Xiao Lu,1,* Dong Zhou,1,* Bing Hou,1 Quan-Xing Liu,1 Qian Chen,2 Xu-Feng Deng,1,3 Zu-Bin Yu,1 Ji-Gang Dai,1 Hong Zheng1
1 Abteilung für Thoraxchirurgie, Xinqiao-Krankenhaus, Dritte Medizinische Militäruniversität, Chongqing, Volksrepublik China
2 Institut für Pathologie und Südwest-Krebszentrum, Südwest-Krankenhaus, Dritte Medizinische Militäruniversität, Chongqing, Volksrepublik China
3 Abteilung für Herz- und Thoraxchirurgie, Erstes Volkskrankenhaus von Zunyi, Guizhou, Volksrepublik China*Diese
Autoren haben gleichermaßen zu dieser Arbeit beigetragen
Korrespondenz: Hong Zheng; Ji-Gang DaiAbteilung
für Thoraxchirurgie, Xinqiao Hospital, Third Military Medical University, Nr. 183, Xinqiao Street, Shapingba District, Chongqing 400037, Volksrepublik ChinaTel
: +86 23 6877 4724Fax
: +86 23 6877 4724Email
: [email protected];
[email protected]: 14. September 2020Akzeptiert
: 4. Dezember 2020Veröffentlicht
: 9. Dezember 2020
Zusammenfassung
Hintergrund: Die Chemotherapie ist nach wie vor die primäre adjuvante Strategie der Krebstherapie; das Auftreten von Multiresistenzen gibt jedoch Anlass zur Sorge. Es wurde nachgewiesen, dass die Autophagie eine schützende Rolle gegen Chemotherapeutika in Krebszellen spielt, und die Hemmung der Autophagie gilt allgemein als vielversprechende therapeutische Strategie. Der Mangel an wirksamen und spezifischen Autophagie-Inhibitoren schränkt ihre Anwendung jedoch ein.
Ziel: Ziel dieser Studie war es, die Wirkung von DCA, einem niedermolekularen Antitumormittel, auf die Autophagie-Regulation und Chemosensibilisierung in NSCLC-Zellen zu untersuchen.
Methoden: Wir untersuchten die Autophagie-Regulierung von Dichloracetat (DCA) durch konfokale Lasermikroskopie und Western Blotting in den Zelllinien A549 und H1975. Der MTT-Assay und die Durchflusszytometrie wurden durchgeführt, um die Chemosensibilisierungseffektivität von DCA zu untersuchen. Die Ergebnisse wurden mit einem subkutanen Tumormodell in Nacktmäusen verifiziert, und die Immunhistochemie wurde angewandt, um den Grad der Zellapoptose und Autophagie in vivo nach der Behandlung zu bewerten.
Ergebnisse: Wir fanden heraus, dass DCA, das in verschiedenen Karzinommodellen antitumorale Eigenschaften aufweist, die Apoptose von nicht-kleinzelligen Lungenkrebszellen (NSCLC) durch Hemmung der Autophagie der Krebszellen auslöste. Darüber hinaus kann Perifosin, ein AKT-Inhibitor, die Fähigkeit, die Apoptose durch DCA auszulösen, stark abschwächen. Die Ergebnisse deuten darauf hin, dass der AKT-mTOR-Signalweg, ein wichtiger negativer Regulator der Autophagie, an der DCA-induzierten Hemmung der Autophagie beteiligt ist. Anschließend untersuchten wir die Wirksamkeit der Autophagie-Hemmung durch DCA. Bei gleichzeitiger Behandlung mit dem Chemotherapeutikum Paclitaxel (PTX) verringerte DCA die Autophagie der Zellen deutlich, verstärkte die Apoptose und hemmte die Proliferation in A549- und H1975-Zellen. Die Ergebnisse des Xenotransplantationsexperiments zeigen, dass die gleichzeitige Behandlung mit PTX und DCA die Zellproliferation in vivo deutlich verringern und das Überleben von Mäusen verlängern kann.
Schlussfolgerung: Unsere Ergebnisse deuten darauf hin, dass DCA die durch Chemotherapeutika induzierte Autophagie von Zellen hemmen kann, was einen neuen Weg für die Sensibilisierung der Chemotherapie bei Krebs eröffnet.
Schlüsselwörter: DCA, Autophagie, Multidrug-Resistenz, nicht-kleinzelliger Lungenkrebs, Paclitaxel, Xenotransplantat-Nacktmäuse, Chemosensibilisierung
EINLEITUNG
Nicht-kleinzelliges Lungenkarzinom (NSCLC) ist weltweit eine der häufigsten Ursachen für Krebssterblichkeit. Er ist die häufigste Krebserkrankung bei Männern und Frauen und tritt häufiger auf als Brust-, Gebärmutterhals- und Darmkrebs zusammengenommen[1,2]. Obwohl die Chemotherapie nach wie vor das wichtigste Mittel der adjuvanten Therapie für inoperable Krebspatienten und Patienten, die sich einer Operation unterziehen, ist der klinische Nutzen der postoperativen Chemotherapien auf Platin- und Paclitaxelbasis bescheiden, insbesondere bei fortgeschrittenem NSCLC. Gleichzeitig haben sich die unerwünschten Arzneimittelwirkungen verschärft, und es hat sich eine Arzneimittelresistenz entwickelt. [3] Daher besteht ein dringender Bedarf an neuartigen Strategien, die die herkömmliche Chemotherapie ersetzen/ergänzen.
In den letzten Jahren wurde immer deutlicher, dass Tumorzellen die Energie für das Zellwachstum und die Zellteilung vorzugsweise durch glykolytische Prozesse und Milchsäuregärung erzeugen. Die Raten des anaeroben Stoffwechsels und der Glykolyse sind in schnell wachsenden bösartigen Tumorzellen deutlich höher als in normalen Zellen. Diese Umprogrammierung des Energiestoffwechsels ist als Warburg-Effekt bekannt und kann als therapeutisches Ziel zur Hemmung des Tumorwachstums genutzt werden. Unter den zahlreichen Medikamenten, die auf den Stoffwechsel abzielen, hat Dichloracetat (DCA) aufgrund seines positiven Beitrags zur Krebsbehandlung ein ausgezeichnetes Potenzial gezeigt.[4,5]
Ein weiterer Mechanismus, der in Krebszellen völlig verändert ist, ist die Autophagie, ein homöostatisches zelluläres Abbausystem, das für den Abbau von beschädigten oder überflüssigen Zellorganellen oder Proteinen verantwortlich ist. [6] Bei der Autophagie wird die zum Abbau bestimmte zelluläre Ladung in einem Autophagosom, einem Bläschen mit doppelter Membran, eingeschlossen. Das beladene Autophagosom verschmilzt nahtlos mit einem Lysosom, um ein Autolysosom zu bilden, in dem das gelieferte Zellmaterial durch verschiedene lysosomale hydrolytische Enzyme abgebaut wird. Der Autophagieprozess ist umfassend erforscht worden. Es wird immer deutlicher, dass eine veränderte Autophagie-Aktivität mit der Entstehung und dem Fortschreiten von Tumoren in Verbindung steht.(7-9) Da die Autophagie in Krebszellen eine schützende Rolle gegen Chemotherapeutika spielt, wird die Unterdrückung der Autophagie während der Chemotherapie als neue therapeutische Strategie angesehen. [10-12] Gegenwärtig wird nur Chloroquin (CQ) als wirksamer Autophagie-Inhibitor in der klinischen Praxis eingesetzt. Obwohl die Wirksamkeit und Durchführbarkeit von Chloroquin in der Krebstherapie nachgewiesen wurde, könnten die unerwünschten Nebenwirkungen ein Problem für die klinische Behandlung darstellen. Die Entdeckung und Verwendung weiterer Autophagie-Inhibitoren in der Krebstherapie wäre von großer klinischer Bedeutung.[13-15]
DCA ist ein auf die Mitochondrien abzielender Wirkstoff, der als Stoffwechselschalter fungiert und den anormalen Stoffwechsel der Krebszellen von der anaeroben Glykolyse zur aeroben Glukoseoxidation umkehrt, indem er die Aktivität der mitochondrialen PDK1 verringert und die Lebensfähigkeit der PDH erhöht. DCA erhöht also die reaktiven Sauerstoffspezies in den Mitochondrien und löst so die Apoptose in bösartigen Tumorzellen aus, ohne die normalen Zellen zu beeinträchtigen.[16,17] Die regulierende Wirkung von DCA auf die Autophagie bei Lungenkrebs ist jedoch noch unklar. In dieser Studie haben wir gezeigt, dass DCA die Zellproliferation hemmt und die Apoptose von Tumorzellen durch die Herabregulierung der Autophagie verstärkt und damit die Effizienz des Zelltods erhöht, wenn es in Co-Behandlung mit Chemotherapeutika eingesetzt wird.
Materialien und Methoden
Zellkultur und Reagenzien
Menschliche Lungenadenokarzinom-Zellen A549 wurden in Dulbecco’s Modified Eagle Medium mit 10 % fötalem Rinderserum und H1975-Zellen in Roswell Park Memorial Institute-1640-Medium mit 10 % fötalem Rinderserum in einem befeuchteten Inkubator mit 95 % Luft und 5 %CO2 bei 37 °C gezüchtet. Die Zelllinien A549 (TCHu150) und H1975 (TCHu193) wurden von der Library of Typical Culture of the Chinese Academy of Sciences (Shanghai, Volksrepublik China) erworben. Nachdem die Zellen an der Wand der Kulturschale gewachsen waren, wurde 0,25 % Trypsin (HyClone, Buckinghamshire, UK) zum Ablösen und Subkultivieren verwendet. Die in dieser Studie verwendeten Reagenzien waren DCA (Sigma-Aldrich, St. Louis, MO, USA), Paclitaxel (PTX; Sigma-Aldrich), Cisplatin (cis-diamminedichloroplatinum [CDDP]; Sigma-Aldrich), Adenovirus (GFP-RFP-LC3; Hanbio, Shanghai, Volksrepublik China), TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling; Promega, Fitchburg, WI, USA), APC-Annexin V, Propidiumjodid (BD Pharmingen, Franklin Lakes, NJ, USA) und MTT-Kit (Sangon Biotech, Shanghai, Volksrepublik China). Die Antikörper waren LC3-I/II, p62, PARP (alle Abcam, Cambridge, UK), β-Actin (Sigma-Aldrich), mTOR/p-mTOR (Sigma-Aldrich) und Ki-67 (Biovisualab, Shanghai, Volksrepublik China).
Immunofluoreszenz-Mikroskopie
Die Zellen (1×105) wurden in eine 24-Well-Platte beimpft und über Nacht inkubiert. Vor dem Experiment wurden die Zellen mit einem Adenovirus infiziert, das Tandem-GFP-RFP-LC3-Strukturen enthält. 24 Stunden nach der Infektion wurden die Medien gewechselt und die Zellen 24 Stunden lang mit 25 mM DCA oder Hank’s balanced salt solution (HBSS; Sigma Aldrich) behandelt. Für die Immunfärbung wurden die Zellen in 4 % Paraformaldehyd fixiert und mit PBS gewaschen. Die Zellen wurden 5 Minuten lang mit DAPI (4′,6-Diamidino-2-Phenylindol; Beyotime Biotechnology, Shanghai, Volksrepublik China) inkubiert und dreimal mit PBS gewaschen. Die Deckgläser wurden auf Objektträger mit Einbettungsmedium montiert. Die Bilder wurden mit einem konfokalen Mikroskop LSM 780 Meta (Carl Zeiss MicroImaging GmbH, Jena, Deutschland) aufgenommen und mit der vom Hersteller bereitgestellten Software bearbeitet.
Apoptose-Assay
Zellen (1×105) wurden in eine 24-Well-Platte geimpft und über Nacht inkubiert. Vor den Experimenten wurden die Zellen 24 Stunden lang mit verschiedenen Konzentrationen von 25 mM DCA, Chemotherapeutika plus DCA oder anderen Reagenzien behandelt. Die Zellen wurden mit APC-Annexin V und Propidiumiodid angefärbt, um die Apoptoserate mittels Durchflusszytometrie zu messen. Jeder Versuch wurde dreimal wiederholt.
Western Blot
Die Zellen wurden auf Eis lysiert, indem sie 15 Minuten lang mit RIPA-Lysepuffer (Sangon Biotech) mit Halt™ Protease- und Phosphatase-Inhibitor-Cocktail (Thermo Fisher Scientific, Waltham, MA, USA) behandelt wurden. Die Überstände wurden nach der Zentrifugation aufgefangen. Die Proteinkonzentrationen wurden mit dem Bicinchoninsäure (BCA) Protein Assay Kit (Beyotime Biotechnology) gemessen. Die Proteinproben wurden auf 10%igen Natriumdodecylsulfat-Polyacrylamid-Gelen elektrophoretisiert und auf eine Immobilon PVDF-Membran übertragen. Die Membranen wurden über Nacht bei 4 °C mit dem angegebenen primären Antikörper getestet und anschließend mit einem Meerrettichperoxidase-konjugierten sekundären Antikörper inkubiert. Die Banden wurden mit Chemilumineszenz-Detektionspuffer (Takara, Shiga, Japan) sichtbar gemacht, und die Bandenintensität wurde mit der vom Hersteller bereitgestellten Software quantifiziert.
Test der Zelllebensfähigkeit
Die Zellen wurden in 96-Well-Platten kultiviert (1.000 Zellen in 100 µL Kulturmedium/Well). Nach der medikamentösen Behandlung wurden 10 µL MTT (0,5 mg/mL) zugegeben und 4 Stunden lang inkubiert. Anschließend wurde das Medium verworfen, und die Formazankristalle wurden durch Zugabe von DMSO (Dimethylsulfoxid; Sigma-Aldrich) aufgelöst. Die Absorption bei 570 nm wurde gemessen. Die Lebensfähigkeiten der Zellen wurden auf die der Kontrollgruppe normiert.
In-vivo-Tumor-Xenograft-Studien
Für diesen Test wurden 5×106 Zellen subkutan in Nacktmäuse (BALB/c, spezifischer pathogenfreier Grad, 4-5 Wochen alt, erworben vom Model Animal Research Center der Universität Nanjing, Nanjing, Volksrepublik China) injiziert. Wenn der Tumor eine Größe von 100 mm3 erreicht hatte (15-20 Tage), wurden die Mäuse nach dem Zufallsprinzip in 4 Gruppen aufgeteilt (PBS, PTX, DCA und PTX plus DCA). Die PTX-Dosis betrug 20 mg/kg/d, die DCA-Dosis 100 mg/kg/d. Länge und Breite der Tumore wurden alle zwei Tage an ihren beiden senkrechten Durchmessern gemessen (berechnetes Volumen = kürzester Durchmesser2 × längster Durchmesser/2). Zur Berechnung der Überlebensrate wurden die Anzahl und das Datum der Todesfälle der Mäuse erfasst. Alle Manipulationen an lebenden Mäusen wurden vom Tierschutz- und Tiernutzungsausschuss des Xinqiao-Krankenhauses genehmigt und entsprachen den chinesischen Richtlinien für das Wohlergehen und die Ethik von Labortieren, und es wurden alle Anstrengungen unternommen, um das Leiden zu minimieren.
Histologische Untersuchung
Alle Tumore wurden nach 24 Tagen für die histopathologische Untersuchung reseziert. Schnitte (4- bis 5-μm dick) des Tumorgewebes wurden mit 10%igem Formalin fixiert und dann mit Ki-67, LC3B-Antikörper und TUNEL-Assay nachgewiesen. Die Schnitte wurden dreimal gewaschen und zur Farbentwicklung mit Diaminobenzidin behandelt. Die histologischen Untersuchungen wurden mit einem NanoZoomer 2.0-RS Lichtmikroskop (Hamamatsu Photonics, Hamamatsu City, Japan) durchgeführt und fotografiert.
Statistische Auswertung
Die Daten sind als Mittelwert ± Standardabweichung angegeben. Zur Analyse der Varianz wurden die Einweg-Varianzanalyse und der t-Test für unabhängige Stichproben verwendet. Die Überlebenskurven wurden mit der Kaplan-Meier-Methode ermittelt, und Vergleiche wurden mit dem Log-Rank-Test durchgeführt. Alle statistischen Analysen wurden mit der Statistiksoftware SPSS 19.0 (SPSS; IBM Corporation, Armonk, NY, USA) durchgeführt. P-Werte<0,05 oder <0,01 wurden als statistisch signifikant angesehen.
Ergebnisse
DCA hemmte die Autophagie in Adenokarzinomzellen der Lunge
Das zytosolische LC3 und SQSTM (p62) sind hochkonservierte Proteine, denen eine wichtige Rolle in den Schlüsselphasen der Autophagie zugeschrieben wird. Um die Rolle von DCA bei der Regulierung der Autophagie zu untersuchen, haben wir die zelluläre Lokalisierung von LC3 mit einem konfokalen Laser-Scanning-Mikroskop untersucht. A549- und H1975-Zellen wurden mit einem Adenovirus infiziert, das für ein Tandem-GFP-RFP-LC3-Konstrukt kodiert, um die Veränderung des Autophagie-Flusses nachzuweisen. Ein Unterschied in der pH-Empfindlichkeit kann den unterschiedlichen Grad der GFP-LC3- und RFP-LC3-Proteinakkumulation in neutralen Autophagosomen und sauren Autolysosomen verursachen. Nach der Behandlung mit HBSS war die Anzahl der GFP- oder RFP-LC3-Punkte signifikant erhöht, was zeigt, dass HBSS-induzierte Hungersnot den Autophagiefluss in A549- und H1975-Zellen signifikant fördert. Die DCA-Behandlung hemmte jedoch erfolgreich die Expression von LC3-Proteinen im Vergleich zur Kontroll- oder HBSS-Behandlung sowohl in A549-Zellen als auch in H1975-Zellen (Abbildung 1A und D, p<0,01).
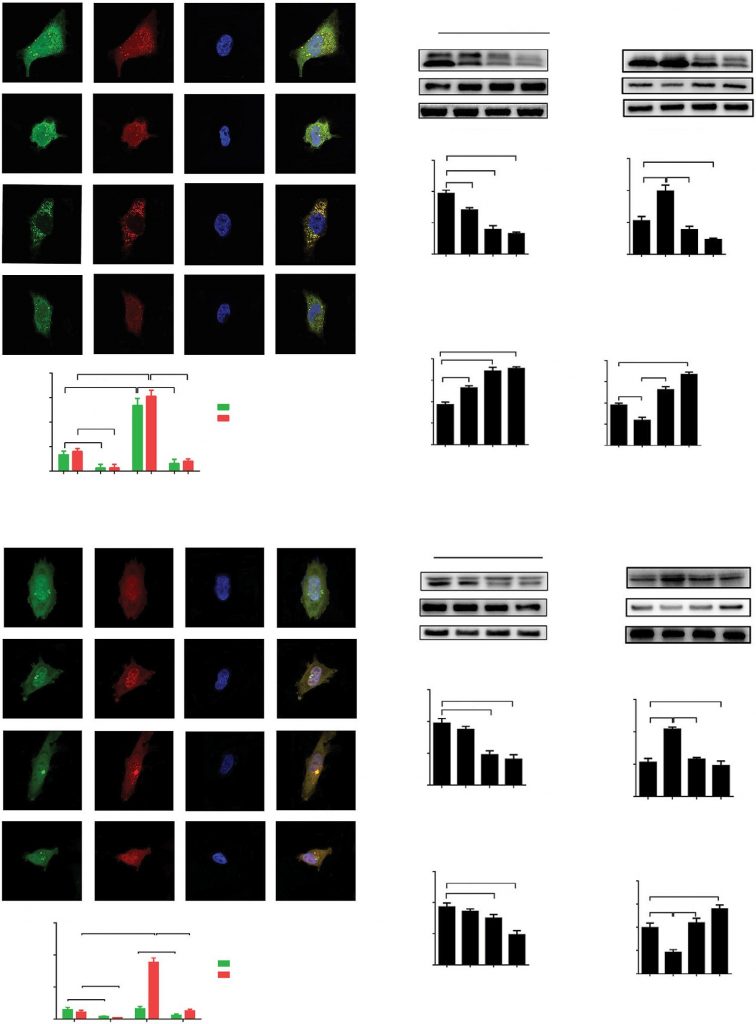
Abkürzungen: DAPI, 4′,6-Diamidino-2-Phenylindol; DCA, Dichloracetat; HBSS, Hank’s balanced salt solution; ns, nicht signifikant.
das p62-Protein sammelt sich an, wenn die Autophagie gehemmt wird, und nimmt ab, wenn die Autophagie induziert wird. Zur Untersuchung der Proteinkonzentrationen von p62 und LC3-II nach DCA-Behandlung wurde eine Western-Blotting-Analyse durchgeführt. Nach der DCA-Behandlung wurde sowohl in A549- als auch in H1975-Zellen ein signifikanter Rückgang der LC3-Konzentration und ein Anstieg von p62 in Abhängigkeit von der Konzentration beobachtet (Abbildung 1B und E, p<0,01). Die Autophagie wurde während der Inkubation mit HBSS verstärkt, was sich bei der Co-Behandlung mit DCA umkehrte (Abbildung 1C und F, p<0,01). Insgesamt deuten diese Ergebnisse darauf hin, dass der autophagische Fluss in A549- und H1975-Zellen durch die DCA-Behandlung deutlich gehemmt wurde.
Die Wirkung der Förderung der Autophagie auf den durch DCA induzierten Zelltod
Es wurde eine systematische Untersuchung der krebshemmenden Wirkung von DCA durchgeführt. Zunächst wurde die krebshemmende Wirkung von DCA in vitro untersucht. Beide Zellen wurden 24 Stunden lang in Gegenwart verschiedener DCA-Konzentrationen inkubiert, und die Apoptose und Lebensfähigkeit der Zellen wurden bewertet. Wir stellten fest, dass die krebshemmende Wirkung von DCA, die sich in der Hemmung der Zelllebensfähigkeit und der Apoptoserate widerspiegelt, bei A549-Zellen und H1975-Zellen konzentrationsabhängig anstieg (Abbildung 2A und B, E und F, p<0,01).
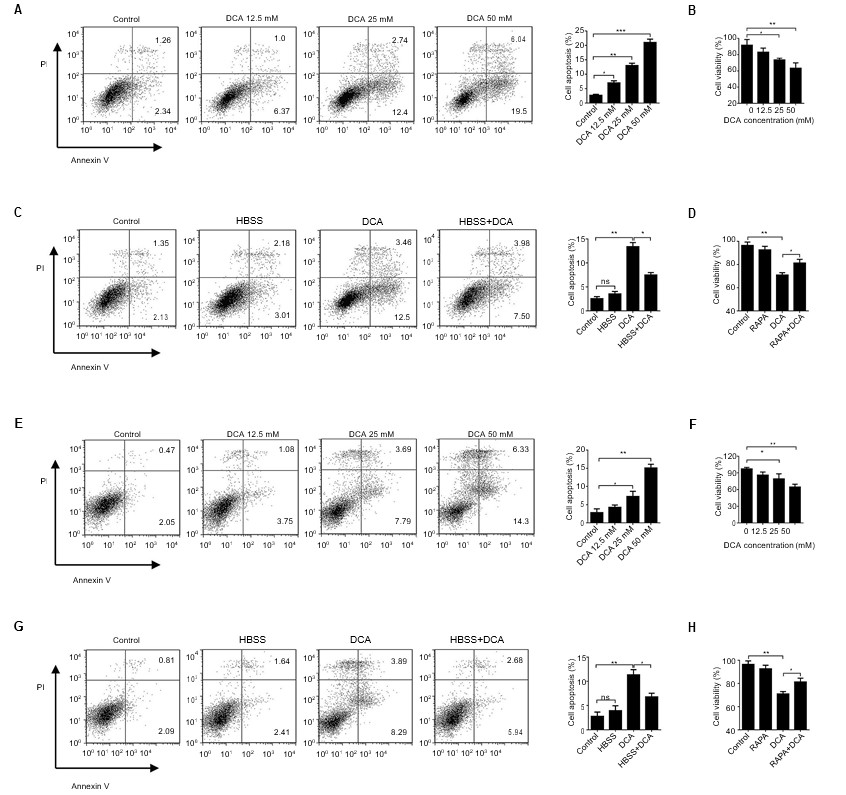
Abkürzungen: DCA, Dichloracetat; FITC/PI, Fluorescein-Isothiocyanat/Propidiumjodid; HBSS, Hank’s balanced salt solution.
DCA aktivierte den metabolischen Schalter, der den anormalen Stoffwechsel in Krebszellen von anaerober Glykolyse zu Glukoseoxidation umkehrte, was zu mitochondrialer Dysfunktion und Zelltod führte. Unsere vorläufigen Experimente zeigten, dass die Autophagie durch die DCA-Behandlung deutlich gehemmt wurde. Um festzustellen, ob die DCA-vermittelte Autophagiehemmung an der Apoptose beteiligt war, wurden A549- und H1975-Zellen 24 Stunden lang mit DCA nach HBSS behandelt. Die durch DCA induzierte Zellapoptose wurde gehemmt, wenn die Zellen mit HBSS koaguliert wurden, verglichen mit der DCA-Behandlung allein. Abgesehen von der Veränderung des anormalen Stoffwechsels bestätigt dieses Ergebnis, dass DCA auch den Zelltod durch Hemmung der Autophagie in Krebszellen aktivieren kann. Die Ergebnisse der Zellviabilität stimmen mit dieser Schlussfolgerung überein (Abbildung 2C und D, G und H, p<0,05).
Die Hemmung des AKT-Signalwegs verringerte die DCA-induzierte Apoptose
In früheren Studien wurde dem ATK-mTOR-Signalweg eine entscheidende Rolle bei der Regulierung der Autophagie zugeschrieben.[18-21] Perifosin,[22] ein chemisches Reagenz, das die AKT-Phosphorylierung und -Aktivierung verhindert, wurde verwendet, um zu untersuchen, ob der ATK-mTOR-Stoffwechselweg an dem Autophagie-hemmenden Prozess von DCA beteiligt ist. Die Zellen wurden 24 Stunden lang mit DCA und Perifosin kotreatiert. Die Zellapoptose war in der Koexpositionsgruppe im Vergleich zur DCA-Behandlungsgruppe fast um die Hälfte reduziert, was mit den Zelllebensfähigkeitstests übereinstimmt (Abbildung 3A und B, D und E, p<0,05). Anschließend untersuchten wir die Konzentrationen der entscheidenden apoptosebezogenen Proteine PARP und Procaspase3. Die AKT-Phosphorylierung wurde gehemmt, und die Spiegel der mit der Apoptose verbundenen Proteine waren in der Mitbehandlungsgruppe deutlich verringert (Abbildung 3C, p<0,05). Diese Ergebnisse wurden auch bei den H1975-Zellen erzielt (Abbildung 3F, p<0,05).
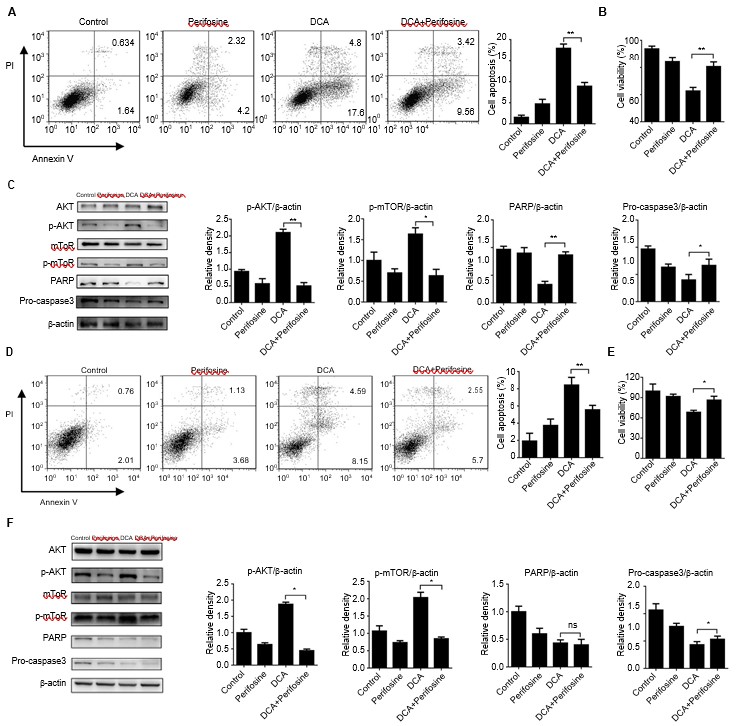
Abkürzungen: DCA, Dichloracetat.
DCA erhöht die PTX- und CDDP-Empfindlichkeit durch Umkehrung der Chemotherapie-induzierten Autophagie
Die Hemmung der Autophagie gilt als neue Strategie zur Verbesserung der Empfindlichkeit von Chemotherapeutika. [23,24] Um zu untersuchen, ob die gleichzeitige Behandlung mit DCA und einem Chemotherapeutikum einen synergistischen Effekt zur Unterdrückung des Zellwachstums hat, wurden A549-Zellen mit DCA und PTX oder CDDP behandelt. Wie in Abbildung 4A-C dargestellt, erhöhte die Behandlung mit Chemotherapeutika und DCA die Apoptosewerte im Vergleich zur alleinigen Behandlung mit dem Medikament oder in Kombination mit CQ, und die Ergebnisse stimmten mit den Zelllebensfähigkeitstests überein(p<0,05). Um festzustellen, ob DCA die durch PTX induzierte Autophagie herunterregulieren kann, wurden die Zellen 24 Stunden lang mit PTX und DCA behandelt und für Western Blotting geerntet. Die Spiegel der mit der Autophagie verbundenen Proteine wurden untersucht. Die Expression von LC3 wurde durch die PTX-Behandlung deutlich erhöht und durch die gleichzeitige Behandlung mit DCA deutlich verringert. Der Spiegel des kritischen, mit der Apoptose verbundenen Proteins PARP war nach der Co-Behandlung deutlich verringert (Abbildung 4D-F, p<0,05). All diese Ergebnisse deuten darauf hin, dass DCA die Arzneimittelresistenz von A549-Zellen gegenüber PTX durch Hemmung der Autophagie verringert.
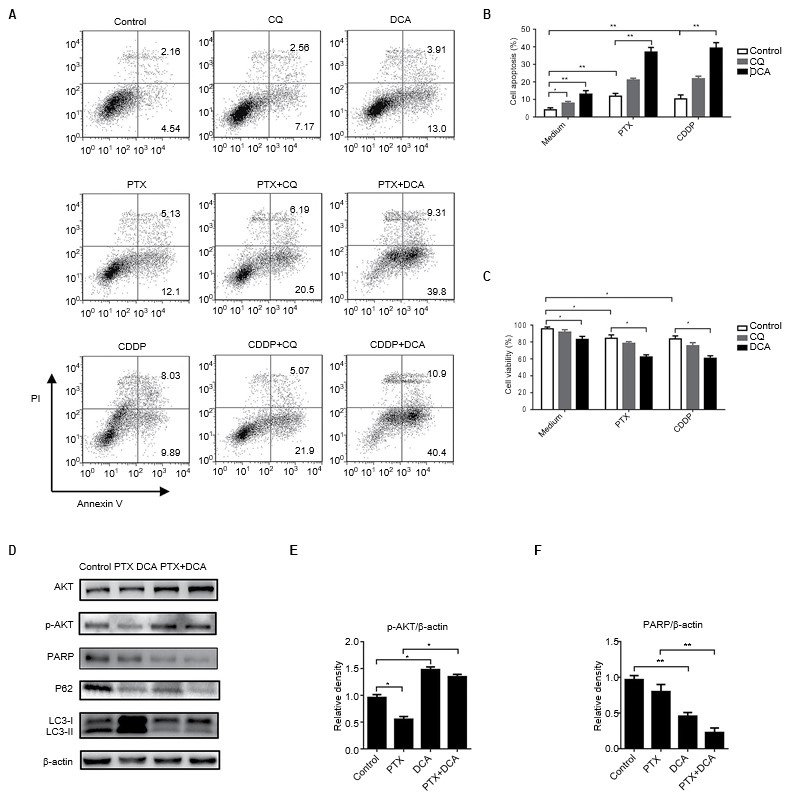
Abkürzungen: CDDP, cis-Diamminedichloroplatin; CQ, Chloroquin; DCA, Dichloracetat; PTX, Paclitaxel.
DCA verstärkte die krebsbekämpfende Wirkung von PTX in vivo durch Hemmung der Autophagie
Da die gemeinsame Behandlung von Zellen mit PTX und DCA die Zelltodrate in vitro im Vergleich zur Behandlung mit einem der beiden Wirkstoffe allein erhöhte, untersuchten wir auch die In-vivo-Wirksamkeit der Kombinationstherapie von DCA und PTX in einem Maus-Xenograft-Tumormodell. Sobald die Größe des subkutanen Xenograft-Tumors 150 mm3 erreicht hatte, wurden die Mäuse in vier Gruppen aufgeteilt: Kontrolle, DCA, PTX und DCA plus PTX. Die Verabreichung von PTX allein hemmte das Tumorwachstum nicht signifikant, aber die Behandlung mit PTX plus DCA hemmte das Tumorwachstum deutlich und verlängerte das Überleben der Mäuse (Abbildung 5A und B, p<0,05). Wie in Abbildung 5C gezeigt, erhöhte sich die Tumorverdopplungszeit der A549-Zellen signifikant von 3 Tagen in der Kontrollgruppe auf 10 Tage in der Mitbehandlungsgruppe (p<0,05), und die Tumorverdopplungszeit der H1975-Zellen stieg von 2,6 auf 7,5 Tage (Abbildung 5D-F, p<0,05). Diese Ergebnisse deuten darauf hin, dass DCA die krebshemmende Wirkung von PTX in vivo verstärken kann.
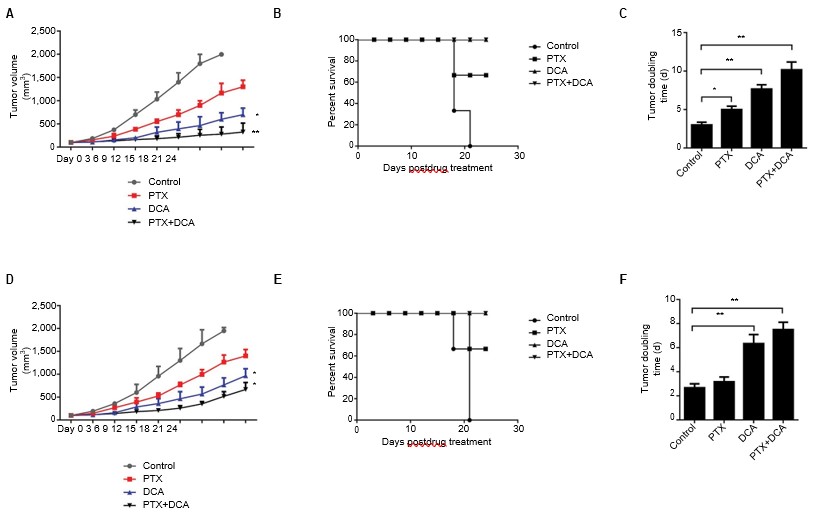
Abkürzungen: DCA, Dichloracetat; PTX, Paclitaxel.
DieBehandlung mit DCA plus PTX hemmte signifikant die Tumorkeimung
Um zu untersuchen, ob die Kombinationstherapie von DCA und PTX zu den für die Apoptose charakteristischen morphologischen Veränderungen in den A549-Zellen in vivo führte, wurden alle Tumore an Tag 24 entnommen, geschnitten, mit TUNEL angefärbt und durch Immunhistochemie analysiert. Die Anzahl der TUNEL-positiven Zellen war in den DCA- oder PTX-Behandlungsgruppen im Vergleich zur Kontrollgruppe minimal erhöht. Der Anteil der TUNEL-positiven Zellen in der Cotreatment-Gruppe stieg jedoch im Vergleich zur Kontroll- oder Einzelbehandlungsgruppe dramatisch an (Abbildung 6A).
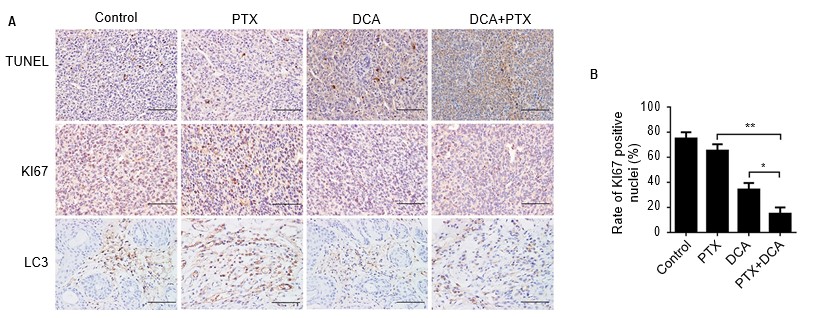
Abkürzungen: DCA, Dichloracetat; PTX, Paclitaxel.
Anschließend untersuchten wir die Expression des proliferationsbezogenen Ki-67 und des entscheidenden Autophagie-Proteins LC3B in den Tumoren durch immunhistochemische Färbung. Die Expression von Ki-67 war in den Kontrollmäusen deutlich höher, während sie in den Tumoren der Tiere, die die Kombinationsbehandlung erhielten, stark reduziert war, und die Expression von LC3B war die gleiche wie die von Ki-67. Zusammenfassend lässt sich sagen, dass die Behandlung mit DCA plus PTX die Apoptose signifikant erhöhte und die Ki-67- und LC3B-Expression in vivo reduzierte (Abbildung 6A und B, p<0,01).
Diskussion
Mit der Entwicklung der Krebsforschung wurde eine zunehmende Anzahl von Behandlungsstrategien bei der Behandlung von Lungenkrebs eingesetzt.(25-27) Obwohl viele Lungenkrebspatienten, insbesondere im Spätstadium oder im fortgeschrittenen Stadium, von EGFR-TKI und PD-1-Antikörpern profitieren, sind die Kosten für die meisten Patienten zu hoch. Die konventionelle Chemotherapie ist in China nach wie vor der wichtigste Ansatz zur Behandlung von fortgeschrittenem Lungenkrebs. Die Multiresistenz ist jedoch ein schwieriges Hindernis bei der Krebsbehandlung. Daher konzentriert sich die aktuelle Forschung auf die Suche nach neuen Möglichkeiten, die Empfindlichkeit der Chemotherapie zu verbessern oder die Arzneimittelresistenz zu verzögern. In jüngster Zeit wurde die Regulierung der Autophagie als ein Ziel für die Krebstherapie angesehen; es gibt viele Hinweise darauf, dass die Hemmung der Autophagie die tumorunterdrückende Wirkung von Chemotherapeutika und Strahlentherapie fördern kann. [23,28-30] Normalerweise erzeugen die meisten soliden Karzinome Energie über den glykolytischen Weg, unabhängig davon, ob ausreichend Sauerstoff vorhanden ist; dies wird als „Warburg-Effekt“ bezeichnet. [29-31] DCA, ein niedermolekulares Antitumormittel, kann diesen Stoffwechselmodus der Mitochondrien von anaerober Atmung auf aerobe Atmung umkehren und so den Tod der Tumorzellen herbeiführen. Ob DCA jedoch in Kombination mit anderen Chemotherapeutika eingesetzt werden kann, ist unbekannt. In dieser Studie fanden wir heraus, dass die gemeinsame Behandlung mit DCA und PTX das Wachstum von Lungenkrebszellen in vitro und in vivo wirksamer unterdrücken kann. DCA hemmte die schützende Autophagie, die von den Krebszellen als Reaktion auf die PTX-Behandlung eingesetzt wird, indem es die AKT-Phosphorylierung aktiviert und den Zelltod fördert. Die synergistische Wirkung dieser beiden Wirkstoffe verstärkt den Zelltod und hemmt dadurch das Tumorwachstum effektiver.
Wie wir wissen, ist der Zellstoffwechsel ein wirksames Ziel für die Krebsbehandlung[32 ], und immer mehr Beweise haben die antitumoralen Eigenschaften von DCA im Zellstoffwechsel gezeigt. Die Wirkungen von DCA sind jedoch umstritten, da in verschiedenen Krebsmodellen unterschiedliche Ergebnisse berichtet wurden. [33,34] In der vorliegenden Studie haben wir festgestellt, dass die Kombination von DCA und PTX die Zellproliferation wirksamer hemmt und die Apoptose fördert als PTX allein. In dem in dieser Studie verwendeten subkutanen Tumor-Xenograft-Modell reduzierte die Behandlung mit DCA plus PTX das Wachstum der in die Mäuse injizierten A549-Zellen um 60 %. Unsere Daten zeigen, dass DCA eine starke krebshemmende Wirkung hat und synergistisch mit PTX auf A549-Zellen wirkt. Eine synergistisch wirkende Mehrfachtherapie kann die therapeutische Wirksamkeit verbessern, indem sie die Medikamententoxizität und die Medikamentenresistenz verringert. [35] In unseren Experimenten kehrte DCA die Multidrug-Resistenz von A549-Zellen um, indem es die durch PTX induzierte Autophagie hemmte und Apoptose auslöste. Daher könnte die Zugabe von DCA zum Chemotherapieschema die Anfälligkeit für Mehrfachresistenzen bei NSCLC verringern.
Die Autophagie ist ein hochkonservierter Stoffwechselweg, bei dem Zellen alternde oder defekte Proteine, Organellen und andere Zellbestandteile zum Abbau an Lysosomen abgeben, um die zelluläre Homöostase zu erhalten. [36,37] Es gibt zahlreiche Belege dafür, dass die meisten Chemotherapeutika sowie ionisierende Strahlung die Autophagie induzieren, was zu einer Multidrug-Resistenz führt. Eine angepasste Autophagie erleichtert das Überleben von Tumorzellen in einer Mikroumgebung mit erhöhtem Stoffwechselstress oder Medikamententoxizität. Die durch Stress ausgelöste Autophagie ist eine wichtige Schutzstrategie, die das Überleben der Zellen aufrechterhalten kann, was letztlich zu einer Arzneimittelresistenz bei verschiedenen Arten von Tumorzellen führt. [38] In dieser Studie hemmte die DCA-Behandlung die zelluläre Autophagie und förderte die Apoptose. RARA, ein Autophagie-Aktivator, kann die Zellen resistenter gegen DCA machen, was darauf hindeutet, dass DCA den Zelltod verursacht, indem es den Stoffwechsel der Krebszellen stört und die Autophagie hemmt. Der PI3K-AKT-mTOR-Signalweg ist ein wichtiger negativer Regulator der Autophagie. Daher kann die Beeinflussung des PI3K-AKT-mTOR-Signalwegs eine neue Strategie für die Krebstherapie darstellen. DCA fördert selektiv die Phosphorylierung der AKT-Kinase und hemmt dadurch die Autophagie. Somit hemmt die Aktivierung des PI3K-AKT-mTOR-Signalwegs durch DCA die PTX-induzierte Autophagie. Die Behandlung mit EGFR-TKIs hängt Berichten zufolge mit dem Autophagie-Niveau von Lungenkrebszellen zusammen, und eine Hemmung der Autophagie könnte die Antitumorwirkung von EGFR-TKIs unterstützen. Dies könnte ein neuer Ansatzpunkt für die Behandlung von Lungenkrebs sein.
Einige Forscher argumentieren jedoch, dass Autophagie ein zweischneidiges Schwert“ für Krebszellen ist und das Überleben und die Proliferation in der Mikroumgebung des Tumors unterdrücken kann.(26,39,40) Darüber hinaus wurde die Anregung der Autophagie bei Krebs auch als Strategie zur Sensibilisierung für die Chemotherapie angesehen. Die unterschiedlichen Funktionen der Autophagie in Krebszellen könnten durch den unterschiedlichen Status der Krebszellen oder Tumortypen verursacht werden; daher wird das Verständnis der zugrundeliegenden Funktionen der Autophagie-Regulierung zwischen verschiedenen Krebszellen ein neues Licht auf die Bestimmung von Strategien gegen Chemotherapieresistenz werfen.[24]
Interessanterweise scheint DCA bei der Autophagie-Regulierung in verschiedenen Tumorzellen doppelte Eigenschaften zu haben. Lin et al.[33] haben berichtet, dass DCA die Apoptose in Darmkrebszellen auslösen und gleichzeitig die Autophagie fördern kann. Daraus folgt, dass ein komplexerer molekularer Mechanismus an der Regulierung der Autophagie durch DCA beteiligt ist. Die Erforschung des Prinzips der unterschiedlichen Auswirkungen von DCA auf die Autophagie würde uns neue Ideen für die Entwicklung neuer adjuvanter Chemotherapeutika durch die Hemmung der Autophagie liefern.
Schlussfolgerung
Unsere Daten zeigen, dass die Autophagie während der Chemotherapie aktiviert werden kann, und dass die Hemmung der Autophagie durch DCA den Zelltod fördern und die Empfindlichkeit der Tumorzellen gegenüber Chemotherapeutika erhöhen kann. Darüber hinaus waren Autophagie und Krebsstoffwechsel an einer Reihe von Tumorereignissen beteiligt, z. B. an der Tumorentwicklung, dem Tumorwachstum und der Chemotherapie. Die Aufklärung des Zusammenhangs zwischen Autophagie und Stoffwechsel[32] in verschiedenen Krebszellen könnte uns neue Möglichkeiten für die Entwicklung von Medikamenten eröffnen, die auf diese wichtigen zellulären Vorgänge abzielen, um Krebs und Arzneimittelresistenz zu bekämpfen.
Danksagung
Die Studie wurde durch Mittel der National Natural Science Foundation of China (81702247) und des Basic Science and Frontier Technology Project of Chongqing für Zheng (cstc2017jcyjAX0048), der National Natural Science Foundation of China für Dai (81472188) und des Clinical Research Project of Xinqiao Hospital für Yu (2015YLC21) unterstützt.
Offenlegung
Die Autoren melden keine Interessenkonflikte im Zusammenhang mit dieser Arbeit.
REFERENZEN
1 Maher AR, Miake-Lye IM, Beroes JM, Shekelle PG. Behandlung des metastasierten nicht-kleinzelligen Lungenkrebses: A Systematic Review of Comparative Effectiveness and Cost-Effectiveness. Washington (DC): Department of Veterans Affairs (US); 2012.2 Spiro SG, Porter JC. Lungenkrebs – wo stehen wir heute? Aktuelle Fortschritte bei der Stadieneinteilung und nicht-chirurgischen Behandlung. Am J Respir Crit Care Med. 2002;166:1166-1196.
3 Song W, Tang Z, Li M, et al. Polypeptide-based combination of paclitaxel and cisplatin for enhanced chemotherapy efficacy and reduced side-effects. Acta Biomater. 2014;10:1392-1402.
4 Ward PS, Thompson CB. Metabolische Reprogrammierung: ein Krebsmerkmal, das selbst Warburg nicht vorausgesehen hat. Cancer Cell. 2012;21:297-308.
5 Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol. 2014;9:1178-1187.
6KimuraT, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3-7.
7 Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42.
8 Gomes LR, Vessoni AT, Menck CF. Mikroumgebung und Autophagie – ein Wechselspiel: Implications in cancer therapy. Pharmacol Res. 2016;107:300-307.
9 Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
10 Koren I, Kimchi A. Zellbiologie. Förderung der Tumorentstehung durch Unterdrückung der Autophagie. Science. 2012;338:889-890.
11 Rosenfeldt MT, Ryan KM. Die Rolle der Autophagie bei der Tumorentwicklung und Krebstherapie. Expert Rev Mol Med. 2009;11:e36.
12 Rubinsztein DC, Codogno P, Levine B. Autophagie-Modulation als potenzielles therapeutisches Ziel für verschiedene Krankheiten. Nat Rev Drug Discov. 2012;11:709-730.
13EggerME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition der Autophagie mit Chloroquin ist wirksam in Melanom. J Surg Res. 2013;184:274-281.
14 Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391-1402.
15 Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refraktär myeloma. Autophagy. 2014;10:1380-1390.
16 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle. 2007;11:37-51.
17 Christofk HR, Vander HM, Harris MH, et al. Die M2-Spleißisoform der Pyruvatkinase ist wichtig für den Krebsstoffwechsel und das Tumorwachstum. Nature. 2008;452:230-233.
18 Tan J, Jiang X, Yin G, et al. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38(3):1373-1382.
19 Chen LM, Song TJ, Xiao JH, Huang ZH, Li Y, Lin TY. Tripchlorolid induziert Autophagie in Lungenkrebszellen durch Hemmung des PI3K/AKT/mTOR-Wegs und verbessert die Cisplatin-Empfindlichkeit in A549/DDP-Zellen. Oncotarget. 2017;8(38):63911–63922.
20 Wang Z, Wang N, Liu P, Xie X. AMPK and cancer. EXS. 2016;107:203-226.
21 Umezawa S, Higurashi T, Nakajima A. AMPK: therapeutic target for diabetes and cancer prevention. Curr Pharm Des. 2017;23(25):3629–3644.
22 Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102-110.
23 Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528-539.
24 Rubinsztein DC, Codogno P, Levine B. Autophagie-Modulation als potenzielles therapeutisches Ziel für verschiedene Krankheiten. Nat Rev Drug Discov. 2012;11:709-730.
25 Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47.
26 Dholaria B, Hammond W, Shreders A, Lou Y. Neu aufkommende therapeutische Wirkstoffe für Lungenkrebs. J Hematol Oncol. 2016;9:138.
27 Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34.
28 White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212-217.
29 Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961-967.
30 Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485-1494.
31 Zhou W, Liotta LA, Petricoin EF. Der Warburg-Effekt und die auf Massenspektrometrie basierende Proteomanalyse. Cancer Genomics Proteomics. 2017;14:211-218.
32 Ho WL, Hsu WM, Huang MC, Kadomatsu K, Nakagawara A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J Hematol Oncol. 2016;9:100.
33 Lin G, Hill DK, Andrejeva G, et al. Dichloracetat induziert Autophagie in kolorektalen Krebszellen und Tumoren. Br J Cancer. 2014;111:375-385.
34 Gong F, Peng X, Sang Y, et al. Dichloracetat induziert schützende Autophagie in LoVo-Zellen: Beteiligung von Cathepsin D/Thioredoxin-ähnlichem Protein 1 und Akt-mTOR-vermittelter Signalübertragung. Cell Death Dis. 2013;4:e913.
35 Chou TC. Theoretische Grundlagen, Versuchsplanung und computergestützte Simulation von Synergismus und Antagonismus in Studien zu Arzneimittelkombinationen. Pharmacol Rev. 2006;58:621-681.
36 Eskelinen EL. Die doppelte Rolle der Autophagie bei Krebs. Curr Opin Pharmacol. 2011;11:294-300.
37 Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383-1435.
38 Naveed S, Aslam M, Ahmad A. Starvation based differential chemotherapy: a novel approach for cancer treatment. Oman Med J. 2014;29:391-398.
39 Yoshida GJ. Therapeutische Strategien, die auf die Autophagie abzielen, um den Tod von Krebszellen herbeizuführen: von der Pathophysiologie zur Behandlung. J Hematol Oncol. 2017;10:67.
40 Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651-662.
Verwandte Inhalte: