Сяо Лу,1,* Дун Чжоу,1,* Бинг Хоу,1 Куань-Син Лю,1 Цянь Чэнь,2 Сюй-Фэн Дэн,1,3 Зу-Бин Юй,1 Джи-Ган Дай,1 Хун Чжэн1
1 Отделение торакальной хирургии, госпиталь Синьцяо, Третий военный медицинский университет, Чунцин, Китайская Народная Республика
2 Институт патологии и Юго-западный онкологический центр, Юго-западный госпиталь, Третий военный медицинский университет, Чунцин, Китайская Народная Республика
Отделение кардиоторакальной хирургии, Первая народная больница Цзуньи, Гуйчжоу, Китайская Народная Республика
авторы внесли равный вклад в эту работу
Переписка: Хун Чжэн; Цзи-Ган Дай
Отделение торакальной хирургии, госпиталь Синьцяо, Третий военно-медицинский университет, № 183, улица Синьцяо, район Шапингба, Чунцин 400037, Китайская Народная Республика
Тел: +86 23 6877 4724
Факс: +86 23 6877 4724
Email: [email protected]; [email protected]
Получено: 14 сентября 2020 г.
Принято: 4 декабря 2020 г.
Опубликовано: 9 декабря 2020 г
Аннотация
Предпосылки: Химиотерапия по-прежнему является основной адъювантной стратегией лечения рака, однако возникновение множественной лекарственной устойчивости вызывает беспокойство. Было продемонстрировано, что аутофагия играет защитную роль против химиотерапевтических препаратов в раковых клетках, и ингибирование аутофагии обычно рассматривается как перспективная терапевтическая стратегия. Однако недостаток эффективных и специфических ингибиторов аутофагии ограничивает ее применение.
Цель: Целью данного исследования было изучить влияние DCA, небольшого молекулярного противоопухолевого препарата, на регуляцию аутофагии и химиосенсибилизацию в клетках NSCLC.
Методы: Мы исследовали регуляцию аутофагии дихлорацетатом (DCA) с помощью лазерной конфокальной микроскопии и вестерн-блоттинга в клеточных линиях A549 и H1975. Для изучения эффективности хемосенсибилизации ДХА был проведен МТТ-анализ и проточная цитометрия. Результаты были проверены на модели подкожной опухоли у мышей nude, а для оценки уровня апоптоза и аутофагии клеток in vivo после лечения применялась иммуногистохимия.
Результаты: Мы обнаружили, что DCA, проявляющий противоопухолевые свойства в различных моделях карциномы, индуцировал апоптоз клеток немелкоклеточного рака легких (NSCLC) путем ингибирования аутофагии раковых клеток. Кроме того, Перифосин, ингибитор AKT, может значительно ослабить способность DCA индуцировать апоптоз. Результаты показывают, что в ингибировании аутофагии, вызванном DCA, участвует AKT-mTOR-путь, основной негативный регулятор аутофагии. Затем мы определили эффективность ингибирования аутофагии с помощью DCA. При совместном применении с химиотерапевтическим препаратом паклитакселом (PTX) DCA заметно снижал аутофагию клеток, усиливал апоптоз и подавлял пролиферацию в клетках A549 и H1975. Результаты эксперимента по ксенотрансплантации показали, что совместное лечение PTX и DCA может значительно снизить пролиферацию клеток in vivo и продлить выживание мышей.
Заключение: Наши результаты показывают, что DCA может ингибировать аутофагию клеток, индуцированную химиотерапевтическими препаратами, открывая новый путь для сенсибилизации химиотерапии рака.
Ключевые слова: DCA, аутофагия, множественная лекарственная устойчивость, немелкоклеточный рак легкого, паклитаксел, ксенотрансплантация на мышах nude, химиосенсибилизация
ВВЕДЕНИЕ
Немелкоклеточный рак легкого (НМКРЛ) является одной из основных причин смертности от рака во всем мире. Это наиболее часто встречающийся рак у мужчин и женщин, частота которого превышает совокупную частоту рака молочной железы, рака шейки матки и колоректального рака.[1,2] Хотя химиотерапия по-прежнему является наиболее важным средством адъювантной терапии для неоперабельных онкологических больных и пациентов, перенесших операцию, клинические преимущества послеоперационной химиотерапии на основе платины и паклитаксела скромны, особенно при распространенном NSCLC. В то же время, побочные реакции на препараты становятся все более серьезными, а также появляется лекарственная устойчивость.[3] Поэтому необходимо срочно разработать новые стратегии для замены/дополнения традиционной химиотерапии.
В последние годы появляется все больше данных о том, что опухолевые клетки преимущественно производят энергию для роста и деления клеток посредством гликолитического процесса и молочнокислого брожения. Скорость анаэробного метаболизма и гликолиза в быстрорастущих клетках злокачественных опухолей значительно выше, чем в нормальных клетках. Такое перепрограммирование энергетического метаболизма известно как эффект Варбурга, и его можно использовать в качестве терапевтической мишени для подавления роста опухоли. Среди многочисленных препаратов, направленных на метаболизм, дихлорацетат (ДХА) показал отличный потенциал благодаря своему положительному вкладу в лечение рака[4,5]
Другим механизмом, который полностью изменяется в раковых клетках, является аутофагия — гомеостатическая система клеточной деградации, которая отвечает за деградацию поврежденных или ненужных клеточных органелл или белков.[6] В процессе аутофагии клеточный груз, предназначенный для деградации, заключен в аутофагосому — двухмембранную везикулу. Загруженная аутофагосома плавно сливается с лизосомой, образуя аутолизосому, где доставленный клеточный материал разрушается различными лизосомальными гидролитическими ферментами. Процесс аутофагии широко изучен. Становится все более очевидным, что изменение активности аутофагии связано с образованием и прогрессированием опухолей.[7-9] Поскольку аутофагия играет защитную роль в раковых клетках против химиотерапевтических препаратов, подавление аутофагии во время химиотерапии рассматривается как новая терапевтическая стратегия.[10-12] В настоящее время только хлорохин (CQ) используется в клинических условиях как эффективный ингибитор аутофагии. Хотя эффективность и целесообразность применения хлорохина в терапии рака были продемонстрированы, нежелательные побочные эффекты могут стать проблемой для клинического лечения. Открытие и использование дополнительных ингибиторов аутофагии в терапии рака имело бы большое клиническое значение.[13-15]
DCA — это препарат, направленный на митохондрии, который действует как метаболический переключатель, обращая аномальный метаболизм раковых клеток от анаэробного гликолиза к аэробному окислению глюкозы путем снижения активности митохондриального PDK1 и повышения жизнеспособности PDH. Таким образом, DCA увеличивает митохондриальные реактивные формы кислорода, тем самым вызывая апоптоз в злокачественных опухолевых клетках, не затрагивая нормальные клетки.[16,17] Однако регуляторное действие DCA на аутофагию при раке легкого до сих пор остается неясным. В данном исследовании мы продемонстрировали, что DCA подавляет пролиферацию клеток и усиливает апоптоз опухолевых клеток через снижение регуляции аутофагии, тем самым повышая эффективность гибели клеток при использовании в котерапии с химиотерапевтическими препаратами.
Материалы и методы
Культура клеток и реактивы
Клетки аденокарциномы легких человека A549 культивировали в среде Дульбекко (Dulbecco’s Modified Eagle Medium) с 10% фетальной бычьей сыворотки, а клетки H1975 — в среде Roswell Park Memorial Institute-1640 с 10% фетальной бычьей сыворотки в увлажненном инкубаторе с 95% воздуха и 5%CO2 при 37°C. Клеточные линии A549 (TCHu150) и H1975 (TCHu193) были приобретены в Библиотеке типовых культур Китайской академии наук (Шанхай, Китайская Народная Республика). После того, как клетки вырастали вдоль стенки культуральной посуды, 0,25% трипсин (HyClone, Бакингемшир, Великобритания) использовался для отделения и субкультивирования. Реагенты, использованные в данном исследовании: DCA (Sigma-Aldrich, St. Louis, MO, USA), паклитаксел (PTX; Sigma-Aldrich), цисплатин (цис-диамминдихлороплатин [CDDP]; Sigma-Aldrich), аденовирус (GFP-RFP-LC3; Hanbio, Shanghai, People’s Republic of China), TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling; Promega, Fitchburg, WI, USA), APC-аннексин V, пропидий йодид (BD Pharmingen, Franklin Lakes, NJ, USA) и набор МТТ (Sangon Biotech, Shanghai, People’s Republic of China). В качестве антител использовались LC3-I/II, p62, PARP (все Abcam, Кембридж, Великобритания), β-актин (Sigma-Aldrich), mTOR/p-mTOR (Sigma-Aldrich) и Ki-67 (Biovisualab, Шанхай, Китайская Народная Республика).
Иммунофлуоресцентная микроскопия
Клетки (1×105) инокулировали в 24-луночный планшет и инкубировали в течение ночи. Перед началом эксперимента клетки инфицировали аденовирусом, содержащим тандемные конструкции GFP-RFP-LC3. Через 24 ч после инфицирования среду меняли, и клетки обрабатывали 25 мМ DCA или сбалансированным солевым раствором Хэнка (HBSS; Sigma Aldrich) в течение 24 ч. Для иммуноокрашивания клетки фиксировали в 4% параформальдегиде и промывали PBS. Клетки инкубировали с DAPI (4′,6-диамидино-2-фенилиндол; Beyotime Biotechnology, Шанхай, Китайская Народная Республика) в течение 5 мин и трижды промывали PBS. Покровные пластины монтировали на предметные стекла с монтажной средой. Изображения получали с помощью конфокального микроскопа LSM 780 Meta (Carl Zeiss MicroImaging GmbH, Йена, Германия) и обрабатывали с помощью программного обеспечения, предоставленного производителем.
Анализ апоптоза
Клетки (1×105) инокулировали в 24-луночный планшет и инкубировали в течение ночи. Перед экспериментами клетки обрабатывали различными концентрациями 25 мМ DCA, химиотерапевтическим агентом плюс DCA или другими реагентами в течение 24 ч. Клетки окрашивали APC-аннексином V и йодистым пропидием для измерения скорости апоптоза методом проточной цитометрии. Каждый эксперимент повторяли три раза.
Вестерн-блот
Клетки лизировали на льду путем обработки буфером для лизиса радиоиммунопреципитации (RIPA) (Sangon Biotech) с коктейлем ингибиторов протеаз и фосфатаз Halt™ (Thermo Fisher Scientific, Waltham, MA, USA) в течение 15 мин. Супернатанты собирали после центрифугирования. Концентрацию белка измеряли с помощью набора для анализа белка на бицинхониновую кислоту (BCA) (Beyotime Biotechnology). Белковые образцы электрофорезировали на 10% додецилсульфат натрия-полиакриламидных гелях и переносили на мембрану Immobilon PVDF. Мембраны зондировали в течение ночи при 4°C с указанным первичным антителом, а затем инкубировали с вторичным антителом, конъюгированным с пероксидазой хрена. Полосы визуализировали с помощью буфера для обнаружения хемилюминесценции (Takara, Shiga, Япония), а интенсивность полос оценивали количественно с помощью программного обеспечения, предоставленного производителем.
Анализ жизнеспособности клеток
Клетки культивировали в 96-луночных планшетах (1 000 клеток в 100 мкл культуральной среды/лунка). После обработки препаратом добавляли 10 мкл МТТ (0,5 мг/мл) и инкубировали в течение 4 ч. Затем среду отбрасывали, а кристаллы формазана солюбилизировали добавлением ДМСО (диметилсульфоксид; Sigma-Aldrich). Измерялась абсорбция при 570 нм. Жизнеспособность клеток была нормализована по отношению к контрольной группе.
Исследования ксенотрансплантации опухолей in vivo
Для этого анализа 5×106 клеток подкожно вводили мышам nude (BALB/c, сорт без специфических патогенов, возраст 4-5 недель, приобретены в Центре исследования модельных животных Нанкинского университета, Нанкин, Китайская Народная Республика). Когда размер опухоли достигал 100 мм3 (15-20 дней), мышей случайным образом делили на 4 группы (PBS, PTX, DCA и PTX плюс DCA). Доза PTX составляла 20 мг/кг/день, а доза DCA — 100 мг/кг/день. Длина и ширина опухолей измерялись каждые два дня по двум перпендикулярным диаметрам (расчетный объем = самый короткий диаметр2 × самый длинный диаметр/2). Количество и даты смерти мышей регистрировали для расчета выживаемости. Все манипуляции с участием живых мышей были одобрены Комитетом по уходу и использованию животных больницы Синьцяо и соответствовали китайскому руководству по благополучию и этике лабораторных животных, и все усилия были направлены на минимизацию страданий.
Гистологическое исследование
Все опухоли были резецированы через 24 дня для гистопатологического исследования. Срезы (толщиной от 4 до 5 мкм) опухолевых тканей фиксировали 10% формалином и затем определяли Ki-67, антитела LC3B и проводили анализ TUNEL. Срезы промывали трижды и обрабатывали диаминобензидином для развития цвета. Проводили гистологические исследования и делали фотографии с помощью оптического микроскопа NanoZoomer 2.0-RS (Hamamatsu Photonics, город Хамамацу, Япония).
Статистический анализ
Данные представлены как среднее ± стандартное отклонение. Для анализа дисперсии использовали односторонний дисперсионный анализ и независимый выборочный t-тест. Кривые выживаемости были получены методом Каплана-Мейера, а сравнение проводилось с помощью теста log-rank. Все статистические анализы проводились с использованием статистического программного обеспечения SPSS 19.0 (SPSS; IBM Corporation, Армонк, Нью-Йорк, США). Р-значения<0,05 или <0,01 считались статистически значимыми.
Результаты
DCA ингибировал аутофагию в клетках аденокарциномы легких
Цитозольный LC3 и SQSTM (p62) — высококонсервативные белки, которые, как считается, играют важную роль на ключевых этапах аутофагии. Чтобы изучить роль DCA в регуляции аутофагии, мы исследовали клеточную локализацию LC3 с помощью конфокального лазерно-сканирующего микроскопа. Клетки A549 и H1975 были инфицированы аденовирусом, кодирующим тандемную конструкцию GFP-RFP-LC3 для обнаружения изменения потока аутофагии. Разница в чувствительности к рН может быть причиной различной степени накопления белков GFP-LC3 и RFP-LC3 в нейтральных аутофагосомах и кислых аутолизосомах. После обработки HBSS количество точек GFP или RFP-LC3 значительно увеличивалось, демонстрируя, что голодание, вызванное HBSS, значительно способствовало потоку аутофагии в клетках A549 и H1975. Однако обработка DCA успешно подавляла экспрессию белков LC3 по сравнению с контролем или обработкой HBSS как в клетках A549, так и в клетках H1975 (Рисунок 1A и D, p<0,01).
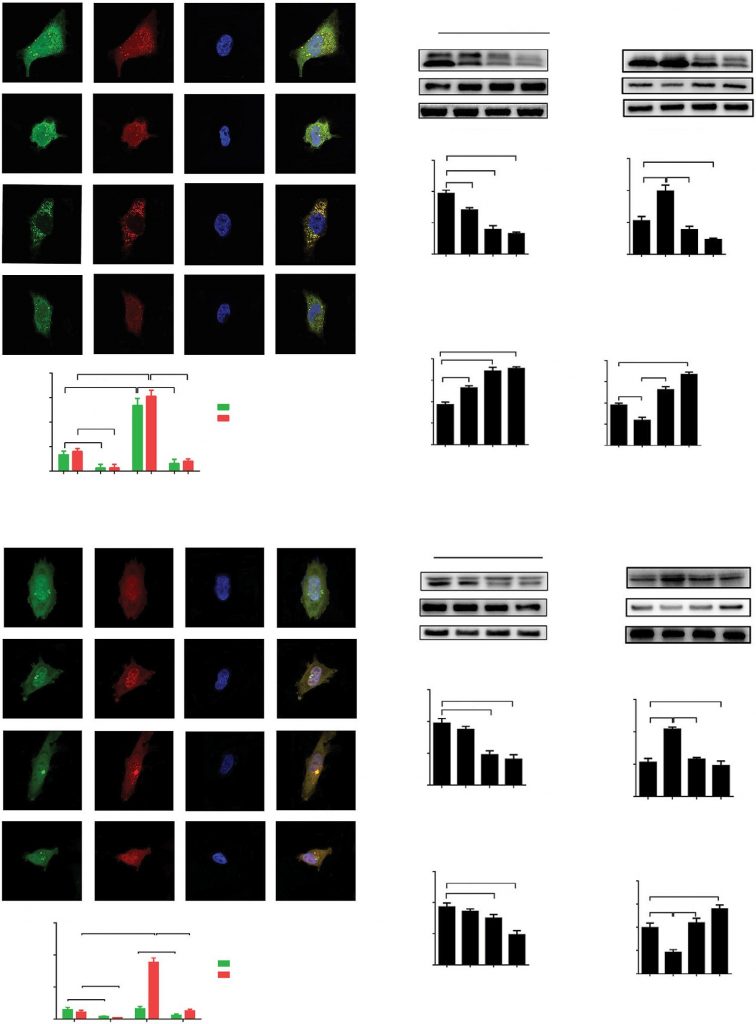
Сокращения: DAPI, 4′,6-диамидино-2-фенилиндол; DCA, дихлорацетат; HBSS, сбалансированный солевой раствор Хэнка; ns, не существенно.
белок p62 накапливается при ингибировании аутофагии и снижается при ее индуцировании. Для изучения уровня белка p62 и LC3-II после обработки DCA был проведен вестерн-блоттинг анализ. Мы наблюдали значительное снижение уровня LC3 и увеличение p62 после обработки DCA в зависимости от концентрации в клетках A549 и H1975 (рис. 1B и E, p<0,01). Аутофагия была усилена во время инкубации с HBSS, что было обращено вспять при совместной обработке с DCA (Рисунок 1С и F, p<0.01). Вместе взятые, эти результаты показывают, что аутофагический поток в клетках A549 и H1975 был значительно подавлен обработкой DCA.
Влияние стимулирования аутофагии на гибель клеток, индуцированную DCA
Было проведено систематическое исследование противораковых эффектов DCA. Сначала противораковую способность DCA исследовали in vitro. Клетки инкубировали в течение 24 ч в присутствии различных концентраций DCA, оценивали апоптоз и жизнеспособность клеток. Мы наблюдали, что противораковая способность DCA, отраженная в показателях ингибирования жизнеспособности и апоптоза клеток, увеличивалась в зависимости от концентрации в клетках A549 и H1975 (Рисунок 2A и B, E и F, p<0,01).
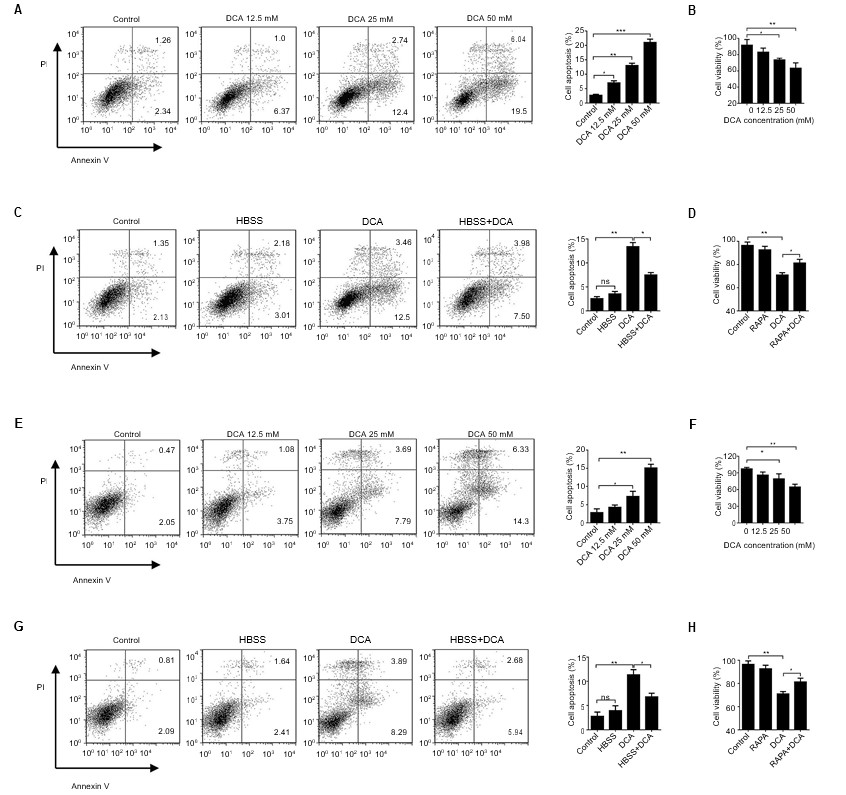
Сокращения: DCA — дихлорацетат; FITC/PI — флуоресцеин изотиоцианат/пропидий йодид; HBSS — сбалансированный солевой раствор Хэнка.
DCA активировал метаболический переключатель, который изменил аномальный метаболизм в раковых клетках с анаэробного гликолиза на окисление глюкозы, что привело к дисфункции митохондрий и гибели клеток. Наши предварительные эксперименты показали, что аутофагия была значительно подавлена обработкой DCA. Чтобы определить, вовлечено ли ингибирование аутофагии, опосредованное DCA, в апоптоз, клетки A549 и H1975 обрабатывали DCA после HBSS в течение 24 ч. Апоптоз клеток, индуцированный DCA, был подавлен, когда клетки совпадали с HBSS, по сравнению с обработкой только DCA. Кроме изменения аномального метаболизма, этот результат подтверждает, что DCA может также активировать гибель клеток путем ингибирования аутофагии в раковых клетках. Результаты жизнеспособности клеток согласуются с этим выводом (Рисунок 2C и D, G и H, p<0,05).
Ингибирование пути AKT уменьшало апоптоз, вызванный DCA
В предыдущих исследованиях считалось, что путь ATK-mTOR играет важную роль в регуляции аутофагии.[18-21] Перифозин,[22] химический реагент, предотвращающий фосфорилирование и активацию AKT, был использован для исследования того, участвует ли путь ATK-mTOR в процессе ингибирования аутофагии DCA. Клетки были обработаны DCA и перифозином в течение 24 ч. Апоптоз клеток был снижен почти наполовину в группе совпадения по сравнению с группой обработки DCA, что согласуется с результатами анализа жизнеспособности клеток (Рисунок 3A и B, D и E, p<0.05). Затем мы исследовали уровни важнейших белков, связанных с апоптозом, PARP и procaspase3. Фосфорилирование AKT было подавлено, а уровни белков, связанных с апоптозом, значительно снизились в группе котерапии (Рисунок 3C, p<0,05). Эти результаты были также получены в клетках H1975 (Рисунок 3F, p<0,05).
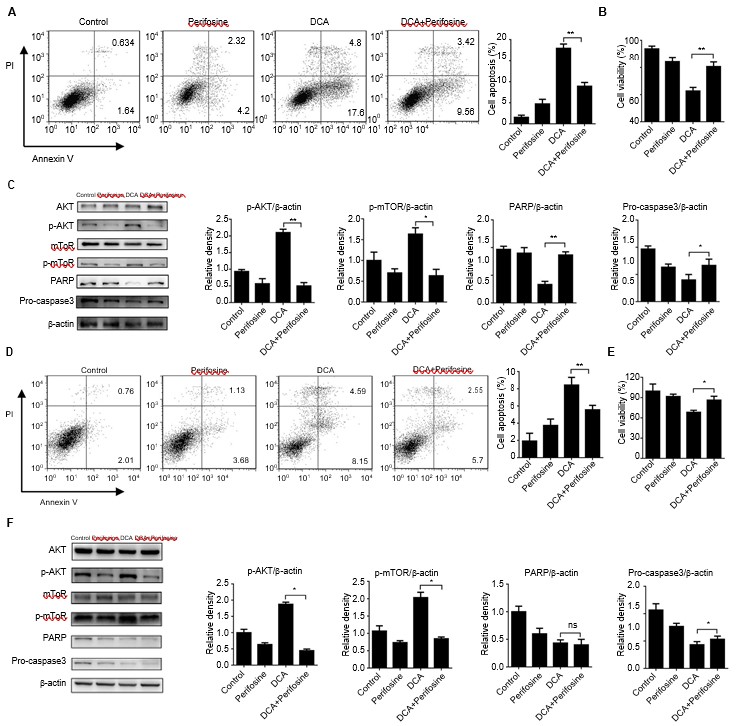
Аббревиатура: ДХА — дихлорацетат.
DCA повышает чувствительность к PTX и CDDP, отменяя индуцированную химиотерапией аутофагию
Ингибирование аутофагии считается новой стратегией для повышения чувствительности химиотерапевтических препаратов.[23,24] Чтобы исследовать, оказывает ли совместная обработка DCA и химиотерапевтическим препаратом синергетический эффект для подавления роста клеток, клетки A549 обрабатывали DCA и PTX или CDDP. Как показано на рисунке 4A-C, лечение химиотерапевтическими препаратами и DCA повысило уровень апоптоза по сравнению с лечением одним препаратом или в комбинации с CQ, и эти результаты соответствовали результатам анализа жизнеспособности клеток (p<0,05). Чтобы определить, может ли DCA снижать уровень аутофагии, индуцированной PTX, клетки обрабатывали PTX и DCA в течение 24 ч и собирали для вестерн-блоттинга. Были исследованы уровни белков, связанных с аутофагией. Экспрессия LC3 заметно повышалась под воздействием PTX, и значительно снижалась при совместной обработке с DCA. Уровень критического белка, связанного с апоптозом, PARP, был значительно снижен после котерапии (Рисунок 4D-F, p<0,05). Все эти результаты свидетельствуют о том, что DCA снижал лекарственную устойчивость клеток A549 к PTX путем ингибирования аутофагии.
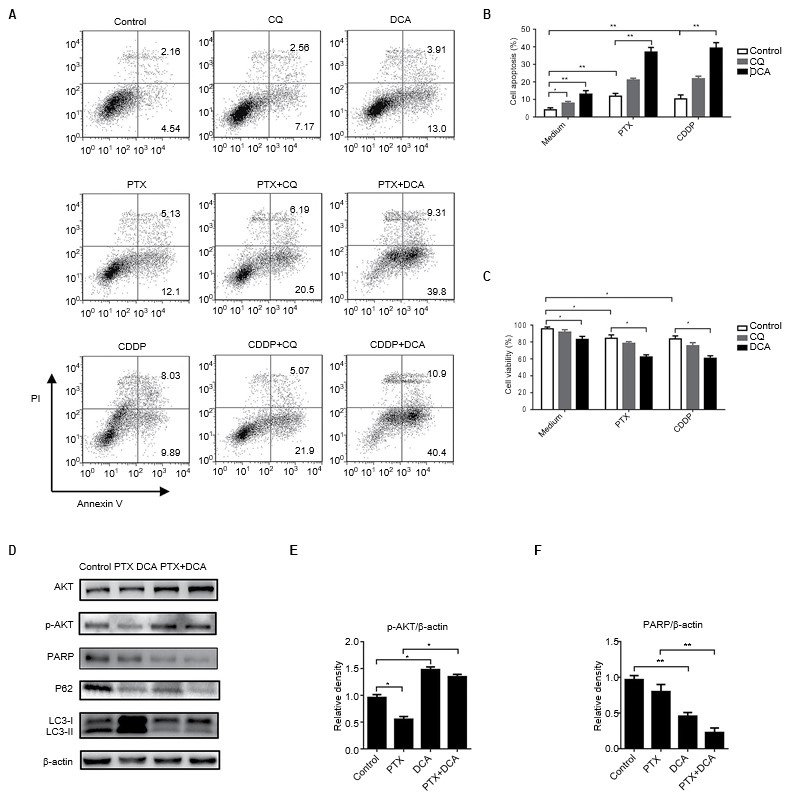
Сокращения: CDDP — цис-диамминдихлорплатин; CQ — хлорохин; DCA — дихлорацетат; PTX — паклитаксел.
DCA усиливал противораковую эффективность PTX in vivo путем ингибирования аутофагии
Поскольку совместное лечение клеток PTX и DCA увеличивало скорость гибели клеток in vitro по сравнению с лечением одним из препаратов, мы также оценили эффективность комбинированной терапии DCA и PTX in vivo на модели ксенотрансплантата опухоли мыши. Когда размер подкожной ксенотрансплантационной опухоли приблизился к 150 мм3, мышей разделили на четыре группы: контрольная, DCA, PTX и DCA плюс PTX. Введение только PTX не приводило к значительному подавлению роста опухоли, но лечение PTX плюс DCA явно подавляло рост опухоли и продлевало выживаемость мышей (Рисунок
, и B, p<0.05). Как показано на рисунке 5C, время удвоения опухоли клеток A549 значительно увеличилось с 3 дней в контрольной группе до 10 дней в группе котерапии (p<0,05), а время удвоения опухоли клеток H1975 увеличилось с 2,6 до 7,5 дней (рисунок 5D-F, p<0,05). Эти результаты показывают, что DCA может усиливать противораковую эффективность PTX in vivo.
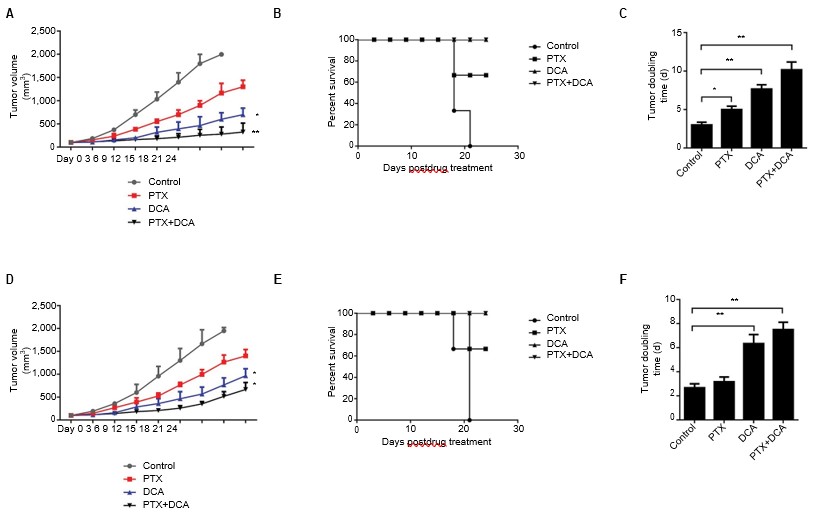
Сокращения: DCA — дихлорацетат; PTX — паклитаксел.
Лечение DCA плюс PTX значительно подавляло прорастание опухоли
Чтобы исследовать, приводит ли комбинированная терапия DCA и PTX к морфологическим изменениям, характерным для апоптоза в клетках A549 in vivo, все опухоли были извлечены на 24-й день, срезаны, окрашены TUNEL и проанализированы с помощью иммуногистохимии. В группах лечения DCA или PTX наблюдалось минимальное увеличение числа TUNEL-позитивных клеток по сравнению с контрольной группой. Однако доля TUNEL-позитивных клеток в группе совместного лечения значительно увеличилась по сравнению с контрольной группой или группой с одним лечением (Рисунок 6A).
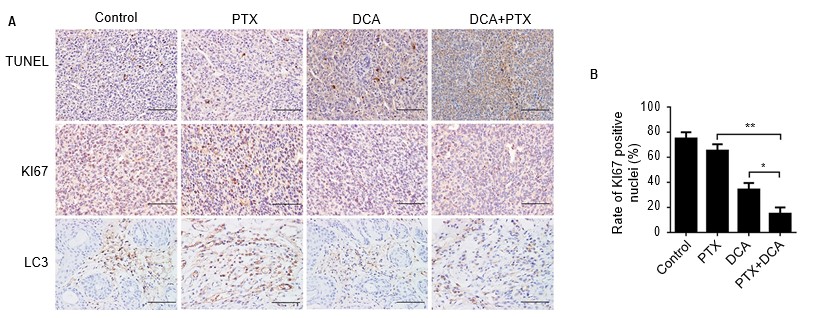
Сокращения: DCA — дихлорацетат; PTX — паклитаксел.
Затем мы исследовали экспрессию связанного с пролиферацией Ki-67 и важнейшего белка аутофагии LC3B в опухолях с помощью иммуногистохимического окрашивания. Экспрессия Ki-67 у контрольных мышей была значительно выше, тогда как в опухолях животных, получавших комбинированное лечение, она была значительно снижена, а экспрессия LC3B была такой же, как и Ki-67. В заключение следует отметить, что лечение DCA плюс PTX значительно усиливало апоптоз и снижало экспрессию Ki-67 и LC3B in vivo (Рисунок 6A и B, p<0,01).
Обсуждение
С развитием онкологических исследований все большее количество лечебных стратегий применяется в лечении рака легких.[25-27] Хотя многим пациентам с раком легких, особенно на поздней или прогрессирующей стадии, помогли EGFR-TKI и антитела PD-1, для большинства пациентов это непомерно дорого. Традиционная химиотерапия по-прежнему является основным методом лечения распространенного рака легких в Китае. Однако множественная лекарственная устойчивость является сложным препятствием в лечении рака, поэтому современные исследования направлены на поиск новых способов повышения химиотерапевтической чувствительности или замедления лекарственной устойчивости. В последнее время регуляция аутофагии рассматривается в качестве мишени для противораковой терапии; многие данные свидетельствуют о том, что ингибирование аутофагии может способствовать подавлению опухоли химиотерапевтическими препаратами и радиотерапией.[23,28-30] Обычно большинство солидных карцином вырабатывают энергию через гликолитический путь независимо от наличия достаточного количества кислорода; это называется «эффектом Варбурга».[29-31] DCA, противоопухолевый препарат с малой молекулой, может изменить этот метаболический режим митохондрий с анаэробного дыхания на аэробное, тем самым вызывая гибель опухолевых клеток. Однако, можно ли использовать DCA в комбинации с другими химиотерапевтическими препаратами, неизвестно. В данном исследовании мы обнаружили, что совместное лечение DCA и PTX может быть более эффективным в подавлении роста клеток рака легких in vitro и in vivo. DCA ингибировал защитную аутофагию, применяемую раковыми клетками в ответ на лечение PTX, активируя фосфорилирование AKT и способствуя гибели клеток. Синергетический эффект этих двух агентов усиливает гибель клеток, тем самым более эффективно подавляя рост опухоли.
Как известно, клеточный метаболизм является эффективной мишенью для лечения рака,[32] и накопленные данные свидетельствуют о противоопухолевых свойствах DCA в клеточном метаболизме. Однако эффекты DCA противоречивы, поскольку в различных моделях рака были получены разные результаты.[33,34] В настоящем исследовании мы обнаружили, что комбинация DCA и PTX более эффективно подавляет пролиферацию клеток и усиливает апоптоз, чем только PTX. В модели подкожной ксенотрансплантации опухоли, использованной в данном исследовании, лечение DCA плюс PTX снизило рост клеток A549, введенных мышам, на 60%. Наши данные показали, что DCA оказывает мощный противораковый эффект и действует синергично с PTX на клетки A549. Многолекарственная терапия, действующая синергично, может повысить терапевтическую эффективность за счет снижения лекарственной токсичности и лекарственной устойчивости.[35] В наших экспериментах DCA обратил вспять многолекарственную устойчивость клеток A549, подавив аутофагию, индуцированную PTX, и вызвав апоптоз. Поэтому добавление DCA в схему химиотерапии может снизить пагубность множественной лекарственной устойчивости в НСКЛК.
Аутофагия — это высококонсервативный метаболический путь, при котором клетки доставляют старые или дефектные белки, органеллы и другие клеточные компоненты в лизосомы для деградации с целью поддержания клеточного гомеостаза.[36,37] Большое количество доказательств показывает, что большинство химиотерапевтических агентов, а также ионизирующее излучение индуцируют аутофагию, что приводит к множественной лекарственной устойчивости. Адаптация аутофагии способствует выживанию опухолевых клеток в микросреде с повышенным метаболическим стрессом или лекарственной токсичностью. Вызванная стрессом аутофагия является важной защитной стратегией, которая может поддерживать выживание клеток, что в конечном итоге приводит к лекарственной устойчивости в различных типах опухолевых клеток.[38] В данном исследовании обработка DCA ингибировала клеточную аутофагию и способствовала апоптозу. RARA, активатор аутофагии, может сделать клетки более устойчивыми к DCA, что указывает на то, что DCA вызывает гибель клеток путем нарушения метаболизма раковых клеток и ингибирования аутофагии. Путь PI3K-AKT-mTOR является важным негативным регулятором аутофагии. Поэтому воздействие на сигналы PI3K-AKT-mTOR может стать новой стратегией лечения рака. DCA избирательно способствует фосфорилированию киназы AKT, тем самым ингибируя аутофагию. Таким образом, активация пути PI3K-AKT-mTOR под действием DCA ингибирует PTX-индуцированную аутофагию. По имеющимся данным, лечение EGFR-TKIs связано с уровнем аутофагии клеток рака легких, и ингибирование аутофагии может способствовать противоопухолевому действию EGFR-TKIs. Это может стать новым направлением в лечении рака легких.
Однако некоторые исследователи утверждают, что аутофагия является «обоюдоострым мечом» для раковых клеток и может подавлять выживание и пролиферацию в микроокружении опухоли.[26,39,40] Кроме того, индуцирование аутофагии в раке также рассматривалось как стратегия сенсибилизации к химиотерапии. Различные функции аутофагии в раковых клетках могут быть вызваны различными статусами раковых клеток или типами опухолей; таким образом, понимание основных функций регуляции аутофагии в различных раковых клетках прольет новый свет на разработку стратегии против резистентности к химиотерапии.[24]
Интересно, что DCA, по-видимому, обладает двойственными характеристиками регуляции аутофагии в различных опухолевых клетках. Lin et al[33] сообщили, что DCA может вызывать апоптоз в клетках колоректального рака и одновременно усиливать аутофагию. Из этого следует, что в регуляции аутофагии DCA участвует более сложный молекулярный механизм. Изучение принципа различных эффектов DCA на аутофагию даст нам новые идеи для разработки новых адъювантных химиотерапевтических препаратов путем ингибирования аутофагии.
Заключение
Наши данные показали, что аутофагия может быть активирована во время химиотерапии, а ингибирование аутофагии с помощью DCA может способствовать гибели клеток и повысить чувствительность опухолевых клеток к химиотерапевтическим препаратам. Кроме того, аутофагия и метаболизм рака вовлечены в ряд опухолевых событий, таких как развитие опухоли, опухолевая прогрессия и химиотерапия. Выяснение взаимосвязи между аутофагией и метаболизмом[32] в различных раковых клетках может предоставить нам новые возможности для разработки лекарств, нацеленных на эти важные клеточные события, против рака и лекарственной устойчивости.
Благодарность
Исследование было поддержано финансированием Национального фонда естественных наук Китая (81702247) и Проектом фундаментальной науки и передовых технологий Чунцина для Чжэн (cstc2017jcyjAX0048), Национального фонда естественных наук Китая для Дай (81472188) и Проектом клинических исследований больницы Синьцяо для Ю (2015YLC21).
Раскрытие информации
Авторы не сообщают о конфликте интересов в данной работе.
ССЫЛКИ
1 Maher AR, Miake-Lye IM, Beroes JM, Shekelle PG. Лечение метастатического немелкоклеточного рака легких: Систематический обзор сравнительной эффективности и экономической целесообразности. Вашингтон (округ Колумбия): Министерство по делам ветеранов (США); 2012.2 Spiro SG, Porter JC. Рак легких — где мы сегодня? Современные достижения в стадировании и нехирургическом лечении. Am J Respir Crit Care Med. 2002;166:1166-1196.
3 Song W, Tang Z, Li M, et al. Комбинация паклитаксела и цисплатина на основе полипептидов для повышения эффективности химиотерапии и снижения побочных эффектов. Acta Biomater. 2014;10:1392-1402.
4 Ward PS, Thompson CB. Метаболическое перепрограммирование: отличительная черта рака, которую не предвидел даже Варбург. Cancer Cell. 2012;21:297-308.
5 Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: молекулярный каркас, нацеленный на митохондрии, для эффективной доставки метаболического модулятора дихлорацетата. ACS Chem Biol. 2014;9:1178-1187.
6KimuraT, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3-7.
7 Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42.
8 Gomes LR, Vessoni AT, Menck CF. Перекрестное взаимодействие микроокружения и аутофагии: Последствия в терапии рака. Pharmacol Res. 2016;107:300-307.
9 Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
10 Koren I, Kimchi A. Cell biology. Продвижение опухолевого генеза путем подавления аутофагии. Science. 2012;338:889-890.
11 Rosenfeldt MT, Ryan KM. Роль аутофагии в развитии опухолей и терапии рака. Expert Rev Mol Med. 2009;11:e36.
12 Rubinsztein DC, Codogno P, Levine B. Модуляция аутофагии как потенциальная терапевтическая мишень для различных заболеваний. Nat Rev Drug Discov. 2012;11:709-730.
13EggerME, Huang JS, Yin W, McMasters KM, McNally LR. Ингибирование аутофагии с помощью хлорохина эффективно при меланоме. J Surg Res. 2013;184:274-281.
14 Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391-1402.
15 Vogl DT, Stadtmauer EA, Tan KS, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380-1390.
16 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51.
17 Christofk HR, Vander HM, Harris MH, et al. M2 сплайс-изоформа пируваткиназы важна для метаболизма рака и роста опухоли. Nature. 2008;452:230-233.
18 Tan J, Jiang X, Yin G, et al. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38(3):1373-1382.
19 Chen LM, Song TJ, Xiao JH, Huang ZH, Li Y, Lin TY. Трипхлоролид индуцирует аутофагию в клетках рака легких путем ингибирования пути PI3K/AKT/mTOR и улучшает чувствительность к цисплатину в клетках A549/DDP. Oncotarget. 2017;8(38):63911–63922.
20 Wang Z, Wang N, Liu P, Xie X. AMPK и рак. EXS. 2016;107:203-226.
21 Umezawa S, Higurashi T, Nakajima A. AMPK: терапевтическая мишень для профилактики диабета и рака. Curr Pharm Des. 2017;23(25):3629–3644.
22 Gills JJ, Dennis PA. Перифозин: обновленная информация о новом ингибиторе Akt. Curr Oncol Rep. 2009;11:102-110.
23 Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528-539.
24 Rubinsztein DC, Codogno P, Levine B. Модуляция аутофагии как потенциальная терапевтическая мишень для различных заболеваний. Nat Rev Drug Discov. 2012;11:709-730.
25 Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47.
26 Dholaria B, Hammond W, Shreders A, Lou Y. Новые терапевтические агенты для лечения рака легких. J Hematol Oncol. 2016;9:138.
27 Wang S, Cang S, Liu D. Ингибиторы третьего поколения, направленные на мутацию EGFR T790M в распространенном немелкоклеточном раке легкого. J Hematol Oncol. 2016;9:34.
28 White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212-217.
29 Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961-967.
30 Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485-1494.
31 Zhou W, Liotta LA, Petricoin EF. Эффект Варбурга и протеомный анализ на основе масс-спектрометрии. Cancer Genomics Proteomics. 2017;14:211-218.
32 Ho WL, Hsu WM, Huang MC, Kadomatsu K, Nakagawara A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J Hematol Oncol. 2016;9:100.
33 Lin G, Hill DK, Andrejeva G, et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br J Cancer. 2014;111:375-385.
34 Gong F, Peng X, Sang Y, et al. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013;4:e913.
35 Chou TC. Теоретические основы, экспериментальный дизайн и компьютерное моделирование синергизма и антагонизма в исследованиях комбинаций лекарственных средств. Pharmacol Rev. 2006;58:621-681.
36 Eskelinen EL. Двойная роль аутофагии при раке. Curr Opin Pharmacol. 2011;11:294-300.
37 Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383-1435.
38 Naveed S, Aslam M, Ahmad A. Дифференцированная химиотерапия на основе голодания: новый подход к лечению рака. Oman Med J. 2014;29:391-398.
39 Yoshida GJ. Терапевтические стратегии репозиционирования лекарств, нацеленные на аутофагию, чтобы вызвать гибель раковых клеток: от патофизиологии к лечению. J Hematol Oncol. 2017;10:67.
40 Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651-662.
Связанный контент: