D.L. Kolesnik1, O.N. Pyaskovskaya1, I.V. Boychuk1, O.I. Dasyukevich1, O.R. Melnikov1, A.S. Tarasov1, G.I. Solyanik1
1R.E. Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology, NAS of Ukraine, Kyiv 03022, Ucraina.
Corrispondenza: D.L. Kolesnik, E-mail: [email protected]
Inviato: 20 marzo 2015
Abstract
Un tratto distintivo delle neoplasie è l’eccessiva glicolisi tumorale, anche in presenza di ossigeno, che causa lattacidosi nel microambiente tumorale e favorisce la proliferazione e la sopravvivenza delle cellule tumorali. Per questo motivo gli agenti antimetabolici che hanno come bersaglio il metabolismo delle cellule tumorali sono oggetto di numerose ricerche come promettenti farmaci antitumorali. Scopo: studiare l’effetto della lattacidosi sulla sopravvivenza delle cellule di carcinoma polmonare di Lewis (LLC) in condizioni di carenza di substrato nutrizionale in vitro e valutare l’attività antitumorale e antimetastatica contro LLC/R9 in vivo. Materiali e metodi: La variante LLC/R9 è stata utilizzata come modello tumorale sperimentale. La vitalità delle cellule tumorali è stata determinata mediante colorazione con blu di tripan. Il livello di apoptosi è stato contato con il colorante Hoechst 33258. Il contenuto di lattato nel tessuto tumorale è stato valutato con il metodo enzimatico della lattato deidrogenasi. Le specie reattive dell’ossigeno sono state determinate utilizzando il 2,7-diclorofluoresceina diacetato. Gli effetti del dicloroacetato (DCA) sulla crescita e sulle metastasi di LLC/R9 sono stati analizzati con procedure di routine. La valutazione dell’effetto del DCA sui componenti della catena di trasporto degli elettroni (ETC) è stata eseguita mediante EPR. Risultati: È stato dimostrato che in condizioni di lattacidosi e carenza di glucosio, la vitalità delle cellule LLC/R9 in vitro era superiore del 30% (р < 0,05) e il livello di apoptosi era triplicato (р < 0,05) rispetto a questi indici in condizioni di sola carenza di glucosio. Nei topi con tumori LLC/R9 trapiantati trattati per 3 settimane per os con DCA alla dose totale di 1,5 g/kg di peso corporeo a partire dal giorno successivo al trapianto del tumore, il volume del tumore primario è risultato inferiore del 30% rispetto al gruppo di controllo. Allo stesso tempo, il numero e il volume delle metastasi polmonari negli animali trattati con DCA sono risultati inferiori rispettivamente del 59% (р < 0,05) e del 94% (р < 0,05) rispetto a questi indici nel gruppo di controllo. Il trattamento con DCA ha determinato un aumento di quasi il 30% (р < 0,05) del contenuto di lattato nel tessuto tumorale rispetto a quello del controllo, ma non ha influenzato in modo significativo i livelli dei complessi del ferro eme con NO (con gmed = 2,007) nelle proteine ETC mitocondriali e delle proteine del cluster Fe-S (con g = 1,94) nelle cellule tumorali. Conclusioni: È stato dimostrato che la lattacidosi ha promosso in modo significativo la sopravvivenza delle cellule LLC/R9 in condizioni di carenza di glucosio in vitro. Se LLC/R9 si sono sviluppate in vivo, il DCA come composto con attività antilattacidosi non ha soppresso in modo significativo la crescita del tumore primario, ma ha esercitato una significativa attività antimetastatica.
Parole chiave: dicloroacetato; radiosensibilità ipossica; cancro al seno; specie reattive dell’ossigeno
Abbreviazioni: DCA – dicloroacetato, ETC – catena di trasporto degli elettroni; LLC/R9 – variante del carcinoma polmonare di Lewis; PDH – piruvato deidrogenasi; PDK – piruvato deidrogenasi chinasi.
INTRODUZIONE
È noto che la lattacidosi, ovvero il grande accumulo di lattato e la diminuzione di рН, è la principale caratteristica del microambiente metabolico delle cellule tumorali in vitro e in vivo. In precedenza la lattacidosi è stata considerata come un prodotto di zavorra del metabolismo delle cellule tumorali. Tuttavia, recentemente è stato dimostrato che potrebbe essere utilizzata dalle cellule tumorali come efficace carburante energetico ed essere tra i fattori responsabili della resistenza tumorale alla carenza di glucosio [1-3]. Come abbiamo dimostrato utilizzando cellule di carcinoma polmonare di Lewis (LLC)/R9, variante di LLC sensibile alla terapia antiangiogenica del cancro [4,5], la lattacidosi potrebbe promuovere la sopravvivenza delle cellule tumorali in condizioni di carenza nutrizionale. Tali condizioni sono state generate mediante incubazione a lungo termine di cellule tumorali senza sostituzione del terreno di coltura (modello “unfed culture”) [6]. Lo studio della cinetica di crescita delle cellule tumorali in condizioni di “coltura non nutrita” ha dimostrato che, in assenza totale di glucosio nel terreno di incubazione, nei giorni 7-8 di crescita delle cellule, la conta delle cellule vitali non scendeva al di sotto della terza parte rispetto al massimo registrato nei giorni 3-4, e rimaneva praticamente a questo livello fino al10° giorno. L’elevato tasso di sopravvivenza delle cellule LLC/R9 nelle condizioni di “coltura non alimentata” era legato in particolare alla capacità di queste cellule di macroautofagia. Tuttavia, non si può escludere che la capacità di adattamento delle cellule LLC/R9 alla carenza di substrati nutrizionali sia stata determinata dalla lattacidosi che si è sviluppata come conseguenza della coltura prolungata delle cellule tumorali senza sostituzione del terreno di incubazione.
Se la lattacidosi è in grado di aumentare la sopravvivenza delle cellule tumorali, allora i composti che sopprimono la formazione di lattacidosi nel microambiente tumorale, in particolare il dicloroacetato (DCA) come composto con attività antilattacida, dovrebbero dimostrare attività antitumorale. Il presente studio si proponeva di verificare tale ipotesi.
È noto che il DCA è un inibitore della piruvato deidrogenasi chinasi (PDK), per cui è considerato un regolatore negativo degli enzimi del complesso della piruvato deidrogenasi (PDH) mitocondriale, che svolge un ruolo chiave nella regolazione dell’acido tricarbossilico e della fosforilazione ossidativa [7]. Se il complesso PDH viene fosforilato, l’ingresso del piruvato nel ciclo di Krebs viene inibito e la glicolisi viene attivata. A causa dell’inibizione della PDK, il DCA è in grado di portare all’attivazione indiretta degli enzimi del complesso PDH e, rispettivamente, provoca lo spostamento dell’equilibrio energetico della cellula dalla glicolisi all’attivazione della fosforilazione ossidativa. Pertanto, il DCA è stato ampiamente utilizzato per correggere la lattiemia causata da un’elevata intensità di glicolisi o da una respirazione cellulare difettosa.
Secondo i dati della letteratura, la capacità del DCA di attivare la fosforilazione ossidativa è alla base della sua attività antitumorale e si esplica, in particolare, attraverso la diminuzione della lattacidosi e l’induzione di specie reattive dell’ossigeno (ROS) [8-12]. Lo scopo del nostro studio è stato quello di analizzare l’influenza della lattacidosi sulla sopravvivenza delle cellule LLC/R9 in condizioni di carenza di nutrienti in vitro e di valutare l’attività antitumorale e antimetastatica del DCA contro le LLC/R9 in vivo.
MATERIALI E METODI
Animali sperimentali, cellule tumorali
Lo studio è stato condotto utilizzando topi C57Bl/6 di 2,0-2,5 mesi di età, del peso di 18-23 g, allevati presso la struttura animale dell’Istituto R.E. Kavetsky di Patologia Sperimentale, Oncologia e Radiobiologia della NAS dell’Ucraina. I protocolli di studio sugli animali e le procedure operative sono stati eseguiti in conformità ai principali requisiti per la detenzione e il lavoro con gli animali da laboratorio e alle regole del Comitato di Bioetica locale.
Nello studio è stata utilizzata la variante LLC/R9 derivata dal ceppo LLC wild-type mediante 9 sessioni sequenziali di chemioterapia in vivo a base di cis-diamminedicloroplatino (cis-DDP) [13]. Le cellule LLC/R9 sono state mantenute in terreno di coltura RPMI (Sigma, USA) integrato con 10% di siero fetale di vitello (FCS) (Sigma, USA) e 40 mg/ml di gentamicina a 37 °C in atmosfera umidificata con il 5% diCO2.
Esperimenti in vitro
La conta delle cellule in sospensione e la loro vitalità è stata analizzata di routine su un emocitometro utilizzando il test di esclusione del blu di tripan.
Per valutare gli effetti della lattacidosi sulla vitalità delle cellule LLC/R9, sono state seminate 1,5-105 cellule/pozzetto in una piastra da 24 pozzetti in terreno RPMI 1640 (Sigma, USA) con un contenuto standard di glucosio. Dopo un’incubazione di una notte, il terreno di incubazione delle cellule è stato sostituito con un terreno fresco a diverso contenuto di glucosio e lattato e a diverso pH per simulare le condizioni di carenza di glucosio, lattacidosi in condizioni di carenza di glucosio e standard (Tabella 1). Il terreno carente di glucosio è stato preparato sulla base del terreno RPMI 1640 senza glucosio (Sigma, USA). La lattacidosi è stata generata aggiungendo acido lattico puro (Sigma, USA) al terreno carente di glucosio fino a raggiungere una concentrazione finale di 14 ± 0,7 mM e pH 6,7.
| Terreno | Contenuto di glucosio, mM | Contenuto di lattato, mM | рН |
|---|---|---|---|
| Standard | 9.0 ± 0.5 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Carenza di glucosio | 3.0 ± 0.1 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Lattacidosi | 3.0 ± 0.1 | 14.0 ± 0.7 | 6.7 ± 0.01 |
L’effetto delle diverse condizioni di incubazione sulla sopravvivenza delle cellule tumorali, sulla produzione di ROS, sul consumo di glucosio e sulla produzione di lattato è stato valutato al2° giorno di incubazione delle cellule tumorali.
Il contenuto di glucosio nei terreni di coltura e negli omogenati di tessuto tumorale è stato determinato con il metodo enzimatico del glucosio-ossidante utilizzando il kit per l’analisi del glucosio nei fluidi biologici (Sigma, USA) secondo le istruzioni del produttore. Il contenuto di lattato nei terreni di incubazione e negli omogenati di tessuto tumorale è stato determinato con il metodo della spettrofotometria enzimatica utilizzando la lattato deidrogenasi (Sigma, USA) [14]. I campioni di terreno e di tessuto tumorale sono stati raccolti e conservati a -20°C o in azoto liquido, rispettivamente, fino al momento della misurazione.
Il livello di apoptosi nelle cellule tumorali è stato analizzato con il colorante Hoechst 33258 (Sigma, USA) e la microscopia a fluorescenza con metodo standard.
La produzione di ROS nelle cellule tumorali è stata determinata con l’uso di 2,7-diclorofluoresceina diacetato (Sigma, USA) mediante spettrofluorometria (eccitazione a 495 nm, emissione a 530 nm) secondo [15].
Tutte le misurazioni sono state ripetute.
Esperimenti in vivo
Per gli esperimenti in vivo, le cellule LLC/R9 sono state propagate in vitro a condizioni standard e sono state inoculate i.m. nei topi (1,0-106 cellule/animale in 0,1 ml di soluzione di Hanks).
Dopo l’inoculazione delle cellule LLC/R9 gli animali sono stati distribuiti in 2 gruppi: gruppo 1 – topi trattati con DCA (Sigma, USA) alla dose totale di 1,5 g/kg (LD50/3) (n = 13); gruppo 2 – topi trattati con acqua allo stesso regime e nello stesso volume (controllo, n = 12).
Il trattamento è stato iniziato il giorno successivo al trapianto di cellule tumorali a regime metronomico, 5 volte a settimana per 3 settimane. Il DCA è stato preparato ex tempore in acqua e somministrato per os in un volume di 0,4 ml/animale.
Il volume del tumore primario è stato calcolato sulla base del suo diametro misurato con un calibro ogni3 giorni a partire dal10° giorno dopo l’inoculazione delle cellule tumorali, mediante la formula:
V = π (d)3/6,
dove d – diametro del tumore (mm).
Il livello di metastasi nei topi portatori di tumore è stato valutato al 21° giorno dopo l’inoculazione delle cellule tumorali mediante il numero e il volume delle metastasi polmonari utilizzando un microscopio binoculare e una scala millimetrica.
Il volume totale delle metastasi è stato calcolato con la formula:
V = Σ niπ(di)3/6,
dove V – volume totale delle metastasi (mm3),ni – numero di metastasi con diametrodi (mm).
L’analisi dell’attività funzionale dei componenti della catena respiratoria mitocondriale nelle cellule tumorali è stata eseguita con l’uso dell’EPR al21° giorno dopo l’inoculazione delle cellule tumorali. Il tessuto tumorale è stato tagliato in campioni di dimensioni particolari (d = 4,0 mm, l = 25-35 mm), congelato e conservato a -70 °C. L’analisi EPR dei campioni è stata eseguita a 77 К utilizzando lo spettrofotometro Е-109 Varian (USA) con velocità di sweep del potenziale di 500 Е/min, ampiezza di modulazione di 1,25×10 Е, potenza di irradiazione SHF di 10. 0 mW, sessione costante dell’apparecchio.i livelli di complessi di ferro eme con NO (a gmed= 2,007) nelle proteine ETC mitocondriali e nelle proteine dei cluster Fe-S (a g = 1,94) nelle cellule tumorali sono stati determinati dai dati degli spettri EPR.
L’analisi statistica dei risultati ottenuti è stata effettuata con metodi descrittivi, analisi di regressione non lineare e t-test di Student con l’uso dei programmi Microsoft Excel e Microcal Origin.
RISULTATI
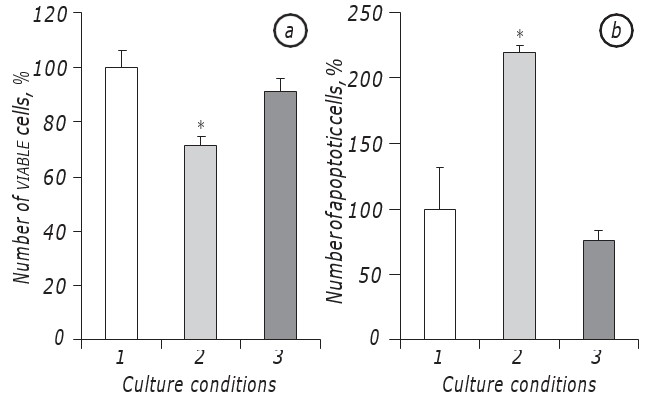
È stato dimostrato che la lattacidosi in condizioni di carenza di glucosio promuoveva significativamente la sopravvivenza delle cellule LLC/R9. Infatti, la cinetica di crescita delle cellule tumorali incubate in condizioni di lattacidosi in condizioni di carenza di glucosio non differiva significativamente da quella delle cellule incubate nel mezzo con contenuto standard di glucosio, almeno nel periodo della loro crescita esponenziale. In particolare, la conta delle cellule vitali al2° giorno di incubazione in condizioni di lattacidosi con carenza di glucosio è stata praticamente la stessa di quella delle cellule incubate nel terreno con contenuto standard di glucosio. Allo stesso tempo, in entrambi i casi (lattacidosi e standard) la conta delle cellule vitali era quasi del 30% superiore (р < 0,05) rispetto al caso di incubazione delle cellule in condizioni di carenza di glucosio (Fig. 1, a).
Inoltre, anche il numero di cellule apoptotiche in condizioni di lattacidosi non differiva statisticamente da quello indice nel caso di incubazione delle cellule nel terreno con contenuto standard di glucosio e al2° giorno era pari all’8,5 ± 0,9%, mentre in condizioni di carenza di glucosio il numero di cellule apoptotiche era quasi tre volte superiore (p < 0,05) rispetto a quello in caso di lattacidosi (Fig. 1, b).
È interessante notare che in condizioni di lattacidosi nelle cellule LLC/R9 il consumo di glucosio era significativamente inferiore. Il basso tasso di consumo di glucosio da parte delle cellule tumorali in caso di lattacidosi, registrato solo al1° giorno di incubazione, è stato ripristinato al2° giorno ed è risultato inferiore del 70% (p < 0,05) rispetto a quello del terreno con contenuto standard di glucosio (Tabella 2). In caso di carenza di glucosio, al contrario della lattacidosi, al2° giorno il livello di glucosio nel terreno di incubazione è sceso a zero, come dimostra anche la diminuzione del consumo di glucosio da parte delle cellule LLC/R9 in condizioni di lattacidosi.
Mentre la lattacidosi ha portato a una diminuzione del tasso di assunzione di glucosio da parte delle cellule LLC/R9, il livello di ROS intracellulare nelle cellule sopravvissute a tali condizioni è aumentato in modo significativo. Questi dati sono presentati nella Tabella 2 e dimostrano che il livello di ROS nelle cellule incubate in condizioni di lattacidosi era quasi del 150% (p < 0,05) e del 230% (p < 0,05) più alto rispetto agli indici corrispondenti per le cellule incubate in mezzi standard e in carenza di glucosio, rispettivamente.
I dati ottenuti hanno quindi dimostrato che la lattacidosi ha promosso in modo significativo la sopravvivenza delle cellule LLC/R9 in condizioni di carenza di glucosio in vitro, come dimostrato dall’elevato numero di cellule sopravvissute in tali condizioni sfavorevoli e dal basso tasso di apoptosi. La sopravvivenza cellulare è stata associata a un inaspettato aumento del livello di ROS intracellulare e a una diminuzione del consumo di glucosio nelle LLC/R9.
| Terreno | Consumo di glucosio, % | ROS, % |
|---|---|---|
| Standard | 100.0 ± 5.9 | 100.0 ± 24.8 |
| Carenza di glucosio | 0.0 ± 0.0* | 75.8 ± 10.7 |
| Lattacidosi | 29.8 ± 1.5* | 248.7 ± 53.2* |
Nota: *p < 0,05.
Questi modelli di sopravvivenza delle cellule LLC/R9 in condizioni di lattacidosi con carenza di glucosio in vitro dimostrano che la diminuzione del contenuto di lattato nel microambiente tumorale può impedire la sopravvivenza delle cellule tumorali in condizioni di stress metabolico, esercitando quindi un effetto antitumorale. Questa ipotesi è stata testata con l’uso del DCA come composto in grado di ridurre la lattacidosi.
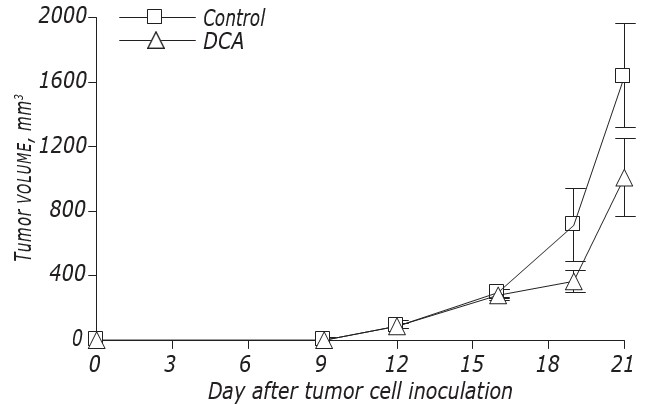
I dati relativi all’influenza del DCA sulla cinetica di crescita e sulle metastasi di LLC/R9 sono mostrati nella Fig. 2 e nella Tabella 3. Secondo questi dati, il DCA non ha influenzato in modo significativo la crescita dei tumori primari, ma ha causato una soppressione espressa delle metastasi. La cinetica di crescita dei tumori primari nei topi con LLC/R9 trattati con DCA non differiva praticamente da quella dei topi di controllo, e al21° giorno dopo il trapianto del tumore il volume dei tumori primari nel gruppo sperimentale era inferiore solo del 39% rispetto a quello del gruppo di controllo (vedi Fig. 2, Tabella 3). Nonostante il DCA non abbia esercitato una notevole soppressione della crescita del tumore primario, la sua attività antimetastatica nei confronti di LLC/R9 è risultata sorprendente. Il numero e il volume delle metastasi polmonari nei topi portatori di tumore trattati con DCA erano inferiori rispettivamente del 59% (р < 0,05) e del 94% (р < 0,05) rispetto a questi indici nel gruppo di controllo (vedi Tabella 3).
| Gruppo di topi | Volume del tumore, mm3 | Numero di metastasi | Volume delle metastasi, mm3 |
|---|---|---|---|
| Controllo (n = 13) | 1702.7 ± 333.9 | 10.9 ± 1.2 | 17.9 ± 5.6 |
| DCA (n = 13) | 1046.0 ± 258.3 | 4.5 ± 1.6* | 1.1 ± 0.4* |
Nota: *р < 0,05, le differenze sono significative rispetto al valore del controllo.
L’analisi del contenuto di lattato nei campioni di tessuto tumorale ha dimostrato che, inaspettatamente, il DCA ha causato un aumento significativo del contenuto di lattato nel tessuto tumorale, almeno al21° giorno dopo il trapianto del tumore. Come si può vedere nella Tabella 4, il contenuto di lattato nel tessuto tumorale dei topi trattati con DCA era superiore di quasi il 30% (р < 0,05) rispetto a quello del controllo. Considerando la capacità del DCA come inibitore della PDH chinasi di riorganizzare il metabolismo energetico del tumore maligno verso la fosforilazione ossidativa, abbiamo considerato la produzione di lattato da parte delle cellule tumorali come un marker surrogato dell’inibizione della glicolisi in seguito all’influenza del DCA. L’aumento del livello di lattato nel tumore causato dal DCA ha indicato che la sua somministrazione a topi con LLC/R9 a una dose totale di 1,5 g/kg di peso corporeo dell’animale potrebbe essere insufficiente per l’attivazione della fosforilazione ossidativa nelle cellule tumorali, il che spiega in parte la sua scarsa efficacia contro i tumori primari.
| Gruppo di topi | Lattato (µmol/1 g di tessuto) |
|---|---|
| Controllo (n = 4) | 11.1 ± 0.6 |
| DCA (n = 5) | 14.4 ± 1.5* |
Nota: р < 0,05, le differenze sono significative rispetto al valore del controllo.
L’analisi degli spettri EPR dei campioni tumorali ha dimostrato che il DCA non ha influenzato in modo significativo lo stato funzionale dei componenti ETC nei mitocondri delle cellule tumorali (Tabella 5). Ad esempio, nei topi con LLC/R9 trattati con DCA, l’intensità dei segnali EPR corrispondenti ai complessi proteici nitrosil-ferro eme (gсер= 2,007) nelle proteine ETC dei mitocondri delle cellule tumorali non era significativamente superiore a quella dei topi di controllo. È noto che l’accumulo di NO-complessi di ferro eme può indicare da un lato lo squilibrio redox verso il dominio dei processi di radicali liberi, in particolare l’iperproduzione di NO, e dall’altro la possibile inibizione della respirazione cellulare attraverso la nitrosilazione delle proteine eme. Tuttavia, il DCA, il cui principale meccanismo d’azione antitumorale si pensa sia legato all’induzione della produzione di ROS da parte dei mitocondri [8, 10, 11], non ha causato un aumento dei complessi ferro-eme con NO nel tessuto tumorale. Quest’ultima osservazione potrebbe essere legata alle caratteristiche delle cellule LLC/R9, ovvero un contenuto estremamente elevato di questi complessi, caratteristici di questo tumore, il cui progressivo accumulo durante lo sviluppo tumorale in vivo è stato da noi registrato anche in assenza di trattamento [16].
| Intensità relativa del segnale EPR | Intensità relativa del segnale EPR | |
| Gruppo di topi | Complessi proteici nitrosil-eme di ferro (g = 2,007) | Proteine Fe-S (g = 1,94) |
| Controllo | 54.3 ± 4.5 | 15.8 ± 0.5 |
| DCA | 97.8 ± 30.1 | 17.8 ± 2.1 |
L’assenza di un effetto significativo del DCA sull’attività funzionale dei componenti dell’ETC mitocondriale nelle cellule tumorali è stata supportata anche dai dati sull’intensità dei segnali EPR corrispondenti alle proteine del cluster Fe-S (g = 1,94) (complessi І, ІІ, ІІІ), che è risultata praticamente uguale in entrambi i gruppi di animali (vedi Tabella 5).
In conclusione, i risultati del nostro studio hanno dimostrato che la lattacidosi ha promosso in modo significativo la sopravvivenza della variante LLC/R9 in condizioni di carenza di glucosio. Allo stesso tempo, se LLC/R9 si sono sviluppate in vivo, il DCA non ha esercitato attività antitumorale contro i tumori primari. La mancata azione antitumorale del DCA contro la crescita di LLC/R9 è in accordo con l’assenza dell’effetto di inibizione del DCA sul contenuto di lattato nel tumore e con l’assenza di un effetto significativo del DCA sulla produzione di ROS delle cellule tumorali. Sebbene il DCA non abbia influenzato la crescita di LLC/R9, ma abbia drasticamente inibito le metastasi, questa osservazione non può essere spiegata dall’azione del DCA all’interno del tumore primario e sono necessari ulteriori studi sulla sua azione antimetastatica.
RIFERIMENTI
1 Feron O. Il piruvato in lattato e viceversa: dall’effetto Warburg allo scambio simbiotico di carburante energetico nelle cellule tumorali. Radiother Oncol 2009; 92: 329-33. doi: 10.1016/j.radonc.2009.06.025.
2 Wu H, Ding Z, Hu D, et al. Ruolo centrale dell’acidosi lattica nella resistenza delle cellule tumorali alla morte cellulare indotta dalla deprivazione di glucosio. J Pathol 2012; 227: 189-99. doi: 10.1002/path.3978.
3 Fiaschi T, Marini A, Giannoni E, et al. La riprogrammazione metabolica reciproca attraverso la navetta del lattato influenza in modo coordinato l’interazione tumore-stroma. Cancer Res 2012; 72: 5130-40.
4 Solyanik GI, Fedorchuk AG, Pyaskovskaya ON, et al. Attività antitumorale dell’estratto di erbe BC1 contenente aconitina. Exp Oncol 2004; 26: 307-11.
5 Pyaskovskaya ON. Azione antiangiogenica della ciclofosfamide nei tumori metastatici sperimentali. J Med Chem 2012; 2: 25-9 (in ucraino).
6 Kolesnik DL, Pyaskovskaya ON, Tregubova NV, Solyanik GI. La variante del carcinoma polmonare di Lewis con un’elevata sensibilità alla terapia antitumorale antiangiogenica presenta un’alta capacità di autofagia. Cytol Genet 2012; 46: 155-60. doi: 10.3103/S009545271203005X.
7 Stacpoole PW. La farmacologia del dicloroacetato. Metabolismo 1989; 38: 1124-44.
8 Bonnet S, Archer SL, Allalunis-Turner J, et al. Un asse mitocondriale-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell 2007; 11: 37-51.
9 Wong JY, Huggins GS, Debidda M, et al. Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol 2008; 109: 394-402. doi: 10.1016/j.ygyno.2008.01.038.
10 Michelakis ED, Sutendra G, Dromparis P, et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci Transl Med 2010; 2: 31-4. doi: 10.1126/scitranslmed.3000677.
11 Stockwin LH, Yu SX, Borgel S, et al. Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale Int J Cancer 2010; 127; 2510-19.
12 Kumar A, Kant S, Singh SM. Nuovi meccanismi molecolari dell’azione antitumorale del dicloroacetato contro il linfoma a cellule T: Implicazione dell’alterazione del metabolismo del glucosio, dell’omeostasi del pH e della regolazione della sopravvivenza cellulare. Chem Biol Interact 2012; 199: 29-37.
13 Pyaskovskaya ON, Dasyukevich OI, Kolesnik DL, et al. Cambiamenti nel livello di VEGF e nelle caratteristiche di crescita tumorale durante la progressione del carcinoma polmonare di Lewis verso la resistenza al cis-DDP. Exp Oncol 2007; 29: 197-202.
14 Metodi biochimici (metabolismo lipidico ed energetico). MI Prohorova, ed. L.: Leningrad Univ, 1982. 272 p.
15 Wang H, Joseph JA. Quantificazione dello stress ossidativo cellulare mediante dosaggio della diclorofluoresceina con lettore di micropiastre. Free Radic Biol Med 1999; 27: 612-6.
16 Pyaskovskaya ON, Sorokina LV, Kolesnik DL, et al. Dynamics of changes of antioxidant system indexes during the growth of two Lewis lung carcinoma variants. Exp Oncol 2014; 36: 29-33.