Xiao Lu,1,* Dong Zhou,1,* Bing Hou,1 Quan-Xing Liu,1 Qian Chen,2 Xu-Feng Deng,1,3 Zu-Bin Yu,1 Ji-Gang Dai,1 Hong Zheng1
1 Departamento de Cirugía Torácica, Hospital Xinqiao, Tercera Universidad Médica Militar, Chongqing, República Popular China
2 Instituto de Patología y Centro Oncológico del Suroeste, Hospital del Suroeste, Tercera Universidad Médica Militar, Chongqing, República Popular China
3 Departamento de Cirugía Cardiotorácica, Primer Hospital Popular de Zunyi, Guizhou, República Popular China
*Estos autores han contribuido a partes iguales a este trabajo
Correspondencia: Hong Zheng; Ji-Gang Dai
Departamento de Cirugía Torácica, Hospital Xinqiao, Tercera Universidad Médica Militar, n.º 183, calle Xinqiao, distrito de Shapingba, Chongqing 400037, República Popular China
Tel.: +86 23 6877 4724
Fax: +86 23 6877 4724
Correo electrónico: [email protected]; [email protected]
Recibido: 14 de septiembre de 2020
Aceptado: 4 de diciembre de 2020
Publicado: 9 de diciembre de 2020
Resumen
Antecedentes: La quimioterapia sigue siendo la principal estrategia adyuvante del tratamiento del cáncer; sin embargo, la aparición de resistencia a múltiples fármacos ha sido motivo de preocupación. Se ha demostrado que la autofagia desempeña una función protectora frente a los fármacos quimioterapéuticos en las células cancerosas y, en general, se considera que la inhibición de la autofagia es una estrategia terapéutica prometedora. Sin embargo, la escasez de inhibidores eficaces y específicos de la autofagia limita su aplicación.
Propósito: El objetivo de este estudio fue explorar el efecto del DCA, pequeño agente antitumoral molecular, sobre la regulación de la autofagia y la quimiosensibilización en células NSCLC.
Métodos: Investigamos la regulación de la autofagia del dicloroacetato (DCA) mediante microscopía confocal láser y western blotting en las líneas celulares A549 y H1975. El ensayo MTT y la citometría de flujo se realizaron para explorar la eficacia quimiosensibilizadora del DCA. Los resultados se verificaron con el modelo de tumor subcutáneo en ratones desnudos y se aplicó la inmunohistoquímica para evaluar el nivel de apoptosis celular y autofagia in vivo tras el tratamiento.
Resultados: Se observó que el DCA, que presentaba propiedades antitumorales en varios modelos de carcinoma, inducía la apoptosis de las células de cáncer de pulmón no microcítico (CPNM) mediante la inhibición de la autofagia de las células cancerosas. Además, la perifosina, un inhibidor de AKT, puede debilitar en gran medida la capacidad de inducción de apoptosis por DCA. Los resultados indican que la vía AKT-mTOR, principal regulador negativo de la autofagia, está implicada en la inhibición de la autofagia inducida por el DCA. A continuación, detectamos la eficacia de la inhibición de la autofagia por el DCA. Cuando se utilizó en co-tratamiento con el fármaco quimioterapéutico paclitaxel (PTX), el DCA disminuyó notablemente la autofagia celular, aumentó la apoptosis e inhibió la proliferación en las células A549 y H1975. Los resultados del experimento con xenoinjertos demuestran que el tratamiento conjunto de PTX y DCA puede disminuir significativamente la proliferación celular in vivo y prolongar la supervivencia de los ratones.
Conclusión: Nuestros resultados sugieren que el DCA puede inhibir la autofagia celular inducida por quimioterápicos, proporcionando una nueva vía para la sensibilización a la quimioterapia del cáncer.
Palabras clave: DCA, autofagia, multirresistencia a fármacos, cáncer de pulmón no microcítico, paclitaxel, xenoinjerto en ratón desnudo, quimiosensibilización
INTRODUCCIÓN
El cáncer de pulmón no microcítico (CPNM) es una de las principales causas de mortalidad por cáncer en todo el mundo. Es el cáncer más frecuente en hombres y mujeres, con una incidencia superior a la incidencia combinada de los cánceres de mama, de cuello uterino y colorrectal[1,2]. Aunque la quimioterapia sigue siendo el medio más importante de tratamiento adyuvante para los pacientes con cáncer inoperable y los pacientes sometidos a cirugía, los beneficios clínicos de las quimioterapias postoperatorias basadas en platino y paclitaxel son modestos, especialmente en el CPNM avanzado. Al mismo tiempo, las reacciones adversas a los fármacos se han agravado y también han aparecido resistencias[3 ], por lo que urge encontrar nuevas estrategias que sustituyan o complementen a la quimioterapia tradicional.
En los últimos años se ha demostrado que las células tumorales producen preferentemente energía para el crecimiento y la división celular a través del proceso glucolítico y la fermentación láctica. Las tasas de metabolismo anaeróbico y de glucólisis en las células tumorales malignas de crecimiento rápido son significativamente superiores a las de las células normales. Esta reprogramación del metabolismo energético se conoce como efecto Warburg, y puede aprovecharse como diana terapéutica para inhibir el crecimiento tumoral. Entre los numerosos fármacos dirigidos al metabolismo, el dicloroacetato (DCA) ha mostrado un excelente potencial por su contribución positiva al tratamiento del cáncer[4,5]
Otro mecanismo que está completamente alterado en las células cancerosas es la autofagia, un sistema homeostático de degradación celular que se encarga de degradar orgánulos o proteínas celulares dañados o innecesarios[6]. Durante la autofagia, la carga celular destinada a la degradación se encierra en un autofagosoma, una vesícula de doble membrana. El autofagosoma cargado se fusiona suavemente con un lisosoma para formar un autolisosoma, donde el material celular entregado es degradado por varias enzimas hidrolíticas lisosómicas. El proceso de autofagia se ha investigado ampliamente. Cada vez está más claro que la alteración de la actividad de la autofagia está relacionada con la formación y progresión de tumores[7-9]. Dado que la autofagia desempeña una función protectora en las células cancerosas frente a los fármacos quimioterapéuticos, la supresión de la autofagia durante la quimioterapia se ha considerado una estrategia terapéutica novedosa[10-12]. En la actualidad, sólo la cloroquina (CQ) se utiliza en entornos clínicos como inhibidor eficaz de la autofagia. Aunque se ha demostrado la eficacia y viabilidad de la cloroquina en la terapia del cáncer, los efectos secundarios no deseados podrían ser un problema para el tratamiento clínico. Descubrir y utilizar otros inhibidores de la autofagia en la terapia del cáncer tendría una gran importancia clínica[13-15]
El DCA es un agente dirigido a las mitocondrias que actúa como un interruptor metabólico, invirtiendo el metabolismo anormal de las células cancerosas de la glucólisis anaeróbica a la oxidación aeróbica de la glucosa mediante la reducción de la actividad de la PDK1 mitocondrial y el aumento de la viabilidad de la PDH. Por lo tanto, el DCA aumenta las especies reactivas de oxígeno mitocondriales, induciendo así la apoptosis en las células tumorales malignas sin afectar a las células normales[16,17] Sin embargo, la acción reguladora del DCA para la autofagia en el cáncer de pulmón aún no está clara. En este estudio, demostramos que el DCA inhibe la proliferación celular y mejora la apoptosis de las células tumorales a través de la regulación a la baja de la autofagia, aumentando así la eficiencia de la muerte celular cuando se utiliza en cotratamiento con agentes quimioterapéuticos.
Materiales y métodos
Cultivo celular y reactivos
Las células de adenocarcinoma pulmonar humano A549 se cultivaron en medio Eagle modificado de Dulbecco con un 10% de suero bovino fetal, y las células H1975 se cultivaron en medio Roswell Park Memorial Institute-1640 con un 10% de suero bovino fetal en una incubadora humidificada con un 95% de aire y un 5% deCO2 a 37°C. Las líneas celulares A549 (TCHu150) y H1975 (TCHu193) se adquirieron en la Biblioteca de Cultivos Típicos de la Academia China de Ciencias (Shanghai, República Popular China). Después de que las células crecieran a lo largo de la pared de la placa de cultivo, se utilizó tripsina al 0,25% (HyClone, Buckinghamshire, Reino Unido) para desprenderlas y subcultivarlas. Los reactivos utilizados en este estudio fueron DCA (Sigma-Aldrich, St. Louis, MO, EE.UU.), paclitaxel (PTX; Sigma-Aldrich), cisplatino (cis-diamminedicloroplatino [CDDP]; Sigma-Aldrich), adenovirus (GFP-RFP-LC3; Hanbio, Shanghai, República Popular China), TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling; Promega, Fitchburg, WI, EE.UU.), APC-Anexina V, yoduro de propidio (BD Pharmingen, Franklin Lakes, NJ, EE.UU.) y kit MTT (Sangon Biotech, Shanghai, República Popular China). Los anticuerpos fueron LC3-I/II, p62, PARP (todos Abcam, Cambridge, Reino Unido), β-actina (Sigma-Aldrich), mTOR/p-mTOR (Sigma-Aldrich) y Ki-67 (Biovisualab, Shanghai, República Popular China).
Microscopía de inmunofluorescencia
Se inocularon células (1×105) en una placa de 24 pocillos y se incubaron durante la noche. Antes del experimento, las células se infectaron con adenovirus que contenían estructuras GFP-RFP-LC3 en tándem. A las 24 h de la infección, se cambiaron los medios y las células se trataron con DCA 25 mM o solución salina equilibrada de Hank (HBSS; Sigma Aldrich) durante 24 h. Para la inmunotinción, las células se fijaron en paraformaldehído al 4% y se lavaron con PBS. Las células se incubaron con DAPI (4′,6-diamidino-2-fenilindol; Beyotime Biotechnology, Shanghai, República Popular China) durante 5 minutos y se lavaron tres veces con PBS. Los cubreobjetos se montaron en portaobjetos con medio de montaje. Las imágenes se capturaron con un microscopio confocal LSM 780 Meta (Carl Zeiss MicroImaging GmbH, Jena, Alemania) y se procesaron con el software proporcionado por el fabricante.
Ensayo de apoptosis
Se inocularon células (1×105) en una placa de 24 pocillos y se incubaron durante la noche. Antes de los experimentos, las células se trataron con diferentes concentraciones de DCA 25 mM, agente quimioterapéutico más DCA u otros reactivos durante 24 h. Las células se tiñeron con APC-Anexina V y yoduro de propidio para medir la tasa de apoptosis mediante citometría de flujo. Cada experimento se repitió tres veces.
Western blot
Las célulasse lisaron en hielo mediante tratamiento con tampón de lisis para ensayo de radioinmunoprecipitación (RIPA) (Sangon Biotech) con Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA, USA) durante 15 min. Los sobrenadantes se recogieron tras centrifugación. Las concentraciones de proteínas se midieron utilizando el kit de ensayo de proteínas con ácido bicinconínico (BCA) (Beyotime Biotechnology). Las muestras de proteínas se electroforizaron en geles de dodecil sulfato de sodio-poliacrilamida al 10% y se transfirieron a una membrana Immobilon PVDF. Las membranas se sondearon durante la noche a 4°C con el anticuerpo primario indicado y luego se incubaron con un anticuerpo secundario conjugado con peroxidasa de rábano picante. Las bandas se visualizaron con un tampón de detección de quimioluminiscencia (Takara, Shiga, Japón) y la intensidad de las bandas se cuantificó con el programa informático proporcionado por el fabricante.
Ensayo de viabilidad celular
Las células se cultivaron en placas de 96 pocillos (1.000 células en 100 µl de medio de cultivo/pocillo). Tras el tratamiento farmacológico, se añadieron 10 µl de MTT (0,5 mg/mL) y se incubaron durante 4 h. A continuación, se desechó el medio y se solubilizaron los cristales de formazán añadiendo DMSO (dimetilsulfóxido; Sigma-Aldrich). Se midió la absorbancia a 570 nm. La viabilidad celular se normalizó con respecto a la del grupo de control.
Estudios de xenoinjerto tumoral in vivo
Para este ensayo, se inyectaron 5×106 células por vía subcutánea en ratones desnudos (BALB/c, grado específico libre de patógenos, 4-5 semanas de edad, adquiridos en el Centro de Investigación de Animales Modelo de la Universidad de Nanjing, Nanjing, República Popular China). Cuando el tamaño del tumor alcanzó los 100 mm3 (15-20 días), los ratones se dividieron aleatoriamente en 4 grupos (PBS, PTX, DCA y PTX más DCA). La dosis de PTX fue de 20 mg/kg/día, y la de DCA de 100 mg/kg/día. La longitud y la anchura de los tumores se midieron cada dos días en sus dos diámetros perpendiculares (volumen calculado = diámetro más corto2 × diámetro más largo/2). Se registraron el número y las fechas de muerte de los ratones para calcular la tasa de supervivencia. Todas las manipulaciones con ratones vivos fueron aprobadas por el Comité de Cuidado y Uso de Animales del Hospital Xinqiao y siguieron las Directrices Chinas de Bienestar y Ética para Animales de Laboratorio, y se hizo todo lo posible por minimizar el sufrimiento.
Examen histológico
Todos los tumores se resecaron a los 24 días para su examen histopatológico. Se fijaron secciones (de 4 a 5 μm de grosor) de los tejidos tumorales con formol al 10% y se detectaron con Ki-67, anticuerpo LC3B y ensayo TUNEL. Las secciones se lavaron tres veces y se trataron con diaminobencidina para revelar el color. Se realizaron exámenes histológicos y se tomaron fotografías con un microscopio óptico NanoZoomer 2.0-RS (Hamamatsu Photonics, Hamamatsu City, Japón).
Análisis estadístico
Los datos se presentan como media ± desviación estándar. Para analizar la varianza se utilizó el análisis de la varianza de una vía y la prueba t de muestras independientes. Las curvas de supervivencia se obtuvieron mediante el método de Kaplan-Meier y las comparaciones se realizaron mediante la prueba de rangos logarítmicos. Todos los análisis estadísticos se realizaron con el programa estadístico SPSS 19.0 (SPSS; IBM Corporation, Armonk, NY, EE.UU.). Los valores p<0,05 o <0,01 se consideraron estadísticamente significativos.
Resultados
El DCAinhibió la autofagia en células de adenocarcinoma de pulmón
La LC3 citosólica y la SQSTM (p62) son proteínas altamente conservadas que se considera que desempeñan papeles esenciales durante las etapas clave de la autofagia. Para investigar el papel del DCA en la regulación de la autofagia, examinamos la localización celular de LC3 con un microscopio confocal de barrido láser. Se infectaron células A549 y H1975 con adenovirus que codificaban un constructo tándem GFP-RFP-LC3 para detectar el cambio en el flujo de autofagia. Una diferencia en la sensibilidad al pH puede causar los distintos grados de acumulación de proteínas GFP-LC3 y RFP-LC3 en autofagosomas neutros y autolisosomas ácidos. Tras el tratamiento con HBSS, el número de puntos GFP o RFP-LC3 aumentó significativamente, lo que demuestra que la inanición inducida por HBSS promovió significativamente el flujo de autofagia en las células A549 y H1975. Sin embargo, el tratamiento con DCA inhibió con éxito la expresión de proteínas LC3 en relación con el control o el tratamiento con HBSS tanto en células A549 como en células H1975 (Figura 1A y D, p<0,01).
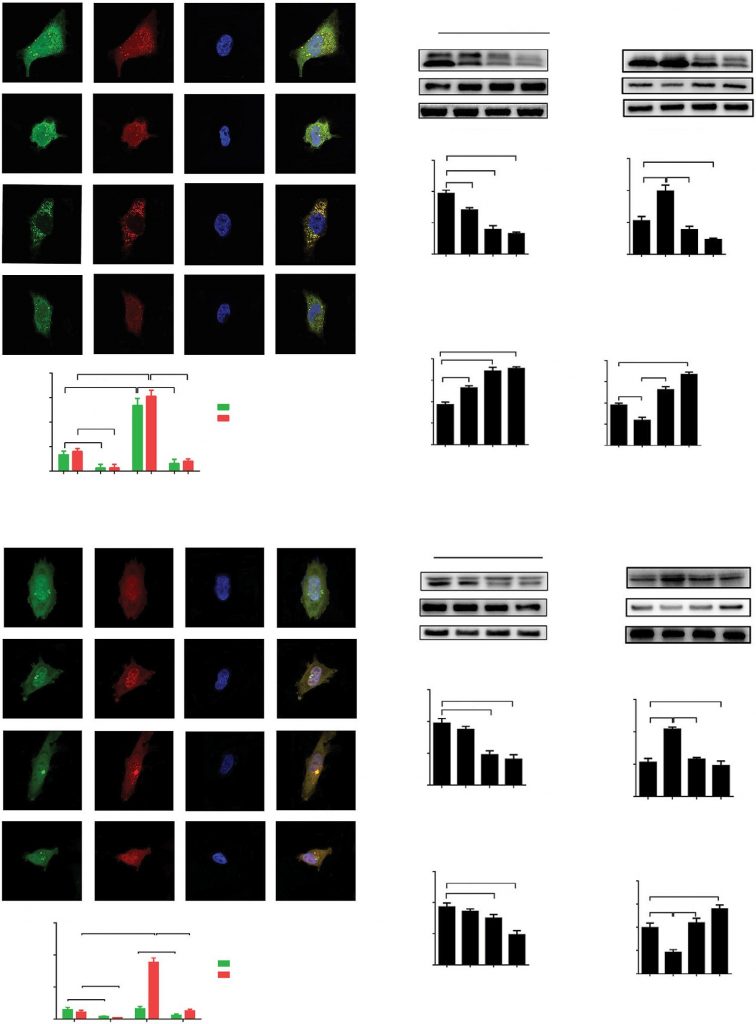
Abreviaturas: DAPI: 4′,6-diamidino-2-fenilindol; DCA: dicloroacetato; HBSS: solución salina equilibrada de Hank; ns: no significativo.
la proteína p62 se acumula cuando se inhibe la autofagia y disminuye cuando se induce la autofagia. Se realizó un análisis de Western blot para examinar los niveles proteicos de p62 y LC3-II tras el tratamiento con DCA. Se observó una disminución significativa de los niveles de LC3 y un aumento de p62 tras el tratamiento con DCA de forma dependiente de la concentración tanto en las células A549 como en las H1975 (Figura 1B y E, p<0,01). La autofagia aumentó durante la incubación con HBSS, lo que se invirtió al cotratarse con DCA (Figura 1C y F, p<0,01). En conjunto, estos resultados indican que el flujo autofágico en las células A549 y H1975 fue inhibido significativamente por el tratamiento con DCA.
El efecto de la promoción de la autofagia sobre la muerte celular inducida por el DCA
Se realizó una investigación sistemática sobre los efectos anticancerígenos del DCA. En primer lugar, se examinó la capacidad anticancerosa del DCA in vitro. Se incubaron ambas células durante 24 h en presencia de diferentes concentraciones de DCA, y se evaluaron la apoptosis y la viabilidad celular. Observamos que la capacidad anticancerosa del DCA, reflejada en los índices de inhibición de la viabilidad celular y de apoptosis, aumentaba de forma dependiente de la concentración en las células A549 y H1975 (Figura 2A y B, E y F, p<0,01).
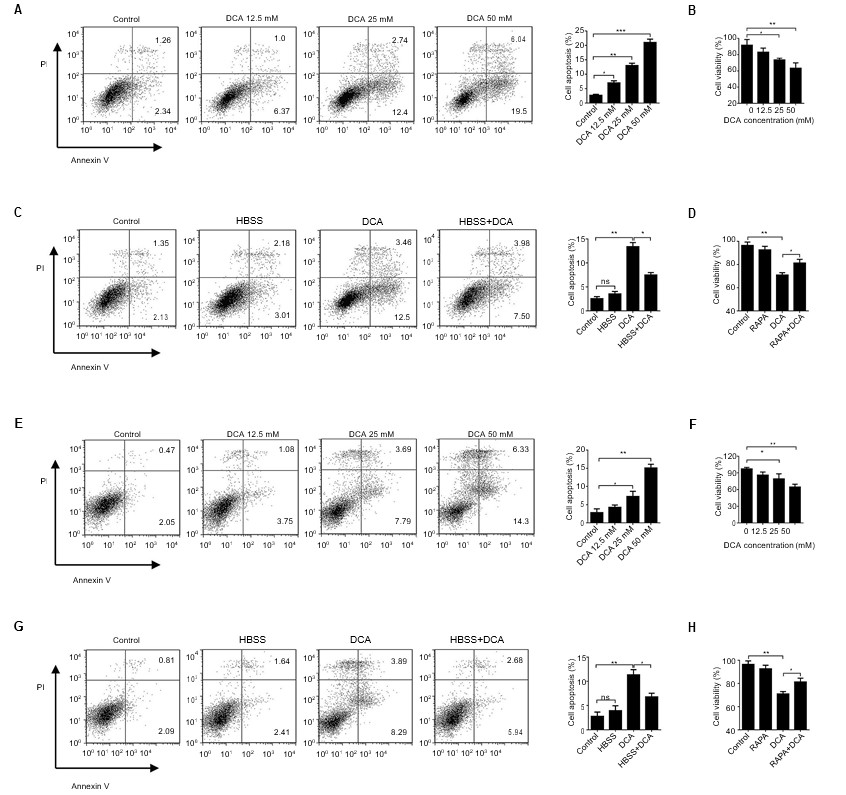
Abreviaturas: DCA: dicloroacetato; FITC/PI: isotiocianato de fluoresceína/yoduro de propidio; HBSS: solución salina equilibrada de Hank.
El DCA activó el interruptor metabólico que invirtió el metabolismo anómalo de las células cancerosas de la glucólisis anaeróbica a la oxidación de la glucosa, lo que provocó la disfunción mitocondrial y la muerte celular. Nuestros experimentos preliminares mostraron que el tratamiento con DCA inhibía significativamente la autofagia. Para determinar si la inhibición de la autofagia mediada por el DCA estaba implicada en la apoptosis, las células A549 y H1975 se trataron con DCA después de HBSS durante 24 h. La apoptosis celular inducida por el DCA se inhibió cuando las células se coincubaron con HBSS en comparación con el tratamiento de DCA solo. Además de alterar el metabolismo anormal, este resultado confirma que el DCA también puede activar la muerte celular mediante la inhibición de la autofagia en las células cancerosas. Los resultados de viabilidad celular concuerdan con esta conclusión (Figura 2C y D, G y H, p<0,05).
Lainhibición de la vía AKT disminuyó la apoptosis inducida por DCA
En estudios anteriores se ha considerado que la vía ATK-mTOR desempeña un papel crítico en la regulación de la autofagia.[18-21] Se utilizó perifosina,[22] un reactivo químico que impide la fosforilación y activación de AKT, para investigar si la vía ATK-mTOR está implicada en el proceso de inhibición de la autofagia por DCA. Las células se trataron conjuntamente con DCA y perifosina durante 24 h. La apoptosis celular se redujo casi a la mitad en el grupo de coincubación en comparación con el grupo de tratamiento con DCA, lo que concuerda con los ensayos de viabilidad celular (Figura 3A y B, D y E, p<0,05). A continuación, examinamos los niveles de proteínas cruciales relacionadas con la apoptosis, PARP y procaspasa3. La fosforilación de AKT se inhibió y los niveles de proteínas relacionadas con la apoptosis disminuyeron significativamente en el grupo de cotratamiento (Figura 3C, p<0,05). Estos resultados también se obtuvieron en las células H1975 (Figura 3F, p<0,05).
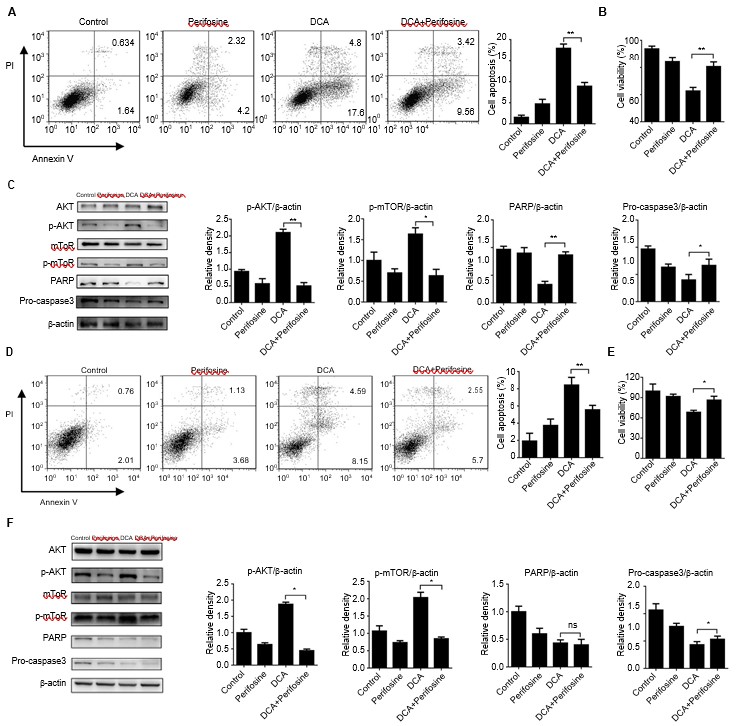
Abreviaturas: DCA, dicloroacetato.
ElDCA aumenta la sensibilidad al PTX y al CDDP al revertir la autofagia inducida por la quimioterapia
La inhibición de la autofagia se considera una nueva estrategia para aumentar la sensibilidad de los quimioterápicos[23,24]. Para investigar si el cotratamiento con DCA y un agente quimioterapéutico ejerce un efecto sinérgico para suprimir el crecimiento celular, se cotrataron células A549 con DCA y PTX o CDDP. Como se muestra en la Figura 4A-C, el tratamiento con fármacos quimioterapéuticos y DCA aumentó los niveles de apoptosis en comparación con el tratamiento farmacológico solo o en combinación con CQ, y los resultados fueron coherentes con los ensayos de viabilidad celular (p<0,05). Para determinar si el DCA puede regular a la baja la autofagia inducida por PTX, se trataron las células con PTX y DCA durante 24 h y se cosecharon para Western blotting. Se examinaron los niveles de proteínas relacionadas con la autofagia. La expresión de LC3 aumentó notablemente con el tratamiento con PTX y disminuyó significativamente con el tratamiento conjunto con DCA. El nivel de la proteína crítica relacionada con la apoptosis, PARP, disminuyó significativamente tras el cotratamiento (Figura 4D-F, p<0,05). Todos estos resultados indican que el DCA disminuyó la resistencia de las células A549 al PTX mediante la inhibición de la autofagia.
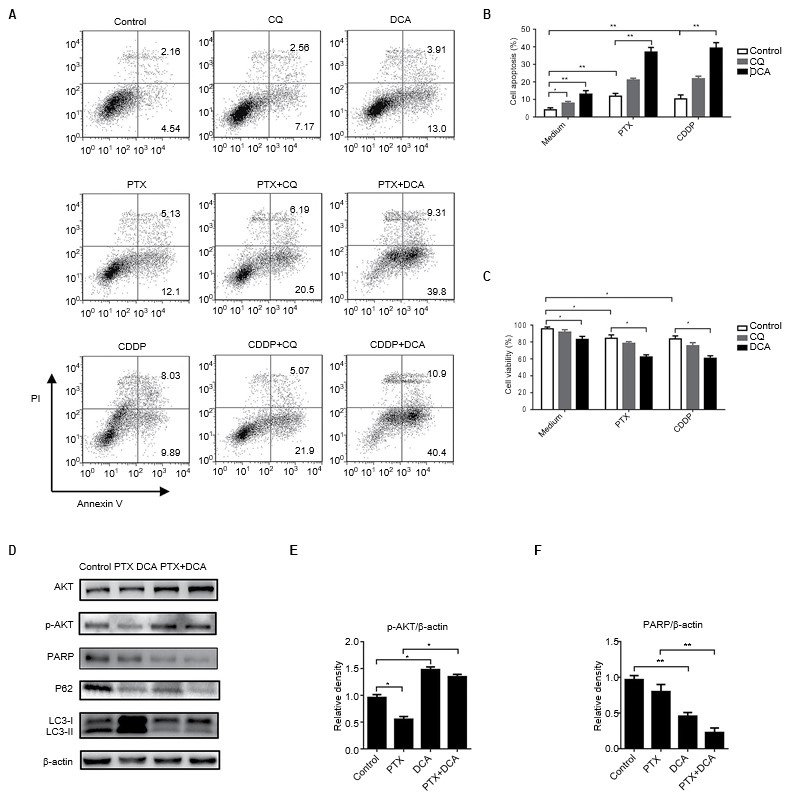
Abreviaturas: CDDP, cis-diamminedicloroplatino; CQ, cloroquina; DCA, dicloroacetato; PTX, paclitaxel.
ElDCA potenció la eficacia anticancerígena del PTX in vivo mediante la inhibición de la autofagia
Dado que el cotratamiento de células con PTX y DCA aumentó la tasa de muerte celular in vitro en comparación con el tratamiento con cualquiera de los fármacos por separado, también evaluamos la eficacia in vivo de la terapia combinada de DCA y PTX en un modelo de tumor xenoinjertado en ratón. Una vez que el tamaño del tumor xenoinjertado subcutáneo se aproximó a 150 mm3, se separó a los ratones en cuatro grupos: control, DCA, PTX y DCA más PTX. La administración de PTX sola no inhibió significativamente el crecimiento tumoral, pero el tratamiento con PTX más DCA inhibió claramente el crecimiento tumoral y prolongó la supervivencia de los ratones (Figura 5A, y B, p<0,05). Como se muestra en la Figura 5C, el tiempo de duplicación tumoral de las células A549 aumentó significativamente de 3 días en el grupo de control a 10 días en el grupo de cotratamiento ( p<0,05), y el tiempo de duplicación tumoral de las células H1975 aumentó de 2,6 a 7,5 días (Figura 5D-F, p<0,05). Estos resultados indican que el DCA puede potenciar la eficacia anticancerígena in vivo del PTX.
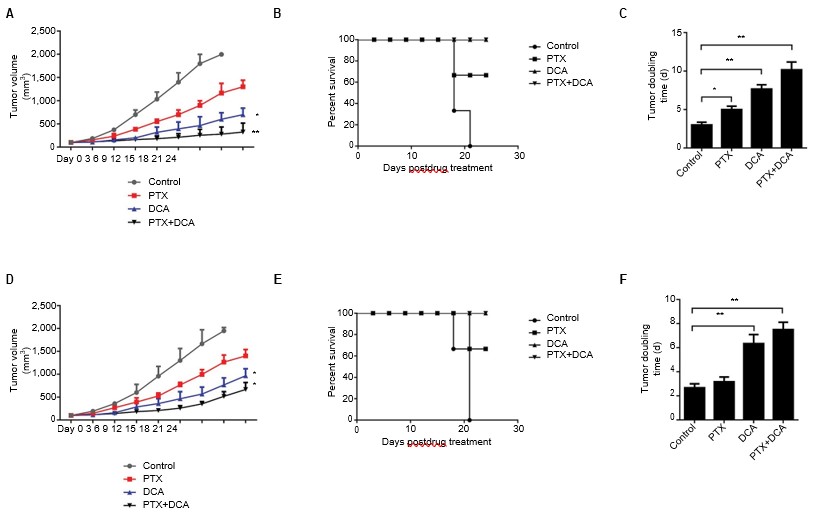
Abreviaturas: DCA, dicloroacetato; PTX, paclitaxel.
Eltratamiento con DCA más PTX inhibió significativamente la germinación tumoral
Para investigar si la terapia combinada de DCA y PTX provocaba los cambios morfológicos característicos de la apoptosis en las células A549 in vivo, se recuperaron todos los tumores el día 24, se seccionaron, se tiñeron con TUNEL y se analizaron mediante inmunohistoquímica. Hubo un aumento mínimo en el número de células TUNEL-positivas en los grupos de tratamiento con DCA o PTX en relación con el grupo de control. Sin embargo, la proporción de células TUNEL positivas en el grupo de cotratamiento aumentó drásticamente en comparación con los grupos de control o de tratamiento único (Figura 6A).
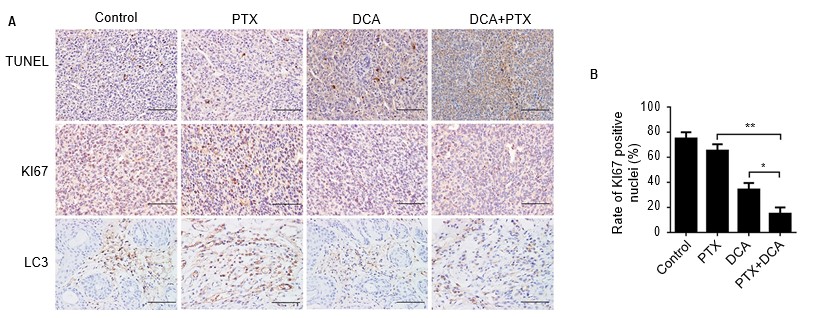
Abreviaturas: DCA, dicloroacetato; PTX, paclitaxel.
A continuación examinamos la expresión de Ki-67, relacionada con la proliferación, y de la proteína LC3B, crucial para la autofagia, en los tumores mediante tinción inmunohistoquímica. La expresión de Ki-67 en los ratones de control fue significativamente mayor, mientras que su expresión se redujo considerablemente en los tumores de los animales que recibieron el tratamiento combinado, y la expresión de LC3B fue la misma que la de Ki-67. En conclusión, el tratamiento con DCA más PTX aumentó significativamente la apoptosis y redujo la expresión de Ki-67 y LC3B in vivo (Figura 6A y B, p<0,01).
Discusión
Con el desarrollo de la investigación oncológica, se ha aplicado un número cada vez mayor de estrategias terapéuticas en el tratamiento del cáncer de pulmón[25-27] Aunque muchos pacientes con cáncer de pulmón, especialmente en la fase avanzada o progresiva, se han beneficiado del EGFR-TKI y del anticuerpo PD-1, su coste es exorbitantemente prohibitivo para la mayoría de los pacientes. La quimioterapia convencional sigue siendo el enfoque principal para tratar el cáncer de pulmón avanzado en China. Sin embargo, la resistencia a múltiples fármacos ha sido un obstáculo difícil en el tratamiento del cáncer; por lo tanto, la investigación actual se centra en la búsqueda de nuevas formas de mejorar la sensibilidad quimioterapéutica o retrasar la resistencia a los fármacos. Recientemente, la regulación de la autofagia se ha considerado como un objetivo para la terapia contra el cáncer; muchas pruebas han indicado que la inhibición de la autofagia puede promover el efecto supresor de tumores de los quimioterapéuticos y la radioterapia [23,28-30].[23,28-30] Por lo general, la mayoría de los carcinomas sólidos generan energía a través de la vía glucolítica independientemente de si hay suficiente oxígeno presente; esto se denomina «efecto Warburg»[29-31] El DCA, un agente antitumoral de molécula pequeña, puede invertir este modo metabólico de las mitocondrias de respiración anaeróbica a respiración aeróbica, provocando así la muerte de las células tumorales. Sin embargo, se desconoce si el DCA puede utilizarse en combinación con otros quimioterápicos. En este estudio, descubrimos que el cotratamiento con DCA y PTX puede ser más eficaz en la supresión del crecimiento de células de cáncer de pulmón in vitro e in vivo. El DCA inhibió la autofagia protectora empleada por las células cancerosas en respuesta al tratamiento con PTX activando la fosforilación de AKT y promoviendo la muerte celular. El efecto sinérgico de estos dos agentes potencia la muerte celular, inhibiendo así el crecimiento tumoral con mayor eficacia.
Como sabemos, el metabolismo celular es un objetivo eficaz para el tratamiento del cáncer,[32 ] y la acumulación de pruebas ha demostrado las propiedades antitumorales del DCA en el metabolismo celular. Sin embargo, los efectos del DCA son controvertidos, ya que se han reportado diferentes resultados en diferentes modelos de cáncer[33,34] En el presente estudio, encontramos que la combinación de DCA y PTX fue más eficaz en la inhibición de la proliferación celular y la mejora de la apoptosis que el PTX solo. En el modelo de xenoinjerto tumoral subcutáneo utilizado en este estudio, el tratamiento con DCA más PTX redujo el crecimiento de las células A549 inyectadas en los ratones en un 60%. Nuestros datos demostraron que el DCA ejercía potentes efectos anticancerígenos y actuaba sinérgicamente con el PTX sobre las células A549.
nuestros experimentos, el DCA revirtió la resistencia a múltiples fármacos de las células A549 mediante la inhibición de la autofagia inducida por PTX y la inducción de apoptosis. Por lo tanto, la adición de DCA al régimen de quimioterapia podría disminuir la perniciosidad de la resistencia a múltiples fármacos en el CPNM.
La autofagia es una vía metabólica altamente conservada en la que las células entregan proteínas seniles o defectuosas, orgánulos y otros componentes celulares a los lisosomas para su degradación con el fin de mantener la homeostasis celular[36,37] Un gran número de pruebas demuestra que la mayoría de los agentes quimioterapéuticos, así como la radiación ionizante, inducen la autofagia, lo que conduce a la resistencia a múltiples fármacos. La adaptación de la autofagia facilita la supervivencia de las células tumorales en un microentorno de mayor estrés metabólico o toxicidad farmacológica. La autofagia inducida por estrés es una importante estrategia protectora que puede mantener la supervivencia celular, lo que en última instancia provoca resistencia a fármacos en varios tipos de células tumorales[38]. En este estudio, el tratamiento con DCA inhibió la autofagia celular y promovió la apoptosis. RARA, un activador de la autofagia, puede hacer que las células sean más resistentes al DCA, lo que indica que el DCA causa la muerte celular al alterar el metabolismo de las células cancerosas e inhibir la autofagia. La vía PI3K-AKT-mTOR es un importante regulador negativo de la autofagia. Por lo tanto, atacar la señalización PI3K-AKT-mTOR puede ser una estrategia novedosa para la terapia del cáncer. El DCA promueve selectivamente la fosforilación de la quinasa AKT, inhibiendo así la autofagia. Así, la activación de la vía PI3K-AKT-mTOR por el DCA inhibe la autofagia inducida por PTX. Según los informes, el tratamiento con EGFR-TKIs está relacionado con el nivel de autofagia de las células de cáncer de pulmón, y la inhibición de la autofagia podría facilitar los efectos antitumorales de los EGFR-TKIs. Este podría ser un nuevo punto focal del tratamiento del cáncer de pulmón.
Sin embargo, algunos investigadores sostienen que la autofagia es un «arma de doble filo» para las células cancerosas, y puede suprimir la supervivencia y la proliferación en el microambiente tumoral[26,39,40] Además, la inducción de la autofagia en el cáncer también se consideró una estrategia para la sensibilización a la quimioterapia. Las diferentes funciones de la autofagia en las células cancerosas podrían deberse a los diferentes estados de las células cancerosas o tipos de tumores; por lo tanto, la comprensión de las funciones subyacentes de la regulación de la autofagia entre las diferentes células cancerosas arrojará nueva luz sobre la designación de la estrategia contra la resistencia a la quimioterapia[24]
Curiosamente, el DCA parece tener características duales de regulación de la autofagia en diferentes células tumorales. Lin et al[33 ] han informado de que el DCA puede inducir la apoptosis en células de cáncer colorrectal y, al mismo tiempo, potenciar la autofagia. De ello se deduce que en la regulación de la autofagia por el DCA interviene un mecanismo molecular más complejo. Explorar el principio de los diferentes efectos sobre la autofagia del DCA nos proporcionaría nuevas ideas para diseñar nuevos fármacos de quimioterapia adyuvante mediante la inhibición de la autofagia.
Conclusión
Nuestros datos mostraron que la autofagia puede activarse durante la quimioterapia, y la inhibición de la autofagia por DCA puede facilitar la muerte celular y aumentar la sensibilidad de las células tumorales a los fármacos quimioterapéuticos. Además, la autofagia y el metabolismo del cáncer estaban implicados en una serie de acontecimientos tumorales, como el desarrollo del tumor, la progresión tumoral y la quimioterapia. Elucidar la relación entre autofagia y metabolismo[32 ] en diferentes células cancerosas podría proporcionarnos nuevas oportunidades para diseñar fármacos dirigidos a estos importantes eventos celulares contra el cáncer y la resistencia a fármacos.
Agradecimientos
El estudio fue financiado por la Fundación Nacional de Ciencias Naturales de China (81702247) y el Proyecto de Ciencia Básica y Tecnología de Frontera de Chongqing para Zheng (cstc2017jcyjAX0048), la Fundación Nacional de Ciencias Naturales de China para Dai (81472188), y el Proyecto de Investigación Clínica del Hospital Xinqiao para Yu (2015YLC21).
Divulgación
Los autores no reportan conflictos de interés en este trabajo.
REFERENCIAS
1 Maher AR, Miake-Lye IM, Beroes JM, Shekelle PG. Tratamiento del cáncer de pulmón metastásico de células no pequeñas: A Systematic Review of Comparative Effectiveness and Cost-Effectiveness. Washington (DC): Departamento de Asuntos de Veteranos (EE. UU.); 2012.2 Spiro SG, Porter JC. Cáncer de pulmón: ¿dónde estamos hoy? Avances actuales en la estadificación y el tratamiento no quirúrgico. Am J Respir Crit Care Med. 2002;166:1166-1196.
3 Song W, Tang Z, Li M, et al. Polypeptide-based combination of paclitaxel and cisplatin for enhanced chemotherapy efficacy and reduced side-effects. Acta Biomater. 2014;10:1392-1402.
4 Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297-308.
5 Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol. 2014;9:1178-1187.
6KimuraT, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3-7.
7 Levine B, Kroemer G. La autofagia en la patogénesis de la enfermedad. Cell. 2008;132:27-42.
8 Gomes LR, Vessoni AT, Menck CF. Microenvironment and autophagy cross-talk: Implicaciones en la terapia del cáncer. Pharmacol Res. 2016;107:300-307.
9 Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
10 Koren I, Kimchi A. Cell biology. Promoción de la tumorigénesis mediante la supresión de la autofagia. Science. 2012;338:889-890.
11 Rosenfeldt MT, Ryan KM. El papel de la autofagia en el desarrollo tumoral y la terapia del cáncer. Expert Rev Mol Med. 2009;11:e36.
12 Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709-730.
13EggerME, Huang JS, Yin W, McMasters KM, McNally LR. La inhibición de la autofagia con cloroquina es eficaz en el melanoma. J Surg Res. 2013;184:274-281.
14 Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391-1402.
15 Vogl DT, Stadtmauer EA, Tan KS, et al. Autofagia combinada e inhibición del proteasoma: un ensayo de fase 1 de hidroxicloroquina y bortezomib en pacientes con mieloma en recaída/refractario. Autophagy. 2014;10:1380-1390.
16 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51.
17 Christofk HR, Vander HM, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-233.
18 Tan J, Jiang X, Yin G, et al. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38(3):1373-1382.
19 Chen LM, Song TJ, Xiao JH, Huang ZH, Li Y, Lin TY. Tripchlorolide induce la autofagia en células de cáncer de pulmón mediante la inhibición de la vía PI3K/AKT/mTOR y mejora la sensibilidad al cisplatino en células A549/DDP. Oncotarget. 2017;8(38):63911–63922.
20 Wang Z, Wang N, Liu P, Xie X. AMPK and cancer (AMPK y cáncer). EXS. 2016;107:203-226.
21 Umezawa S, Higurashi T, Nakajima A. AMPK: diana terapéutica para la diabetes y la prevención del cáncer. Curr Pharm Des. 2017;23(25):3629–3644.
22 Gills JJ, Dennis PA. Perifosina: actualización sobre un nuevo inhibidor de Akt. Curr Oncol Rep. 2009;11:102-110.
23 Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528-539.
24 Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709-730.
25 Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47.
26 Dholaria B, Hammond W, Shreders A, Lou Y. Agentes terapéuticos emergentes para el cáncer de pulmón. J Hematol Oncol. 2016;9:138.
27 Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34.
28 White E, Karp C, Strohecker AM, Guo Y, Mathew R. Papel de la autofagia en la supresión de la inflamación y el cáncer. Curr Opin Cell Biol. 2010;22:212-217.
29 Mathew R, Karantza-Wadsworth V, White E. Papel de la autofagia en el cáncer. Nat Rev Cancer. 2007;7:961-967.
30 Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485-1494.
31 Zhou W, Liotta LA, Petricoin EF. El efecto Warburg y el análisis proteómico basado en la espectrometría de masas. Proteómica genómica del cáncer. 2017;14:211-218.
32 Ho WL, Hsu WM, Huang MC, Kadomatsu K, Nakagawara A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J Hematol Oncol. 2016;9:100.
33 Lin G, Hill DK, Andrejeva G, et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br J Cancer. 2014;111:375-385.
34 Gong F, Peng X, Sang Y, et al. El dicloroacetato induce la autofagia protectora en células LoVo: participación de la catepsina D/proteína similar a la tioredoxina 1 y la señalización mediada por Akt-mTOR. Cell Death Dis. 2013;4:e913.
35 Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-681.
36 Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol. 2011;11:294-300.
37 Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383-1435.
38 Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
39 Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10:67.
40 Choi AM, Ryter SW, Levine B. Autofagia en la salud y la enfermedad humanas. N Engl J Med. 2013;368:651-662.
Contenido relacionado: