D.L. Kolesnik1, O.N. Pyaskovskaya1, I.V. Boychuk1, O.I. Dasyukevich1, O.R. Melnikov1, A.S. Tarasov1, G.I. Solyanik1
1R.E. Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology, NAS of Ukraine, Kyiv 03022, Ukraine.
Correspondence: D.L. Kolesnik, E-mail: [email protected]
Submitted: 20 March 2015
Abstract
A hallmark of malignancy is excessive tumor glycolysis, even in the presence of oxygen, which causes lactacidosis in the tumor microenvironment and favors tumor cell proliferation and survival. For this reason antimetabolic agents which target tumor cell metabolism are being researched extensively as promising anticancer drugs. Aim: To study the effect of lactacidosis on survival of Lewis lung carcinoma (LLC) cells at the conditions of nutritional substrate deficiency in vitro and evaluate antitumor and antimetastatic activity against LLC/R9 in vivo. Materials and Methods: LLC variant LLC/R9 was used as experimental tumor model. Tumor cell viability was determined using trypan blue staining. Apoptosis level was counted with the use of Hoechst 33258 dye. Lactate content in the tumor tissue was evaluated by enzyme method with the use of lactate dehydrogenase. Reactive oxygen species was determined using 2.7-dichlorofluorescein diacetate. Effects of dichloroacetate (DCA) on the growth and metastasis of LLC/R9 were analyzed by routine procedures. Evaluation of DCA effect toward electron-transport chain (ETC) components was performed using EPR. Results: It has been shown that at the conditions of lactacidosis and glucose deficiency, LLC/R9 cell viability in vitro was higher by 30% (р < 0.05) and apoptosis level was triply lower (р < 0.05) than these indices at the conditions of glucose deficiency only. In mice with transplanted LLC/R9 tumors treated for 3 weeks per os with DCA at the total dose of 1.5 g/kg of body weight starting from the next day after tumor transplantation, the primary tumor volume was just by 30% lower than that in control group. At the same time, the number and volume of lung metastases in animals treated with DCA were by 59% (р < 0.05) and 94% (р < 0.05) lower, respectively, than these indices in the control group. DCA treatment resulted in nearly 30% increase (р < 0.05) of lactate content in tumor tissue compared to that in the control, but did not affect significantly the levels of heme iron complexes with NO (at gmed = 2.007) in mitochondrial ETC proteins and Fe-S cluster proteins (at g = 1.94) in tumor cells. Conclusions: It has been shown that lactacidosis significantly promoted LLC/R9 cell survival at the conditions of glucose deficiency in vitro. If LLC/R9 developed in vivo, DCA as the compound with antilactacidosis activity did not suppress significantly the primary tumor growth but exerted significant antimetastatic activity.
Keywords: dichloroacetate; hypoxic radiosensitivity; breast cancer; reactive oxygen species
Abbreviations: DCA — dichloroacetate, ETC — electron-transport chain; LLC/R9 — Lewis lung carcinoma variant; PDH — pyruvate dehydrogenase; PDK — pyruvate dehydrogenase kinase.
INTRODUCTION
It is well known that lactacidosis, large accumulation of lactate and decrease of рН, is the main characteristics of metabolic tumor cell microenvironment in vitro and in vivo. Earlier lactacidosis has been considered as a ballast product of tumor cell metabolism. However, recently it has been shown that it could be used by tumor cells as an effective energetic fuel and be among the factors responsible for tumor resistance to glucose deficiency [1–3]. As we have shown using Lewis lung carcinoma (LLC)/R9 cells, LLC variant sensitive to antiangiogenic cancer therapy [4,5], lactacidosis could promote tumor cell survival at the conditions of nutritional deficiency. Such conditions were generated via long-term incubation of tumor cells without replacement of culture medium (“unfed culture” model) [6]. The study of tumor cell growth kinetics at the conditions of “unfed culture” has demonstrated that at the background of complete absence of glucose in incubation medium at days 7–8 of cell growth the viable cell counts did not fall below the third part from their maximum registered at days 3–4, and remained practically at this level till the 10th day. High survival rate of LLC/R9 cells at the conditions of “unfed culture” was related in particular to the ability of these cells to macroautophagy. However, it could not be excluded that the ability of LLC/R9 cells for adaptation toward nutritional substrate deficiency was determined by lactacidosis which developed as a consequence of prolonged tumor cell culturing without replacement of incubation medium.
If lactacidosis is capable to increase tumor cell survival, then the compounds suppressing lactacidosis formation in tumor microenvironment, in particular, dichloroacetate (DCA) as a compound with antilactacidosis activity should demonstrate antitumor activity. The present study was aimed on the testing of such assumption.
It is known that DCA is an inhibitor of pyruvate dehydrogenase kinase (PDK), that’s why it is considered as a negative regulator of enzymes of mitochondrial pyruvate dehydrogenase (PDH) complex which plays a key role in the regulation of tricarboxylic acid and oxidative phosphorylation [7]. If PDH complex is phosphorylated, an entry of pyruvate in Krebs cycle is inhibited, so glycolysis is being activated. Due to PDK inhibition, DCA is capable to lead for indirect activation of PDH complex enzymes and respectively causes the shift of cell energetic balance from glycolysis toward activation of oxidative phosphorylation. Therefore, DCA has been widely used for correction of lacticemia caused by high intensity of glycolysis or defective cell respiration.
According to the data of literature, an ability of DCA to activate oxidative phosphorylation underlies its antitumor activity and is realized, in particular, via the decrease of lactacidosis and induction of reactive oxygen species (ROS) [8–12]. The aim of our study was to analyze an influence of lactacidosis on survival of LLC/R9 cells at the conditions of nutrient deficiency in vitro and evaluate antitumor and antimetastatic activity of DCA against LLC/R9 in vivo.
MATERIALS AND METHODS
Experimental animals, tumor cells
The study was carried out using 2.0–2.5 months old C57Bl/6 mice weighting 18–23 g, bred at animal facility of R.E. Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology of the NAS of Ukraine. Animal study protocols and operation procedures were carried out in accordance with the main requirements to keeping and working with laboratory animals and to the rules of local Bioethics Committee.
In the study LLC variant LLC/R9 derived from wild-type LLC strain by 9 sequential chemotherapy in vivo sessions based on cis-diamminedichloroplatinum (cis-DDP), was used [13]. LLC/R9 cells were maintained in RPMI culture medium (Sigma, USA) supplemented with 10% fetal calf serum (FCS) (Sigma, USA), and 40 mg/ml gentamycine at 37 °C in humidified atmosphere with 5% CO2.
Experiments in vitro
Cell counts in suspension and their viabilitywas routinely analyzed on a hemocytometer using trypan blue exclusion test.
For evaluation of effects of lactacidosis on viability of LLC/R9 cells, 1.5•105 cells/well were seeded in 24-well plate in RPMI 1640 medium (Sigma, USA) with standard glucose content. After overnight incubation cell incubation medium was replaced with the fresh media with different content of glucose, lactate and with different pH for simulation of the conditions of glucose deficiency, lactacidosis at the background of glucose deficiency, as well as standard (Table 1). Glucose deficient medium was prepared on the basis of RPMI 1640 medium without glucose (Sigma, USA). Lactacidosis was generated by adding pure lactic acid (Sigma, USA) to glucose deficient medium to a final concentration of 14 ± 0.7 mM and pH 6.7.
| Medium | Glucose content, mM | Lactate content, mM | рН |
|---|---|---|---|
| Standard | 9.0 ± 0.5 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Glucose deficiency | 3.0 ± 0.1 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Lactacidosis | 3.0 ± 0.1 | 14.0 ± 0.7 | 6.7 ± 0.01 |
Effect of different incubation conditions upon tumor cell survival, ROS production, glucose consumption and lactate production were estimated on the 2nd day of tumor cell incubation.
Glucose content in culture media and in tumor tissue homogenates was determined by enzyme glucose-oxidant method using the kit for glucose analysis in biologic fluids (Sigma, USA) according to instructions of the manufacturer. Lactate content in incubation media and in tumor tissue homogenates was determined by enzyme spectrophotometry method using lactate dehydrogenase (Sigma, USA) [14]. Medium and tumor tissues samples were collected and stored at −20 °С or in liquid nitrogen, correspondently, until the measurement performance.
Apoptosis level in tumor cells was analyzed with the use of Hoechst 33258 dye (Sigma, USA) and fluorescent microscopy by standard method.
Production of ROS in tumor cells was determined with the use of 2.7-dichlorofluorescein diacetate (Sigma, USA) by spectrofluorometry (excitation at 495 nm, emission at 530 nm) according to [15].
All measurements were repeated.
Experiments in vivo
For in vivo experiments, LLC/R9 cells were propagated in vitro at standard conditions and were inoculated i.m. to mice (1.0•106 cell/animal in 0.1 ml of Hanks’ solution).
After LLC/R9 cell inoculation the animals were distributed into 2 groups: group 1 — mice treated with DCA (Sigma, USA) at the total dose of 1.5 g/kg (LD50/3) (n = 13); group 2 — mice treated with water at the same regimen and in the same volume (control, n = 12).
The treatment has been initiated on the following day after tumor cell transplantation at metronomic regimen, 5 times per week for 3 weeks. DCA was prepared ex tempore in water, and was administered per os in a volume of 0.4 ml/animal.
Primary tumor volume was calculated on the basis of its diameter measured using caliper each 3rd day starting from the 10th day after tumor cell inoculation, by the formula:
V = π (d)3/6,
where d — diameter of tumor (mm).
Metastasis level in tumor bearing mice was evaluated at the 21st day after tumor cell inoculation by the number and volume of lung metastases using binocular microscope and millimeter scale.
Total volume of metastasis was calculated by the formula:
V = Σ niπ(di)3/6,
where V — total volume of metastases (mm3), ni — number of metastases with the diameter of di (mm).
Analysis of functional activity of mitochondrial respiratory chain components in tumor cells was performed with the use of EPR at the 21st day after tumor cell inoculation. Tumor tissue was cut into the samples of special size (d = 4.0 mm, l = 25–35 mm), frozen and stored at −70 °C. EPR analysis of the samples was performed at 77 К using spectrophotometer Е-109 Varian (USA) at potential sweep speed of 500 Е/min, modulation amplitude of 1.25×10 Е, power of SHF-irradiation of 10.0 mW, constant session of apparatus of 1.0 s. The levels of heme iron complexes with NO (at gmed = 2.007) in mitochondrial ETC proteins and Fe-S cluster proteins (at g = 1.94) in tumor cells were determined by the data of EPR spectra.
Statistical analysis of obtained results was carried out by descriptive methods, nonlinear regression analysis and Student’s t-test with the useof Microsoft Excel and Microcal Origin programs.
RESULTS
It has been shown that lactacidosis at the conditions of glucose deficiency significantly promoted survival of LLC/R9 cells. Indeed, growth kinetics of tumor cells incubated at the conditions of lactacidosis at the background of glucose deficiency did not differ significantly of that of the cells incubated in the medium with standard glucose content, at least at the period of their exponential growth. In particular, viable cell counts at the 2nd day of incubation at the conditions of lactacidosis at the background of glucose deficiency was practically the same as in the case of cell incubation in the medium with standard glucose content. At the same time, in both cases (lactacidosis and standard) the viable cell counts were nearly by higher 30% (р < 0.05) than in the case of cell incubation at the conditions of glucose deficiency itself (Fig. 1, a).
Apart from this, the number of apoptotic cells at the conditions of lactacidosis also did not differ statistically from that index in the case of cell incubation in the medium with standard glucose content and at the 2nd day was equal to 8.5 ± 0.9%, while at the conditions of glucose deficiency the number of apoptotic cells was nearly three times higher (p < 0.05) than that in the case of lactacidosis (Fig. 1, b).
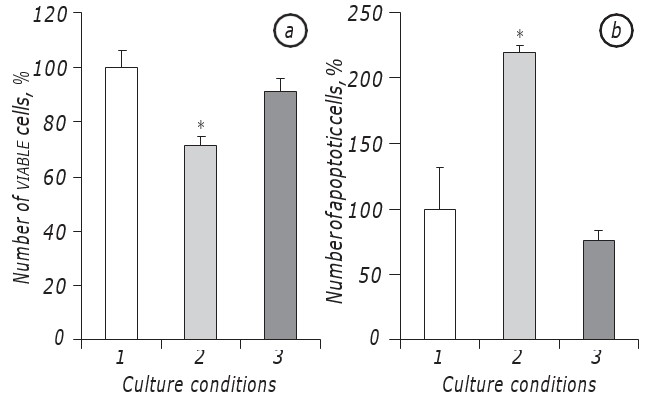
Interestingly, at the conditions of lactacidosis in LLC/R9 cells glucose consumption was significantly lower. Low rate of glucose consumption by tumor cells upon lactacidosis registered just at 1st day of their incubation, has been restored at 2nd day and was by 70% lower (p < 0.05) than that in the case of the medium with standard glucose content (Table 2). In the case of glucose deficiency in contrary to lactacidosis at 2nd day the level of glucose in incubation medium fall to zero, what additionally evidenced on decreased glucose consumption by LLC/R9 cells at the conditions of lactacidosis.
While lactacidosis led to decreased rate of glucose intake by LLC/R9 cells, the level of intracellular ROS in the cells that survived at such conditions significantly increased. These data are presented in Table 2 and demonstrate that ROS level in the cells incubated at the conditions of lactacidosis was nearly by 150% (p < 0.05) and 230% (p < 0.05) higher than corresponding indices for the cells incubated in standard and glucose-deficient media, respectively.
So, the obtained data have demonstrated that lactacidosis significantly promoted LLC/R9 cell survival at the conditions of glucose deficiency in vitro what is supported by high counts of the cells survived at such unfavorable conditions, and by low apoptosis rate. The cell survival was associated with unexpected increase of intracellular ROS level and decreased glucose consumption in LLC/R9.
| Medium | Glucose consumption, % | ROS, % |
|---|---|---|
| Standard | 100.0 ± 5.9 | 100.0 ± 24.8 |
| Glucose deficiency | 0.0 ± 0.0* | 75.8 ± 10.7 |
| Lactacidosis | 29.8 ± 1.5* | 248.7 ± 53.2* |
Note: *p < 0,05.
These patterns of survival of LLC/R9 cells at the conditions of lactacidosis at the background of glucose deficiency in vitro evidenced on the fact that decreased lactate content in tumor microenvironment may prevent tumor cell survival at the conditions of metabolic stress therefore exerting antitumor effect. Such hypothesis was tested by us with the use of DCA as a compound capable to reduce lactacidosis.
The data on DCA influence on LLC/R9 growth kinetics and metastasis are shown on Fig. 2 and Table 3. According to these data, DCA did not affect significantly the growth of primary tumors, but caused an expressed suppression of metastasis. The growth kinetics of primary tumors in mice with LLC/R9 treated with DCA did not practically differ from that in control mice, and at the 21st day after tumor transplantation the volume of primary tumors in experimental group was just by 39% lower than that in the control group (see Fig. 2, Table 3). Despite the fact that DCA exerted no notable suppression of primary tumor growth, its antimetastatic activity toward LLC/R9 was found to be striking. The number and volume of lung metastases in tumor bearing mice treated with DCA were by 59% (р < 0.05) and 94% (р < 0.05) lower than these indices in control group, respectively (see Table 3).
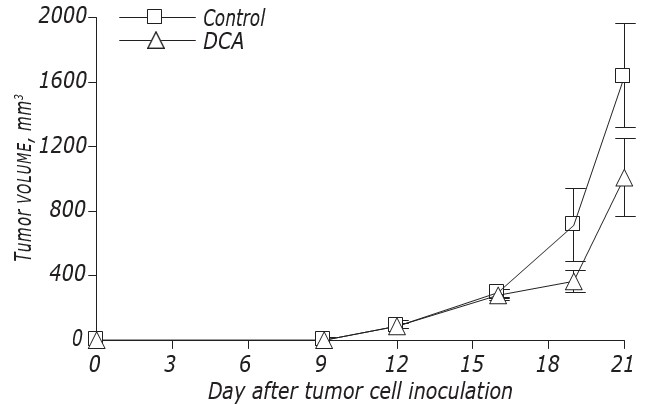
| Group of mice | Tumor volume,mm3 | Number of metastasis | Volume of metastasis, mm3 |
|---|---|---|---|
| Control (n = 13) | 1702.7 ± 333.9 | 10.9 ± 1.2 | 17.9 ± 5.6 |
| DCA (n = 13) | 1046.0 ± 258.3 | 4.5 ± 1.6* | 1.1 ± 0.4* |
Note: *р < 0.05, differences are significant as compared to the value for control.
An analysis of lactate content in tumor tissue samples has demonstrated that unexpectedly DCA caused significant increase of lactate content in tumor tissue, at least at the 21st day after tumor transplantation. As one may see in Table 4, lactate content in tumor tissue of mice treated with DCA, was nearly by 30% higher (р < 0.05) than that in the control. Accounting an ability of DCA as PDH kinase inhibitor reorganize energetic metabolism of malignant tumor toward oxidative phosphorylation, we have considered the lactate production by tumor cells as a surrogate marker of glycolysis inhibition upon DCA influence. The increase of lactate level in tumor caused by DCA indicated that its administration to mice with LLC/R9 at a total dose of 1.5 g/kg of animal body weight could be insufficient for activation of oxidative phosphorylation in tumor cells what explains in part its low efficacy against primary tumors.
| Group of mice | Lactate (µmol/1 g tissue) |
|---|---|
| Control (n = 4) | 11.1 ± 0.6 |
| DCA (n = 5) | 14.4 ± 1.5* |
Note: р < 0.05, differences are significant as compared to the value for control.
An analysis of EPR spectra of tumor samples has shown that DCA did not affect significantly the functional state of the ETC components in tumor cell mitochondria (Table 5). For example, in mice with LLC/R9 treated with DCA, an intensity of EPR signals corresponding to nitrosyl-heme iron protein complexes (gсер = 2.007) in ETC proteins of tumor cell mitochondria was not significantly higher than that in control mice. It is known that accumulation of NO-complexes of hem iron may indicate from one side the redox imbalance toward domination of free radical processes, in particular, NO hyperproduction, and from other side on possible inhibition of cell respiration via nitrosylation of heme proteins. However, DCA, the main mechanism of antitumor action of which is thought to be related to the induction of ROS production by mitochondria [8, 10, 11], did not cause elevation of heme iron complexes with NO in tumor tissue. The latest observation could be possibly related to the features of LLC/R9 cells, namely, an extremely high content of these complexes characteristic for this tumor and progressive accumulation of which during tumor development in vivo has been registered by us even in the absence of treatment [16].
| Relative EPR signal intensity | Relative EPR signal intensity | |
| Group of mice | Nitrosyl-heme iron protein complexes (g = 2.007) | Fe-S protein(g = 1.94) |
| Control | 54.3 ± 4.5 | 15.8 ± 0.5 |
| DCA | 97.8 ± 30.1 | 17.8 ± 2.1 |
An absence of significant effect of DCA on functional activity of mitochondrial ETC components in tumor cells was also supported by the data on intensity of EPR signals corresponding to Fe-S cluster proteins (g = 1.94) (complexes І, ІІ, ІІІ), that was practically equal in both animal groups (see Table 5).
In conclusion, the results of our study have shown that lactacidosis significantly promoted the survival of LLC variant LLC/R9 at the conditions of glucose deficiency. At the same time, if LLC/R9 developed in vivo DCA did not exert antitumor activity against primary tumors. The failure of antitumor action of DCA against LLC/R9 growth was in agreement with the absence of DCA inhibition effect on lactate content in the tumor as well as an absence of notable DCA effect on tumor cell ROS production. Although DCA did not affect LLC/R9 growth but drastically inhibited metastasis; this observation could not be explained by DCA action within primary tumor and further additional studies of its antimetastatic action are required.
REFERENCES
1 Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol 2009; 92: 329–33. doi: 10.1016/j.radonc.2009.06.025.
2 Wu H, Ding Z, Hu D, et al. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J Pathol 2012; 227: 189–99. doi: 10.1002/path.3978.
3 Fiaschi T, Marini A, Giannoni E, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 2012; 72: 5130–40.
4 Solyanik GI, Fedorchuk AG, Pyaskovskaya ON, et al. Anticancer activity of aconitine-containing herbal extract BC1. Exp Oncol 2004; 26: 307–11.
5 Pyaskovskaya ON. Antiangiogenic action of cyclophosphamide to experimental metastatic tumors. J Med Chem 2012; 2: 25–9 (in Ukrainian).
6 Kolesnik DL, Pyaskovskaya ON, Tregubova NV, Solyanik GI. Lewis lung carcinoma variant with a high sensitivity to antitumor antiangiogenic therapy exhibits a high capacity for autophagy. Cytol Genet 2012; 46: 155–60. doi: 10.3103/S009545271203005X.
7 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism 1989; 38: 1124–44.
8 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37–51.
9 Wong JY, Huggins GS, Debidda M, et al. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008; 109: 394–402. doi: 10.1016/j.ygyno.2008.01.038.
10 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2: 31–4. doi: 10.1126/scitranslmed.3000677.
11 Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC Int J Cancer 2010; 127; 2510–19.
12 Kumar A, Kant S, Singh SM. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 2012; 199: 29–37.
13 Pyaskovskaya ON, Dasyukevich OI, Kolesnik DL, et al. Changes in VEGF level and tumor growth characteristics during Lewis lung carcinoma progression towards cis-DDP resistance. Exp Oncol 2007; 29: 197–202.
14 Biochemical methods (lipid and energy metabolism). MI Prohorova, ed. L.: Leningrad Univ, 1982. 272 p.
15 Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med 1999; 27: 612–6.
16 Pyaskovskaya ON, Sorokina LV, Kolesnik DL, et al. Dynamics of changes of antioxidant system indexes during the growth of two Lewis lung carcinoma variants. Exp Oncol 2014; 36: 29–33.