Tiziana Tataranni 1, Francesca Agriesti 1, Consiglia Pacelli 2, Vitalba Ruggieri 1, Ilaria Laurenzana 1, Carmela Mazzoccoli 1 , Gerardo Della Sala 1, Concetta Panebianco 3, Valerio Pazienza 3, Nazzareno Capitanio 2 e Claudia Piccoli 1,2,*
1 Laboratorio di Ricerca Pre-Clinica e Traslazionale, IRCCS-CROB, Centro Tumori di Riferimento della Basilicata, 85028 Rionero in Vulture (Pz), Italia; [email protected] (T.T.); [email protected] (F.A.); [email protected] (V.R.); [email protected] (I.L.); [email protected] (C.M.); [email protected] (G.D.S.))
2 Dipartimento di Medicina Clinica e Sperimentale, Università di Foggia, 71100 Foggia, Italia; [email protected] (C.P.); [email protected] (N.C.)
3 Divisione di Gastroenterologia, Ospedale IRCCS “Casa Sollievo della Sofferenza”, 71013 San Giovanni Rotondo, Italia; [email protected] (C.P.); [email protected] (V.P.)
* Corrispondenza: [email protected]; Tel.: +39-0881-588-060
Ricevuto: 21 febbraio 2019
Accettato: 15 maggio 2019
Pubblicato: 18 maggio 2019
Abstract
Prendere di mira il metabolismo rappresenta un possibile approccio di successo per il trattamento del cancro. Il dicloroacetato (DCA) è un farmaco noto per deviare il metabolismo dalla glicolisi anaerobica alla fosforilazione ossidativa mitocondriale attraverso la stimolazione della PDH. In questo studio abbiamo analizzato la risposta di due linee cellulari di cancro al pancreas al DCA, in colture cellulari bidimensionali e tridimensionali e in un modello murino. PANC-1 e BXPC-3 trattate con DCA hanno mostrato una marcata riduzione della proliferazione e della migrazione cellulare, non correlata a un aumento dell’apoptosi, indicando un effetto citostatico piuttosto che citotossico. Nonostante l’attivazione della PDH, il trattamento con DCA ha determinato una riduzione del consumo di ossigeno mitocondriale senza influenzare la glicolisi. Inoltre, il DCA ha provocato un aumento della produzione di ROS, del mtDNA e del marcatore di mitofagia LC3B-II in entrambe le linee cellulari, ma ha ridotto i marcatori di fusione mitocondriale solo nelle BXPC-3. In particolare, il DCA ha ridotto l’espressione dei marcatori delle cellule staminali tumorali CD24/CD44/EPCAM solo nel PANC-1, ma ha inibito la formazione di sferoidi e la vitalità in entrambe le linee cellulari. In un modello murino di tumore pancreatico xenotrapiantato, il trattamento con DCA ha ritardato la progressione del tumore. Nel complesso, i nostri risultati indicano chiaramente che l’efficacia del DCA nell’inibire la crescita del cancro dipende meccanicamente dal fenotipo cellulare e da molteplici vie off-target. In questo contesto, la novità che il DCA potrebbe influenzare il comparto delle cellule staminali del cancro è terapeuticamente rilevante.
Parole chiave: metabolismo; mitocondri; cellule staminali tumorali
© 2019 degli autori. Licenziatario MDPI, Basilea, Svizzera. Questo articolo è un articolo ad accesso libero distribuito secondo i termini e le condizioni della licenza Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
INTRODUZIONE
L’adenocarcinoma duttale del pancreas (PDAC) è un tumore molto aggressivo, con una bassa percentuale di pazienti affetti idonei alla resezione chirurgica e altamente refrattario alle terapie convenzionali [1,2]. Pertanto, per migliorare gli attuali schemi di trattamento, è necessario ricorrere a farmaci più efficaci. Oltre alla crescita cellulare, alla riparazione del DNA, all’invasività e all’angiogenesi, le cellule del PDAC sono caratterizzate da mutazioni in geni coinvolti nel metabolismo [1,3]. Nuove strategie terapeutiche mirate al metabolismo stanno emergendo come approcci promettenti per superare la chemioresistenza [4]. Tuttavia, l’eterogeneità inter- e intra-tumorale spesso si traduce in fenotipi metabolici diversi, anche come conseguenza delle molteplici interazioni con il microambiente tumorale [5]. Ciò pone dei limiti terapeutici e sottolinea l’importanza di una caratterizzazione metabolica preliminare dei lignaggi tumorali, propedeutica alla somministrazione di farmaci efficaci. Recentemente abbiamo dimostrato che due linee cellulari di tumore del pancreas, caratterizzate da un diverso profilo metabolico, producono una risposta dissimile alla deprivazione di glucosio/sostituzione di galattosio, un approccio in grado di ricablare il metabolismo energetico [6]. Inoltre, il nostro gruppo aveva già dimostrato l’efficacia del dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi (PDK), per uccidere cellule in coltura derivate da carcinomi orali umani, un effetto inversamente correlato alla capacità respiratoria mitocondriale delle cellule tumorali [7]. Diversi studi in vivo e in vitro descrivono la capacità del DCA di aumentare la fosforilazione ossidativa mitocondriale (OxPhos), invertendo l’effetto Warburg e colpendo selettivamente le cellule tumorali [8,9]. Inoltre, un’ampia letteratura dimostra l’efficacia del DCA nel migliorare la chemio-sensibilità in diversi tipi di cancro [10,11]. Il trattamento con DCA è stato utilizzato sia per studi in vitro che in vivo anche nel tumore del pancreas [8,12,13,14]. Tuttavia, sono necessarie ulteriori indagini per definire meglio l’efficacia del farmaco in questo tipo di tumore, per chiarire potenziali meccanismi aggiuntivi che portano alla morte cellulare e per esplorare ulteriori modi per limitare gli effetti collaterali riscontrati. In questo studio abbiamo analizzato gli effetti del DCA su due linee cellulari di PDAC, PANC-1 e BXPC-3, scelte tra le altre per le loro condizioni di crescita simili e per il loro geno/fenotipo ben caratterizzato [6,15,16]. Un’ampia profilazione dei metaboliti e del trascrittoma delle linee cellulari di PDAC ha identificato tre sottotipi tumorali, con PANC-1 e BXPC-3 appartenenti a un cluster lipogenico caratterizzato da un’evidente dipendenza dall’ossidazione del glucosio e dal metabolismo legato ai mitocondri [17]. Testando il DCA su colture 2D e 3D delle linee cellulari PDAC, abbiamo dimostrato che il farmaco influisce negativamente sui parametri vitali riducendo l’attività respiratoria mitocondriale e, soprattutto, il comparto delle cellule staminali tumorali. Inoltre, abbiamo dimostrato che il DCA è anche in grado di attenuare la crescita tumorale in vivo in un modello di xenotrapianto di PDAC nei topi.
Materiali e metodi
Coltura cellulare
Le cellulePANC-1e BXPC-3 sono state acquistate dall’American Type Culture Collection (ATCC, Manassas, VA, USA) e coltivate a 37 °C in un’atmosfera umidificata al 5%di CO2 in terreno RPMI completo integrato con 10% di siero fetale bovino, penicillina-streptomicina (100 U/mL) e 2 mM di glutammina; la concentrazione di glucosio era tipicamente di 10 mM o 1 mM quando indicato. Il dicloroacetato (DCA) è stato acquistato da Sigma-Aldrich (St. Louis, MO, USA). Per ogni esperimento in vitro, le cellule sono state trattate con DCA 4 mM e 10 mM ai tempi indicati.
Curve di crescita cellulare
Le curve di crescita cellulare sono state eseguite come precedentemente descritto [18].
Monitoraggio della proliferazione cellulare in tempo reale mediante il sistema xCELLigence
Gli esperimenti xCELLigence sono stati eseguiti utilizzando lo strumento RTCA (real-time cell analyzer), secondo le istruzioni del produttore (ACEA Biosciences, San Diego, CA, USA). Il numero di semina ottimale è stato determinato in precedenza mediante esperimenti di titolazione e crescita cellulare (dati non mostrati). Sono state quindi seminate 2500 cellule/pozzetto e la loro proliferazione è stata monitorata automaticamente ogni 30 minuti; 24 ore dopo la semina, le cellule sono state trattate con DCA. L’indice cellulare è stato monitorato fino a 90 ore dalla semina. I dati sono stati analizzati con il software xCELLigence (versione 2.0, Acea biosciences, San Diego, CA, USA) ed espressi come media ± SD dell’indice cellulare normalizzato all’ultimo indice cellulare registrato prima dell’aggiunta di DCA.
Saggio di apoptosi
Dopo l’incubazione con DCA, le cellule sono state colorate con Annexin-V-FITC e PI (BD Biosciences). Le cellule vive, apoptotiche e necrotiche sono state rilevate mediante citometria a flusso (Navios, Beckman Coulter, Brea, CA, USA). Sono stati eseguiti tre esperimenti indipendenti. Sono stati acquisiti in totale104 eventi per ciascun campione.
Saggio di migrazione
Gli effetti del DCA sulla capacità di migrazione di PANC-1 e BXPC-3 sono stati valutati utilizzando un saggio di ferita da graffio. In breve, le cellule sono state seminate in piastre di coltura a sei pozzetti e coltivate fino alla completa confluenza. Successivamente, in ogni piastra sono state prodotte tre ferite parallele e lineari con un puntale di plastica da 200 μL. Le cellule sono state poi trattate con DCA e la capacità di guarigione delle ferite, monitorata in diversi punti temporali, è stata quantificata dopo 48 h. Tre immagini rappresentative delle aree graffiate di ciascun piatto sono state fotografate per stimare la migrazione delle cellule. Il tasso di migrazione cellulare è stato calcolato con la seguente formula: [1 – (larghezza del graffio 48 h/0 h)] × 100%.
Misurazioni del lattato
È stato utilizzatounkit per il dosaggio colorimetrico del lattato (Abcam, Cambridge, MA, USA) seguendo il protocollo del produttore e la concentrazione di lattato rilevata (intracellulare o rilasciata) è stata normalizzata al numero di cellule.
Analisi del flusso metabolico e attività enzimatica del complesso respiratorio mitocondriale
Il tasso di consumo di ossigeno (OCR) e il tasso di acidificazione extracellulare (ECAR) sono stati misurati in cellule PANC-1 e BXPC-3 aderenti con un analizzatore di flusso extracellulare XF96 (Seahorse Bioscience, Billerica, MA, USA) come precedentemente descritto [19]. In breve, per l’analisi OCR, dopo aver misurato la respirazione basale, sono stati iniettati in ciascun pozzetto, in sequenza, oligomicina (1 μM), FCCP (1 μM) e rotenone + antimicina A (1 μM + 1 μM) per valutare, rispettivamente, l’accoppiamento della catena respiratoria e il consumo di ossigeno massimo e non mitocondriale. Per l’analisi ECAR, il flusso glicolitico (glicolisi basale, capacità glicolitica e riserva glicolitica) è stato analizzato mediante l’aggiunta sequenziale di 10 mM di glucosio, 1 μM di oligomicina e 100 mM di 2-deossiglucosio. I valori di OCR ed ECAR sono stati normalizzati rispetto al contenuto proteico di ciascun pozzetto, determinato con il saggio BCA (Thermo Scientific, Waltham, MA, USA).
Quantificazione del DNA mitocondriale
La misurazione del numero di copie di mtDNA, rispetto al numero di copie di DNA nucleare, è stata determinata come precedentemente descritto [6].
Imaging di mtΔΨ e ROS
Le cellule coltivate a bassa densità su piatti da 35 mm con fondo in vetro rivestiti di fibronectina (Eppendorf, Amburgo, Germania) sono state incubate per 20 minuti a 37 °C con 2 μM di TMRE e 10 μM di DCF (Molecular Probes, Eugene, OR, USA) per monitorare mtΔΨ e ROS, rispettivamente. Le cellule colorate sono state lavate con PBS ed esaminate con un microscopio confocale a scansione laser Leica TCS SP8. L’acquisizione, la memorizzazione e l’analisi dei dati sono state eseguite con un software strumentale dedicato di Leica (LAS-X, Wetzlar, Germania).
Analisi di Western Blotting
Aliquote, contenenti 40 μg di proteine da ogni lisato cellulare, sono state sottoposte a elettroforesi su gel di poliacrilammide SDS e trasferite su una membrana di polivinilidene difluoruro utilizzando un Trans Blot Turbo Transfer System. Le membrane (Bio-Rad Laboratories, Hercules, CA, USA) sono state testate con i seguenti anticorpi primari: piruvato deidrogenasi E1-alfa (PDH) e pPDHSer293 (1:500, Abcam, Cambridge, UK), LC3B (1:1000 Cell Signaling Technology), TOM20 (1:1000, Santa Cruz Biotechnology, Cambridge, UK):1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), DRP1 (1:1000, BD Bioscences), OPA-1 (1:1000, BD Bioscences), MFN1 (1:1000, Santa Cruz), MFN2 (1:1000, Abnova, Tapei, Taiwan) e CASPASE 3 (1:1000, Cell Signaling Technology, Danvers, MA, USA). Dopo l’incubazione con un anticorpo secondario coniugato con perossidasi di rafano (1:2500; Cell Signaling Technology), i segnali sono stati sviluppati utilizzando il kit di chemiluminescenza potenziata (ClarityTM Western ECL Substrate, Bio-Rad) e il sistema di imaging ChemiDoc XRS + (BioRad), quindi analizzati con il software Image Lab (versione 4.1, Bio-Rad, Hercules, CA, USA). L’intensità delle bande LC3B-II (corrispondente alla frazione clivata), TOM20, DRP1, OPA-1, MFN1 e MFN2 è stata normalizzata rispetto al segnale della β-actina, mentre la fosforilazione di PDH è stata normalizzata rispetto alle proteine totali.
Rilevazione citometrica a flusso dei marcatori di superficie
L’espressione dei marcatori di superficie CD44, CD24 ed EPCAM è stata valutata mediante analisi citofluorimetrica in PANC-1 e BXPC-3 trattate con DCA per 24 ore. In breve, dopo la tripsinizzazione, le cellule sono state incubate al buio a temperatura ambiente per 15 minuti con anticorpi monoclonali CD44-APC, EPCAM-FITC e CD24-PE direttamente coniugati (BDB). L’analisi citofluorimetrica è stata eseguita con Navios (Beckman Coulter). Il segnale fluorescente emesso di 10.000 eventi per ciascun campione è stato acquisito e analizzato con il software Kaluza Analysis (versione 1.3, Beckman Coulter, Brea, CA, USA).
Analisi della trascrizione inversa e della PCR in tempo reale
Un microgrammo di RNA totale, isolato con il reagente Trizol (Life Technologies, Paisley, Regno Unito), secondo le istruzioni del produttore e quantificato con uno spettrofotometro Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA), è stato utilizzato in una reazione di trascrizione inversa (RT) utilizzando il kit di sintesi di cDNA a primo filamento Transcriptor (Roche Diagnostic, Penzberg, Germania) secondo le istruzioni del produttore. La reazione a catena della polimerasi (PCR) quantitativa in tempo reale è stata eseguita in duplicato, utilizzando il QuantiTect Primer Assay (Qiagen, Basilea, Svizzera) per rilevare l’mRNA di Lin28. La quantificazione dei livelli di mRNA è stata eseguita su uno strumento LightCycler® 480 per la PCR in tempo reale. Le quantità relative di Lin28 sono state normalizzate con l’espressione di GAPDH mediante Light Cycler® 480 Software versione 1.5 (ROCHE) utilizzando il metodo 2ΔΔCt.
coltura 3D
Le cellulePANC-1e BXPC-3 sono state staccate con tripsina-EDTA e contate. Quindi, 1000 cellule/pozzetto sono state seminate in piastre a 96 pozzetti a fondo tondo a bassissimo attacco e coltivate in RPMI. Per valutare l’effetto del DCA sugli sferoidi preformati, le colture 3D sono state mantenute per 7 giorni, ottenendo sferoidi. Quindi, il terreno è stato sostituito con terreno fresco e gli sferoidi sono stati trattati con DCA 4 mM e 10 mM per 72 h. Per valutare l’effetto del DCA sulla formazione di sferoidi, il DCA è stato aggiunto alla sospensione cellulare quando è stata seminata in piastre a bassissimo attacco e la coltura è stata mantenuta per 7 giorni. Gli sferoidi sono stati fotografati su un microscopio ottico invertito (Axio Vert A1, Zeiss, Oberkochen, Germania) e il loro diametro è stato misurato con il software di imaging ZEISS ZEN. La vitalità degli sferoidi è stata valutata con un saggio MTS. Una soluzione di cellTiter 96® Aqueous MTS Reagent Powder (Promega, Madison, WI, USA) e PMS (Sigma Aldrich, Saint Louis, MO, USA) è stata aggiunta a ciascun pozzetto della coltura di sferoidi 3D. Dopo 2 ore di incubazione a 37 °C è stata misurata l’assorbanza a 490 nm e la percentuale di vitalità in ogni pozzetto è stata calcolata utilizzando gli sferoidi non trattati come 100%.
Studi sugli animali
I test sugli animali sono stati eseguiti in una struttura sperimentale accreditata AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International, Frederick, MD USA) con il numero di approvazione ANM14_002/468862. Un numero totale di 5 ×106 cellule tumorali BxPC-3-luc sono state coltivate, risospese in 0,1 mL di miscela PBS/matrigel (1:1) e quindi iniettate s.c. nel fianco destro di topi nudi Nu/Nu di 5-6 settimane. Quando le dimensioni del tumore hanno raggiunto un volume medio di 100 mm3, i topi nudi portatori di tumore BxPC-3-luc sono stati assegnati a caso in 2 gruppi (6 topi/gruppo). Gruppo 1 (soluzione salina normale, i.p, qw), gruppo 2 (DCA, mg/kg, i.p, qw). Gli animali avevano libero accesso all’acqua. Il DCA è stato disciolto per ottenere una concentrazione finale di 100 mg/kg/die (s.c: sottocutaneo; i.p: intraperitoneale; qw: una volta alla settimana).
Analisi statistica
I dati sperimentali sono espressi come media ± errore standard medio (SEM) o media ± deviazione standard (SD). I dati sono stati confrontati utilizzando il test t di Student non accoppiato o l’Anova a una via, seguita dal test di Bonferroni. Un valore p < 0,05 è stato accettato come statisticamente significativo.
Risultati
IlDCA influenza negativamente la proliferazione, la sopravvivenza e la migrazione cellulare nelle linee cellulari PANC-1 e BXPC-3
Le due linee cellulari di PDAC selezionate per questo studio erano PANC-1 e BXPC-3. La PANC-1 è una linea cellulare derivata dal carcinoma pancreatico di origine duttale. È in grado di metastatizzare, ma ha una scarsa capacità di differenziazione e presenta mutazioni in KRAS e TP53 e una delezione omozigote in CDKN2A/p16 [16]. La BxPC-3 è una linea cellulare primaria derivata da adenocarcinoma con moderata differenziazione e morfologia epiteliale. Esprime mucina ed elevati livelli di fattori angiogenici e marcatori di cellule staminali tumorali [16,20], è priva di mutazioni KRAS ma presenta mutazioni in TP53 e delezioni omozigoti in CDKN2A/p16 e SMAD4/DPC4 [16]. L’effetto del DCA sui parametri di vitalità delle linee cellulari PANC-1 e BXPC-3 è stato valutato alle concentrazioni di 4 mM e 10 mM, già testate e dimostrate efficaci come dimostrato nel nostro precedente studio [7]. In primo luogo, abbiamo eseguito un saggio di crescita cellulare per 72 ore, che ha rivelato una significativa sensibilità dose- e tempo-dipendente di entrambe le linee cellulari al trattamento con DCA (Figura 1A,B). In particolare, PANC-1 e BXPC-3 hanno mostrato un blocco simile della crescita cellulare quando sono state trattate con 10 mM DCA a partire dal primo giorno di incubazione; al contrario, alla dose inferiore di 4 mM testata, la linea cellulare PANC-1 è apparsa significativamente più sensibile al farmaco.
L’osservazione sopra riportata, particolarmente interessante a causa della ben nota chemioresistenza dimostrata dalla linea cellulare PANC-1 [21,22], ci ha spinto a verificare l’inibizione della crescita cellulare mediata dal DCA con un approccio diverso. A tal fine, abbiamo monitorato in tempo reale le variazioni dinamiche della proliferazione e della vitalità cellulare mediante la tecnologia impedenziometrica. Come mostrato nella Figura 1C-F, il trattamento con 10 mM di DCA ha ridotto drasticamente la proliferazione cellulare in entrambe le linee cellulari, mentre il trattamento con 4 mM di DCA ha causato un effetto inibitorio molto più forte nelle linee cellulari PANC-1 rispetto a quelle BXPC-3. Da notare che gli effetti del DCA erano chiaramente visibili già dopo 24 ore di incubazione con il farmaco. L’analisi della crescita cellulare in tempo reale è stata effettuata anche con un basso contenuto di glucosio nel terreno di coltura (cioè 1 mM in RPMI). Come previsto, il tasso di crescita di entrambe le linee cellulari di PDAC è stato fortemente ridotto, data la loro dipendenza metabolica dall’ossidazione del glucosio [17]. Tuttavia, la diversa sensibilità al trattamento con DCA 4 mM è stata confermata anche con un regime a basso contenuto di glucosio (Figura S1 supplementare). Per valutare i parametri vitali, abbiamo utilizzato il saggio dell’annexina V-FITC/PI e valutato con la citometria a flusso la quantità relativa di cellule necrotiche, apoptotiche tardive e precoci. I risultati ottenuti hanno mostrato che dopo 24 ore di incubazione con DCA entrambe le linee cellulari PANC-1 e BXPC-3 hanno mostrato un leggero ma significativo aumento dose-dipendente dell’apoptosi rispetto alle cellule non trattate. Tuttavia, le quantità di cellule apoptotiche erano relativamente basse (cioè < 10% alla concentrazione più alta di DCA testata) e non aumentavano ulteriormente a 48 ore di trattamento con DCA (Figura 2A). Di conseguenza, l’espressione della caspasi 3 non salvata non è cambiata in seguito al trattamento con DCA e non è stata rilevata alcuna quantità apprezzabile della sua forma clivata (Figura 2B). Questo risultato suggerisce un’attività citostatica piuttosto che citotossica del farmaco in entrambe le linee cellulari per spiegare la marcata riduzione del tasso di crescita mostrata nelle Figure 1A-D.
Successivamente, abbiamo valutato l’effetto del DCA sulla motilità cellulare, eseguendo il saggio scratch wound-healing. La capacità di migrazione, osservata in diversi momenti, è stata misurata dopo 48 ore dal trattamento con DCA 4 mM e 10 mM. Sia le cellule PANC-1 che le BXPC-3 hanno diminuito la loro motilità quando sono state trattate con la dose più alta di DCA, mentre la capacità migratoria delle BXPC-3 non è stata influenzata dal trattamento con DCA 4 mM, che invece ha causato un ritardo nella capacità di chiusura della ferita nelle cellule PANC-1, confermando la loro maggiore sensibilità al farmaco rilevabile a una concentrazione più bassa (Figura supplementare S2).
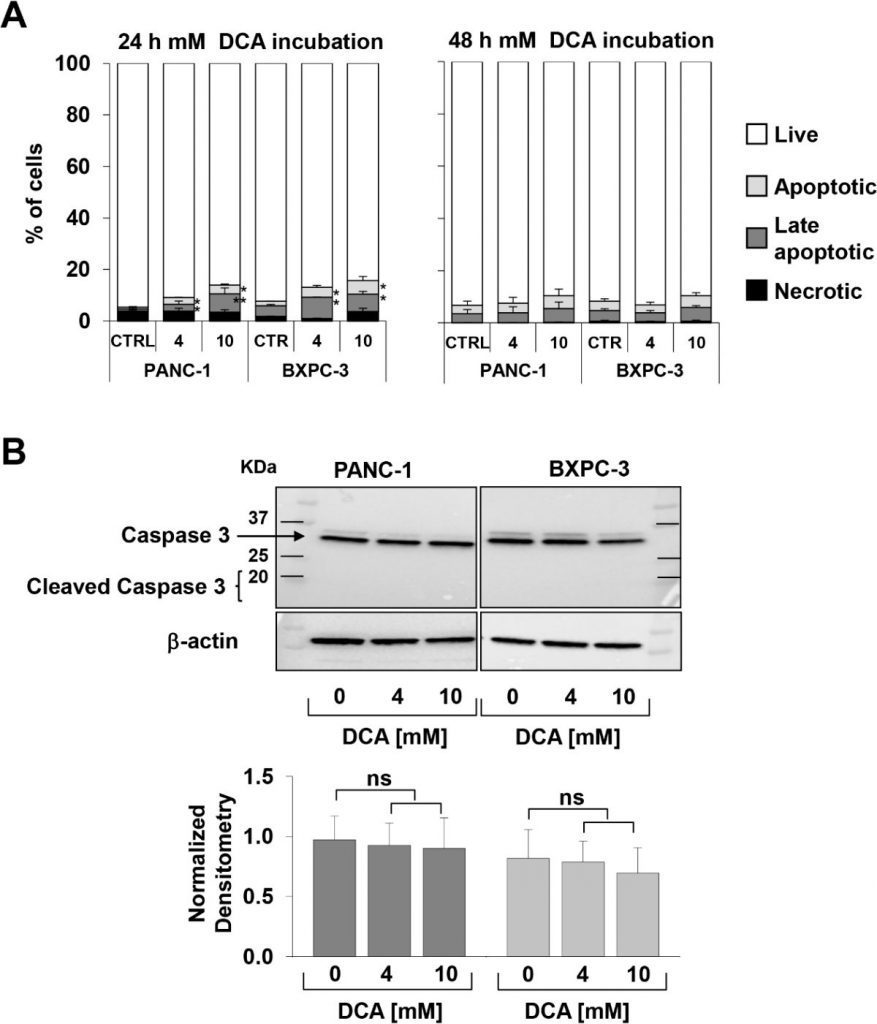
IlDCA altera il metabolismo energetico cellulare nelle linee cellulari di PDAC
Per indagare il legame tra l’effetto antiproliferativo indotto dal DCA e le alterazioni del metabolismo delle linee cellulari di PDAC, abbiamo valutato l’efficacia del composto nell’inibire il suo bersaglio riconosciuto, la piruvato deidrogenasi chinasi (PDK), valutando lo stato di fosforilazione della subunità E1α (residuo S293) del complesso della piruvato deidrogenasi (PDC) mediante Western blotting nelle PANC-1 e BXPC-3. Come mostrato nella Figura 3A,B, il livello normalizzato di P-PDH-E1 è risultato significativamente ridotto in entrambe le linee cellulari, mentre il livello di espressione della PDH totale era comparabile e, come previsto, non modificato dal trattamento farmacologico. Inaspettatamente, la produzione di lattato extra- e intracellulare è rimasta apparentemente inalterata in entrambe le linee cellulari di PDAC trattate con DCA (Figura S3 supplementare).
Quindi, abbiamo analizzato i principali flussi metabolici misurando l’acidificazione extracellulare e il consumo di ossigeno utilizzando la tecnologia SeaHorse. Quando i flussi metabolici sono stati valutati dopo 24 ore di incubazione di DCA, non sono stati osservati cambiamenti significativi in entrambe le linee cellulari di PDAC (Figura S4 supplementare). Un’esposizione più lunga (48 ore) al DCA ha causato una diminuzione dose-dipendente dei tassi di consumo di ossigeno mitocondriale (OCR) in entrambe le linee cellulari PANC-1 e BXPC-3 sia al basale che in presenza dell’inibitore dell’ATP-sintasi oligomicina o del disaccoppiatore FCCP (cioè la capacità respiratoria massima) (Figura 3C,D). Va notato che l’attività respiratoria mitocondriale era significativamente più alta in PANC-1 rispetto a BXPC-3, indicando un fenotipo metabolico più dipendente da OxPhos. I tassi di acidificazione extracellulare (ECAR), che sono legati al flusso glicolitico, non hanno subito cambiamenti significativi in seguito al trattamento con DCA di PANC-1, mentre è stata osservata un’inibizione per l’ECAR basale in BXPC-3 alla concentrazione più elevata di DCA e in modo dipendente dalla dose per la capacità glicolitica (Figura 3E,F). Di conseguenza, i profili bioenergetici complessivi delle capacità di flusso basali e stimolate di entrambe le cellule PDAC sono stati influenzati dal DCA, con la PANC-1 che ha mostrato una diminuzione della capacità OxPhos e la BXPC-3 che ha mostrato una compromissione più grave di entrambi i flussi metabolici (Figura 3G,H). La misurazione dei flussi metabolici in condizioni di crescita a basso contenuto di glucosio ha evidenziato una riduzione dell’OCR in entrambe le linee cellulari (più consistente in BXPC-3) e un aumento dell’ECAR in BXPC-3. Il trattamento con DCA (48 ore) ha causato in entrambe le linee cellulari un effetto inibitorio significativamente inferiore sull’OCR (in particolare a 4 mM) a basso glucosio rispetto al regime ad alto glucosio. Il trattamento con DCA non ha causato alcun cambiamento significativo sull’ECAR in PANC-1, mentre in BxPC-3 è stata osservata un’inibizione del 40-50%, tuttavia indipendente dalla disponibilità di glucosio (Figura S5). Nel complesso, queste osservazioni inaspettate indicano che nelle cellule di PDAC, nonostante il DCA sia apparentemente in grado di attivare la PDH, non è stata raggiunta un’inversione dell’effetto Warburg. Al contrario, il trattamento con DCA ha causato una crisi bioenergetica, che ha portato a un rallentamento della crescita cellulare attraverso un meccanismo fuori bersaglio.
IlDCA induce la produzione di ROS nelle linee cellulari di PDAC e influenza in modo differenziato la biogenesi e la dinamica mitocondriale
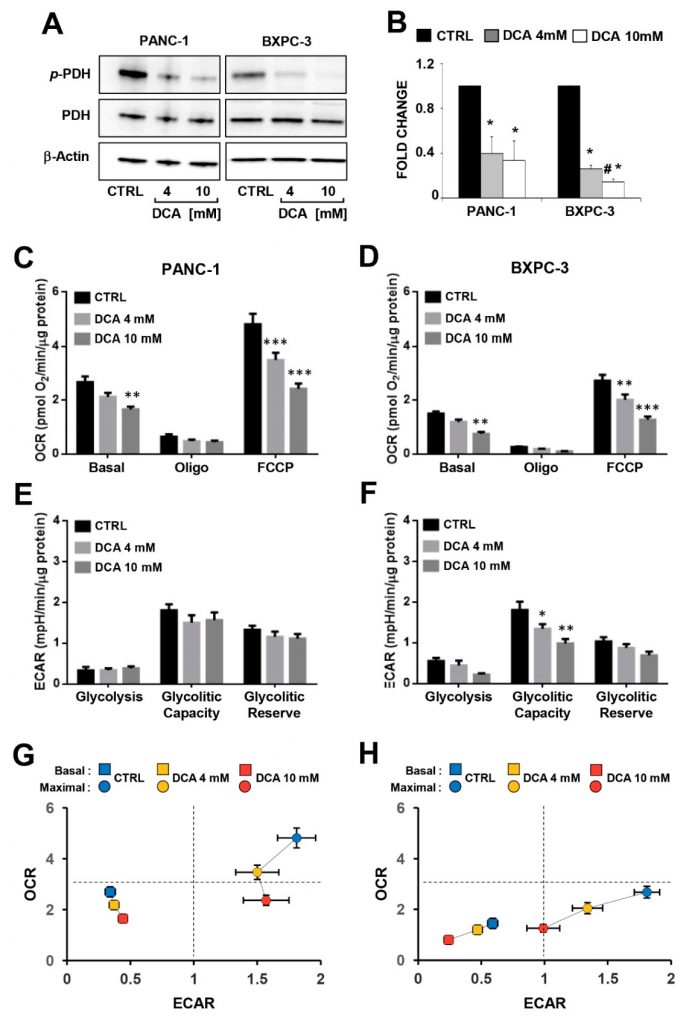
La depressione della respirazione mitocondriale causata dal DCA ci ha indotto a indagare ulteriormente sulle funzioni mitocondriali. In primo luogo, abbiamo valutato l’architettura morfo-funzionale del compartimento mitocondriale mediante immagini in microscopia confocale utilizzando la sonda fluorescente ΔΨ TMRE, che si accumula nei mitocondri in respirazione. La Figura 4A mostra che nel PANC-1 il segnale legato a TMRE ha assunto un aspetto particellare diffuso, largamente diffuso nel citoplasma, indicativo di una struttura prevalentemente frammentata piuttosto che interconnessa. Una caratteristica simile è stata riscontrata anche nelle cellule più piccole BXPC-3, che tuttavia hanno mostrato una compartimentazione peri-nucleare anulare. Il trattamento con DCA per 24 ore non ha provocato cambiamenti rilevanti né nell’intensità del segnale fluorescente di TMRE né nel suo aspetto morfologico.
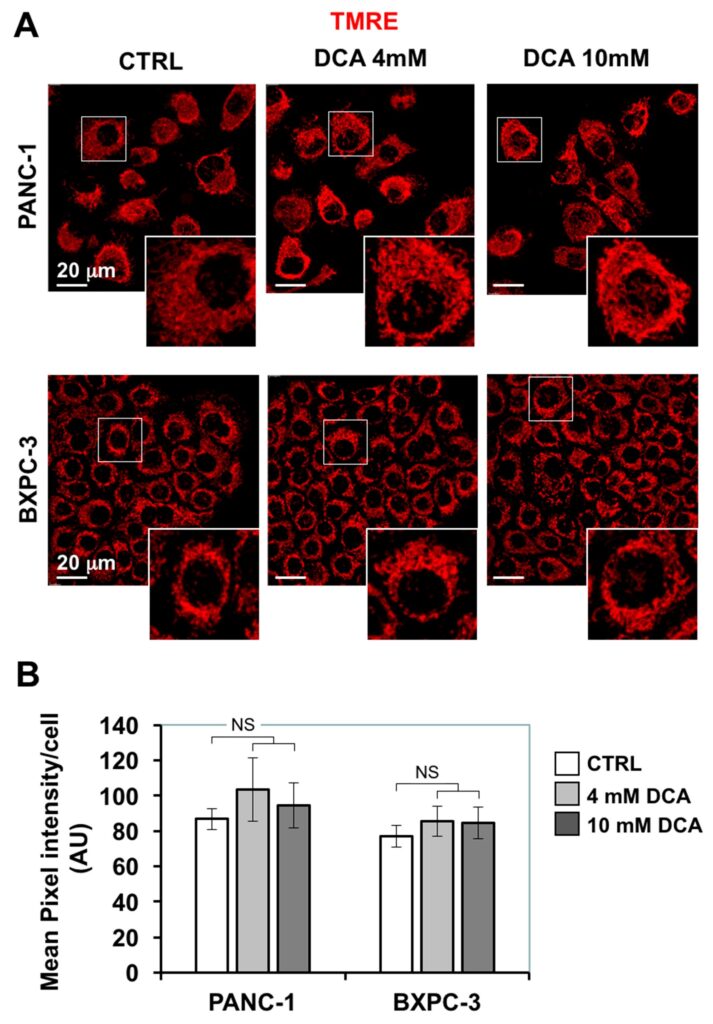
Successivamente, abbiamo analizzato le due linee cellulari PDAC per verificarne il tono redox utilizzando la sonda perossido DCF. Le Figure 5A e B mostrano che il trattamento con DCA 10 mM per 24 ore ha causato un aumento significativamente elevato del segnale DCF in entrambe le linee cellulari PANC-1 e BXPC-3 rispetto ai livelli basali non trattati. Questo risultato indica uno squilibrio pro-ossidativo o lo stato redox causato dall’esposizione al DCA.
L’analisi sopra riportata è stata integrata con la misurazione del DNA mitocondriale (mtDNA). Come mostrato nella Figura 6A, il numero di copie di mtDNA per cellula era significativamente più alto in PANC-1 rispetto a BXPC-3, coerentemente con l’attività respiratoria più attiva. Dopo il trattamento con DCA, è stato osservato un aumento progressivo dose-dipendente del mtDNA in entrambe le linee cellulari di PDAC. Ciò è probabilmente dovuto a una risposta compensatoria alla disfunzione dell’OxPhos causata dal DCA.
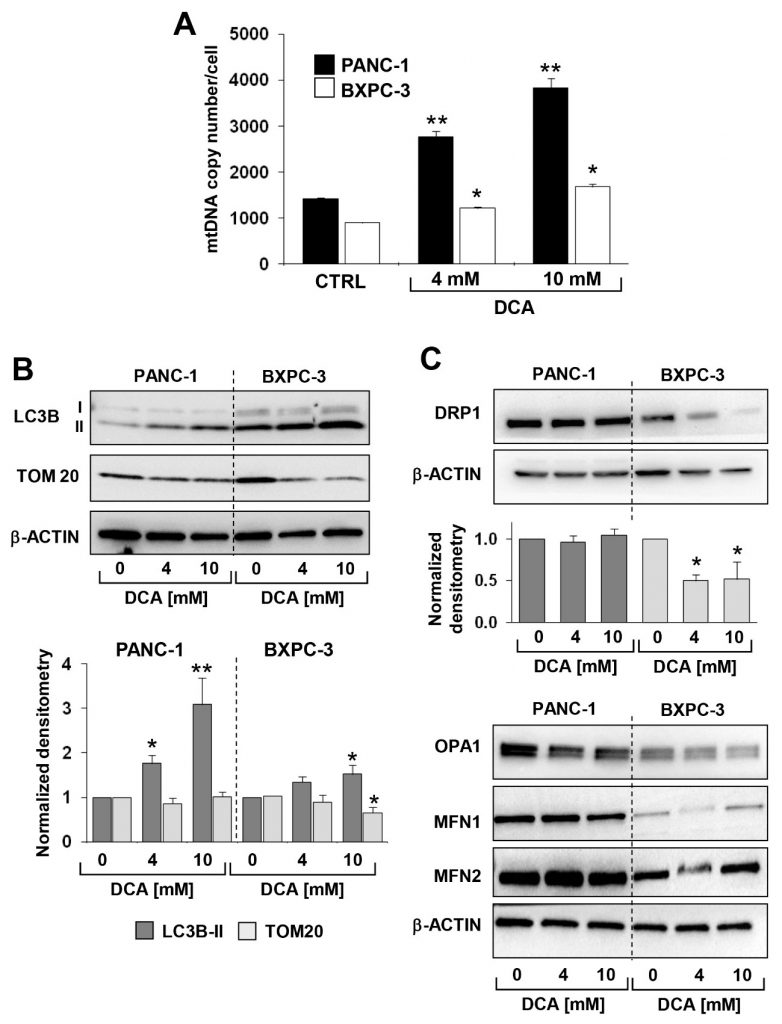
Successivamente, abbiamo valutato mediante immunoblotting il livello di espressione di proteine note per essere coinvolte nella clearance mitocondriale (cioè la mitofagia) e nella dinamica. Le Figure 6B e C mostrano che l’espressione del marcatore di autofagosoma LC3B-II era significativamente più alta nelle BXPC-3 rispetto alle PANC-1 e che il DCA ha causato un aumento progressivo dose-dipendente del marcatore in entrambe le linee cellulari. Tuttavia, TOM20, un marcatore della membrana mitocondriale esterna, è diminuito significativamente solo nella linea cellulare BXPC-3. L’analisi dei fattori coinvolti nel processo di fusione/fissione dei mitocondri ha rivelato che tutti erano espressi a livelli più elevati nella PANC-1 rispetto alla BXPC-3, ma con un effetto differenziale su di essi causato dal trattamento con DCA (Figura 6C). In particolare, solo DRP1, un fattore coinvolto nella fissione mitocondriale, è diminuito a seguito del trattamento con DCA in BXPC-3. Non sono stati rilevati cambiamenti significativi nell’espressione di DRP1 in PANC-1 e dei fattori che inducono la fusione mitocondriale OPA1, MFN1 e MFN2 in entrambe le linee cellulari in seguito al trattamento con DCA (Figura S6 supplementare). Al contrario, le cellule BXPC-3 sembrano fenotipicamente più inclini a perseguire il controllo di qualità dell’organello. Ciò potrebbe essere coerente con l’aspetto più frammentario della rete mitocondriale nelle cellule BXPC-3. Il trattamento con DCA ha provocato un evidente aumento del marcatore mitofagico LC3B in entrambe le linee cellulari, che tuttavia è rimasto molto più grande in BXPC-3.
IlDCA influisce in modo differenziato sul comparto delle cellule staminali del cancro nelle linee cellulari di PDAC
Per indagare sui possibili meccanismi aggiuntivi dell’effetto citostatico del DCA, abbiamo deciso di verificarne l’impatto sulla frazione delle cellule staminali del cancro (CSC) delle linee cellulari di PDAC. A questo scopo, abbiamo valutato l’espressione del fattore delle cellule staminali embrionali Lin28, dimostrato essere coinvolto nel metabolismo cellulare [23] e noto per essere un biomarcatore di prognosi sfavorevole della progressione del cancro [24,25]. La Figura 7A mostra che il trattamento con DCA per 48 ore ha causato una riduzione significativa dell’espressione di Lin28 in entrambe le linee cellulari, con la BXPC-3 che appare più sensibile della PANC-1. Un risultato simile è stato ottenuto nelle colture 3D ottenute da entrambe le linee cellulari (Figura 7B).
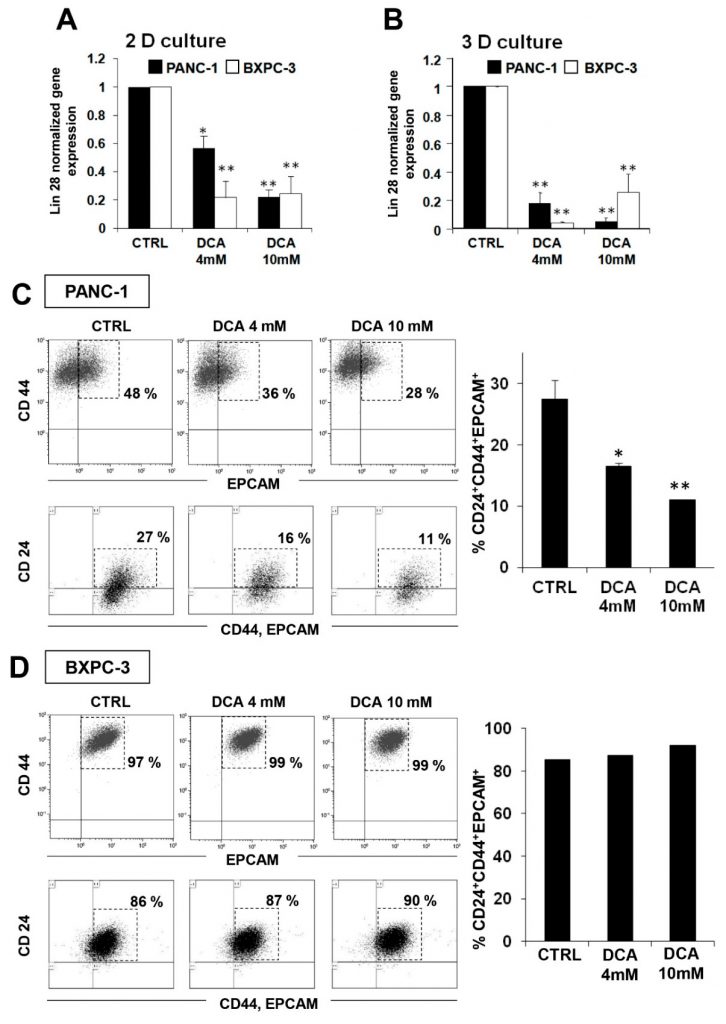
Questa osservazione ci ha spinto ad approfondire l’effetto del DCA sulle CSC mediante analisi FACS dei marcatori di superficie specifici delle CSC pancreatiche, CD24, CD44 ed EPCAM [26,27]. La Figura 7C mostra una significativa riduzione dose-dipendente delle cellule CD24+/CD44+/EPCAM+ nelle cellule PANC-1 trattate per 48 ore con DCA. Da notare che l’intensità della fluorescenza è stata ridotta di quasi il 50% dopo il trattamento con DCA a 4 mM nelle cellule PANC-1, mentre a 10 mM è stata osservata una riduzione più marcata dell’intensità della fluorescenza già dopo 24 ore di trattamento e non è cambiata dopo 48 ore di trattamento (dati non mostrati). Al contrario, sebbene i marcatori CSC fossero espressi in circa il 90% delle cellule BXPC-3, la loro espressione non sembrava essere influenzata dal trattamento con DCA (Figura 7D). È stato recentemente riportato che, più che il livello assoluto di espressione di determinati marcatori, è il loro rapporto a identificare la sottopopolazione con caratteristiche di staminalità più genuine. In particolare, il rapporto CD44/CD24 sembra essere il marcatore più affidabile di CSC nella tumorigenesi e nelle metastasi [28]. In linea con questo concetto, è rilevante che il rapporto di espressione CD44/CD24 in PANC-1 sia risultato 8 volte superiore a quello di BXPC-3 (Figura S7 supplementare), indicando così che, sebbene meno popolato, il comparto CSC è qualitativamente più simile alle cellule staminali. Al contrario, il basso livello di espressione dei marcatori CSC ampiamente diffusi nella popolazione di BXPC-3 li fenotizza come progenitori precoci.
Effetto del DCA su colture 3D
Per approfondire le proprietà antitumorali del DCA, abbiamo analizzato i suoi effetti biologici su un banco di prova alternativo costituito da colture 3D di PANC-1 e BXPC-3. Un numero crescente di evidenze suggerisce che gli sferoidi derivati da cellule tumorali sono arricchiti in CSC o in cellule con caratteristiche correlate alle cellule staminali [29,30]. La Figura 8A mostra le micrografie degli sferoidi ottenuti dopo 7 giorni di coltura, da cui si può notare chiaramente una differenza nelle dimensioni e nella compattezza dello strato limite tra le linee cellulari PANC-1 e BXPC-3. In particolare, gli sferoidi derivati dalle cellule PANC-1 sono apparsi più grandi e con bordi irregolari rispetto a quelli derivati dalle cellule BXPC-3. Il trattamento con DCA per 72 ore ha alterato la morfologia degli sferoidi, che sono diventati progressivamente meno definiti. Ciò è stato particolarmente evidente negli sferoidi derivati da PANC-1 al trattamento con DCA 10 mM. Coerentemente con questa osservazione, è stata chiaramente rilevata una progressiva riduzione dose-dipendente della vitalità cellulare negli sferoidi di PANC-1 e BXPC-3.
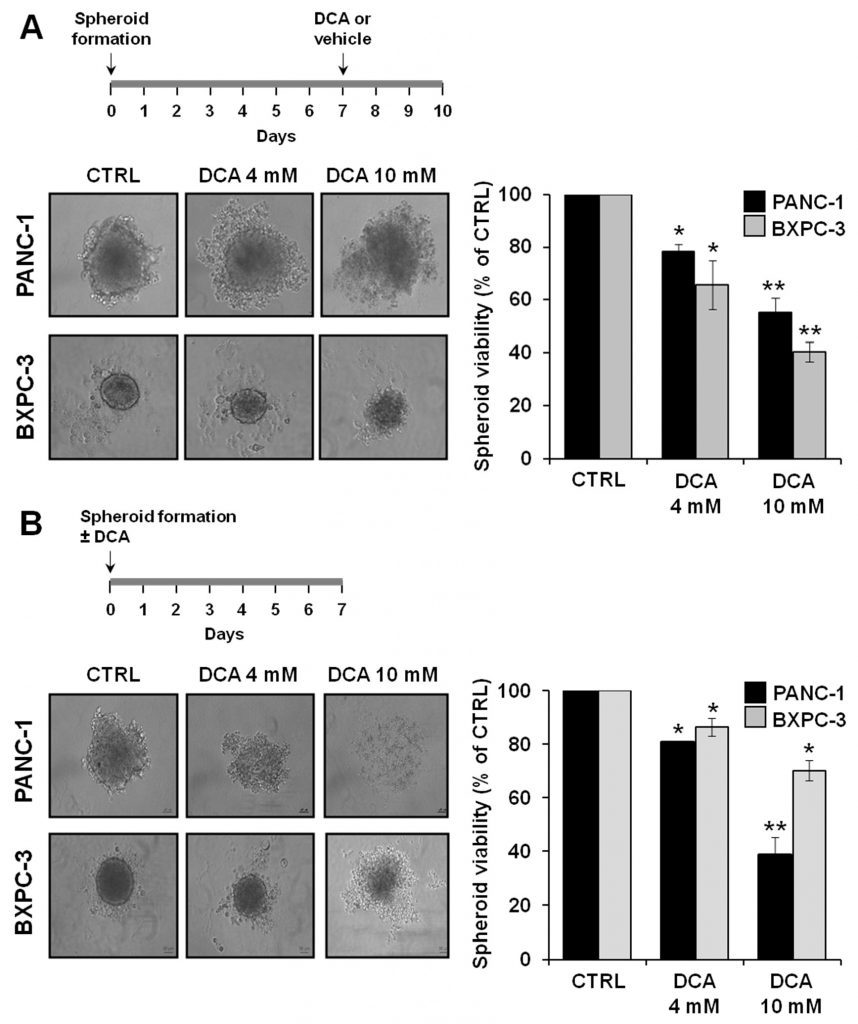
Abbiamo anche studiato la capacità del DCA di influenzare la formazione di sferoidi trattando la sospensione cellulare al momento della semina. Come mostrato nella Figura 8B, il DCA ha fortemente influenzato la formazione di sferoidi in entrambe le linee cellulari di PDAC, con la PANC-1 che è risultata più sensibile al farmaco. Di conseguenza, la vitalità cellulare degli sferoidi di PANC-1 è risultata significativamente più compromessa di quella degli sferoidi di BXPC-3.
IlDCA attenua la progressione del tumore in un modello murino di xenotrapianto di PC
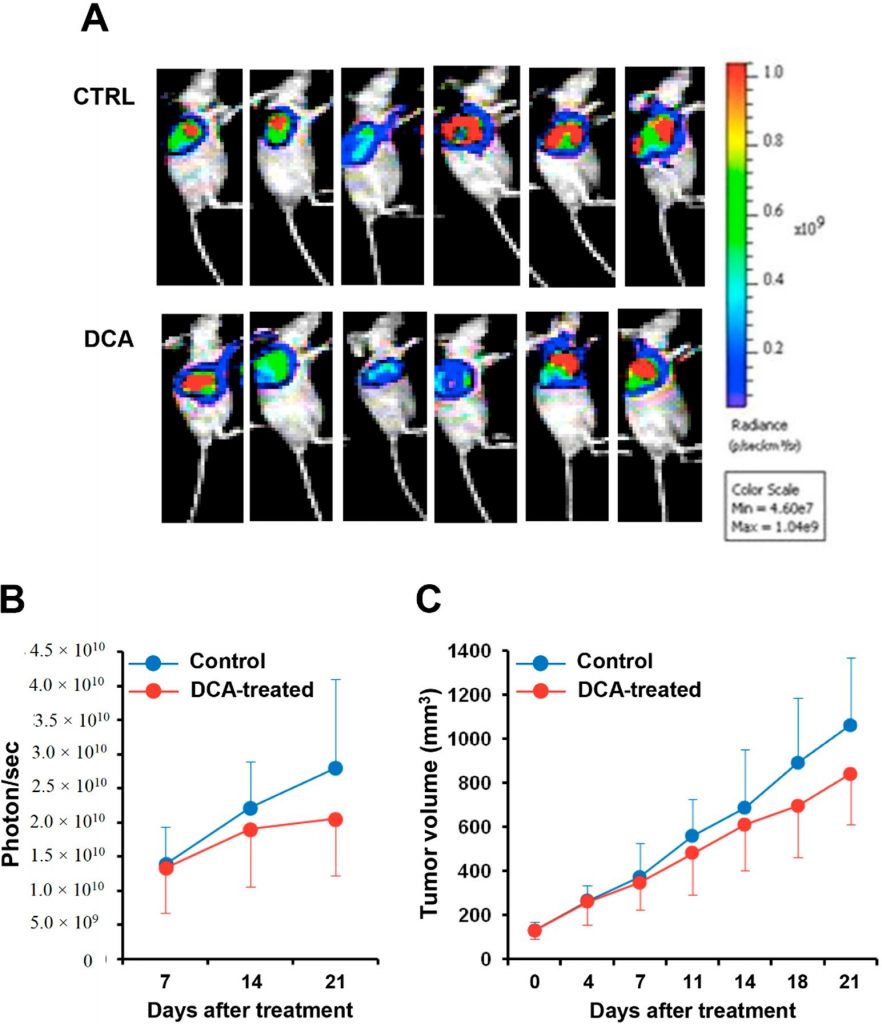
Abbiamo infine valutato l’effetto della somministrazione di DCA in un modello murino di xenotrapianto di tumore del pancreas. La linea cellulare BXPC-3 esprimente luciferasi è stata iniettata in topi nudi e, dopo aver raggiunto un volume di 100 mm3, è stata trattata con DCA o veicolo per tre settimane. La Figura 9A mostra l’imaging della bioluminescenza della massa tumorale nei topi trattati con DCA e in quelli di controllo. La quantificazione del segnale di bioluminescenza ha rivelato un ritardo nella progressione del tumore pancreatico nei topi trattati con DCA, documentato da una riduzione del 25-30% sia dell’intensità del segnale di bioluminescenza della massa tumorale sia del suo volume rispetto ai topi trattati con il veicolo (Figura 9B,C). Tuttavia, a causa della grande variabilità interindividuale, le differenze non hanno raggiunto la significatività statistica.
4. Discussione
Nelle cellule tumorali si verificano generalmente processi metabolici aberranti e, pertanto, il bersaglio del metabolismo rappresenta una strategia emergente per il trattamento dei tumori, compreso il cancro al pancreas [31,32,33]. L’eterogeneità del tumore può portare a cellule maligne con un fenotipo metabolico distinto e, di conseguenza, una diversa sensibilità ai farmaci metabolici, come nel caso della gemcitabina verso la quale la maggior parte dei pazienti con tumore del pancreas sviluppa resistenza [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34].
Nel presente studio, abbiamo testato l’efficacia del farmaco metabolico DCA in colture 2D e 3D di due diverse linee cellulari di cancro al pancreas ben caratterizzate (PANC-1 e BXPC-3) e in un modello di xenotrapianto di cancro al pancreas. Entrambe le linee cellulari, coltivate in monostrato, hanno mostrato una marcata sensibilità al DCA alla più alta concentrazione testata (10 mM), che ha arrestato la proliferazione cellulare e inibito gravemente la capacità di migrazione. Testando il farmaco a una concentrazione più bassa (cioè 4 mM di DCA) si è evidenziata una maggiore sensibilità della linea cellulare PANC-1. Questa osservazione è interessante perché la PANC-1 è segnalata come una linea cellulare aggressiva e resistente alla chemioterapia [21,22,35].
L’analisi dei parametri di vitalità in entrambe le linee cellulari di PDAC trattate con DCA ha evidenziato una percentuale limitata di cellule apoptotiche/necrotiche, suggerendo così un effetto citostatico piuttosto che citotossico esercitato dal farmaco, a conferma di precedenti segnalazioni [36,37].
Un effetto importante del DCA è generalmente attribuito alla sua capacità di indurre un passaggio metabolico dalla glicolisi all’ossidazione mitocondriale del glucosio. Ciò avviene attraverso l’inibizione della chinasi PDH PDK, spostando così la PDH verso il suo stato non fosforilato, più attivo [38,39]. Di conseguenza, il piruvato viene convertito in acetil-CoA che entra nel ciclo degli acidi tricarbossilici e alimenta la fosforilazione ossidativa mitocondriale.
Tuttavia, in questo studio abbiamo riscontrato che, nonostante la sostanziale de-fosforilazione della PDH indotta dal DCA, non è stata osservata alcuna attivazione dell’attività respiratoria mitocondriale in entrambe le linee cellulari di PDAC trattate con il farmaco. Al contrario, il DCA ha causato una riduzione dose-dipendente dell’OxPhos mitocondriale, che è stata accoppiata all’inibizione della capacità glicolitica nelle BXPC-3.
Questo risultato è stato piuttosto sorprendente se si considera che in uno studio precedente con linee cellulari di cancro orale abbiamo dimostrato che le cellule PE15, caratterizzate da una sostenuta OxPhos, erano resistenti al trattamento con DCA, mentre le cellule HSC2/3, che mostrano un profilo glicolitico, apparivano più sensibili al farmaco con un effetto marcato anche sui parametri morfo-funzionali mitocondriali [7]. Inoltre, in un altro studio condotto su linee cellulari PANC-1 e BXPC-3, abbiamo dimostrato una sensibilità differenziale alla deprivazione di glucosio/sostituzione di galattosio, una condizione che favorisce anche il metabolismo ossidativo, con le cellule BXPC-3, più glicolitiche, più vulnerabili [6]. Questo ci ha portato a ipotizzare che la diversa sensibilità di diverse linee cellulari a farmaci o condizioni che promuovono un cambiamento metabolico pro-ossidativo dipenda dal loro profilo metabolico basale, con quelle che si basano maggiormente sulla glicolisi e/o con una bassa capacità respiratoria più vulnerabili.
L’effetto deprimente del DCA sulla respirazione mitocondriale non si è apparentemente tradotto in cambiamenti della morfologia mitocondriale, sebbene sia stata osservata una riduzione significativa del fattore di promozione della fissione Drp1 nelle cellule BXPC-3 trattate con DCA. È probabile che il fenotipo frammentato della rete mitocondriale in BXPC-3 sia nascosto per apprezzare un’ulteriore frammentazione mitocondriale. Tuttavia, il numero di copie di mtDNA/cellula è risultato significativamente aumentato in entrambe le linee cellulari di PDAC, probabilmente a causa di un meccanismo di compensazione come conseguenza della disfunzione mitocondriale che porta all’attivazione della mitofagia, come dimostrato dall’aumento della forma clivata di LC3B-II. L’aumentata produzione di ROS osservata nelle linee cellulari di PDAC trattate con DCA potrebbe indurre il controllo di qualità dell’organello per rimuovere i mitocondri danneggiati. Lo squilibrio dell’omeostasi dei ROS è comunemente correlato alla disfunzione della catena respiratoria mitocondriale, anche se spesso la relazione non è chiara (cioè, causa, effetto, circolo vizioso). Generalmente considerata un meccanismo pro-sopravvivenza che protegge le cellule in condizioni di stress (funzione oncogena) [40], più recentemente è stato dimostrato che la disregolazione della mitofagia contribuisce alla resistenza ai farmaci (ruolo soppressivo del tumore) [41]. In ogni caso, l’induzione e l’inibizione della mitofagia nella progressione del cancro sono ancora controverse.
L’insieme delle osservazioni citate non ci permette di razionalizzare l’effetto citostatico del DCA come semplicemente legato al ricablaggio metabolico delle cellule di PDAC. Occorre considerare che il DCA può colpire altre vie cellulari oltre alla PDK. Infatti, è stato riportato che il DCA influisce sulla via biosintetica del CoA [42], attiva la via di segnalazione AMPK [43], si antagonizza con l’acetato [44] e disturba il catabolismo della tirosina [45]. Inoltre, il confronto dei profili dei metaboliti nelle cellule trattate con DCA o con nuovi inibitori più selettivi della PDK ha dato risultati diversi [46]. Questo ci ha portato a indagare su ulteriori potenziali effetti off-target del DCA per spiegare la sua efficacia nel colpire le cellule tumorali.
Le cellule staminali tumorali (CSC) rappresentano una frazione dell’intera massa tumorale e si sono rivelate responsabili della refrattarietà alla terapia, della diffusione delle metastasi e della recidiva del tumore [47], suscitando così un crescente interesse come bersaglio per lo sviluppo di nuove terapie antitumorali [48]. Per quanto ne sappiamo, non esiste alcun rapporto sull’effetto del DCA sulle cellule staminali del cancro del pancreas. Per approfondire questo intrigante aspetto, abbiamo innanzitutto valutato l’effetto del trattamento con DCA sull’espressione di Lin 28, rivelando una significativa downregulation dose-dipendente rilevabile in entrambe le linee cellulari. L’espressione di Lin28 è strettamente legata al metabolismo, poiché è in grado di regolare la progressione delle cellule tumorali tramite PDK1 e di indurre uno switch energetico [49]. Lin28 è coinvolto nella formazione delle CSC [50] e la sua espressione aberrante è associata a molte malattie neoplastiche umane, tra cui il cancro al pancreas [51,52]. L’analisi FACS dell’espressione degli antigeni di superficie CD44, CD24 ed EPCAM, che caratterizzano tipicamente le CSC pancreatiche [53], ha rivelato che il trattamento con DCA ha ridotto la percentuale della frazione triplamente positiva nel PANC-1. Al contrario, non abbiamo riscontrato alcuna modulazione indotta dal DCA nelle BXPC-3, che erano costituite principalmente da cellule triplo positive. Va tenuto presente che, sebbene oltre il 90% delle BXPC-3 fosse positivo per i marcatori di staminalità, il loro livello di espressione era relativamente basso. Al contrario, il PANC-1 ha espresso livelli più elevati di marcatori di staminalità, anche se solo in meno del 30% della popolazione cellulare, suggerendo un fenotipo di CSC più giovane che caratterizza questo sottogruppo cellulare. Coerente con questa osservazione è l’idea che più che l’espressione assoluta dei marcatori CSC sia il loro rapporto a “qualificare” la propensione alla staminalità delle cellule tumorali [28]. Considerando che il rapporto di espressione CD44/CD24 in PANC-1 è molto più alto rispetto a BXPC-3, ciò indicherebbe che, sebbene meno popolato, il comparto CSC in PANC-1 è qualitativamente più simile alle cellule staminali. Al contrario, il basso livello di espressione dei marcatori CSC ampiamente diffusi nella popolazione di cellule BXPC-3 le fenotipizzerebbe come progenitori precoci. Questa differenza tra le due linee cellulari di carcinoma pancreatico potrebbe spiegare i loro distinti fenotipi metabolici e la sensibilità ai farmaci chemioterapici e al DCA.
La tecnologia delle colture cellulari tridimensionali (3D) è diventata il fulcro della ricerca nella biologia delle cellule tumorali. Rispetto al 2D, le colture 3D di linee cellulari comportano un arricchimento in CSC [30,54] e, imitando i gradienti metabolici e proliferativi dei tumori in vivo, forniscono una previsione più affidabile della risposta a un eventuale trattamento [55,56]. In questa prospettiva, abbiamo testato l’efficacia del DCA su colture 3D ottenute da PANC-1 e BXPC-3 e abbiamo dimostrato che il trattamento con DCA comprometteva la struttura e la vitalità degli sferoidi già formati e comprometteva la formazione di sferoidi da entrambe le linee cellulari. In particolare, alla dose più elevata il DCA è stato in grado di inibire quasi completamente la formazione di sferoidi da PANC-1. Coerentemente con le nostre osservazioni nelle colture 2D, gli sferoidi di BXPC-3 erano meno sensibili di quelli di PANC-1 al trattamento con DCA. Allo stesso modo, nelle colture 2D è stata osservata una significativa downregulation di Lin28 negli sferoidi di entrambe le linee cellulari. Sebbene i cambiamenti sopra riportati nel livello di espressione di marcatori CSC ampiamente riconosciuti in colture 2D e 3D non implichino prove conclusive dell’effetto del DCA sul comparto staminale del PDAC, tuttavia forniscono indizi finora non apprezzati che meritano ulteriori indagini. A livello in vivo, il trattamento con DCA ha causato una crescita tumorale più lenta, anche se non significativa, nei topi portatori di tumore BxPC-3-luc rispetto ai topi di controllo, come valutato dalla riduzione del conteggio dei fotoni e del volume del tumore.
Per razionalizzare gli effetti sconcertanti del DCA sulle linee cellulari di PDAC riportati nel nostro studio, abbiamo proposto la seguente sequenza ipotetica di eventi con lo scopo di stimolare ulteriori indagini (Figura 10). Suggeriamo che, a seguito di una maggiore ossidazione del piruvato, un maggior numero di equivalenti riducenti venga trasferito alla catena respiratoria mitocondriale con generazione di ROS. I complessi respiratori sono sia produttori che bersaglio di ROS [57], favorendo così un circolo vizioso che porta alla progressiva inibizione del trasferimento funzionale di elettroni lungo la catena respiratoria, promuovendo un’ulteriore deviazione di elettroni verso l’O2. Inoltre, lo smorzamento dell’attività respiratoria può causare l’accumulo di intermedi del ciclo degli acidi tricarbossilici e di acetil-CoA. Quando quest’ultimo si accumula, è noto che provoca l’acetilazione della lisina e l’inibizione della funzione di numerose proteine mitocondriali, compresi i complessi della catena respiratoria [58,59]. Il progressivo danno/disfunzione mitocondriale che ne consegue è contrastato dalla regolazione della mitofagia. Come e se queste alterazioni mitocondriali mediate dai DCA causino l’arresto della crescita e il ricablaggio del fenotipo cellulare, in particolare del compartimento delle cellule staminali tumorali, resta da stabilire. Tuttavia, una serie di evidenze riportate indicano che uno stato pro-ossidativo causa l’uscita delle cellule staminali dallo stato indifferenziato e induce/favorisce il commitment [60,61]. Inoltre, le modifiche epigenetiche, come quelle che causano il rimodellamento della cromatina, regolano l’equilibrio tra pluripotenza e differenziazione delle cellule staminali [62,63]. È possibile che il ciclo TCA in stallo causi l’efflusso del citrato nel citosol, dove rilascia acetil-CoA, aumentando così la sua disponibilità per l’acetilazione degli istoni. Ovviamente, altri bersagli non caratterizzati del DCA possono contribuire o addirittura dominare gli effetti del farmaco osservati.
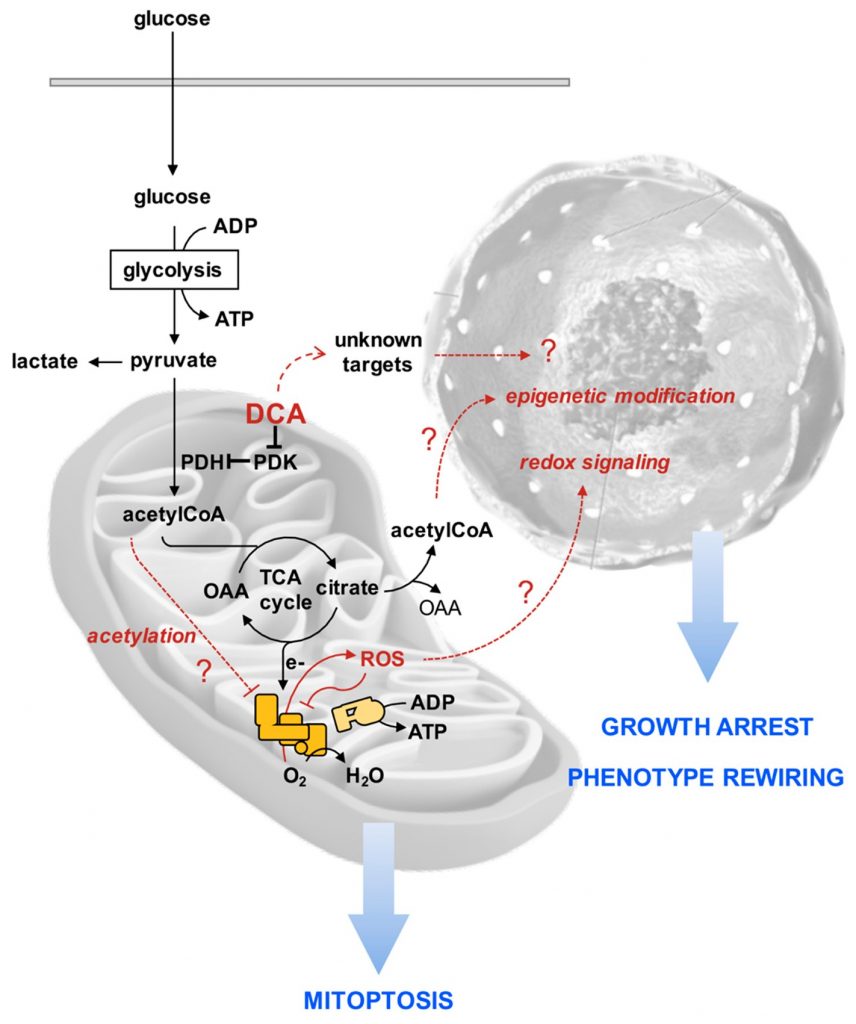
In conclusione, i nostri risultati indicano chiaramente che l’efficacia del DCA nell’inibire la crescita delle cellule tumorali non è sempre causalmente legata al suo documentato effetto stimolante sull’attività della PDH e di conseguenza all’effetto Warburg inverso. Altri off-target devono essere considerati meccanicamente a seconda del fenotipo cellulare. In questo contesto, è rilevante l’evidenza, emersa da questo studio, che il compartimento delle CSC nelle linee cellulari derivate dal PDAC potrebbe essere influenzato dal trattamento con DCA. Sarebbe opportuno verificare se ciò si verifica in altri tipi di cellule tumorali e nel nostro laboratorio si sta lavorando in questa direzione. I recenti progetti di ricerca per lo sviluppo di nanoparticelle caricate con dicloroacetato e funzionalizzate in superficie [64] e di farmaci multifunzione ottenuti da agenti chemioterapici con DCA come ligando [65] potrebbero aiutare a progettare la giusta formulazione farmacologica mirata per lo sviluppo di nuove strategie terapeutiche efficaci per combattere il pancreas e altri tipi di cancro.
Materiali supplementari
I seguenti sono disponibili online su https://www.mdpi.com/2073-4409/8/5/478/s1: Figura S1: Effetto del DCA sulla proliferazione cellulare valutata mediante XCELLigence in terreno contenente basso glucosio; Figura S2: Guarigione da graffio; Figura S3: Effetto del DCA sulla produzione di lattato; Figura S4: Figura S4: Effetto del trattamento con DCA per 24 ore sui flussi metabolici; Figura S5: Effetto comparativo del DCA sui flussi metabolici in linee cellulari di PDAC in condizioni di coltura a basso glucosio (LG) e ad alto glucosio (HG); Figura S6: Espressione proteica dei fattori coinvolti nella fusione-fissione dei mitocondri (OPA1, MFN1/2); Figura S7: Valutazione della staminalità nelle linee cellulari di PDAC.
Contributi degli autori
T.T. pianificazione, indagine e scrittura; F.A., C.P. (Consiglia Pacelli) e C.M. indagine; V.R. e I.L. cura dei dati; G.D.S. visualizzazione; C.P. (Concetta Panebianco) e V.P. modello animale; N.C. revisione ed editing e C.P. (Claudia Piccoli) concettualizzazione, supervisione.
Finanziamento
Questa ricerca è stata finanziata da fondi di ricerca corrente del Ministero della Salute all’IRCCS CROB e da borse di studio del Ministero della Salute attraverso la Divisione di Gastroenterologia (RC1703GA31 e RC1803GA30) dell’IRCCS “Casa Sollievo della Sofferenza”.
Conflitti di interesse
Gli autori non dichiarano alcun conflitto di interesse.
RIFERIMENTI
1 Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetica e biologia dell’adenocarcinoma duttale pancreatico. Genes Dev. 2016, 30, 355-385. [CrossRef]2 Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605-1617. [CrossRef] [PubMed]
3 Camelo, F.; Le, A. The Intricate Metabolism of Pancreatic Cancers. Adv. Exp. Med. Biol. 2018, 1063, 73-81. [PubMed]
4 Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, E1338. [CrossRef] [PubMed]
5 Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: Nuovi concetti in un vecchio campo. Antioxid. Redox Signal. 2017, 26, 462-485. [CrossRef]
6 Tataranni, T.; Agriesti, F.; Ruggieri, V.; Mazzoccoli, C.; Simeone, V.; Laurenzana, I.; Scrima, R.; Pazienza, V.; Capitanio, N.; Piccoli, C. Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cells lines with diverse metabolic profiles. Oncotarget 2017, 8, 41265-41281. [CrossRef]
7 Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Lo Muzio, L.; Capitanio, N.; Piccoli, C. Dicloroacetato, un farmaco selettivo mirato ai mitocondri per il carcinoma orale a cellule squamose: una prospettiva metabolica del trattamento. Oncotarget 2015, 6, 1217-1230. [CrossRef]
8 Anderson, K.M.; Jajeh, J.; Guinan, P.; Rubenstein, M. Effetti in vitro del dicloroacetato e della CO2 su cellule HeLa ipossiche. Anticancer Res. 2009, 29, 4579-4588.
9 Chen, Y.; Cairns, R.; Papandreou, I.; Koong, A.; Denko, N.C. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS ONE 2009, 4, e7033. [CrossRef]
10 Lu, X.; Zhou, D.; Hou, B.; Liu, Q.X.; Chen, Q.; Deng, X.F.; Yu, Z.B.; Dai, J.G.; Zheng, H. Il dicloroacetato potenzia l’efficacia antitumorale degli agenti chemioterapici attraverso l’inibizione dell’autofagia nel tumore del polmone non a piccole cellule. Cancer Manag. Res. 2018, 10, 1231-1241. [CrossRef]
11 Yang, C.; Wu, T.; Qin, Y.; Qi, Y.; Sun, Y.; Kong, M.; Jiang, X.; Qin, X.; Shen, Y.; Zhang, Z. A easy doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery. Int. J. Nanomed. 2018, 13, 1281-1293. [CrossRef]
12 Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017, 23, 5639-5647. [CrossRef]
13 Khan, A.; Marier, D.; Marsden, E.; Andrews, D.; Eliaz, I. Una nuova forma di terapia con dicloroacetato per pazienti con cancro avanzato: Un rapporto di 3 casi. Altern. Ther. Health Med. 2014, 20 (Suppl. 2), 21-28.
14 Hanberry, B.S.; Berger, R.; Zastre, J.A. La vitamina B1 ad alte dosi riduce la proliferazione in linee cellulari tumorali analogamente al dicloroacetato. Cancer Chemother. Pharm. 2014, 73, 585-594. [CrossRef]
15 Lowe, A.W.; Olsen, M.; Hao, Y.; Lee, S.P.; Taek Lee, K.; Chen, X.; van de Rijn, M.; Brown, P.O. Gene expression patterns in pancreatic tumors, cells and tissues. PLoS ONE 2007, 2, e323. [CrossRef]
16 Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425-435. [CrossRef]
17 Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifica gli adenocarcinomi duttali pancreatici in sottotipi con sensibilità distinte agli inibitori metabolici. Proc. Natl. Acad. Sci. USA 2015, 112, E4410-E4417. [CrossRef]
18 Mazzoccoli, C.; Ruggieri, V.; Tataranni, T.; Agriesti, F.; Laurenzana, I.; Fratello, A.; Capitanio, N.; Piccoli, C. N-acetilaspartato (NAA) induce la differenziazione neuronale della linea cellulare di neuroblastoma SH-SY5Y e la sensibilizza agli agenti chemioterapici. Oncotarget 2016, 7, 26235-26246. [CrossRef]
19 Scrima, R.; Menga, M.; Pacelli, C.; Agriesti, F.; Cela, O.; Piccoli, C.; Cotoia, A.; De Gregorio, A.; Gefter, J.V.; Cinnella, G.; et al. Il para-idrossifenilpiruvato inibisce la stimolazione pro-infiammatoria dei macrofagi prevenendo lo squilibrio nitro-ossidativo mediato da LPS e lo shift immunometabolico. PLoS ONE 2017, 12, e0188683. [CrossRef]
20 Fredebohm, J.; Boettcher, M.; Eisen, C.; Gaida, M.M.; Heller, A.; Keleg, S.; Tost, J.; Greulich-Bode, K.M.; Hotz-Wagenblatt, A.; Lathrop, M.; et al. Creazione e caratterizzazione di una linea cellulare di cancro al pancreas altamente tumorigenica e arricchita di cellule staminali tumorali come sistema modello ben definito. PLoS ONE 2012, 7, e48503. [CrossRef]
21 Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Meccanismi alla base della resistenza alla gemcitabina nel tumore del pancreas e sensibilizzazione da parte della lenalidomide iMiD™. Anticancer Res. 2011, 31, 3747-3756.
22 Yin, T.; Wei, H.; Gou, S.; Shi, P.; Yang, Z.; Zhao, G.; Wang, C. Le cellule simil-cancro arricchite nelle sfere Panc-1 possiedono una maggiore capacità di migrazione e resistenza alla gemcitabina. Int. J. Mol. Sci. 2011, 12, 1595-
23 Jun-Hao, E.T.; Gupta, R.R.; Shyh-Chang, N. Lin28 e let-7 nella fisiologia metabolica dell’invecchiamento. Trends Endocrinol. Metab. 2016, 27, 132-141. [CrossRef] [PubMed]
24 Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66-80. [CrossRef] [PubMed]
25 Wang, X.; Weng, M.; Jin, Y.; Yang, W.; Wu, D.; Wang, T.; Li, X. Beyond an oncogene, Lin28 is a master regulator of cancer progression. Histol. Histopathol. 2018, 33, 327-334. [PubMed]
26 Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identificazione delle cellule staminali del cancro del pancreas. Cancer Res. 2007, 67, 1030-1037. [CrossRef]
27 Ohara, Y.; Oda, T.; Sugano, M.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Akashi, Y.; Miyamoto, R.; Kobayashi, A.; Fukunaga, K.; et al. Importanza istologica e prognostica dell’espressione di CD44(+)/CD24(+)/EpCAM(+) nel cancro pancreatico clinico. Cancer Sci. 2013, 104, 1127-1134. [CrossRef]
28 Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Correzione dell’autore: Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2018, 8, 4276. [CrossRef] [PubMed]
29 Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 16. [CrossRef] [PubMed]
30 Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Rilevanza delle cellule staminali del cancro e applicazioni cliniche. Cancer Sci. 2017, 108, 283-289. [CrossRef]
31 Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Dipendenza da oncogeni e non-oncogeni. Cell 2009, 136, 823-837. [CrossRef]
32 Vivanco, I. Targeting molecular addictions in cancer. Br. J. Cancer 2014, 111, 2033-2038. [CrossRef]
33 Belizário, J.E.; Sangiuliano, B.A.; Perez-Sosa, M.; Neyra, J.M.; Moreira, D.F. Using Pharmacogenomic Databases for Discovering Patient-Target Genes and Small Molecule Candidates to Cancer Therapy. Front. Pharmacol. 2016, 7, 312. [CrossRef]
34 Grasso, C.; Jansen, G.; Giovannetti, E. Resistenza ai farmaci nel cancro del pancreas: impatto dell’alterazione del metabolismo energetico. Crit. Rev. Oncol. Hematol. 2017, 114, 139-152. [CrossRef]
35 Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. La chemioresistenza intrinseca alla gemcitabina è associata alla fosforilazione costitutiva e indotta dalla laminina di FAK in linee cellulari di cancro del pancreas. Mol. Cancer 2009, 8, 125. [CrossRef]
36 Fedorchuk, A.G.; Pyaskovskaya, O.N.; Gorbik, G.V.; Prokhorova, I.V.; Kolesnik, D.L.; Solyanik, G.I. L’efficacia del dicloroacetato di sodio contro il glioma C6 dipende dallo schema di somministrazione e dal dosaggio. Exp. Oncol. 2016, 38, 80-83. [CrossRef]
37 Ma, W.; Zhao, X.; Wang, K.; Liu, J.; Huang, G. L’acido dicloroacetico (DCA) sinergizza con l’inibitore SIRT2, il sirtinolo e l’AGK2 per aumentare l’efficacia antitumorale nel carcinoma polmonare non a piccole cellule. Cancer Biol. Ther. 2018, 19, 835-846. [CrossRef]
38 Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci. Transl. Med. 2010, 2, 31ra34. [CrossRef]
39 Papandreou, I.; Goliasova, T.; Denko, N.C. Farmaci antitumorali che agiscono sul metabolismo: il dicloroacetato è il nuovo paradigma? Int. J. Cancer 2011, 128, 1001-1008. [CrossRef]
40 Yan, C.; Li, T.S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617-621.
41 Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell Mol. Life Sci. 2018. [CrossRef]
42 Dubuis, S.; Ortmayr, K.; Zampieri, M. A framework for large-scale metabolome drug profiling links coenzyme A metabolism to the toxicity of anti-cancer drug dichloroacetate. Commun. Biol. 2018, 1, 101. [CrossRef]
43 Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604-617. [CrossRef]
44 El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.; El-Sawy, S.A.; Ayat, M.; Elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; et al. Il dicloroacetato è un antimetabolita che antagonizza l’acetato e priva le cellule tumorali dei suoi benefici: Una nuova ipotesi medica basata sull’evidenza. Med. Hypotheses 2019, 122, 206-209. [CrossRef] [PubMed]
45 Guo, X.; Dixit, V.; Liu, H.; Shroads, A.L.; Henderson, G.N.; James, M.O.; Stacpoole, P.W. Inibizione e recupero della glutatione S-transferasi zeta epatica di ratto e alterazione del metabolismo della tirosina in seguito all’esposizione e alla sospensione del dicloroacetato. Drug Metab. Dispos. 2006, 34, 36-42. [CrossRef]
46 Zhang, W.; Hu, X.; Zhou, W.; Tam, K.Y. Liquid Chromatography-Tandem Mass Spectrometry Method Revealed that Lung Cancer Cells Exhibited Distinct Metabolite Profiles upon the Treatment with Different Pyruvate Dehydrogenase Kinase Inhibitors. J. Proteome Res. 2018, 17, 3012-3021. [CrossRef]
47 Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [CrossRef] [PubMed]
48 Chen, L.S.; Wang, A.X.; Dong, B.; Pu, K.F.; Yuan, L.H.; Zhu, Y.M. Una nuova prospettiva nella terapia del cancro: Prendere di mira le cellule staminali del cancro per debellarlo. Chin. J. Cancer 2012, 31, 564-572. [CrossRef]
49 Ma, X.; Li, C.; Sun, L.; Huang, D.; Li, T.; He, X.; Wu, G.; Yang, Z.; Zhong, X.; Song, L.; et al. L’asse Lin28/let-7 regola la glicolisi aerobica e la progressione del cancro tramite PDK1. Nat. Commun. 2014, 5, 5212. [CrossRef]
50 Zhou, J.; Ng, S.B.; Chng, W.J. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol. 2013, 45, 973-978. [CrossRef] [PubMed]
51 Wang, Y.; Li, J.; Guo, S.; Ouyang, Y.; Yin, L.; Liu, S.; Zhao, Z.; Yang, J.; Huang, W.; Qin, H.; et al. Lin28B facilita la progressione e le metastasi dell’adenocarcinoma duttale pancreatico. Oncotarget 2017, 8, 60414-60428. [CrossRef] [PubMed]
52 Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [CrossRef] [PubMed]
53 Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Caratteristiche e metodi di rilevamento. Pathol. Oncol. Res. 2018, 24, 797-805. [CrossRef]
54 Ravi, M.; Ramesh, A.; Pattabhi, A. Contributi delle colture cellulari 3D per la ricerca sul cancro. J. Cell Physiol. 2017, 232, 2679-2697. [CrossRef]
55 Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Screening di farmaci basato su sferoidi: Considerazioni e approccio pratico. Nat. Protoc. 2009, 4, 309-324. [CrossRef]
56 Gutierrez-Barrera, A.M.; Menter, D.G.; Abruzzese, J.L.; Reddy, S.A. Establishment of three-dimensional cultures of human pancreatic duct epithelial cells. Biochem. Biophys. Res. Commun. 2007, 358, 698-703. [CrossRef]
57 Dröse, S.; Brandt, U.; Wittig, I. I complessi della catena respiratoria mitocondriale come fonti e bersagli della redox-regolazione basata sui tioli. Biochim. Biophys. Acta 2014, 1844, 1344-1354. [CrossRef]
58 Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomica, regolazione da parte delle sirtuine e implicazioni metaboliche e patologiche. Cell Metab. 2018, 27, 497-512. [CrossRef]
59Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. L’acetilazione dipendente dai geni dell’orologio del complesso I regola l’attività ritmica dell’OxPhos mitocondriale. Biochim. Biophys. Acta 2016, 1863, 596-606. [CrossRef]
60 Chaudhari, P.; Ye, Z.; Jang, Y.Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 2014, 20, 1881-1890. [CrossRef]
61 Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015. [CrossRef] [PubMed]
62 Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018, 2018, 8908751. [CrossRef]
63 Yadav, T.; Quivy, J.P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332-1336. [CrossRef]
64 Abánades Lázaro, I.; Haddad, S.; Rodrigo-Muñoz, J.M.; Orellana-Tavra, C.; Del Pozo, V.; Fairen-Jimenez, D.; Forgan, R.S. Mechanistic Investigation into the Selective Anticancer Cytotoxicity and Immune System Response of Surface-Functionalized, Dichloroacetate-Loaded, UiO-66 Nanoparticles. ACS Appl. Mater Interfaces 2018, 10, 5255-5268. [CrossRef] [PubMed]
65 Petruzzella, E.; Sirota, R.; Solazzo, I.; Gandin, V.; Gibson, D. Triple action Pt(iv) derivatives of cisplatin: A new class of potent anticancer agents that overcome resistance. Chem. Sci. 2018, 9, 4299-4307. [CrossRef]
Contenuti correlati: