клетках HCT-8/F и HCT116/F, что проявлялось в измерении роста клеток (Дополнительный рис. 1E). Способность к образованию колоний была значительно подавлена (рис. 1c), а апоптоз был заметно индуцирован при комбинированном лечении (рис. 1d), все из которых были дополнительно оценены количественно. Эти результаты свидетельствуют о том, что DCA может восстанавливать химиочувствительность клеток CRC, устойчивых к 5-FU.
miR-149-3p играет решающую роль в химиочувствительности клеток CRC
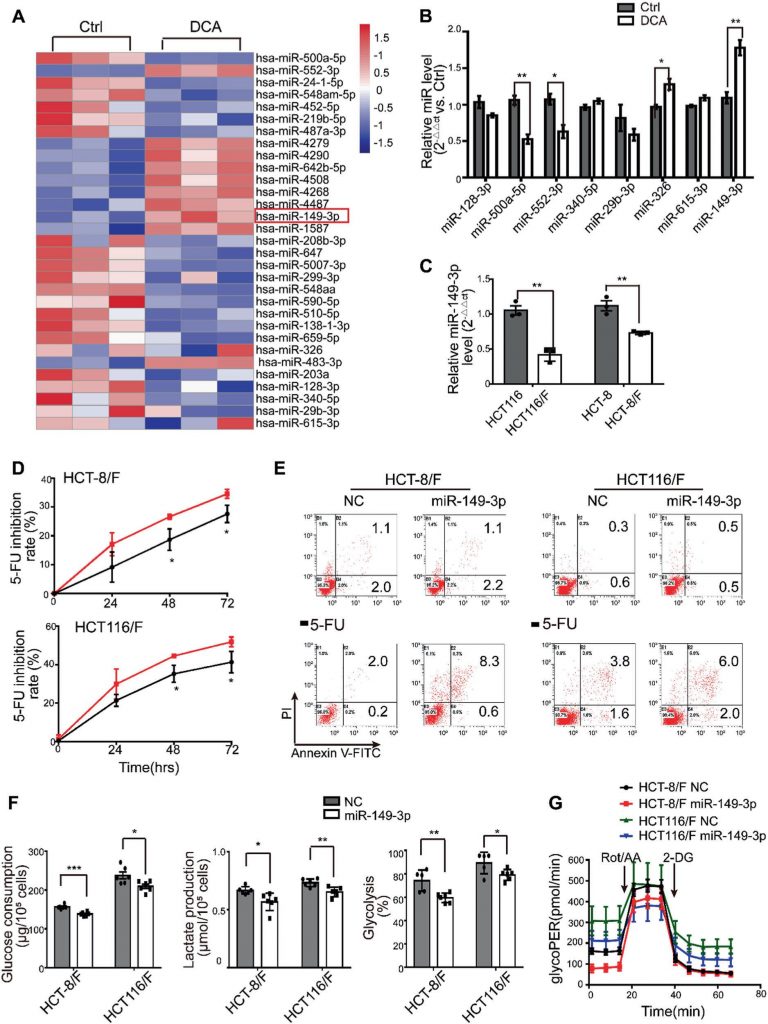
миРНК считаются перспективным терапевтическим инструментом благодаря их влиянию на подавление опухоли [35]. В связи с этим мы сначала определили профили экспрессии миРНК с помощью массива миРНК, содержащего 2059 человеческих миРНК. В общей сложности 119 миРНК дифференциально экспрессировались в ответ на DCA в клетках HCT116 (рис. 2a и дополнительный рис. S2A). Среди них уровни экспрессии восьми миРНК были дополнительно подтверждены количественной ПЦР в реальном времени (рис. 2b), и было установлено, что miR-149-3p повышается под действием ДКА в дозозависимой манере (дополнительный рис. S2B).
Далее мы обнаружили, что клетки HCT116 с более высоким базальным уровнем miR-149-3p обладают большей чувствительностью к 5-FU и L-OHP, как показано на дополнительном рис. 2C. Более того, трансфекция клеток HCT116 анти-miR-149-3p значительно снижала химиотерапевтический эффект 5-FU (Дополнительный рис. 2D-E). Примечательно, что уровни miR-149-3p в химиочувствительных клетках CRC были значительно выше, чем в химиорезистентных клетках CRC (рис. 2c). Поэтому трансфекция мимиков miR-149-3p значительно увеличивала скорость ингибирования; способствовала апоптозу клеток, индуцированному 5-FU; снижала потребление глюкозы, производство лактата и гликолиз в клетках HCT-8/F и HCT116/F (рис. 2d-f). Анализ гликолитической скорости Seahorse XF показал, что экспрессия miR-149-3p снижала базальный гликопер в устойчивых к 5-ФУ клетках CRC, что согласуется с вышеприведенными результатами (рис. 2g и дополнительный рис. 2F). Эти результаты позволяют предположить, что miR-149-3p благоприятствует преодолению химиорезистентности в клетках CRC.
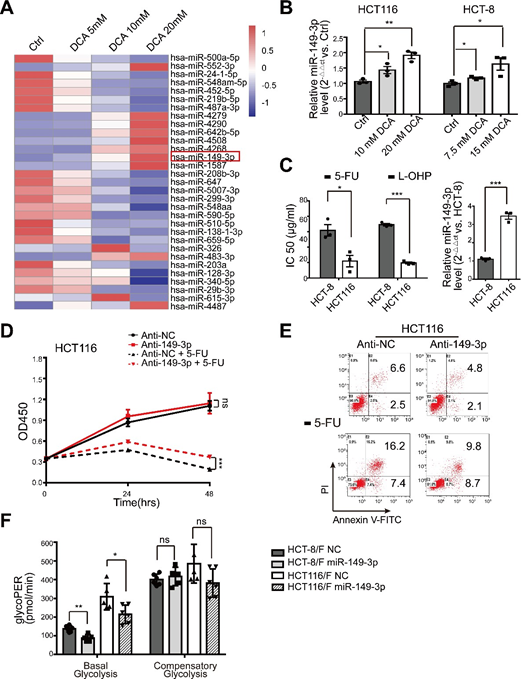
(A) Тепловая карта профиля дифференциально экспрессированных микроРНК в клетках HCT116, обработанных контролем, 5 мМ, 10 мМ, 20 мМ DCA в течение 24 часов. (B) Тотальная РНК была получена через 24 часа после обработки 10/7,5 мМ и 20/15 мМ DCA из клеток HCT116 и HCT-8. Уровень miR-149-3p анализировали с помощью количественной ПЦР в реальном времени. (C) Клетки HCT-8 и HCT116 обрабатывали различными концентрациями 5-FU и оксалиплатина (L-OHP) соответственно в течение 24 часов и рассчитывали половину максимальной ингибирующей концентрации (IC50) (левая панель). Базальные уровни miR-149-3p определяли с помощью количественной ПЦР в реальном времени в клетках HCT-8 и HCT116 (правая панель). (D-E) Клетки HCT116 были трансфицированы Anti-NC или ингибитором miR-149-3p. После трансфекции клетки обрабатывали 25 мкг/мл 5-ФУ в течение 24 часов. Рост и апоптоз клеток определяли с помощью CCK8 и проточной цитометрии соответственно. (F) Определение скорости гликолиза, включая базальный гликопер и компенсаторный гликопер в 5-FU-резистентных CRC-клетках, трансфицированных NC или мимиком miR-149-3p, рассчитывали с помощью Seahorse Glycolytic Rate Assay Report Generator. Данные трех независимых экспериментов представлены как среднее ± SEM. Каждый эксперимент проводился в 3-6 биологических репликах. *p < 0,05; **, P < 0,01; ***, P < 0,001; ns, нет значимости.
DCA индуцирует экспрессию miR-149-3p через p53 дикого типа (wt)
Учитывая, что несколько недавних исследований показали, что miR-149-3p регулируется несколькими препаратами [36,37], мы определили, как miR-149-3p регулируется DCA. Мы обнаружили, что DCA значительно увеличивает экспрессию wt p53 и его нижележащих сигналов, включая экспрессию p21, PUMA и MDM2, в клетках CRC (рис. 3a, b). Более того, мы отметили, что изменения в экспрессии wt p53 способны значительно модулировать экспрессию miR-149-3p, как показано на рис. 3c, d, что указывает на то, что miR-149-3p положительно регулируется wt p53. Поэтому, используя р53-нулевую клеточную линию HCT116 (TP53-/-), мы обнаружили, что miR-149-3p не повышалась при обработке DCA, но эктопическая экспрессия р53 отменяла этот эффект. Более того, мы использовали nutlin-3, мощный ингибитор, который подавляет взаимодействие MDM2-p53, что приводит к активации p53 в качестве положительного контроля, при этом экспрессия miR-149-3p была повышена в wt линии клеток HCT116 (TP53+/+) (рис. 3e-g). С помощью программного обеспечения для биоинформационного анализа (IGV) были предсказаны четыре предполагаемых сайта связывания р53 с фланкирующим участком геномной ДНК miR-149. Затем в клетках были проведены анализы ChIP с использованием антитела против wt p53. Вытянутая ДНК была амплифицирована с помощью обычной ПЦР с праймерами, которые были разработаны на основе этих сайтов. Наши результаты показали, что по сравнению с регионом 4, регион 3 был заметно обогащен после обработки DCA в хроматине HCT116, иммунопреципитированном wt p53 (рис. 3h), что указывает на то, что только регион 3 содержит специфический сайт связывания, активируемый DCA. Эти результаты показывают, что DCA модулирует miR-149-3p через wt p53.
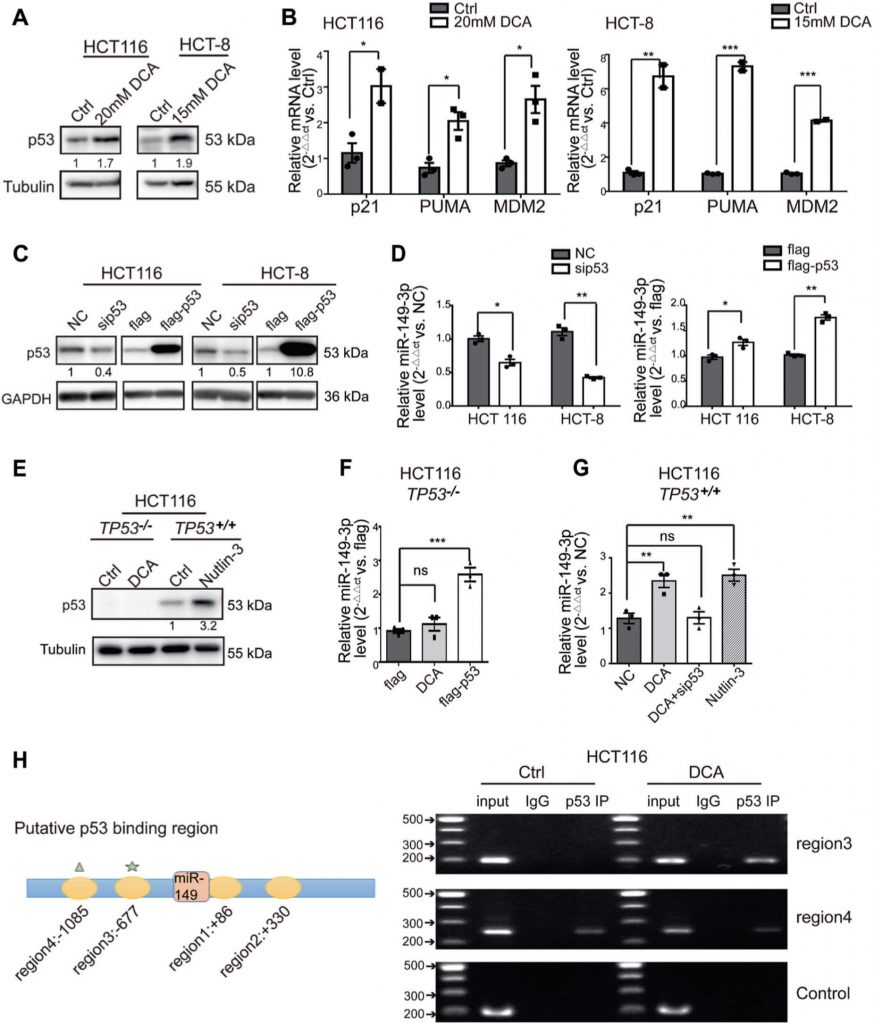
PDK2 является прямой мишенью miR-149-3p
Для выяснения механизмов, посредством которых miR-149-3p регулирует химиочувствительность клеток CRC, мы проанализировали гены, связанные с энергетическим метаболизмом, которые регулируются miR-149-3p, используя две общедоступные платформы (TargetScan и miRDB). Наконец, мы определили, что киназа пируватдегидрогеназы 2 (PDK2) и гексокиназа 1 (HK1) являются потенциальными кандидатами (рис. 4a). Для подтверждения этих выводов мимикр или ингибитор miR-149-3p был трансфецирован в клетки CRC. Мы обнаружили, что уровни мРНК PDK2 негативно регулировались miR-149-3p, но не HK1 (рис. 4b и дополнительный рис. 3A). Было обнаружено два возможных сайта связывания miR-149-3p в 3′-UTR PDK2, а двойной люциферазный репортерный анализ показал, что miR-149-3p связывается с предсказанным сайтом (686-693) 3′-UTR PDK2 (рис. 4c, d). Затем мы подтвердили, что уровень белка PDK2 отрицательно регулируется miR-149-3p (рис. 4e).
PDK имеет четыре изофермента PDK1, 2, 3 и 4, все из которых, как сообщалось, регулируются DCA [26, 38, 39]. Затем мы проанализировали экспрессию мРНК PDK1-4 в клетках HCT8 и HCT116 после обработки ДКА. DCA значительно подавлял экспрессию мРНК PDK2, но не других изоферментов PDK (Дополнительный рис. S3B). Кроме того, трансфекция анти-149-3p частично отменяла снижение PDK2 под действием DCA (рис. 4f). Далее мы определили уровни белка PDK2 и его субъединицы пируватдегидрогеназы E1-альфа (PDHA1) в чувствительных к 5-ФУ и устойчивых к 5-ФУ клетках CRC. Базальные уровни PDK2 были повышены в химиорезистентных клетках CRC по сравнению с химиочувствительными клетками. В соответствии с этим повышением, фосфорилирование PDHA1 также было повышено (рис. 4g).
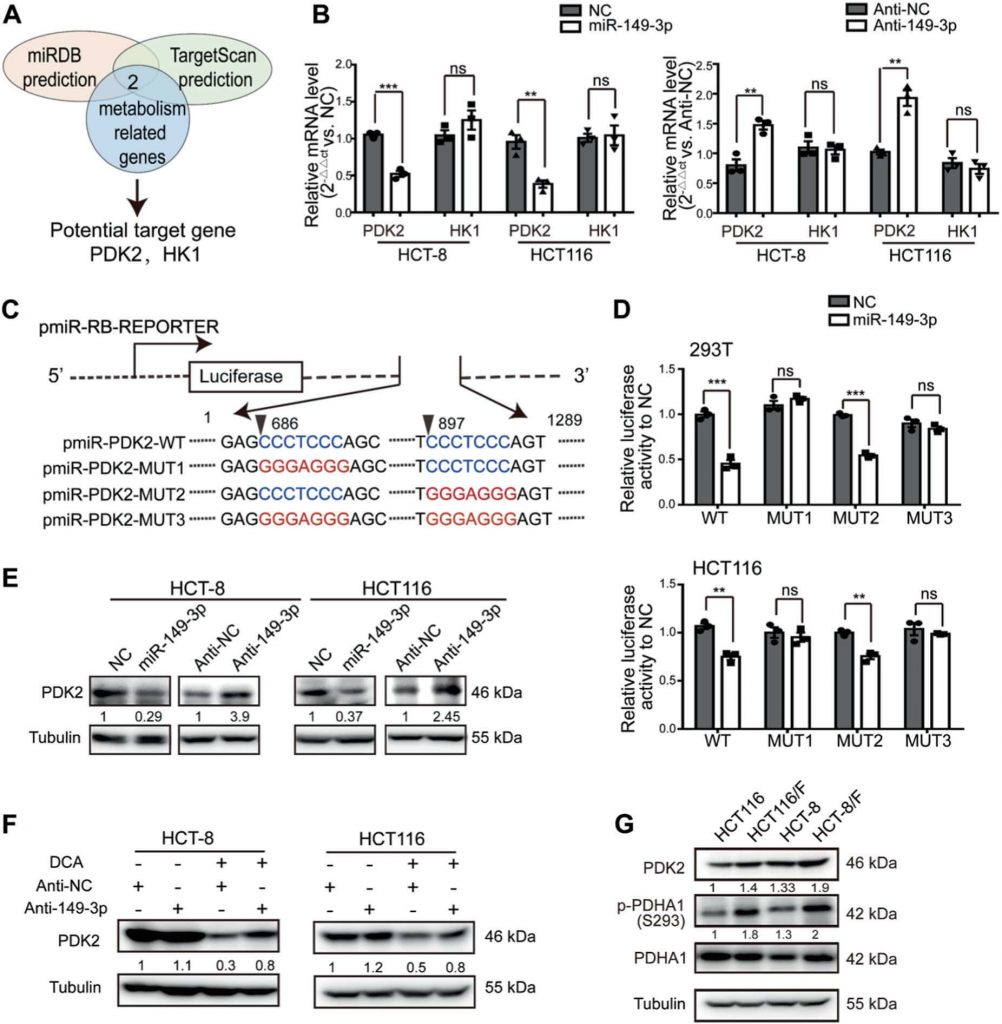
Путь miR-149-3p/PDK2 регулирует химиочувствительность
Чтобы понять, как путь miR-149-3p/PDK2 регулирует ответ клеток CRC на 5-FU, клеточная линия HCT-8/F была выбрана в качестве репрезентативной клеточной линии для исследования того, влияют ли уровни экспрессии miR-149-3p и PDK2 на ответ клеток на 5-FU. Нокдаун PDK2 в клетках HCT-8/F подавлял фосфорилирование PDHA1 (рис. 5a) и снижал маркеры энергетического метаболизма, такие как потребление глюкозы, производство лактата и гликолиз (рис. 5b). Более того, снижение PDK2 усиливало действие 5-ФУ на повышение уровней расщепленного PARP (c-PARP) и Bax, которые являются признанными биомаркерами апоптоза клеток HCT-8/F (рис. 5c). Кроме того, нокдаун PDK2 повышал химиочувствительность к 5-ФУ в клетках HCT-8/F, что определялось с помощью анализа CCK8 и колониеобразования (рис. 5d, e). Нокдаун PDK2 способствовал 5-ФУ-индуцированному апоптозу в клетках HCT-8/F и HCT116/F, что показано на дополнительном рис. S4A, тогда как сверхэкспрессия PDK2 способствовала фосфорилированию PDHA1 и смягчала апоптоз клеток, индуцированный 5-ФУ в клетках HCT-8 и HCT116 (дополнительные рис. S4B-S4C).
Кроме того, сверхэкспрессия PDK2 отменяла ингибирующий эффект miR-149-3p на PDK2 (рис. 5f) и частично отменяла ингибирующий эффект miR-149-3p на потребление глюкозы, производство лактата и гликолиз (рис. 5g). Эктопическая экспрессия PDK2 заметно отменяла ингибирующий эффект miR-149-3p на рост клеток и образование колоний в клетках HCT-8/F, обработанных 5-ФУ (рис. 5h, i). В целом, наши результаты свидетельствуют о том, что путь miR-149-3p/PDK2 восстанавливает химиочувствительность, по крайней мере, частично регулируя метаболизм глюкозы в химиорезистентных клетках CRC.
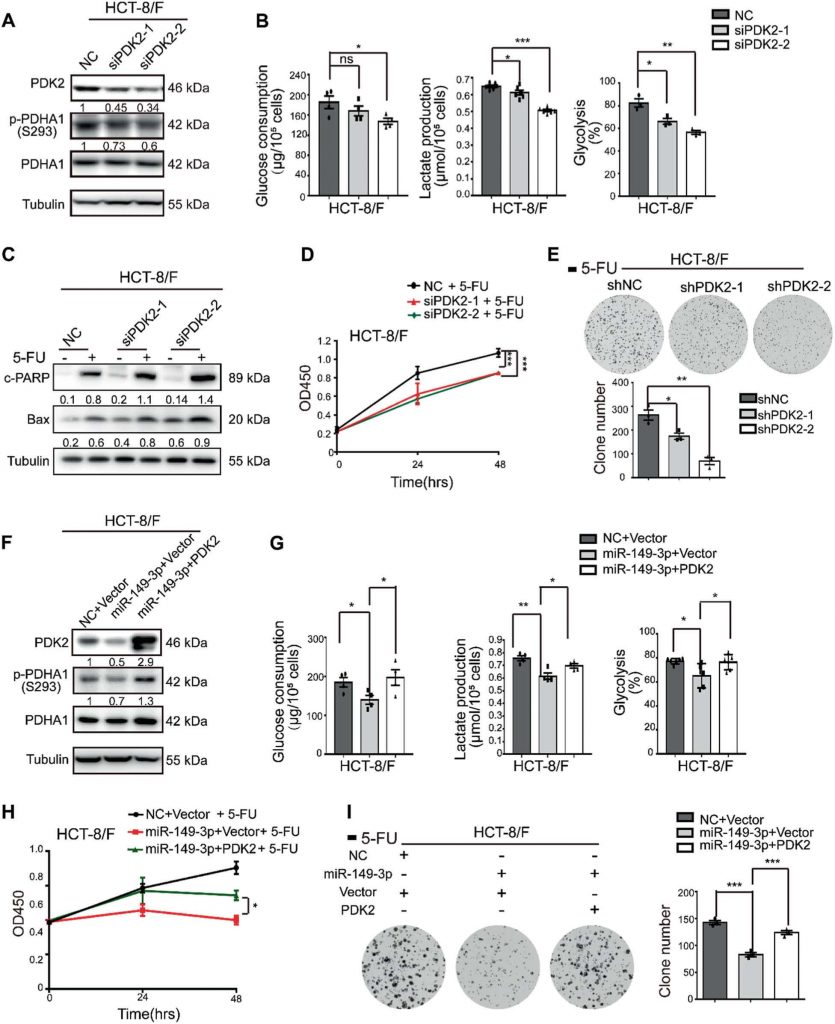
DCA повышает химиочувствительность 5-FU in vivo
Далее 5-FU, DCA или комбинацию 5-FU с DCA вводили внутрибрюшинно в модель подкожного ксенотрансплантата (отмечена как внутрибрюшинная группа). Учитывая недостаточное кровоснабжение центральной части подкожных опухолей, мы также интратуморально вводили DCA или PBS плюс интраперитонеальную инъекцию 5-ФУ (отмечена как интратуморальная группа). Комбинации DCA и 5-FU показали лучший ингибирующий эффект на рост опухоли, чем DCA или 5-FU по отдельности после четырех недель инъекций в группе внутрибрюшинного введения (рис. 6a, b). Экспрессия Ki-67 была снижена после комбинированного лечения в группе внутрибрюшинного введения (рис. 6c). DCA значительно подавлял рост опухоли в интратуморальной группе по сравнению с группой PBS (рис. 6d), а miR-149-3p был повышен в группе интратуморального введения DCA, что согласуется с результатами in vitro (рис. 6e). Интратуморальное введение DCA также способствовало апоптозу опухоли (рис. 6F).
Для дальнейшей оценки влияния miR-149-3p на ответ клеток CRC на 5-ФУ in vivo, в модели подкожного ксенотрансплантата проводили интратуморальное введение agomiR-149-3p или agomiR-NC плюс внутрибрюшинное введение 5-ФУ. miR-149-3p значительно подавлял рост опухоли в интратуморальной группе по сравнению с контрольной группой, в которой интратуморально вводили agomiR-отрицательный контроль (NC) (рис. 6g, h). Сверхэкспрессия miR-149-3p была подтверждена количественной ПЦР в реальном времени (рис. 6i), а сверхэкспрессия miR-149-3p способствовала апоптозу (рис. 6j) и снижала экспрессию PDK2 и p-PDHA1 (рис. 6k, l). С помощью микропозитронно-эмиссионной томографии (ПЭТ)-КТ с 18F-фтордезоксиглюкозой (ФДГ) визуализацию опухоли проводили через 3 недели лечения. в местах имплантации опухоли наблюдалось поглощение 18F-ФДГ, измерялись максимальное стандартизированное поглощение (SUVmax) и метаболические объемы опухоли (MTVs). Разницы в SUVmax между двумя группами не наблюдалось, в то время как MTV значительно уменьшился в группе miR-149-3p (рис. 6m).
miR-149-3p обратно коррелирует с PDK2 у пациентов с CRC
Значительная обратная корреляция между уровнями мРНК miR-149-3p и PDK2 наблюдалась в тканях CRC человека (рис. 7a, b). Среди них восемь пациентов в стабильном состоянии/болезни (SD) в течение 3 лет после химиотерапии экспрессировали более высокий уровень miR-149-3p, чем пять пациентов с прогрессирующей болезнью (PD) (Дополнительный рис. S5A). Были проанализированы пять пар пациентов с PD и SD с одинаковой патологией и стадией TNM, и окрашивание PDK2 значительно различалось в образцах от пациентов с PD и SD (Дополнительный рис. S5B). Пациенты CRC из базы данных TCGA с высокой экспрессией PDK2 также характеризовались худшей общей выживаемостью (OS) (рис. 7c). База данных TCGA также показала, что экспрессия PDK2 в группе wt p53 была снижена по сравнению с группой мутантных p53 (рис. 7d). Эти результаты позволили предположить, что PDK2 негативно регулируется miR-149-3p у пациентов с CRC.
Обсуждение
CRC характеризуется туморигенными аномалиями и измененными метаболическими путями и является одной из основных причин смерти от рака [2]. Резистентность к химиотерапии является основной причиной неудачного лечения [7]. В этом исследовании мы обнаружили, что DCA может увеличить химиотерапевтический эффект 5-FU в химиорезистентных клетках CRC и что активация пути p53/miR-149-3p/PDK2 способна увеличить химиочувствительность in vitro и in vivo.
Все больше данных указывают на то, что повышенный гликолиз тесно связан с устойчивостью к химиотерапии [15, 17, 40]. Здесь мы также обнаружили, что по сравнению с родительскими клеточными линиями, химиорезистентные клетки CRC показали повышенное потребление глюкозы, выработку лактата и гликолиз, что говорит о том, что метаболические нарушения являются типичной особенностью, но молекулярные механизмы все еще остаются неясными в клетках CRC.
Было отмечено, что DCA снижает уровень лактата в крови in vivo у грызунов в дозах ~25-50 мг/кг/24 ч [41], а также применение DCA в широком диапазоне доз от 1 до 50 мМ [26]. Примечательно, что неблагоприятное действие DCA у людей обычно ограничивается обратимой сенсорной и моторной периферической нейропатией, которая зависит от возраста и генотипа [42]. Недавно DCA был определен в качестве новой метаболической терапии для различных онкологических больных [26, 29].
Сообщается, что DCA способен ингибировать активность PDK и преобразовывать пируват в ацетил-КоА, что приводит к переходу выработки энергии от гликолиза к митохондриальному окислительному фосфорилированию [43,44]. Также было показано, что DCA ослабляет вызванную гипоксией устойчивость к 5-ФУ при раке желудка [45], преодолевает устойчивость к сорафенибу при гепатоцеллюлярной карциноме [46] и ослабляет устойчивость к цисплатину при раке головы и шеи [47]. Наше исследование показало, что DCA способен ослаблять химиорезистентность клеток CRC к 5-ФУ. Более того, мы продемонстрировали, что DCA снижает потребление глюкозы и выработку лактата в химиорезистентных клетках CRC до базового уровня химиочувствительных клеток CRC. Учитывая, что аутофагия способна подпитывать метаболизм рака [48], было определено влияние DCA и miR-149-3p на аутофагию. Мы обнаружили, что DCA активирует аутофагию, а miR-149-3p не влияет на аутофагию. Это позволило предположить, что аутофагия не вовлечена в эффект DCA/miR-149-3p в регуляции метаболизма глюкозы (Дополнительный рисунок S6).
PDKs, как ключевые регуляторы гликолиза при раке, вызвали большую озабоченность в связи с результатами многих исследований [49]. Существует четыре изоформы PDK (PDK1-4), и каждая из них проявляется в тканеспецифической манере следующим образом: PDK1 высоко экспрессируется в сердце, PDK2 экспрессируется повсеместно, PDK3 имеет относительно ограниченное тканевое распределение, а PDK4 экспрессируется в сердце и скелетных мышцах [27]. PDK2 экспрессируется на более высоких уровнях по сравнению с другими изоферментами, что позволяет предположить, что он может быть основной изоформой, ответственной за регуляцию ферментативной активности пируватдегидрогеназного комплекса (ПДГК) [50]. Кроме того, изоферменты PDK различаются по остроте регуляции метаболитами [51]. Здесь мы сосредоточились на PDK2, наиболее чувствительной изоформе к ДКА [31]. Хотя молекулярные взаимодействия между ДКА и PDK были изучены, потенциальный механизм транскрипционной регуляции PDK остается неясным. В недавнем исследовании сообщалось, что miR-182 играет регуляторную роль в метаболических путях рака легких, нацеливаясь на PDK4 [52]. Настоящее исследование показало, что PDK2 регулируется miR-149-3p в CRC и что уровни PDK2 в первичных опухолях пациентов CRC обратно коррелируют с экспрессией miR-149-3p. Поскольку PDK2 широко присутствует в большинстве тканей, воздействие на PDK2 может стать более важным и эффективным способом уничтожения опухолевых клеток и преодоления химиорезистентности.
Сообщалось, что miR-149-3p играет важную роль в различных видах рака и индуцируется некоторыми противоопухолевыми препаратами [36, 37, 53]. В частности, мы обнаружили, что обработка DCA может индуцировать связывание p53 с восходящей областью (от -677 до -477) miR-149 и что miR-149-3p повышается под воздействием обработки DCA в зависимости от p53. TP53, классический опухолевый супрессор, часто инактивируется в опухолях [54], и недавно сообщалось, что он регулирует метаболизм глюкозы при раке. Было показано, что Wt p53 может подавлять «эффект Варбурга», контролируя PDK2. Однако частота мутаций TP53 в CRC составляет ~40-50% [55, 56], что приводит к потере его супрессивной функции. Сообщалось, что пациенты CRC с wt TP53 получают преимущество в выживании после химиотерапии на основе 5-FU, а пациенты с мутантным TP53 — нет [57]. Наши результаты показывают новый механизм между р53 и PDK2, который модулируется miR-149-3p. Эти данные позволяют предположить, что пациенты с мутантным TP53 могут получить больше пользы от смежной химиотерапии с miR-149-3p, а не с DCA. Учитывая высокую частоту мутаций TP53 в CRC, мы считаем, что miR-149-3p играет важную роль в мониторинге и модуляции химиочувствительности в CRC.
Раковые клетки потребляют большое количество глюкозы и демонстрируют высокий уровень аэробного гликолиза, поэтому снижение поглощения глюкозы является перспективной стратегией для ограничения роста рака [58]. Мы наблюдали, что повышение уровня miR-149-3p значительно подавляло гликолиз в химиорезистентных клетках CRC; однако, по сравнению с контрольной группой, в группе ксенотрансплантатов, которым интратуморально вводили мимикрирующий miR-149-3p, SUVmax оставался неизменным. Возможно, этот противоречивый результат может быть связан с развитием некроза в основной части подкожных опухолевых тканей, что требует изучения.
В целом, мы обнаружили, что сигнальный путь p53/miR-149-3p/PDK2 потенциально может быть направлен на преодоление химиорезистентности CRC при лечении DCA, что обеспечивает потенциальную стратегию лечения CRC с точки зрения вмешательства в метаболизм опухоли (рис. 7e).
Материалы и методы
Раковая ткань
В период с 2013 по 2016 год в исследование были включены 28 пациентов с раком CRC из Девятой народной больницы при Медицинской школе Шанхайского университета Цзяо Тун. Тринадцать из этих пациентов получили послеоперационную химиотерапию на основе 5-ФУ и наблюдались в течение не менее 3 лет. Все ткани были собраны после получения информированного согласия, а все процедуры с участием пациентов проводились в соответствии с правилами, установленными Этическим комитетом Девятой народной больницы при Медицинском колледже Шанхайского университета Цзяо Тун. Клиническая информация о пациентах с КРК представлена в Дополнительной таблице 1.
| Характеристика пациентов с КРР | |
| Характеристика | Всего (n=28) |
| Возраст-год | 65.4±10.5 |
| Пол — нет (%) | |
| Мужчина | 17(60.7) |
| Женщина | 11(39.3) |
| Локализация — нет (%) | |
| Прямая кишка | 10(35.7) |
| Толстая кишка | 18(64.3) |
| Стадия — нет (%) | |
| Т1 или Т2 | 6(21.4) |
| Т3 или Т4 | 22(78.6) |
| Вовлеченные лимфатические узлы — нет (%) | |
| N0 | 12(42.9) |
| N1 или N2 | 16(57.1) |
| Метастазы — нет (%) | |
| M0 | 27(96.4) |
| M1 | 1(3.6) |
| Послеоперационная химиотерапия с использованием схемы на основе флуороурацила | |
| Да | 21(75) |
| Наблюдение в течение 3 лет | |
| Да | 13(61.9) |
| Нет | 8(38.1) |
| Нет | 7(25) |
Культура клеток
Устойчивая к 5-ФУ клеточная линия HCT-8/F и ее родительская клеточная линия HCT-8 были приобретены у iCell Bioscience, Inc. (Шанхай, Китай). Устойчивая к 5-ФУ клеточная линия HCT116/F и ее родительская клеточная линия HCT116 были любезно предоставлены доктором Гу (Yanhong Gu, Нанкинский медицинский университет, Цзянсу, Китай). Клетки HCT116-/- были подарены доктором Лу (Hua Lu, Университет Фудань, Шанхай, Китай). Линия клеток эмбриональной почки человека 293T была получена из American Type Culture Collection (ATCC, Manassas, VA, USA). Клетки линий 293T, HCT116, HCT-8 культивировали в среде Дульбекко (Dulbecco’s Modified Eagle’s Medium, HyClone, Utah, US) или среде RPMI-1640 (HyClone, Utah, US), содержащей 10% фетальной бычьей сыворотки (Gemini, California, US), 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина (HyClone, Utah, US) в увлажненном инкубаторе при 37 °C, 5%CO2. В культуральную среду клеточных линий HCT-8/F и HCT116/F добавляли 15 мкг/мл 5-ФУ и 5 мкг/мл 5-ФУ, соответственно. Все клеточные линии были подтверждены путем секвенирования коротких тандемных повторов компанией Genetic Testing Biotechnology Corporation (Сучжоу, Цзянсу, Китай). DCA был приобретен у компании Sigma-Aldrich Co. Ltd. (МО, США).
Иммунофлуоресцентное окрашивание Edu и ROS
Клетки HCT-8/F и HCT116/F высевали в 96-луночные планшеты в количестве 15 000 клеток/лунку. После ночной инкубации клетки обрабатывали 15 мМ и 20 мМ DCA, соответственно, в течение 24 ч. Окрашивание Edu проводили в соответствии с инструкцией производителя (Ribobio, Гуанчжоу, Китай). Уровень ROS измеряли в клетках, инкубированных с 10 мкМ 2′,7′-дихлорфлуоресцеин диацетата (DCF-DA) (Beyotime, Шанхай, Китай) в течение 30 мин при 37 °C. Затем планшеты промывали дважды, и клетки анализировали с помощью флуоресцентного микроскопа.
Рост клеток
Клетки высевали в 96-луночные планшеты по 5000 клеток/лунку на ночь, обрабатывали препаратами в течение 24 ч, после чего каждую лунку заменяли смесью из 10
CCK8 (Dojindo, Япония) и 90
мклкультуральной среды. Абсорбцию измеряли при значении OD 450 нм с помощью энзимного микропланшетного ридера (BioTeck, Вермонт, США) через два часа. Коэффициент ингибирования препарата рассчитывали по следующей формуле: 1-ODdrug/ODctrl. IC50 каждой клетки рассчитывали с помощью GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Анализ апоптоза клеток
Клетки HCT-8/F и HCT116/F высевали в шестилуночные планшеты в концентрации 2 ×105 клеток/лунку. Клетки обрабатывали DCA (15 мМ) / 5-ФУ (50 мкг/мл) и DCA (20 мМ) / 5-ФУ (25 мкг/мл), соответственно, в течение 48 ч. Затем клетки трипсинизировали, промывали и окрашивали антителами Annexin V-FITC/PI или Annexin V-PE/7-AAD в соответствии с протоколом производителя (BD, CA, США). Апоптоз измеряли методом проточной цитометрии (BD, CA, США).
Анализ образования колоний
Клетки HCT-8/F и HCT116/F обрабатывали DCA (15 мМ)/5-FU (50 мкг/мл) и DCA (20 мМ)/5-FU (25
), соответственно, в течение 24 часов. Затем клетки высевали в шестилуночные планшеты по 1000 клеток в лунку и культивировали в свежей среде при 37 °C в течение 1-2 недель, после чего фиксировали 4% параформальдегидом в течение 30 мин; затем клетки окрашивали 1% кристаллическим фиолетовым и подсчитывали количество колоний клеток с помощью счетчика (Gelcount, Optronix, Oxford).
Переходная трансфекция генов
Мимики miR-149-3p, ингибиторы, siPDK2, sip53 и соответствующие им последовательности олигонуклеотидов NC были синтезированы компанией GenePharma (Шанхай, Китай). Плазмида Flag-p53 была любезно предоставлена доктором Лу (Хуа Лу, Университет Фудань, Шанхай, Китай). Трансфекцию проводили с помощью Lipofectamine 3000 (Invitrogen, CA, США) при конечной концентрации 50 нмоль/л (мимики и siRNAs) или 100 нмоль/л (ингибиторы). Клетки собирали для анализа через 24 или 48 ч после трансфекции. Последовательности siRNA, мимиков и ингибиторов представлены в Дополнительной таблице 2.
| последовательность siRNA, мимика и ингибитора | |
| siPDK2-1 | смысл: 5′-GACCGAUGCUGUCAUCUAUU-3′ |
| антисмысл: 5′-AAUAGAUGAUGACAGCAUCGGUC-3′ | |
| siPDK2-2 | смысл: 5′-GACUCUUCAGCUACAUGUA-3′ |
| антисмысл: 5′-UACAUGUAGCUGAAGAGAGUC-3′ | |
| NC | смысл: 5′-UUCUCCGAACGUGUCACGUTT-3′ |
| антисмысл: 5′-ACGUGUGACACGUUCGGAGAATT-3′ | |
| мимик-149-3p | смысл: 5′-AGGGAGGGAGGACGGGGGCUGUGC-3′ |
| антисмысл: 5′-ACAGCCCCCGUCCCUCCCUUU-3′ | |
| ингибитор-NC | 5′-CAGUACUUUUGUGUAGUACAA-3′ |
| ингибитор-149-3p | 5′-GCACACAGCCCCCCGUCCCUCCCU-3′ |
| sip53 | смысл: 5′-GUAAUCUACUGGGACGGACGGAAtt-3′ |
| антисмысл: 5′-UUCCGUCCCAGUAGAUUACca-3′ |
Трансфекция стабильных генов
LV-PDK2 и соответствующий NC-вирус были приобретены у компании GeneChem (Шанхай, Китай). shPDK2-1, shPDK2-2 и контрольная плазмида были приобретены у компании GeneChem (Шанхай, Китай). Вирусный супернатант собирали из 293 Т-клеток. Затем клетки CRC были инфицированы вирусом и обследованы с помощью пуромицина. Эффективность инфекции была подтверждена проточной цитометрией и флуоресцентной микроскопией. Экспрессию мРНК и белка PDK2 далее анализировали с помощью количественной ПЦР в реальном времени и Вестерн-блота.
репортерные люциферазные анализы 3′-UTR
wt или мутантные последовательности связывания miR-149-3p в 3′UTR PDK2 человека были клонированы в downstream люциферазы pmiR-RB-Reporter (Ribobio, Guangzhou, China), обозначенные как WT, MUT1, MUT2 и MUT3 на рис. 3c. клетки 293 T и HCT116 высевали в 24-луночные планшеты с последующей котрансфекцией 500 нг репортерных конструкций и либо 50 нмоль/л мимика miR-149-3p, либо NC с помощью Lipofectamine 3000 (Thermo Fisher Scientific, Waltham MA). Активность люциферазы измеряли после 48 ч инкубации с помощью системы Dual-Luciferase Reporter Assay System (Promega, Мэдисон, США) в соответствии с протоколом производителя.
Потребление глюкозы и выработка лактата
Клетки HCT-8/F и HCT116/F высевали в 24-луночные планшеты в количестве 1 ×105 клеток/лунку на ночь, затем обрабатывали 15 мМ и 20 мМ DCA, соответственно, в течение 24 ч. После обработки клетки культивировали в среде без фенолового красного, содержащей 10% фетальной бычьей сыворотки, в течение 24 ч. Культурную среду собирали, измеряли потребление глюкозы и выработку лактата. Лактат измеряли с помощью набора Lactate Assay Kit (Njjcbio, Нанкин, Китай), а глюкозу — с помощью набора Glucose Assay Kit (Rsbio, Шанхай, Китай). Все значения были стандартизированы путем подсчета равного количества клеток. Для оценки состояния гликолиза в культивируемые клетки добавляли 100 нг/мл олигомицина (ингибитор АТФ-синтазы; Sangon Biotech) на 6 ч. Соотношение концентрации лактата в присутствии и отсутствии олигомицина измеряли и определяли, как описано ранее [46].
Seahorse XF-96 glycolytic rate assay
Клетки высевали в 96-луночный культуральный планшет при плотности 25 000 клеток/лунку и инкубировали в течение ночи в ростовой среде, содержащей 10% фетальной бычьей сыворотки. Сенсорный картридж увлажняли в течение ночи. На следующий день среду для клеток меняли на безбикарбонатную низкобуферную среду анализа, дополненную глюкозой, пируватом натрия и глутамином. После того как клетки инкубировали в течение 1 ч при 37 °C в инкубаторе без CO2, скорость потребления кислорода и скорость внеклеточного закисления измеряли до и после введения DCA/ctrl, ротенона (Rot) + антимицина A (AA) и 2-дезокси-d-глюкозы (2-DG) с помощью прибора Seahorse XF (Agilent, Santa Clara, CA), как описано ранее [59, 60]. Эксперименты проводились в режиме реального времени в пяти-шести повторных лунках. Гликоперы, включая базальный гликопер, индуцированный гликопер и компенсаторный гликопер, рассчитывались автоматически с помощью программного обеспечения Wave (Agilent, Santa Clara, CA).
Количественная ПЦР в реальном времени и микрочипы миРНК
Общая РНК была выделена из тканей или клеток CRC с помощью TRIzol Reagent (Life, CA, USA). кДНК была синтезирована с помощью PrimeScript RT Reagent Kit (TaKaRa, Tokyo, Japan). Анализ микрочипов проводили с тремя репликами клеток HCT116, обработанных 5 мМ, 10 мМ или 20 мМ DCA в течение 12, 24 или 48 ч. Исходные данные были загружены в базу данных GEO (GSE125309). Количественную ПЦР в реальном времени проводили с использованием премикса Ex Taq 420 A (TaKaRa, Токио, Япония) на платформе ABI-7500. Актин и U6 использовали в качестве внутренних контролей. Последовательности праймеров представлены в Дополнительной таблице 3.
| последовательности праймеров для количественной ПЦР с обратной транскрипцией в реальном времени | |
| Актин | F: 5′-CTCCATCATCCTGGCCTCGCTGT-3′ |
| R: 5′-GCTGTCACCTCACCTTCACCGTTCC-3′ | |
| PDK2 | F: 5′-CGCTGCTGGCTGGCTTTGGTTTTATG-3′ |
| R: 5′-ACAGGGCCTTGAGATAGATG-3′ | |
| PDK1 | F: 5′-GCTGTATGGCCTGCAAGATG-3′ |
| R: 5′-GCTGTTCCTGTGTGATTTTGCA-3′ | |
| PDK3 | F: 5′-GGTTTGCCAATTTCCCGTCTG-3′ |
| R: 5′-CATCGGCTTCAGGCGTGGTGTC-3′ | |
| PDK4 | F: 5′-GAGAATTATTGACCGCCTCT-3′ |
| R: 5′-CGAGAAATTGGCAAGCCGTAA-3′ | |
| HK1 | F: 5′-CTTACTAAGGGATGCGATAA-3′ |
| R: 5′-TCCCAACAATGAGTCCAACC-3′ | |
| TP53 | F: 5′-CCCAAGCAATGGATGATGATTTGA-3′ |
| R: 5′-GGCATTCTCTGGAGGAGCTTCATCT-3′ | |
| p21 | F: 5′-CTGGACTGTTTTTTCTCGGCTC-3′ |
| R: 5′-TGTATATTCAGCATTGTGGGAGGA-3′ | |
| MDM2 | F: 5′-ATGAATCCCCCCCCCTTCCAT-3′ |
| R: 5′-CAGGAAGCCAATTCTCACGAA-3′ | |
| PUMA | F: 5′-ACAGTACGAGCGGCGAGACAA-3′ |
| R: 5′-GGCGGGTGCAGGCACACCTAATT-3′ | |
| miR-149-3p | RT: 5′-GTCGTATCCAGTCCAGGGTATTCGCACTGGTCCGAGTATTCGCACTGG ATACGACGCACAG-3′ |
| F: 5′-ACAGGGAGGGACGGGGG-3′ | |
| R: 5′-ATCCAGTGGCAGGTCCGAGG-3′ | |
| miR-128-3p | RT: 5′-TCGTATATCCAGTGCAGGTCCGAGGTATTCGCACTGGA TACGACAAAGAG-3′ |
| F: 5′-CGCGTCACAGTGAACCGGT-3′ | |
| R: 5′-AGTGCAGGGTCCGAGGTATT-3′ | |
| miR-500a-5p | RT: 5’-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCAC TGGATACGACTCTCACC-3’ |
Вестерн-блоты
Загружали тридцать микрограммов лизатов общего белка и наносили первичные антитела: анти-PDK2 (sc-100534, Santa Cruz, California, US), анти-p-PDHA1 (S293) (ABS204, Merck, Darmstadt, Germany), анти-PDHA1 (ab168379, Abcam, Cambridge, UK), анти-p53 (sc-126, Santa Cruz, California, US), anti-c-PARP (D64E10, CST, Массачусетс, США), anti-Bax (D2E11, CST, Массачусетс, США), anti-LC3B (L7543, sigma, MO, США), anti-GAPDH (Proteintech, Ухань, Китай) и anti-α-tubulin (Proteintech, Ухань, Китай). Вторичные антитела были приобретены у компании Sungene (Тяньцзинь, Китай). Блот-анализы визуализировали с помощью системы хемилюминесценции (Bioshine, Шанхай, Китай).
Иммунопреципитация хроматина (ChIP)
Клетки HCT116, посеянные в 10-см планшеты, обрабатывали 20 мМ DCA или без него в течение 24 ч, затем проводили фиксацию клеток и фрагментацию хромосом в соответствии с инструкциями производителя (Pierce Agarose ChIP Kit, Thermo). Хроматин инкубировали с антителами IgG и анти-р53 (Sigma, MO, США) при 4°C в течение ночи. После инкубации добавляли 60 ul протеина А агарозы/ДНК спермы лосося. Затем осажденный комплекс промывали буферами для промывки IP 1, 2, 3 и элюировали буфером для элюирования. Сшивание было отменено добавлением 6 мкл 5 М NaCl и 2 мл протеиназы К при 65°C в течение 1,5 ч. Иммунопреципитированная ДНК и ДНК цельноклеточного экстракта (входная) были очищены и затем использованы для ПЦР-анализа с использованием соответствующих праймеров. Для контроля эксперимента использовался контрольный праймер. Последовательности праймеров для ПЦР представлены в Дополнительной таблице 3.
Подкожная ксенотрансплантация опухоли у голых мышей и микро-ПЭТ/КТ визуализация
Сначала 1 ×107 клеток HCT-8/F, разведенных в 100
PBS, подкожно имплантировали голым мышам (самцы, 6 недель). Через 12 дней мышей случайным образом разделили на шесть групп (по шесть на группу). Мыши из групп I — IV получали ежедневную внутрибрюшинную инъекцию PBS, DCA (50 мг/кг)/PBS, 5-FU (10 мг/кг)/PBS и 5-FU (10 мг/кг)/DCA (50 мг/кг), соответственно. Мышам из группы V и группы VI интратуморально вводили PBS или DCA (50 мг/кг), соответственно, каждый второй день и внутрибрюшинную инъекцию 5-FU (10 мг/кг) каждый второй день. Объем опухоли измеряли вслепую каждые 3 дня. Мышей приносили в жертву после 3 недель лечения, опухоли иссекали, взвешивали и замораживали при -80 °C для дальнейшего изучения.
Чтобы оценить, оказывает ли miR-149-3p эффект химиосенсибилизации, были созданы еще две группы животных моделей. Вкратце, мышам подкожно имплантировали 6 ×106 клеток HCT-8/F. После того, как размеры опухоли достигали ~50 мм3, мыши получали внутрибрюшинную дозу 5-FU (10 мг/кг) каждый второй день, а также интратуморальную инъекцию 5 нмоль холестерин-конъюгированных мимиков miR-149-3p или NC каждые 3 дня в течение 3 недель. Затем трех мышей из каждой группы постили на ночь и внутривенно вводили 0,15 мКи 18F-ФДГ. через 60 мин проводилось микро-ПЭТ-КТ сканирование с 18F-ФДГ (Siemens, Берлин, Германия). Изображения, полученные в ходе ПЭТ, были представлены с использованием псевдоцветной карты, где красный цвет указывал на высокое поглощение 18F-ФДГ. SUVmax и MTV использовались для определения активности 18F-ФДГ-ПЭТ. Все эксперименты и уход за животными были одобрены Этическим комитетом Девятой народной больницы при медицинском колледже Шанхайского университета Цзяо Тун.
Иммуногистохимическое и иммунофлуоресцентное окрашивание
Вкратце, срезы тканей инкубировали с первичными антителами Ki67 (Servicebio, Ухань, Китай) и PDK2 (Proteintech, Ухань, Китай) при 4 °C в течение ночи, а затем инкубировали со вторичным антителом. Хромогенную реакцию проводили с 3,3-диаминобензидином и контрастировали гематоксилином. Иммунореактивный балл (IRS) рассчитывался двумя исследователями, слепыми к распределению по группам. IRS = SI (интенсивность окрашивания) × PP (процент положительных клеток). SI присваивался следующим образом: 0 × отрицательный; 1 × слабый; 2 × умеренный; 3 × сильный. PP определяется как 0 × 0%; 1 × 0-25%; 2 × 25-50%; 3 × 50-75%; 4 × 75-100%. Шестимиллиметровые замороженные срезы окрашивали с помощью набора для реакции TUNEL (Roche, Базель, Швейцария) и контрастировали DAPI. Изображения получали с помощью флуоресцентного микроскопа с соответствующими фильтрами возбуждения и испускания.
Статистический анализ
Данные анализировали с помощью программы GraphPad Prism 6.0. Данные представлены как среднее ± SD/SEM из трех независимых экспериментов. Каждый эксперимент проводился как минимум в трех повторах. Для сравнения различий между двумя группами использовали двухфакторный t-тест Стьюдента. Для множественных сравнений использовался односторонний ANOVA с последующим пост-хок тестом Бонферрони. Кривые Каплана-Мейера для анализа выживаемости определялись с помощью теста log-rank. Связь между miR-149-3p и PDK2 оценивали с помощью анализа коэффициента ранговой корреляции Спирмена. Значение P <0,05 считалось статистически значимым.
Благодарности
Эта работа была поддержана Национальным фондом естественных наук Китая (81272745, 81872419 и 81272404) и Программой для профессора специального назначения (Восточный стипендиат JW) в Шанхайских высших учебных заведениях. Мы благодарим д-ра Yanhong Gu за предоставление клеточных линий HCT116, устойчивых к 5-FU, и д-ра Hua Lu за предоставление плазмиды p53.
Соблюдение этических норм
Конфликт интересов
Авторы заявляют, что у них нет конфликта интересов.
Примечание издателя
Springer Nature сохраняет нейтралитет в отношении юрисдикционных претензий в опубликованных картах и институциональной принадлежности.
Открытый доступ
Эта статья лицензирована по лицензии Creative Commons Attribution 4.0 International License, которая разрешает использование, обмен, адаптацию, распространение и воспроизведение на любом носителе или в любом формате при условии, что вы отдаете должное оригинальному автору (авторам) и источнику, даете ссылку на лицензию Creative Commons и указываете, были ли внесены изменения. Изображения или другие материалы третьих лиц в этой статье включены в статью по лицензии Creative Commons, если иное не указано в кредитной строке к материалу. Если материал не включен в лицензию Creative Commons статьи, а предполагаемое вами использование не разрешено законом или выходит за рамки разрешенного использования, вам необходимо получить разрешение непосредственно у правообладателя. Чтобы ознакомиться с копией этой лицензии, посетите сайт http://creativecommons. org/licenses/by/4.0/.
ССЫЛКИ
1 Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, et al. Cancer incidence and mortality in China, 2014. Chin J Cancer Res. 2018;30:1-12.2 Siegel RL, Miller KD, Jemal A. Статистика рака, 2018. CA: Cancer J Clin. 2018;68:7-30.
3 Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB 3rd, et al. Dichloroacetate alters Warburg metabolism, inhibits cell growth, and increases the X-ray sensitivity of human A549 and H1299 NSC lung cancer cells. Free Radic Biol Med. 2015;89:263-73.
4 Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167: 1540-54 e12.
5 Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Сообщение о серьезном нежелательном явлении после введения Т-клеток, трансдуцированных химерным антигенным рецептором, распознающим ERBB2. Mol Ther: J Am Soc Gene Ther. 2010;18:843-51. Дихлорацетат восстанавливает химиочувствительность колоректального рака через p53/miR-149-3p/PDK2-опосредованный. . . 483
6 Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA: Cancer J Clin. 2016;66:271-89.
7 Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Therapeutic Adv Med Oncol. 2016;8:57-84.
8 Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041-7.
9 Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Исследовательская группа по иринотекану. New Engl J Med. 2000;343:905-14.
10 Hanahan D, Weinberg RA. Признаки рака: следующее поколение. Cell. 2011;144:646-74.
11 Matthew G, Vander Heiden LCC, Craig BT. Понимание эффекта Варбурга: метаболические требования клеточной пролиферации. Science. 2009;324:1029-33.
12 Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
13 Shaw RJ. Метаболизм глюкозы и рак. Curr Opin Cell Biol. 2006;18:598-608.
14 Ван Данг С, Поллак М. Почему рак и метаболизм? Почему сейчас? Cancer Metab. 2013;1:1.
15 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532.
16 Cairns RA, Harris IS, Mak TW. Регуляция метаболизма раковых клеток. Nat Rev Cancer. 2011;11:85-95.
17 Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613-21.
18 Guo WQZ, Wang Z, et al. MiR-199a-5p негативно связан со злокачественными опухолями и регулирует гликолиз и производство лактата путем нацеливания на гексокиназу 2 при раке печени. Hepatology. 2015;62:1132-44.
19 Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. МикроРНК-124 снижает пентозофосфатный путь и пролиферацию путем нацеливания на мРНК PRPS1 и RPIA в клетках колоректального рака человека. Gastroenterology. 2015;149:1587-98 e11.
20 Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. МикроРНК-129- 5p регулирует гликолиз и пролиферацию клеток путем нацеливания на транспортер глюкозы SLC2A3 в клетках рака желудка. Front Pharmacol. 2018;9:502.
21 Bartel DP. МикроРНК: геномика, биогенез, механизм и функция. Cell. 2004;116:281-97.
22 Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167-79.
23 Huang S, He X. МикроРНК: крошечные молекулы РНК, огромные движущие силы для движения клетки. Protein Cell. 2010;1:916-26.
24 Zhang Y, Wang J. МикроРНК являются важными регуляторами лекарственной устойчивости при колоректальном раке. Biol Chem. 2017;398:929-38.
25 Stacpoole PW, Nagaraja NV, Hutson AD. Эффективность дихлорацетата как лактат-снижающего препарата. J Clin Pharmacol. 2003;43:683-91.
26 Kankotia S, Stacpoole PW. Дихлорацетат и рак: новый дом для сиротского препарата? Biochim Biophys Acta. 2014;1846:617-29.
27 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Доказательства существования тканеспецифической регуляции комплекса пируватдегидрогеназы млекопитающих. Biochemical J. 1998;329(Pt 1):191-6.
28 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell 2007;11:37-51.
29 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra4.
30 Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33:603-10.
31 Papandreou I, Goliasova T, Denko NC. Противораковые препараты, направленные на метаболизм: является ли дихлорацетат новой парадигмой? Int J Cancer. 2011;128:1001-8.
32 Michelakis ED, Webster L, Mackey JR. Дихлорацетат (DCA) как потенциальная метаболически-таргетная терапия рака. Br J Cancer. 2008;99:989-94.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Дихлорацетат индуцирует апоптоз и остановку клеточного цикла в клетках колоректального рака. Br J Cancer. 2010;102:1746-52.
34 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Дихлорацетат натрия (DCA) снижает апоптоз при гипоксии колоректальных опухолей. Cancer Lett. 2010;297:75-83.
35 Bertoli G, Cava C, Castiglioni I. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015;5:1122-43.
36 Cao D, Jia Z, You L, Wu Y, Hou Z, Suo Y, et al. 18betaglycyrrhetinic acid suppresses gastric cancer by activation of miR149-3p-Wnt-1 signaling. Oncotarget. 2016;7:71960-73.
37 Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, et al. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br J Pharm. 2017;174:553-68.
38 Kato M, Li J, Chuang JL, Chuang DT. Различные структурные механизмы ингибирования изоформ киназы пируватдегидрогеназы AZD7545, дихлорацетатом и радициколом. Structure. 2007;15:992-1004.
39 Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Дивергентная регуляция экспрессии генов изоформ киназы пируватдегидрогеназы в культивируемых мышечных клетках человека. FEBS J. 2005;272:3004-14.
40 Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, BelouecheBabari M, Koh KX, et al. Повышенная лекарственная устойчивость связана со снижением уровня глюкозы и усиленным фенотипом гликолиза. Br J Pharm. 2014;171:3255-67.
41 Stacpoole PW. The pharmacology of dichloroacetate. Metab: Clin Exp. 1989;38:1124-44.
42 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Зависимая от возраста кинетика и метаболизм дихлорацетата: возможное отношение к токсичности. J Pharmacol Exp Therap. 2008;324:1163-71.
43 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Комбинированное воздействие на PDK1 и EGFR вызывает регрессию глиобластомы, обращая вспять эффект Варбурга. Cancer Res. 2013;73:7277-89.
44 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. Inactivation of the HIF-1alpha/ PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035-47.
45 Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5- fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res. 2014;321:219-30.
46 Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72-81. 484 Y. Liang et al.
47 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016;371:20-9.
48 Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab 2017;25:1037-43.
49 Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32:1638-50.
50 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Разнообразие семейства генов киназы пируватдегидрогеназы у человека. J Biol Chem. 1995;270:28989-94.
51 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003; 284: E855-62.
52 Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182- PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98.
53 Bellazzo A, Di Minin G, Valentino E, Sicari D, Torre D, Marchionni L, et al. Клеточно-автономное и клеточно-неавтономное снижение регуляции опухолевого супрессора DAB2IP микроРНК-149- 3p способствует агрессивности раковых клеток. Cell Death Differ. 2018;25:1224-38.
54 Vazquez A, Bond EE, Levine AJ, Bond GL. Генетика пути р53, апоптоз и терапия рака. Nat Rev Drug Discov. 2008;7:979-87.
55 Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. Супрессор опухоли p53 в контроле метаболизма и ферроптоза. Front Endocrinol. 2018;9:124.
56 Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560-7.
57 Iacopetta B. Мутация TP53 при колоректальном раке. Hum Mutat. 2003;21:271-6.
58 Kim JW, Dang CV. Молекулярная сладость рака и эффект Варбурга. Cancer Res. 2006;66:8927-30.
59 Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, Tempera I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by EpsteinBarr virus latent membrane protein 1. PLoS Pathog. 2018;14: e1007394.
60 Hlouschek J, Ritter V, Wirsdorfer F, Klein D, Jendrossek V, Matschke J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018;439:24-38.
Связанный контент: