Xiao Lu,1,* Dong Zhou,1,* Bing Hou,1 Quan-Xing Liu,1 Qian Chen,2 Xu-Feng Deng,1,3 Zu-Bin Yu,1 Ji-Gang Dai,1 Hong Zheng1
1 Dipartimento di Chirurgia Toracica, Ospedale Xinqiao, Terza Università Medica Militare, Chongqing, Repubblica Popolare Cinese
2 Istituto di Patologia e Centro Tumori Sud-Ovest, Ospedale Sud-Ovest, Terza Università Medica Militare, Chongqing, Repubblica Popolare Cinese
3 Dipartimento di Chirurgia Cardiotoracica, Primo Ospedale del Popolo di Zunyi, Guizhou, Repubblica Popolare Cinese
*Questi autori hanno contribuito ugualmente a questo lavoro
Corrispondenza: Hong Zheng; Ji-Gang Dai
Dipartimento di Chirurgia Toracica, Ospedale Xinqiao, Terza Università Medica Militare, No. 183, Xinqiao Street, Distretto di Shapingba, Chongqing 400037, Repubblica Popolare Cinese
Tel.: +86 23 6877 4724
Fax: +86 23 6877 4724
Email: [email protected]; [email protected]
Ricevuto: 14 settembre 2020
Accettato: 4 dicembre 2020
Pubblicato: 9 dicembre 2020
Abstract
Premessa: La chemioterapia è ancora la principale strategia adiuvante della terapia del cancro; tuttavia, l’emergere di una resistenza multifarmaco è stata fonte di preoccupazione. È stato dimostrato che l’autofagia ha un ruolo protettivo contro i farmaci chemioterapici nelle cellule tumorali e l’inibizione dell’autofagia è generalmente considerata una strategia terapeutica promettente. Tuttavia, la scarsità di inibitori dell’autofagia efficaci e specifici ne limita l’applicazione.
Scopo: l’obiettivo di questo studio è stato quello di esplorare l’effetto del DCA, piccolo agente molecolare antitumorale, sulla regolazione dell’autofagia e sulla chemiosensibilizzazione in cellule di NSCLC.
Metodi: Abbiamo studiato la regolazione dell’autofagia del dicloroacetato (DCA) mediante microscopia confocale laser e western blotting nelle linee cellulari A549 e H1975. Il saggio MTT e la citometria a flusso sono stati eseguiti per esplorare l’efficacia chemiosensibilizzante del DCA. I risultati sono stati verificati con un modello tumorale sottocutaneo in topi nudi e l’immunoistochimica è stata applicata per valutare il livello di apoptosi cellulare e di autofagia in vivo dopo il trattamento.
Risultati: Abbiamo scoperto che il DCA, che ha mostrato proprietà antitumorali in vari modelli di carcinoma, ha indotto l’apoptosi delle cellule di carcinoma polmonare non a piccole cellule (NSCLC) inibendo l’autofagia delle cellule tumorali. Inoltre, la perifosina, un inibitore di AKT, può indebolire notevolmente la capacità di indurre apoptosi da parte del DCA. I risultati indicano che la via AKT-mTOR, un principale regolatore negativo dell’autofagia, è coinvolta nell’inibizione dell’autofagia indotta dal DCA. Poi, abbiamo rilevato l’efficacia dell’inibizione dell’autofagia da parte del DCA. Quando è stato utilizzato in co-trattamento con il farmaco chemioterapico paclitaxel (PTX), il DCA ha ridotto notevolmente l’autofagia cellulare, ha aumentato l’apoptosi e ha inibito la proliferazione nelle cellule A549 e H1975. I risultati dell’esperimento di xenotrapianto dimostrano che il co-trattamento di PTX e DCA può ridurre significativamente la proliferazione cellulare in vivo e prolungare la sopravvivenza dei topi.
Conclusioni: I nostri risultati suggeriscono che il DCA può inibire l’autofagia cellulare indotta dai chemioterapici, fornendo una nuova via per la sensibilizzazione alla chemioterapia del cancro.
Parole chiave: DCA, autofagia, resistenza multifarmaco, carcinoma polmonare non a piccole cellule, paclitaxel, xenotrapianto di topi nudi, chemiosensibilizzazione
INTRODUZIONE
Il carcinoma polmonare non a piccole cellule (NSCLC) è una delle principali cause di mortalità per cancro a livello mondiale. Sebbene la chemioterapia sia ancora il mezzo più importante di terapia adiuvante per i pazienti oncologici non operabili e per quelli sottoposti a intervento chirurgico, i benefici clinici delle chemioterapie post-operatorie a base di platino e paclitaxel sono modesti, soprattutto nel NSCLC avanzato. Allo stesso tempo, le reazioni avverse ai farmaci sono diventate più gravi ed è emersa anche la resistenza ai farmaci.[3] Pertanto, è urgente la necessità di nuove strategie per sostituire/integrare la chemioterapia tradizionale.
Negli ultimi anni è emerso che le cellule tumorali producono preferenzialmente energia per la crescita e la divisione cellulare attraverso il processo glicolitico e la fermentazione lattica. I tassi di metabolismo anaerobico e di glicolisi nelle cellule tumorali maligne in rapida crescita sono significativamente più alti di quelli delle cellule normali. Questa riprogrammazione del metabolismo energetico è nota come effetto Warburg e può essere sfruttata come bersaglio terapeutico per inibire la crescita tumorale. Tra i numerosi farmaci che hanno come bersaglio il metabolismo, il dicloroacetato (DCA) ha mostrato un eccellente potenziale per il suo contributo positivo al trattamento del cancro.[4,5]
Un altro meccanismo completamente alterato nelle cellule tumorali è l’autofagia, un sistema di degradazione cellulare omeostatico responsabile della degradazione di organelli cellulari o proteine danneggiati o non necessari.[6] Durante l’autofagia, il carico cellulare destinato alla degradazione è racchiuso in un autofagosoma, una vescicola a doppia membrana. L’autofagosoma carico si fonde senza problemi con un lisosoma per formare un autolisosoma, dove il materiale cellulare consegnato viene degradato da vari enzimi idrolitici lisosomiali. Il processo di autofagia è stato ampiamente studiato. È sempre più chiaro che l’alterazione dell’attività dell’autofagia è associata alla formazione e alla progressione dei tumori.[7-9] Poiché l’autofagia svolge un ruolo protettivo nelle cellule tumorali contro i farmaci chemioterapici, la soppressione dell’autofagia durante la chemioterapia è stata considerata una nuova strategia terapeutica.[10-12] Attualmente, solo la clorochina (CQ) viene utilizzata in ambito clinico come efficace inibitore dell’autofagia. Sebbene siano state dimostrate l’efficacia e la fattibilità della clorochina nella terapia del cancro, gli effetti collaterali indesiderati potrebbero rappresentare un problema per il trattamento clinico. La scoperta e l’utilizzo di ulteriori inibitori dell’autofagia nella terapia del cancro sarebbe di grande importanza clinica.[13-15]
Il DCA è un agente mirato ai mitocondri che agisce come interruttore metabolico, invertendo il metabolismo anormale delle cellule tumorali dalla glicolisi anaerobica all’ossidazione aerobica del glucosio, riducendo l’attività della PDK1 mitocondriale e potenziando la vitalità della PDH. Pertanto, il DCA aumenta le specie reattive dell’ossigeno mitocondriale, inducendo così l’apoptosi nelle cellule tumorali maligne senza influenzare le cellule normali.[16,17] Tuttavia, l’azione regolatrice del DCA per l’autofagia nel cancro del polmone non è ancora chiara. In questo studio abbiamo dimostrato che il DCA inibisce la proliferazione cellulare e aumenta l’apoptosi delle cellule tumorali attraverso la downregulation dell’autofagia, aumentando così l’efficienza della morte cellulare quando viene utilizzato in cotrattamento con agenti chemioterapici.
Materiali e metodi
Coltura cellulare e reagenti
Le cellule di adenocarcinoma polmonare umano A549 sono state coltivate in Dulbecco’s Modified Eagle Medium con il 10% di siero fetale bovino, mentre le cellule H1975 sono state coltivate in Roswell Park Memorial Institute-1640 medium con il 10% di siero fetale bovino in un incubatore umidificato con il 95% di aria e il 5% diCO2 a 37°C. Le linee cellulari A549 (TCHu150) e H1975 (TCHu193) sono state acquistate dalla Library of Typical Culture of the Chinese Academy of Sciences (Shanghai, Repubblica Popolare Cinese). Dopo che le cellule sono cresciute lungo la parete della piastra di coltura, è stata utilizzata la tripsina allo 0,25% (HyClone, Buckinghamshire, Regno Unito) per il distacco e la subcultura. I reagenti utilizzati in questo studio sono stati DCA (Sigma-Aldrich, St. Louis, MO, USA), Paclitaxel (PTX; Sigma-Aldrich), Cisplatino (cis-diamminedicloroplatino [CDDP]; Sigma-Aldrich), adenovirus (GFP-RFP-LC3; Hanbio, Shanghai, Repubblica Popolare Cinese), TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling; Promega, Fitchburg, WI, USA), APC-Annexin V, ioduro di propidio (BD Pharmingen, Franklin Lakes, NJ, USA) e kit MTT (Sangon Biotech, Shanghai, Repubblica Popolare Cinese). Gli anticorpi erano LC3-I/II, p62, PARP (tutti Abcam, Cambridge, UK), β-actina (Sigma-Aldrich), mTOR/p-mTOR (Sigma-Aldrich) e Ki-67 (Biovisualab, Shanghai, Repubblica Popolare Cinese).
Microscopia di immunofluorescenza
Le cellule (1×105) sono state inoculate in una piastra a 24 pozzetti e incubate per una notte. Prima dell’esperimento, le cellule sono state infettate con adenovirus contenenti strutture tandem GFP-RFP-LC3. A 24 ore dall’infezione, i mezzi di coltura sono stati cambiati e le cellule sono state trattate con DCA 25 mM o soluzione salina bilanciata di Hank (HBSS; Sigma Aldrich) per 24 ore. Per l’immunocolorazione, le cellule sono state fissate in paraformaldeide al 4% e lavate con PBS. Le cellule sono state incubate con DAPI (4′,6-diamidino-2-fenilindolo; Beyotime Biotechnology, Shanghai, Repubblica Popolare Cinese) per 5 minuti e lavate tre volte con PBS. I coprioggetti sono stati montati su vetrini con mezzo di montaggio. Le immagini sono state acquisite con un microscopio confocale LSM 780 Meta (Carl Zeiss MicroImaging GmbH, Jena, Germania) ed elaborate con il software fornito dal produttore.
Saggio di apoptosi
Le cellule (1×105) sono state inoculate in una piastra a 24 pozzetti e incubate per una notte. Prima degli esperimenti, le cellule sono state trattate con diverse concentrazioni di DCA 25 mM, agente chemioterapico più DCA o altri reagenti per 24 ore. Le cellule sono state colorate con APC-Annexin V e ioduro di propidio per misurare il tasso di apoptosi mediante citometria a flusso. Ogni esperimento è stato ripetuto tre volte.
Western blot
Le cellule sono state lisate in ghiaccio mediante trattamento con il tampone di lisi RIPA (Sangon Biotech) con Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA, USA) per 15 minuti. I surnatanti sono stati raccolti dopo centrifugazione. Le concentrazioni proteiche sono state misurate utilizzando il kit per il dosaggio delle proteine con acido bicinchoninico (BCA) (Beyotime Biotechnology). I campioni di proteine sono stati elettroforesi su gel di sodio dodecil solfato-poliacrilammide al 10% e trasferiti su una membrana Immobilon PVDF. Le membrane sono state testate per una notte a 4°C con l’anticorpo primario indicato e poi incubate con un anticorpo secondario coniugato con perossidasi di rafano. Le bande sono state visualizzate utilizzando il tampone di rilevazione della chemiluminescenza (Takara, Shiga, Giappone) e l’intensità delle bande è stata quantificata con il software fornito dal produttore.
Saggio di vitalità cellulare
Le cellule sono state coltivate in piastre da 96 pozzetti (1.000 cellule in 100 µL di terreno di coltura/pozzetto). Dopo il trattamento con il farmaco, sono stati aggiunti 10 µL di MTT (0,5 mg/mL) e sono stati incubati per 4 ore. Il terreno è stato quindi scartato e i cristalli di formazan sono stati solubilizzati con l’aggiunta di DMSO (dimetilsolfossido; Sigma-Aldrich). È stata misurata l’assorbanza a 570 nm. La vitalità cellulare è stata normalizzata rispetto a quella del gruppo di controllo.
Studi di xenotrapianto tumorale in vivo
Per questo saggio, 5×106 cellule sono state iniettate per via sottocutanea in topi nudi (BALB/c, Specific pathogen free grade, 4-5 settimane di età, acquistati dal Model Animal Research Center dell’Università di Nanjing, Nanjing, Repubblica Popolare Cinese). Quando le dimensioni del tumore hanno raggiunto i 100 mm3 (15-20 giorni), i topi sono stati divisi a caso in 4 gruppi (PBS, PTX, DCA e PTX più DCA). Il dosaggio di PTX era di 20 mg/kg/d, mentre quello di DCA era di 100 mg/kg/d. La lunghezza e la larghezza dei tumori sono state misurate ogni due giorni attraverso i loro due diametri perpendicolari (volume calcolato = diametro più corto2 × diametro più lungo/2). Il numero e le date di morte dei topi sono stati registrati per calcolare il tasso di sopravvivenza. Tutte le manipolazioni che coinvolgevano topi vivi sono state approvate dal Comitato per la cura e l’uso degli animali dell’ospedale di Xinqiao e hanno seguito le linee guida cinesi per il benessere e l’etica degli animali da laboratorio; sono stati compiuti tutti gli sforzi per ridurre al minimo le sofferenze.
Esame istologico
Tutti i tumori sono stati asportati dopo 24 giorni per l’esame istopatologico. Sezioni (spessore da 4 a 5μm) dei tessuti tumorali sono state fissate con formalina al 10% e quindi analizzate con Ki-67, anticorpo LC3B e saggio TUNEL. Le sezioni sono state lavate tre volte e trattate con diaminobenzidina per lo sviluppo del colore. Sono stati eseguiti esami istologici e sono state scattate fotografie con un microscopio ottico NanoZoomer 2.0-RS (Hamamatsu Photonics, Hamamatsu City, Giappone).
Analisi statistica
I dati sono presentati come media ± deviazione standard. Per l’analisi della varianza sono stati utilizzati l’analisi della varianza a senso unico e il test t a campione indipendente. Le curve di sopravvivenza sono state ottenute con il metodo Kaplan-Meier e i confronti sono stati effettuati con il test log-rank. Tutte le analisi statistiche sono state condotte utilizzando il software statistico SPSS 19.0 (SPSS; IBM Corporation, Armonk, NY, USA). I valori P<0,05 o <0,01 sono stati considerati statisticamente significativi.
Risultati
IlDCA ha inibito l’autofagia nelle cellule di adenocarcinoma polmonare
LC3 citosolico e SQSTM (p62) sono proteine altamente conservate che si ritiene svolgano ruoli essenziali durante le fasi chiave dell’autofagia. Per indagare il ruolo del DCA nella regolazione dell’autofagia, abbiamo esaminato la localizzazione cellulare di LC3 con un microscopio confocale a scansione laser. Le cellule A549 e H1975 sono state infettate con un adenovirus che codifica un costrutto GFP-RFP-LC3 in tandem per rilevare la variazione del flusso di autofagia. Una differenza nella sensibilità al pH può causare il diverso grado di accumulo delle proteine GFP-LC3 e RFP-LC3 negli autofagosomi neutri e negli autolisosomi acidi. Dopo il trattamento con HBSS, il numero di punti GFP o RFP-LC3 è aumentato significativamente, dimostrando che l’inedia indotta da HBSS ha promosso in modo significativo il flusso di autofagia nelle cellule A549 e H1975. Tuttavia, il trattamento con DCA ha inibito con successo l’espressione delle proteine LC3 rispetto al controllo o al trattamento con HBSS sia nelle cellule A549 che nelle cellule H1975 (Figura 1A e D, p<0,01).
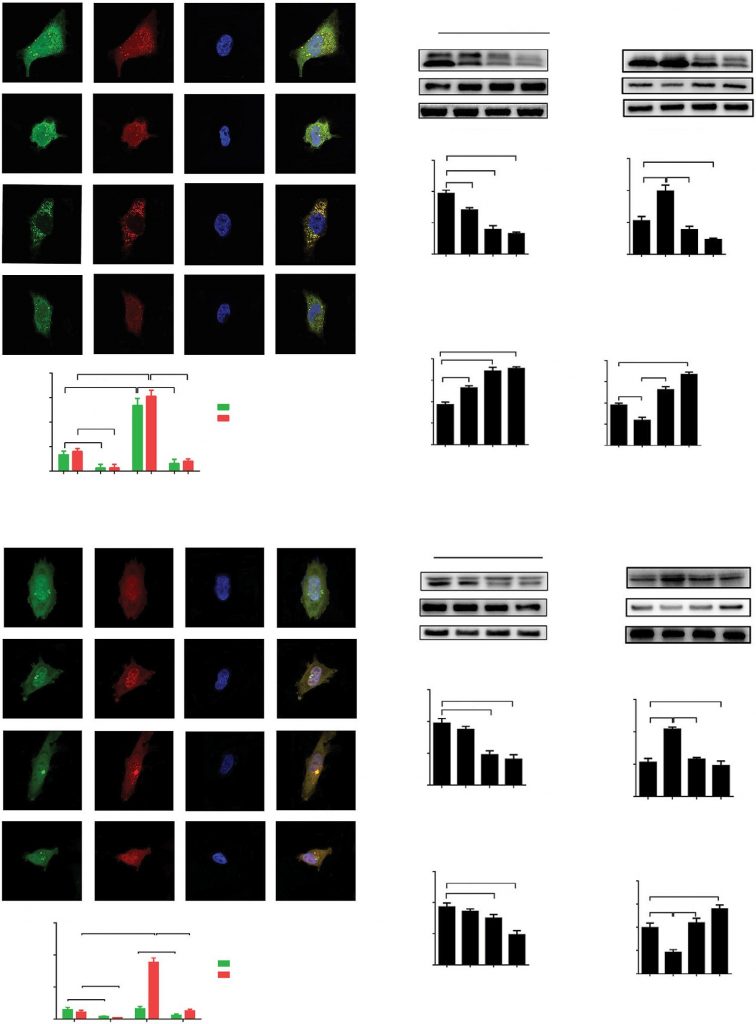
Abbreviazioni: DAPI, 4′,6-diamidino-2-fenilindolo; DCA, dicloroacetato; HBSS, soluzione salina bilanciata di Hank; ns, non significativo.
la proteina p62 si accumula quando l’autofagia è inibita e diminuisce quando l’autofagia è indotta. L’analisi di Western blotting è stata eseguita per esaminare i livelli proteici di p62 e LC3-II dopo il trattamento con DCA. Abbiamo osservato una diminuzione significativa dei livelli di LC3 e un aumento di p62 in seguito al trattamento con DCA in modo dipendente dalla concentrazione sia nelle cellule A549 che in quelle H1975 (Figura 1B e E, p<0,01). L’autofagia è stata potenziata durante l’incubazione con HBSS, invertita quando sono state trattate con DCA (Figura 1C e F, p<0,01). L’insieme di questi risultati indica che il flusso autofagico nelle cellule A549 e H1975 è stato significativamente inibito dal trattamento con DCA.
L’effetto della promozione dell’autofagia sulla morte cellulare indotta dal DCA
Sono state condotte ricerche sistematiche sugli effetti antitumorali del DCA. In primo luogo, la capacità antitumorale del DCA è stata esaminata in vitro. Entrambe le cellule sono state incubate per 24 ore in presenza di diverse concentrazioni di DCA e sono state valutate l’apoptosi e la vitalità cellulare. Abbiamo osservato che la capacità antitumorale del DCA, riflessa nei tassi di inibizione della vitalità cellulare e di apoptosi, aumentava in modo dipendente dalla concentrazione nelle cellule A549 e H1975 (Figura 2A e B, E e F, p<0,01).
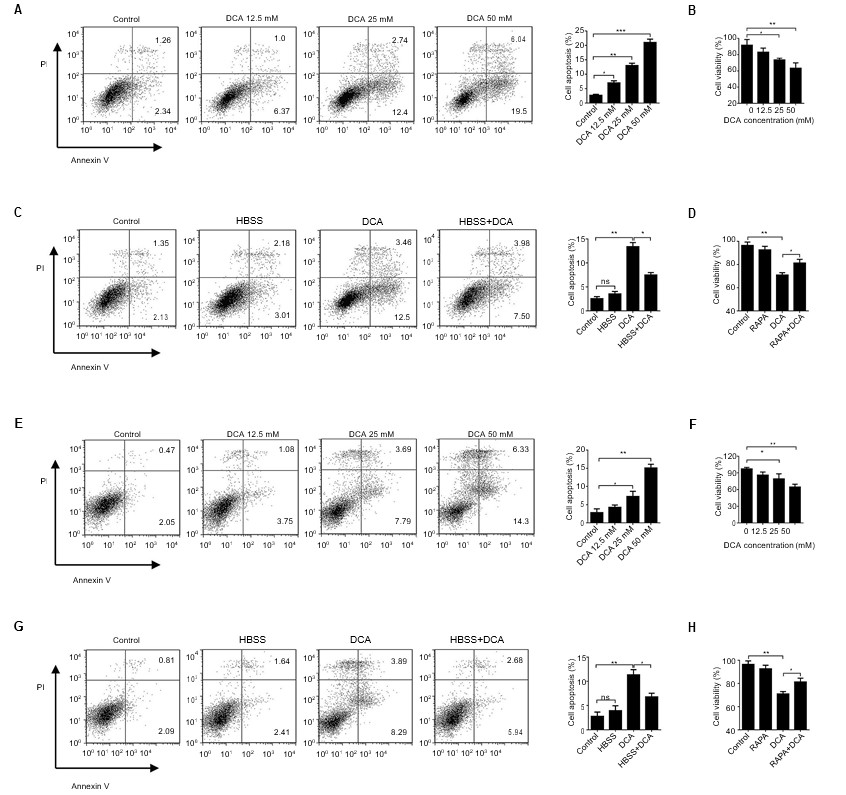
Abbreviazioni: DCA, dicloroacetato; FITC/PI, isotiocianato di fluoresceina/ioduro di pidio; HBSS, soluzione salina bilanciata di Hank.
Il DCA ha attivato l’interruttore metabolico che ha invertito il metabolismo anormale delle cellule tumorali dalla glicolisi anaerobica all’ossidazione del glucosio, con conseguente disfunzione mitocondriale e morte cellulare. I nostri esperimenti preliminari hanno mostrato che l’autofagia era significativamente inibita dal trattamento con DCA. Per determinare se l’inibizione dell’autofagia mediata dal DCA fosse coinvolta nell’apoptosi, le cellule A549 e H1975 sono state trattate con DCA dopo HBSS per 24 ore. L’apoptosi cellulare indotta dal DCA è stata inibita quando le cellule sono state fatte coincidere con HBSS rispetto al solo trattamento con DCA. Oltre ad alterare il metabolismo anomalo, questo risultato conferma che il DCA può anche attivare la morte cellulare inibendo l’autofagia nelle cellule tumorali. I risultati della vitalità cellulare sono coerenti con questa conclusione (Figura 2C e D, G e H, p<0,05).
L’inibizione della via AKT ha diminuito l’apoptosi indotta dal DCA
In studi precedenti si è ritenuto che la via ATK-mTOR svolga un ruolo critico nella regolazione dell’autofagia.[18-21] È stata utilizzata la perifosina,[22] un reagente chimico che impedisce la fosforilazione e l’attivazione di AKT, per verificare se la via ATK-mTOR sia coinvolta nel processo di inibizione dell’autofagia del DCA. Le cellule sono state co-trattate con DCA e perifosina per 24 ore. L’apoptosi cellulare è stata quasi dimezzata nel gruppo di coincubazione rispetto al gruppo di trattamento con DCA, il che è coerente con i saggi di vitalità cellulare (Figura 3A e B, D ed E, p<0,05). Abbiamo poi esaminato i livelli di proteine cruciali legate all’apoptosi, PARP e procaspasi3. La fosforilazione di AKT è stata inibita e i livelli di proteine correlate all’apoptosi sono diminuiti significativamente nel gruppo di cotrattamento (Figura 3C, p<0,05). Questi risultati sono stati ottenuti anche nelle cellule H1975 (Figura 3F, p<0,05).
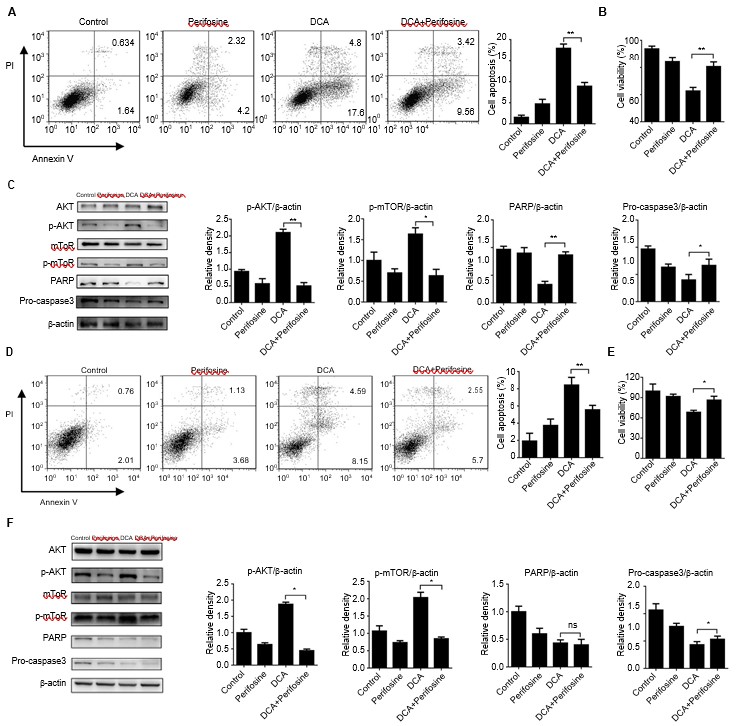
Abbreviazioni: DCA, dicloroacetato.
IlDCA aumenta la sensibilità di PTX e CDDP invertendo l’autofagia indotta dalla chemioterapia
L’inibizione dell’autofagia è considerata una nuova strategia per aumentare la sensibilità dei chemioterapici.[23,24] Per verificare se il co-trattamento con DCA e un agente chemioterapico eserciti un effetto sinergico per sopprimere la crescita cellulare, le cellule A549 sono state co-trattate con DCA e PTX o CDDP. Come mostrato nella Figura 4A-C, il trattamento con farmaci chemioterapici e DCA ha aumentato i livelli di apoptosi rispetto al trattamento con il solo farmaco o in combinazione con CQ, e i risultati sono stati coerenti con i saggi di vitalità cellulare (p<0,05). Per determinare se il DCA può downregolare l’autofagia indotta dalla PTX, le cellule sono state trattate con PTX e DCA per 24 ore e raccolte per il Western blotting. Sono stati esaminati i livelli di proteine correlate all’autofagia. L’espressione di LC3 è stata marcatamente aumentata dal trattamento con PTX ed è stata significativamente ridotta quando sono state trattate con DCA. Il livello della proteina critica legata all’apoptosi, PARP, è stato significativamente ridotto dopo il cotrattamento (Figura 4D-F, p<0,05). Tutti questi risultati indicano che il DCA ha ridotto la resistenza delle cellule A549 al PTX inibendo l’autofagia.
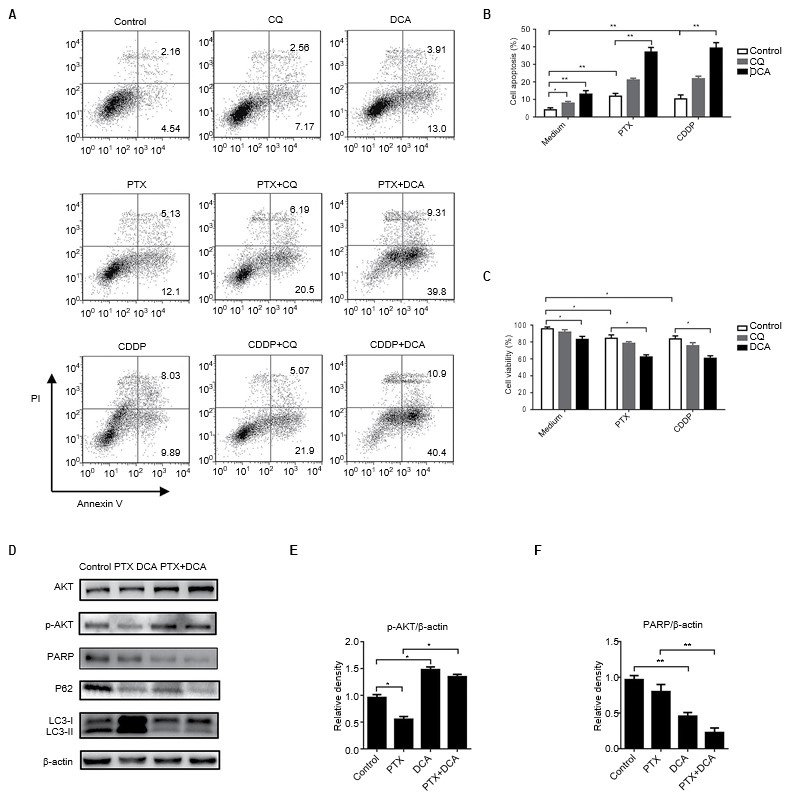
Abbreviazioni: CDDP, cis-diamminedicloroplatino; CQ, clorochina; DCA, dicloroacetato; PTX, paclitaxel.
IlDCA ha potenziato l’efficacia antitumorale del PTX in vivo inibendo l’autofagia
Poiché il cotrattamento delle cellule con PTX e DCA ha aumentato il tasso di morte cellulare in vitro rispetto al trattamento con uno dei due farmaci da solo, abbiamo valutato anche l’efficacia in vivo della terapia combinata di DCA e PTX in un modello di xenotrapianto tumorale di topo. Una volta che le dimensioni del tumore xenograft sottocutaneo si sono avvicinate a 150 mm3, i topi sono stati separati in quattro gruppi: controllo, DCA, PTX e DCA più PTX. La somministrazione di PTX da sola non ha inibito in modo significativo la crescita del tumore, ma il trattamento con PTX più DCA ha chiaramente inibito la crescita del tumore e prolungato la sopravvivenza dei topi (Figura 5A, e B, p<0,05). Come mostrato nella Figura 5C, il tempo di raddoppiamento del tumore delle cellule A549 è aumentato significativamente da 3 giorni nel gruppo di controllo a 10 giorni nel gruppo di cotrattamento (p<0,05), e il tempo di raddoppiamento del tumore delle cellule H1975 è aumentato da 2,6 a 7,5 giorni (Figura 5D-F, p<0,05). Questi risultati indicano che il DCA può potenziare l’efficacia antitumorale in vivo della PTX.
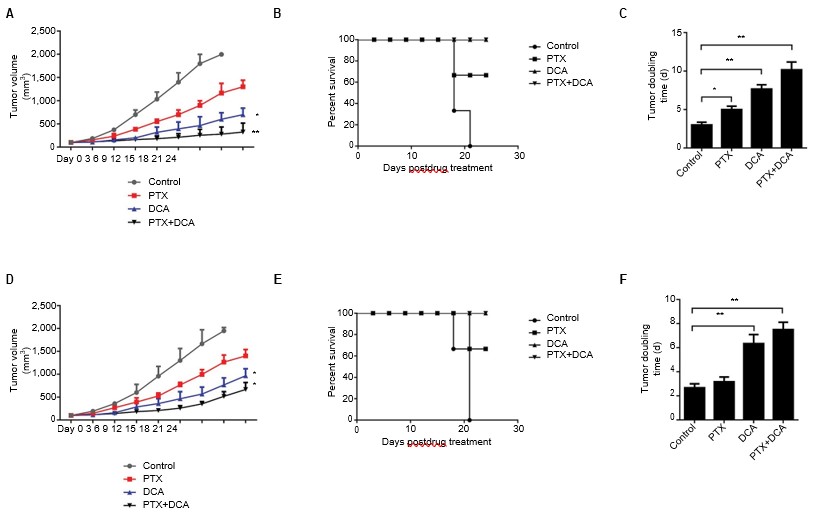
Abbreviazioni: DCA, dicloroacetato; PTX, paclitaxel.
Iltrattamento con DCA e PTX ha inibito significativamente la germinazione tumorale
Per verificare se la terapia combinata di DCA e PTX abbia portato a cambiamenti morfologici caratteristici dell’apoptosi nelle cellule A549 in vivo, tutti i tumori sono stati recuperati il giorno 24, sezionati, colorati con TUNEL e analizzati mediante immunoistochimica. Il numero di cellule TUNEL-positive nei gruppi di trattamento con DCA o PTX è aumentato in misura minima rispetto al gruppo di controllo. Tuttavia, la proporzione di cellule TUNEL-positive nel gruppo di cotrattamento è aumentata drasticamente rispetto ai gruppi di controllo o di singolo trattamento (Figura 6A).
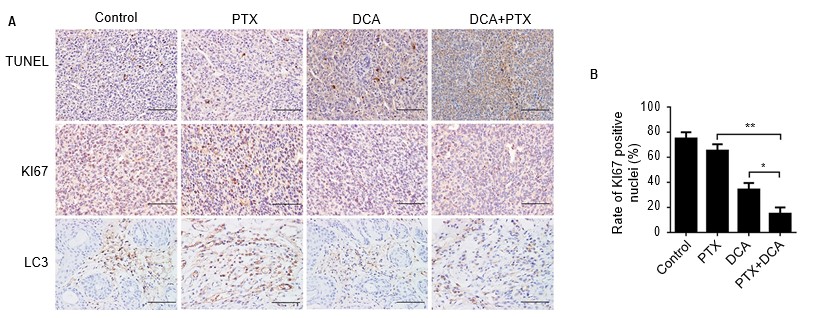
Abbreviazioni: DCA, dicloroacetato; PTX, paclitaxel.
Abbiamo poi esaminato l’espressione del Ki-67, legato alla proliferazione, e della proteina LC3B, cruciale per l’autofagia, nei tumori mediante colorazioni immunoistochimiche. L’espressione di Ki-67 nei topi di controllo era significativamente più alta, mentre la sua espressione era notevolmente ridotta nei tumori degli animali sottoposti al trattamento combinato, mentre l’espressione di LC3B era uguale a quella di Ki-67. In conclusione, il trattamento con DCA più PTX ha aumentato significativamente l’apoptosi e ridotto l’espressione di Ki-67 e LC3B in vivo (Figura 6A e B, p<0,01).
Discussione
Con lo sviluppo della ricerca sul cancro, un numero crescente di strategie terapeutiche è stato applicato al trattamento del tumore del polmone.[25-27] Sebbene molti pazienti affetti da tumore del polmone, soprattutto in fase avanzata o progressiva, abbiano beneficiato dell’EGFR-TKI e dell’anticorpo PD-1, per la maggior parte dei pazienti i costi sono esorbitanti. La chemioterapia convenzionale è ancora l’approccio principale per il trattamento del carcinoma polmonare avanzato in Cina. Tuttavia, la resistenza multifarmaco è un ostacolo difficile da superare nel trattamento del cancro; pertanto, la ricerca attuale si concentra sulla ricerca di nuovi modi per migliorare la sensibilità chemioterapica o ritardare la resistenza ai farmaci. Recentemente, la regolazione dell’autofagia è stata considerata un bersaglio per la terapia antitumorale; molte prove hanno indicato che l’inibizione dell’autofagia può promuovere l’effetto antitumorale dei chemioterapici e della radioterapia.[23,28-30] Di solito, la maggior parte dei carcinomi solidi genera energia attraverso la via glicolitica, indipendentemente dalla presenza di ossigeno sufficiente; questo fenomeno è chiamato “effetto Warburg”.[29-31] Il DCA, un agente antitumorale a piccola molecola, può invertire questa modalità metabolica dei mitocondri, passando dalla respirazione anaerobica a quella aerobica, causando così la morte delle cellule tumorali. Tuttavia, non è noto se il DCA possa essere utilizzato in combinazione con altri chemioterapici. In questo studio abbiamo scoperto che il cotrattamento con DCA e PTX può essere più efficace nel sopprimere la crescita delle cellule di cancro al polmone in vitro e in vivo. Il DCA ha inibito l’autofagia protettiva impiegata dalle cellule tumorali in risposta al trattamento con PTX, attivando la fosforilazione di AKT e promuovendo la morte cellulare. L’effetto sinergico di questi due agenti aumenta la morte cellulare, inibendo così la crescita tumorale in modo più efficace.
Come sappiamo, il metabolismo cellulare è un bersaglio efficace per il trattamento del cancro[32] e le prove che si accumulano hanno dimostrato le proprietà antitumorali del DCA nel metabolismo cellulare. Tuttavia, gli effetti del DCA sono controversi, poiché sono stati riportati risultati diversi in diversi modelli di cancro.[33,34] Nel presente studio, abbiamo riscontrato che la combinazione di DCA e PTX era più efficace nell’inibire la proliferazione cellulare e nell’aumentare l’apoptosi rispetto al solo PTX. Nel modello di xenotrapianto tumorale sottocutaneo utilizzato in questo studio, il trattamento con DCA e PTX ha ridotto del 60% la crescita delle cellule A549 iniettate nei topi. I nostri dati hanno dimostrato che il DCA ha esercitato potenti effetti antitumorali e ha agito in modo sinergico con il PTX sulle cellule A549. Una terapia multifarmaco che agisce in modo sinergico può migliorare l’efficacia terapeutica diminuendo la tossicità e la resistenza ai farmaci.[35] Nei nostri esperimenti, il DCA ha invertito la resistenza multifarmaco delle cellule A549 inibendo l’autofagia indotta dal PTX e inducendo l’apoptosi. Pertanto, l’aggiunta di DCA al regime chemioterapico potrebbe ridurre la pericolosità della resistenza ai farmaci multipli nel NSCLC.
L’autofagia è una via metabolica altamente conservata in cui le cellule trasportano proteine senili o difettose, organelli e altri componenti cellulari ai lisosomi per la degradazione al fine di mantenere l’omeostasi cellulare.[36,37] Numerose evidenze dimostrano che la maggior parte degli agenti chemioterapici e le radiazioni ionizzanti inducono l’autofagia, portando alla resistenza ai farmaci multipli. L’adattamento dell’autofagia facilita la sopravvivenza delle cellule tumorali in un microambiente caratterizzato da un maggiore stress metabolico o dalla tossicità dei farmaci. L’autofagia indotta dallo stress è un’importante strategia protettiva in grado di mantenere la sopravvivenza cellulare, che in ultima analisi si traduce in resistenza ai farmaci in vari tipi di cellule tumorali.[38] In questo studio, il trattamento con DCA ha inibito l’autofagia cellulare e promosso l’apoptosi. RARA, un attivatore dell’autofagia, può rendere le cellule più resistenti al DCA, indicando che il DCA causa la morte cellulare interrompendo il metabolismo delle cellule tumorali e inibendo l’autofagia. La via PI3K-AKT-mTOR è un importante regolatore negativo dell’autofagia. Pertanto, il bersaglio della segnalazione PI3K-AKT-mTOR può essere una nuova strategia per la terapia del cancro. Il DCA promuove selettivamente la fosforilazione della chinasi AKT, inibendo così l’autofagia. Pertanto, l’attivazione della via PI3K-AKT-mTOR da parte del DCA inibisce l’autofagia indotta dalla PTX. Il trattamento con EGFR-TKIs è correlato al livello di autofagia delle cellule di cancro al polmone e l’inibizione dell’autofagia potrebbe facilitare gli effetti antitumorali degli EGFR-TKIs. Questo potrebbe essere un nuovo punto focale del trattamento del cancro al polmone.
Tuttavia, alcuni ricercatori sostengono che l’autofagia sia una “spada a doppio taglio” per le cellule tumorali e che possa sopprimere la sopravvivenza e la proliferazione nel microambiente tumorale.[26,39,40] Inoltre, l’induzione dell’autofagia nel cancro è stata considerata anche una strategia di sensibilizzazione alla chemioterapia. Le diverse funzioni dell’autofagia nelle cellule tumorali potrebbero essere causate dai diversi stati delle cellule tumorali o dai tipi di tumore; pertanto, la comprensione delle funzioni sottostanti la regolazione dell’autofagia tra le diverse cellule tumorali getterà nuova luce sulla designazione della strategia contro la resistenza alla chemioterapia.[24]
È interessante notare che il DCA sembra avere una duplice caratteristica di regolazione dell’autofagia in diverse cellule tumorali. Lin et al[33] hanno riportato che il DCA può indurre l’apoptosi nelle cellule di cancro del colon-retto e allo stesso tempo potenziare l’autofagia. Ne consegue che nella regolazione dell’autofagia da parte del DCA è coinvolto un meccanismo molecolare più complesso. Esplorare il principio dei diversi effetti del DCA sull’autofagia ci fornirebbe nuove idee per progettare nuovi farmaci chemioterapici adiuvanti attraverso l’inibizione dell’autofagia.
Conclusioni
I nostri dati hanno dimostrato che l’autofagia può essere attivata durante la chemioterapia e l’inibizione dell’autofagia da parte del DCA può facilitare la morte cellulare e migliorare la sensibilità delle cellule tumorali ai farmaci chemioterapici. Inoltre, l’autofagia e il metabolismo del cancro sono stati coinvolti in una serie di eventi tumorali, come lo sviluppo del tumore, la progressione del tumore e la chemioterapia. La comprensione della relazione tra autofagia e metabolismo[32] in diverse cellule tumorali potrebbe fornirci nuove opportunità per la progettazione di farmaci mirati a questi importanti eventi cellulari contro il cancro e la resistenza ai farmaci.
Riconoscimento
Lo studio è stato finanziato dalla National Natural Science Foundation of China (81702247) e dal Basic Science and Frontier Technology Project of Chongqing per Zheng (cstc2017jcyjAX0048), dalla National Natural Science Foundation of China per Dai (81472188) e dal Clinical Research Project of Xinqiao Hospital per Yu (2015YLC21).
Divulgazione
Gli autori non segnalano conflitti di interesse in questo lavoro.
RIFERIMENTI
1 Maher AR, Miake-Lye IM, Beroes JM, Shekelle PG. Trattamento del carcinoma polmonare non a piccole cellule metastatico: A Systematic Review of Comparative Effectiveness and Cost-Effectiveness. Washington (DC): Dipartimento degli Affari dei Veterani (USA); 2012.2 Spiro SG, Porter JC. Cancro del polmone: a che punto siamo oggi? Gli attuali progressi nella stadiazione e nel trattamento non chirurgico. Am J Respir Crit Care Med. 2002;166:1166-1196.
3 Song W, Tang Z, Li M, et al. Combinazione polipeptidica di paclitaxel e cisplatino per una maggiore efficacia chemioterapica e riduzione degli effetti collaterali. Acta Biomater. 2014;10:1392-1402.
4 Ward PS, Thompson CB. Riprogrammazione metabolica: un segno distintivo del cancro che nemmeno Warburg aveva previsto. Cancer Cell. 2012;21:297-308.
5 Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: un’impalcatura molecolare mirata ai mitocondri per la somministrazione efficace del modulatore metabolico dicloroacetato. ACS Chem Biol. 2014;9:1178-1187.
6KimuraT, Takabatake Y, Takahashi A, Isaka Y. Clorochina nella terapia del cancro: un’arma a doppio taglio dell’autofagia. Cancer Res. 2013;73:3-7.
7 Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42.
8 Gomes LR, Vessoni AT, Menck CF. Il cross-talk tra microambiente e autofagia: Implicazioni nella terapia del cancro. Pharmacol Res. 2016;107:300-307.
9 Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosi, autofagia, necroptosi e metastasi del cancro. Mol Cancer. 2015;14:48.
10 Koren I, Kimchi A. Biologia cellulare. Promuovere la tumorigenesi sopprimendo l’autofagia. Science. 2012;338:889-890.
11 Rosenfeldt MT, Ryan KM. Il ruolo dell’autofagia nello sviluppo dei tumori e nella terapia del cancro. Expert Rev Mol Med. 2009;11:e36.
12 Rubinsztein DC, Codogno P, Levine B. La modulazione dell’autofagia come potenziale bersaglio terapeutico per diverse malattie. Nat Rev Drug Discov. 2012;11:709-730.
13EggerME, Huang JS, Yin W, McMasters KM, McNally LR. L’inibizione dell’autofagia con la clorochina è efficace nel melanoma. J Surg Res. 2013;184:274-281.
14 Rangwala R, Chang YC, Hu J, et al. Inibizione combinata di MTOR e autofagia: studio di fase I di idrossiclorochina e temsirolimus in pazienti con tumori solidi avanzati e melanoma. Autofagia. 2014;10:1391-1402.
15 Vogl DT, Stadtmauer EA, Tan KS, et al. Inibizione combinata dell’autofagia e del proteasoma: studio di fase 1 su idrossiclorochina e bortezomib in pazienti con mieloma recidivato/refrattario. Autofagia. 2014;10:1380-1390.
16 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51.
17 Christofk HR, Vander HM, Harris MH, et al. L’isoforma splice M2 della piruvato chinasi è importante per il metabolismo del cancro e la crescita tumorale. Nature. 2008;452:230-233.
18 Tan J, Jiang X, Yin G, et al. L’acido anacardico induce l’apoptosi cellulare del cancro prostatico attraverso l’autofagia tramite la via di segnalazione ER stress/DAPK3/Akt. Oncol Rep. 2017;38(3):1373-1382.
19 Chen LM, Song TJ, Xiao JH, Huang ZH, Li Y, Lin TY. Il triclorolide induce l’autofagia nelle cellule di cancro al polmone inibendo la via PI3K/AKT/mTOR e migliora la sensibilità al cisplatino nelle cellule A549/DDP. Oncotarget. 2017;8(38):63911–63922.
20 Wang Z, Wang N, Liu P, Xie X. AMPK e cancro. EXS. 2016;107:203-226.
21 Umezawa S, Higurashi T, Nakajima A. AMPK: target terapeutico per il diabete e la prevenzione del cancro. Curr Pharm Des. 2017;23(25):3629–3644.
22 Gills JJ, Dennis PA. Perifosina: aggiornamento su un nuovo inibitore di Akt. Curr Oncol Rep. 2009;11:102-110.
23 Janku F, McConkey DJ, Hong DS, Kurzrock R. L’autofagia come bersaglio della terapia antitumorale. Nat Rev Clin Oncol. 2011;8:528-539.
24 Rubinsztein DC, Codogno P, Levine B. La modulazione dell’autofagia come potenziale bersaglio terapeutico per diverse malattie. Nat Rev Drug Discov. 2012;11:709-730.
25 Ma W, Gilligan BM, Yuan J, Li T. Stato attuale e prospettive della ricerca traslazionale sui biomarcatori per la terapia di blocco del checkpoint immunitario PD-1/PD-L1. J Hematol Oncol. 2016;9:47.
26 Dholaria B, Hammond W, Shreders A, Lou Y. Agenti terapeutici emergenti per il cancro del polmone. J Hematol Oncol. 2016;9:138.
27 Wang S, Cang S, Liu D. Inibitori di terza generazione mirati alla mutazione T790M di EGFR nel carcinoma polmonare non a piccole cellule in stadio avanzato. J Hematol Oncol. 2016;9:34.
28 White E, Karp C, Strohecker AM, Guo Y, Mathew R. Ruolo dell’autofagia nella soppressione dell’infiammazione e del cancro. Curr Opin Cell Biol. 2010;22:212-217.
29 Mathew R, Karantza-Wadsworth V, White E. Ruolo dell’autofagia nel cancro. Nat Rev Cancer. 2007;7:961-967.
30 Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Il blocco dell’autofagia sensibilizza le cellule di carcinoma resistenti alla radioterapia. Cancer Res. 2008;68:1485-1494.
31 Zhou W, Liotta LA, Petricoin EF. L’effetto Warburg e l’analisi proteomica basata sulla spettrometria di massa. Cancer Genomics Proteomics. 2017;14:211-218.
32 Ho WL, Hsu WM, Huang MC, Kadomatsu K, Nakagawara A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J Hematol Oncol. 2016;9:100.
33 Lin G, Hill DK, Andrejeva G, et al. Il dicloroacetato induce l’autofagia nelle cellule e nei tumori del cancro colorettale. Br J Cancer. 2014;111:375-385.
34 Gong F, Peng X, Sang Y, et al. Il dicloroacetato induce l’autofagia protettiva nelle cellule LoVo: coinvolgimento della catepsina D/terodossina-like protein 1 e della segnalazione mediata da Akt-mTOR. Cell Death Dis. 2013;4:e913.
35 Chou TC. Basi teoriche, disegno sperimentale e simulazione computerizzata di sinergismo e antagonismo negli studi di combinazione di farmaci. Pharmacol Rev. 2006;58:621-681.
36 Eskelinen EL. Il duplice ruolo dell’autofagia nel cancro. Curr Opin Pharmacol. 2011;11:294-300.
37 Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383-1435.
38 Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
39 Yoshida GJ. Strategie terapeutiche di riposizionamento di farmaci mirati all’autofagia per indurre la morte delle cellule tumorali: dalla fisiopatologia al trattamento. J Hematol Oncol. 2017;10:67.
40 Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651-662.
Contenuti correlati: