D.L. Kolesnik1, O.N. Pyaskovskaya1i.V. Boychuk1o.I. Dasyukevich1, O.R. Melnikov1, A.S. Tarasov1, G.I. Solyanik1
1R.E. Kavetsky Instituto de Patología Experimental, Oncología y Radiobiología, NAS de Ucrania, Kyiv 03022, Ucrania.
Correspondencia: D.L. Kolesnik, Correo electrónico: [email protected]
Enviado: 20 de marzo de 2015
Resumen
Un sello distintivo de la malignidad es la glucólisis tumoral excesiva, incluso en presencia de oxígeno, lo que provoca lactacidosis en el microambiente tumoral y favorece la proliferación y supervivencia de las células tumorales. Por este motivo, los agentes antimetabólicos dirigidos contra el metabolismo de las células tumorales se están investigando intensamente como prometedores fármacos contra el cáncer. Objetivo: Estudiar el efecto de la lactacidosis sobre la supervivencia de células de carcinoma pulmonar de Lewis (LLC) en condiciones de deficiencia de sustrato nutricional in vitro y evaluar la actividad antitumoral y antimetastásica frente a LLC/R9 in vivo. Materiales y métodos: Se utilizó la variante LLC/R9 como modelo tumoral experimental. La viabilidad de las células tumorales se determinó mediante tinción con azul tripán. El nivel de apoptosis se contabilizó con el colorante Hoechst 33258. El contenido de lactato en el tejido tumoral se evaluó mediante el método enzimático con lactato deshidrogenasa. Las especies reactivas de oxígeno se determinaron utilizando diacetato de 2,7-diclorofluoresceína. Los efectos del dicloroacetato (DCA) sobre el crecimiento y la metástasis de LLC/R9 se analizaron mediante procedimientos rutinarios. La evaluación del efecto del DCA sobre los componentes de la cadena de transporte de electrones (ETC) se realizó mediante EPR. Resultados: Se ha demostrado que en las condiciones de lactacidosis y deficiencia de glucosa, la viabilidad in vitro de las células LLC/R9 fue superior en un 30% (р < 0,05) y el nivel de apoptosis fue tres veces inferior (р < 0,05) a estos índices en las condiciones de deficiencia de glucosa únicamente. En ratones con tumores LLC/R9 trasplantados tratados durante 3 semanas por vía oral con DCA a una dosis total de 1,5 g/kg de peso corporal a partir del día siguiente al trasplante del tumor, el volumen del tumor primario fue un 30% inferior al del grupo de control. Al mismo tiempo, el número y el volumen de las metástasis pulmonares en los animales tratados con DCA fueron un 59% (р < 0,05) y un 94% (р < 0,05) menores, respectivamente, que estos índices en el grupo de control. El tratamiento con DCA produjo un aumento de casi el 30% (р < 0,05) del contenido de lactato en el tejido tumoral en comparación con el del grupo de control, pero no afectó significativamente a los niveles de complejos de hierro hemo con NO (en gmed = 2,007) en las proteínas ETC mitocondriales ni a las proteínas del cluster Fe-S (en g = 1,94) en las células tumorales. Conclusiones: Se ha demostrado que la lactacidosis promovió significativamente la supervivencia de las células LLC/R9 en las condiciones de deficiencia de glucosa in vitro. Si LLC/R9 se desarrollaba in vivo, el DCA como compuesto con actividad antilactacidosis no suprimía significativamente el crecimiento del tumor primario pero ejercía una actividad antimetastásica significativa.
Palabras clave: dicloroacetato; radiosensibilidad hipóxica; cáncer de mama; especies reactivas del oxígeno
Abreviaturas: DCA: dicloroacetato; ETC: cadena de transporte de electrones; LLC/R9: variante de carcinoma pulmonar de Lewis; PDH: piruvato deshidrogenasa; PDK: piruvato deshidrogenasa cinasa.
INTRODUCCIÓN
Es bien sabido que la lactacidosis, gran acumulación de lactato y disminución de рН, es la principal característica del microambiente metabólico de las células tumorales in vitro e in vivo. Anteriormente, la lactacidosis se consideraba un producto de lastre del metabolismo de las células tumorales. Sin embargo, recientemente se ha demostrado que podría ser utilizada por las células tumorales como un eficaz combustible energético y figurar entre los factores responsables de la resistencia tumoral a la deficiencia de glucosa [1-3]. Como hemos demostrado utilizando células de carcinoma pulmonar de Lewis (LLC)/R9, variante de LLC sensible a la terapia antiangiogénica del cáncer [4,5], la lactacidosis podría promover la supervivencia de las células tumorales en condiciones de deficiencia nutricional. Tales condiciones se generaron mediante la incubación a largo plazo de células tumorales sin sustitución del medio de cultivo (modelo de «cultivo sin alimentación») [6]. El estudio de la cinética de crecimiento de las células tumorales en condiciones de «cultivo no alimentado» ha demostrado que en ausencia total de glucosa en el medio de incubación en los días 7-8 de crecimiento celular el recuento de células viables no caía por debajo de la tercera parte de su máximo registrado en los días 3-4, y se mantenía prácticamente en este nivel hasta el10º día. La alta tasa de supervivencia de las células LLC/R9 en las condiciones de «cultivo sin alimentación» estaba relacionada en particular con la capacidad de estas células para la macroautofagia. Sin embargo, no se pudo excluir que la capacidad de adaptación de las células LLC/R9 a la deficiencia de sustratos nutricionales estuviera determinada por la lactacidosis que se desarrolló como consecuencia del cultivo prolongado de células tumorales sin sustitución del medio de incubación.
Si la lactacidosis es capaz de aumentar la supervivencia de las células tumorales, entonces los compuestos que suprimen la formación de lactacidosis en el microambiente tumoral, en particular, el dicloroacetato (DCA) como compuesto con actividad antilactacidosis debería demostrar actividad antitumoral. El presente estudio tiene por objeto comprobar esta hipótesis.
Se sabe que el DCA es un inhibidor de la piruvato deshidrogenasa quinasa (PDK), por lo que se considera un regulador negativo de las enzimas del complejo piruvato deshidrogenasa mitocondrial (PDH), que desempeña un papel clave en la regulación de la fosforilación oxidativa y del ácido tricarboxílico [7]. Si se fosforila el complejo PDH, se inhibe la entrada de piruvato en el ciclo de Krebs, por lo que se activa la glucólisis. Debido a la inhibición de la PDK, el DCA es capaz de llevar a la activación indirecta de las enzimas del complejo PDH y respectivamente causa el cambio del balance energético celular de la glicólisis hacia la activación de la fosforilación oxidativa. Por lo tanto, el DCA ha sido ampliamente utilizado para la corrección de la lacticemia causada por la alta intensidad de la glucólisis o la respiración celular defectuosa.
Según los datos de la literatura, la capacidad del DCA para activar la fosforilación oxidativa subyace a su actividad antitumoral y se realiza, en particular, a través de la disminución de la lactacidosis y la inducción de especies reactivas de oxígeno (ROS) [8-12]. El objetivo de nuestro estudio era analizar la influencia de la lactacidosis en la supervivencia de las células LLC/R9 en condiciones de deficiencia de nutrientes in vitro y evaluar la actividad antitumoral y antimetastásica del DCA contra LLC/R9 in vivo.
MATERIALES Y MÉTODOS
Animales experimentales, células tumorales
El estudio se llevó a cabo utilizando ratones C57Bl/6 de 2,0-2,5 meses de edad y un peso de 18-23 g, criados en las instalaciones para animales del Instituto R.E. Kavetsky de Patología Experimental, Oncología y Radiobiología de la NAS de Ucrania. Los protocolos de estudio con animales y los procedimientos operativos se llevaron a cabo de acuerdo con los principales requisitos para mantener y trabajar con animales de laboratorio y con las normas del Comité de Bioética local.
En el estudio se utilizó la variante LLC/R9 derivada de la cepa LLC de tipo salvaje mediante 9 sesiones secuenciales de quimioterapia in vivo basadas en cis-diamminedicloroplatino (cis-DDP) [13]. Las células LLC/R9 se mantuvieron en medio de cultivo RPMI (Sigma, EE.UU.) suplementado con un 10% de suero fetal de ternera (FCS) (Sigma, EE.UU.), y 40 mg/ml de gentamicina a 37 °C en atmósfera humidificada con un 5% deCO2.
Experimentos in vitro
El recuento de células en suspensión y su viabilidad se analizaron rutinariamente en un hemocitómetro mediante la prueba de exclusión con azul tripán.
Para evaluar los efectos de la lactacidosis sobre la viabilidad de las células LLC/R9, se sembraron 1,5-105 células/pocillo en una placa de 24 pocillos en medio RPMI 1640 (Sigma, EE.UU.) con contenido estándar de glucosa. Tras una noche de incubación, el medio de incubación de las células se sustituyó por medio fresco con diferente contenido de glucosa, lactato y con diferente pH para simular las condiciones de deficiencia de glucosa, lactacidosis en el fondo de deficiencia de glucosa, así como estándar (Tabla 1). El medio con deficiencia de glucosa se preparó a partir del medio RPMI 1640 sin glucosa (Sigma, EE.UU.). La lactacidosis se generó añadiendo ácido láctico puro (Sigma, EE.UU.) al medio con deficiencia de glucosa hasta una concentración final de 14 ± 0,7 mM y pH 6,7.
| Medio | Contenido de glucosa, mM | Contenido en lactato, mM | рН |
|---|---|---|---|
| Estándar | 9.0 ± 0.5 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Deficiencia de glucosa | 3.0 ± 0.1 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Lactacidosis | 3.0 ± 0.1 | 14.0 ± 0.7 | 6.7 ± 0.01 |
El efecto de las diferentes condiciones de incubación sobre la supervivencia de las células tumorales, la producción de ROS, el consumo de glucosa y la producción de lactato se estimó el2º día de incubación de las células tumorales.
El contenido de glucosa en los medios de cultivo y en los homogeneizados de tejido tumoral se determinó mediante el método enzimático glucosa-oxidante utilizando el kit para el análisis de glucosa en fluidos biológicos (Sigma, EE.UU.) de acuerdo con las instrucciones del fabricante. El contenido de lactato en los medios de incubación y en los homogeneizados de tejido tumoral se determinó por el método de espectrofotometría enzimática utilizando lactato deshidrogenasa (Sigma, EE.UU.) [14]. Las muestras de medio y tejidos tumorales se recogieron y almacenaron a -20 °С o en nitrógeno líquido, correspondientemente, hasta la realización de la medición.
El nivel de apoptosis en las células tumorales se analizó con el colorante Hoechst 33258 (Sigma, EE.UU.) y microscopía fluorescente por el método estándar.
La producción de ROS en células tumorales se determinó con el uso de diacetato de 2,7-diclorofluoresceína (Sigma, EE.UU.) por espectrofluorometría (excitación a 495 nm, emisión a 530 nm) según [15].
Se repitieron todas las mediciones.
Experimentos in vivo
Para los experimentos in vivo, se propagaron células LLC/R9 in vitro en condiciones estándar y se inocularon i.m. a ratones (1,0-106 células/animal en 0,1 ml de solución de Hanks).
Tras la inoculación de células LLC/R9, los animales se distribuyeron en 2 grupos: grupo 1 – ratones tratados con DCA (Sigma, EE.UU.) a la dosis total de 1,5 g/kg (LD50/3) (n = 13); grupo 2 – ratones tratados con agua al mismo régimen y en el mismo volumen (control, n = 12).
El tratamiento se inició al día siguiente del trasplante de células tumorales en régimen metronómico, 5 veces por semana durante 3 semanas. El DCA se preparó ex tempore en agua, y se administró per os en un volumen de 0,4 ml/animal.
El volumen del tumor primario se calculó sobre la base de su diámetro medido con un calibrador cada3 días a partir del10º día tras la inoculación de las células tumorales, mediante la fórmula:
V = π (d)3/6,
donde d – diámetro del tumor (mm).
El nivel de metástasis en los ratones portadores de tumores se evaluó a los 21 días de la inoculación de las células tumorales mediante el número y el volumen de metástasis pulmonares utilizando un microscopio binocular y una escala milimétrica.
El volumen total de metástasis se calculó mediante la fórmula:
V = Σ niπ(di)3/6,
donde V – volumen total de metástasis (mm3),ni – número de metástasis con el diámetrodi (mm).
El análisis de la actividad funcional de los componentes de la cadena respiratoria mitocondrial en las células tumorales se llevó a cabo con el uso de EPR en el día21 después de la inoculación de células tumorales. El tejido tumoral se cortó en muestras de tamaño especial (d = 4,0 mm, l = 25-35 mm), se congeló y se almacenó a -70 °C. El análisis EPR de las muestras se realizó a 77 К utilizando el espectrofotómetro Е-109 Varian (EE.UU.) a una velocidad de barrido potencial de 500 Е/min, amplitud de modulación de 1,25×10 Е, potencia de irradiación SHF de 10.0 mW, sesión constante del aparato de 1,0 s. Los niveles de complejos de hierro hemo con NO (a gmed = 2,007) en proteínas ETC mitocondriales y proteínas de clúster Fe-S (a g = 1,94) en células tumorales se determinaron mediante los datos de los espectros EPR.
Elanálisis estadístico de los resultados obtenidos se llevó a cabo mediante métodos descriptivos, análisis de regresión no lineal y prueba t de Student con el uso de los programas Microsoft Excel y Microcal Origin.
RESULTADOS
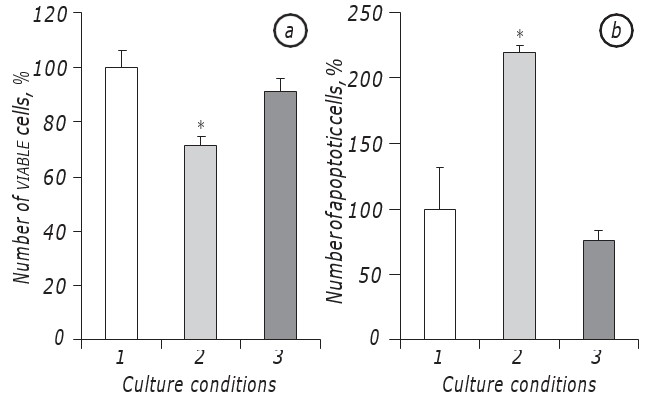
Se ha demostrado que la lactacidosis en las condiciones de deficiencia de glucosa promovió significativamente la supervivencia de las células LLC/R9. De hecho, la cinética de crecimiento de las células tumorales incubadas en las condiciones de lactacidosis en el fondo de la deficiencia de glucosa no difirió significativamente de la de las células incubadas en el medio con contenido de glucosa estándar, al menos en el período de su crecimiento exponencial. En particular, el recuento de células viables en elsegundo día de incubación en condiciones de lactacidosis en el fondo de la deficiencia de glucosa fue prácticamente el mismo que en el caso de la incubación de células en el medio con contenido estándar de glucosa. Al mismo tiempo, en ambos casos (lactacidosis y estándar) los recuentos de células viables fueron casi un 30% más altos (р < 0,05) que en el caso de incubación de células en condiciones de deficiencia de glucosa (Fig. 1, a).
Aparte de esto, el número de células apoptóticas en las condiciones de lactacidosis tampoco difirió estadísticamente de ese índice en el caso de incubación de células en el medio con contenido estándar de glucosa y en el2º día fue igual a 8,5 ± 0,9%, mientras que en las condiciones de deficiencia de glucosa el número de células apoptóticas fue casi tres veces mayor (p < 0,05) que en el caso de lactacidosis (Fig. 1, b).
Curiosamente, en las condiciones de lactacidosis en las células LLC/R9 el consumo de glucosa fue significativamente menor. La baja tasa de consumo de glucosa por las células tumorales en lactacidosis, registrada justo alprimer día de su incubación, se restableció alsegundo día y fue un 70% menor (p < 0,05) que en el caso del medio con contenido estándar de glucosa (Tabla 2). En el caso de deficiencia de glucosa, al contrario que en la lactacidosis, en elsegundo día el nivel de glucosa en el medio de incubación descendió a cero, lo que también se evidenció en la disminución del consumo de glucosa por parte de las células LLC/R9 en condiciones de lactacidosis.
Mientras que la lactacidosis condujo a una disminución de la tasa de consumo de glucosa por las células LLC/R9, el nivel de ROS intracelular en las células que sobrevivieron en tales condiciones aumentó significativamente. Estos datos se presentan en la Tabla 2 y demuestran que el nivel de ROS en las células incubadas en condiciones de lactacidosis fue casi un 150% (p < 0,05) y un 230% (p < 0,05) superior a los índices correspondientes para las células incubadas en medios estándar y deficientes en glucosa, respectivamente.
Por lo tanto, los datos obtenidos han demostrado que la lactacidosis promovió significativamente la supervivencia de las células LLC/R9 en condiciones de deficiencia de glucosa in vitro, lo que se ve apoyado por los altos recuentos de células que sobrevivieron en condiciones tan desfavorables, y por la baja tasa de apoptosis. La supervivencia celular se asoció a un aumento inesperado del nivel intracelular de ROS y a un menor consumo de glucosa en LLC/R9.
| Medio | Consumo de glucosa, % ROS | ROS, % |
|---|---|---|
| Estándar | 100.0 ± 5.9 | 100.0 ± 24.8 |
| Deficiencia de glucosa | 0.0 ± 0.0* | 75.8 ± 10.7 |
| Lactacidosis | 29.8 ± 1.5* | 248.7 ± 53.2* |
Nota: *p < 0,05.
Estos patrones de supervivencia de las células LLC/R9 en condiciones de lactacidosis en un contexto de deficiencia de glucosa in vitro evidencian el hecho de que la disminución del contenido de lactato en el microambiente tumoral puede impedir la supervivencia de las células tumorales en condiciones de estrés metabólico, ejerciendo así un efecto antitumoral. Hemos comprobado esta hipótesis utilizando DCA como compuesto capaz de reducir la lactacidosis.
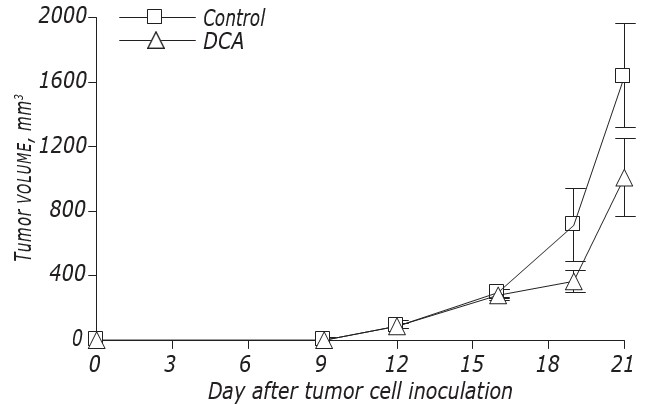
Los datos sobre la influencia del DCA en la cinética de crecimiento y metástasis de LLC/R9 se muestran en la Fig. 2 y en la Tabla 3. De acuerdo con estos datos, el DCA no afectó a la supervivencia de las células LLC/R9. Según estos datos, el DCA no afectó significativamente al crecimiento de los tumores primarios, pero causó una supresión expresa de la metástasis. La cinética de crecimiento de los tumores primarios en ratones con LLC/R9 tratados con DCA no difería prácticamente de la de los ratones de control, y al día21 después del trasplante tumoral el volumen de los tumores primarios en el grupo experimental era sólo un 39% inferior al del grupo de control (véanse la Fig. 2 y la Tabla 3). A pesar de que el DCA no ejerció una supresión notable del crecimiento del tumor primario, su actividad antimetastásica hacia LLC/R9 resultó ser sorprendente. El número y el volumen de metástasis pulmonares en los ratones portadores de tumores tratados con DCA fueron un 59% (р < 0,05) y un 94% (р < 0,05) inferiores a estos índices en el grupo de control, respectivamente (véase la Tabla 3).
| Grupo de ratones | Volumen tumoral,mm3 | Número de metástasis | Volumen de metástasis, mm3 |
|---|---|---|---|
| Control (n = 13) | 1702.7 ± 333.9 | 10.9 ± 1.2 | 17.9 ± 5.6 |
| DCA (n = 13) | 1046.0 ± 258.3 | 4.5 ± 1.6* | 1.1 ± 0.4* |
Nota: *р < 0,05, las diferencias son significativas en comparación con el valor del control.
Un análisis del contenido de lactato en las muestras de tejido tumoral ha demostrado que, inesperadamente, el DCA provocó un aumento significativo del contenido de lactato en el tejido tumoral, al menos en el día21 tras el trasplante del tumor. Como se puede ver en la Tabla 4, el contenido de lactato en el tejido tumoral de los ratones tratados con DCA, fue casi un 30% mayor (р < 0,05) que en el control. Teniendo en cuenta la capacidad del DCA como inhibidor de la PDH quinasa para reorganizar el metabolismo energético del tumor maligno hacia la fosforilación oxidativa, hemos considerado la producción de lactato por las células tumorales como un marcador sustituto de la inhibición de la glucólisis bajo la influencia del DCA. El aumento del nivel de lactato en el tumor causado por el DCA indicó que su administración a ratones con LLC/R9 a una dosis total de 1,5 g/kg de peso corporal animal podría ser insuficiente para la activación de la fosforilación oxidativa en las células tumorales, lo que explica en parte su baja eficacia contra los tumores primarios.
| Grupo de ratones | Lactato (µmol/1 g de tejido) |
|---|---|
| Control (n = 4) | 11.1 ± 0.6 |
| DCA (n = 5) | 14.4 ± 1.5* |
Nota: р < 0,05, las diferencias son significativas en comparación con el valor del control.
Un análisis de los espectros EPR de las muestras tumorales ha demostrado que el DCA no afectó significativamente al estado funcional de los componentes ETC en las mitocondrias de las células tumorales (Tabla 5). Por ejemplo, en ratones con LLC/R9 tratados con DCA, la intensidad de las señales EPR correspondientes a complejos proteicos nitrosil-hierro hemo (gсер= 2,007) en proteínas ETC de mitocondrias de células tumorales no fue significativamente mayor que en ratones control. Se sabe que la acumulación de NO-complejos de hem hierro puede indicar de un lado el desequilibrio redox hacia la dominación de los procesos de radicales libres, en particular, la hiperproducción de NO, y de otro lado en la posible inhibición de la respiración celular a través de nitrosilación de hem proteínas. Sin embargo, el DCA, cuyo principal mecanismo de acción antitumoral se cree que está relacionado con la inducción de la producción de ROS por las mitocondrias [8, 10, 11], no causó una elevación de los complejos de hierro hemo con NO en el tejido tumoral. Esta última observación podría estar posiblemente relacionada con las características de las células LLC/R9, a saber, un contenido extremadamente elevado de estos complejos característico de este tumor y cuya acumulación progresiva durante el desarrollo tumoral in vivo ha sido registrada por nosotros incluso en ausencia de tratamiento [16].
| Intensidad relativa de la señal EPR | Intensidad relativa de la señal EPR | |
| Grupo de ratones | Complejos proteicos de hierro-nitrosil-heme (g = 2,007) | Proteína Fe-S (g = 1,94) |
| Control | 54.3 ± 4.5 | 15.8 ± 0.5 |
| DCA | 97.8 ± 30.1 | 17.8 ± 2.1 |
La ausencia de un efecto significativo del DCA sobre la actividad funcional de los componentes del ETC mitocondrial en las células tumorales también fue apoyada por los datos sobre la intensidad de las señales EPR correspondientes a las proteínas del cluster Fe-S (g = 1,94) (complejos І, ІІ, ІІІ), que fue prácticamente igual en ambos grupos de animales (ver Tabla 5).
En conclusión, los resultados de nuestro estudio han demostrado que la lactacidosis promovió significativamente la supervivencia de LLC variante LLC/R9 en las condiciones de deficiencia de glucosa. Al mismo tiempo, si LLC/R9 se desarrollaba in vivo DCA no ejercía actividad antitumoral contra los tumores primarios. El fracaso de la acción antitumoral del DCA contra el crecimiento de LLC/R9 concordaba con la ausencia de efecto inhibidor del DCA sobre el contenido de lactato en el tumor, así como con la ausencia de un efecto notable del DCA sobre la producción de ROS de las células tumorales. Aunque el DCA no afectó al crecimiento de LLC/R9 pero inhibió drásticamente la metástasis, esta observación no podría explicarse por la acción del DCA dentro del tumor primario y se requieren estudios adicionales de su acción antimetastásica.
REFERENCIAS
1 Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol 2009; 92: 329-33. doi: 10.1016/j.radonc.2009.06.025.
2 Wu H, Ding Z, Hu D, et al. Papel central de la acidosis láctica en la resistencia de las células cancerosas a la muerte celular inducida por la privación de glucosa. J Pathol 2012; 227: 189-99. doi: 10.1002/path.3978.
3 Fiaschi T, Marini A, Giannoni E, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 2012; 72: 5130-40.
4 Solyanik GI, Fedorchuk AG, Pyaskovskaya ON, et al. Anticancer activity of aconitine-containing herbal extract BC1. Exp Oncol 2004; 26: 307-11.
5 Pyaskovskaya ON. Antiangiogenic action of cyclophosphamide to experimental metastatic tumors. J Med Chem 2012; 2: 25-9 (en ucraniano).
6 Kolesnik DL, Pyaskovskaya ON, Tregubova NV, Solyanik GI. Lewis lung carcinoma variant with a high sensitivity to antitumor antiangiogenic therapy exhibits a high capacity for autophagy. Cytol Genet 2012; 46: 155-60. doi: 10.3103/S009545271203005X.
7 Stacpoole PW. La farmacología de dicloroacetato. Metabolism 1989; 38: 1124-44.
8 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37-51.
9 Wong JY, Huggins GS, Debidda M, et al. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008; 109: 394-402. doi: 10.1016/j.ygyno.2008.01.038.
10 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2: 31-4. doi: 10.1126/scitranslmed.3000677.
11 Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC Int J Cancer 2010; 127; 2510-19.
12 Kumar A, Kant S, Singh SM. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implicación de la alteración del metabolismo de la glucosa, la homeostasis del pH y la regulación de la supervivencia celular. Chem Biol Interact 2012; 199: 29-37.
13 Pyaskovskaya ON, Dasyukevich OI, Kolesnik DL, et al. Changes in VEGF level and tumor growth characteristics during Lewis lung carcinoma progression towards cis-DDP resistance. Exp Oncol 2007; 29: 197-202.
14 Métodos bioquímicos (metabolismo lipídico y energético). MI Prohorova, ed. L.: Universidad de Leningrado, 1982. 272 p.
15 Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med 1999; 27: 612-6.
16 Pyaskovskaya ON, Sorokina LV, Kolesnik DL, et al. Dinámica de los cambios de los índices del sistema antioxidante durante el crecimiento de dos variantes de carcinoma de pulmón de Lewis. Exp Oncol 2014; 36: 29-33.