D.L. Kolesnik1, O.N. Pyaskovskaya1, I.V. Boychuk1, O.I. Dasyukevich1, O.R. Melnikov1, A.S. Tarasov1, G.I. Soljanik1
1R.E. Kavetsky Institut für experimentelle Pathologie, Onkologie und Radiobiologie, NAS der Ukraine, Kiew 03022, Ukraine.
Korrespondenz: D.L. Kolesnik, E-Mail: [email protected]
Eingereicht: 20. März 2015
Zusammenfassung
Ein Kennzeichen bösartiger Erkrankungen ist die übermäßige Glykolyse im Tumor, selbst in Gegenwart von Sauerstoff, die eine Laktazidose in der Mikroumgebung des Tumors verursacht und die Vermehrung und das Überleben der Tumorzellen begünstigt. Aus diesem Grund werden antimetabolische Wirkstoffe, die auf den Stoffwechsel der Tumorzellen abzielen, als vielversprechende Krebsmedikamente intensiv erforscht. Ziel: Untersuchung der Wirkung von Laktazidose auf das Überleben von Zellen des Lewis-Lungenkarzinoms (LLC) unter den Bedingungen eines Nährstoffmangels in vitro und Bewertung der antitumoralen und antimetastatischen Aktivität gegen LLC/R9 in vivo. Materialien und Methoden: Die LLC-Variante LLC/R9 wurde als experimentelles Tumormodell verwendet. Die Lebensfähigkeit der Tumorzellen wurde mittels Trypanblau-Färbung bestimmt. Das Apoptose-Niveau wurde mit Hilfe des Farbstoffs Hoechst 33258 ermittelt. Der Laktatgehalt im Tumorgewebe wurde mit der Enzymmethode unter Verwendung von Laktatdehydrogenase bestimmt. Reaktive Sauerstoffspezies wurden mit 2,7-Dichlorfluorescein-Diacetat bestimmt. Die Auswirkungen von Dichloracetat (DCA) auf das Wachstum und die Metastasierung von LLC/R9 wurden mit Routineverfahren analysiert. Die Bewertung der Wirkung von DCA auf die Komponenten der Elektronentransportkette (ETC) erfolgte mit Hilfe der EPR. Ergebnisse: Es wurde gezeigt, dass die Lebensfähigkeit von LLC/R9-Zellen in vitro unter den Bedingungen der Laktazidose und des Glukosemangels um 30 % höher (р < 0,05) und das Apoptose-Niveau um das Dreifache (р < 0,05) niedriger war als diese Indizes unter den Bedingungen des reinen Glukosemangels. Bei Mäusen mit transplantierten LLC/R9-Tumoren, die 3 Wochen lang per os mit DCA in einer Gesamtdosis von 1,5 g/kg Körpergewicht ab dem nächsten Tag nach der Tumortransplantation behandelt wurden, war das Volumen des Primärtumors nur um 30% geringer als in der Kontrollgruppe. Gleichzeitig waren die Anzahl und das Volumen der Lungenmetastasen bei den mit DCA behandelten Tieren um 59% (р < 0,05) bzw. 94% (р < 0,05) geringer als diese Indizes in der Kontrollgruppe. Die DCA-Behandlung führte zu einem Anstieg des Laktatgehalts im Tumorgewebe um fast 30 % (р < 0,05) im Vergleich zu dem der Kontrollgruppe, hatte jedoch keinen signifikanten Einfluss auf den Gehalt an Häm-Eisen-Komplexen mit NO (bei gmed = 2,007) in mitochondrialen ETC-Proteinen und Fe-S-Cluster-Proteinen (bei g = 1,94) in Tumorzellen. Schlussfolgerungen: Es wurde gezeigt, dass Laktazidose das Überleben von LLC/R9-Zellen unter den Bedingungen des Glukosemangels in vitro signifikant fördert. Wenn sich LLC/R9 in vivo entwickelte, unterdrückte DCA als Verbindung mit Antilaktazidose-Aktivität das Wachstum des Primärtumors nicht signifikant, übte aber eine signifikante antimetastatische Aktivität aus.
Schlüsselwörter: Dichloracetat; hypoxische Radiosensitivität; Brustkrebs; reaktive SauerstoffspeziesAbkürzungen
: DCA – Dichloracetat, ETC – Elektronentransportkette; LLC/R9 – Lewis-Lungenkarzinom-Variante; PDH – Pyruvatdehydrogenase; PDK – Pyruvatdehydrogenase-Kinase.
EINLEITUNG
Es ist bekannt, dass die Laktazidose, d. h. die starke Anhäufung von Laktat und die Abnahme von рН, die Hauptmerkmale der metabolischen Mikroumgebung von Tumorzellen in vitro und in vivo sind. Früher wurde die Laktazidose als ein Ballastprodukt des Tumorzellstoffwechsels betrachtet. Kürzlich wurde jedoch gezeigt, dass sie von Tumorzellen als effektiver Energieträger genutzt werden könnte und zu den Faktoren gehört, die für die Resistenz von Tumoren gegenüber Glukosemangel verantwortlich sind [1-3]. Wie wir anhand von Lewis-Lungenkarzinom (LLC)/R9-Zellen gezeigt haben, ist die LLC-Variante empfindlich gegenüber einer antiangiogenen Krebstherapie [4,5]gezeigt, dass Laktazidose das Überleben von Tumorzellen unter den Bedingungen eines Nährstoffmangels fördern kann. Solche Bedingungen wurden durch langfristige Inkubation von Tumorzellen ohne Austausch des Kulturmediums („unfed culture“-Modell) geschaffen [6]. Die Untersuchung der Kinetik des Tumorzellwachstums unter den Bedingungen der „Unfed Culture“ hat gezeigt, dass bei völligem Fehlen von Glukose im Inkubationsmedium an den Tagen 7 bis 8 des Zellwachstums die Zahl der lebensfähigen Zellen nicht unter ein Drittel des an den Tagen 3 bis 4 registrierten Maximums fiel und praktisch bis zum10. Die hohe Überlebensrate der LLC/R9-Zellen unter den Bedingungen der „ungefütterten Kultur“ hing insbesondere mit der Fähigkeit dieser Zellen zur Makroautophagie zusammen. Es konnte jedoch nicht ausgeschlossen werden, dass die Fähigkeit der LLC/R9-Zellen zur Anpassung an den Mangel an Nährsubstraten durch die Laktazidose bestimmt wurde, die sich als Folge der langen Kultivierung der Tumorzellen ohne Austausch des Inkubationsmediums entwickelte.
Wenn Laktazidose in der Lage ist, das Überleben von Tumorzellen zu erhöhen, dann sollten die Verbindungen, die die Bildung von Laktazidose in der Mikroumgebung des Tumors unterdrücken, insbesondere Dichloracetat (DCA) als Verbindung mit Antilaktazidose-Aktivität, eine Antitumor-Aktivität aufweisen. Die vorliegende Studie zielte darauf ab, diese Annahme zu überprüfen.
Es ist bekannt, dass DCA ein Inhibitor der Pyruvat-Dehydrogenase-Kinase (PDK) ist, weshalb es als negativer Regulator von Enzymen des mitochondrialen Pyruvat-Dehydrogenase (PDH)-Komplexes gilt, der eine Schlüsselrolle bei der Regulierung der Tricarbonsäure und der oxidativen Phosphorylierung spielt [7]. Wenn der PDH-Komplex phosphoryliert ist, wird der Eintritt von Pyruvat in den Krebszyklus gehemmt, so dass die Glykolyse aktiviert wird. Aufgrund der PDK-Hemmung ist DCA in der Lage, die Enzyme des PDH-Komplexes indirekt zu aktivieren, was zu einer Verschiebung des Energiegleichgewichts der Zelle von der Glykolyse zur Aktivierung der oxidativen Phosphorylierung führt. Daher wurde DCA häufig zur Korrektur von Laktämie eingesetzt, die durch eine hohe Intensität der Glykolyse oder eine gestörte Zellatmung verursacht wird.
Den Angaben in der Literatur zufolge liegt die Fähigkeit von DCA, die oxidative Phosphorylierung zu aktivieren, seiner Antitumoraktivität zugrunde und wird insbesondere durch die Verringerung der Laktazidose und die Induktion reaktiver Sauerstoffspezies (ROS) realisiert [8-12]. Ziel unserer Studie war es, den Einfluss der Laktazidose auf das Überleben von LLC/R9-Zellen unter den Bedingungen des Nährstoffmangels in vitro zu analysieren und die antitumorale und antimetastatische Aktivität von DCA gegen LLC/R9 in vivo zu bewerten.
MATERIALIEN UND METHODEN
Versuchstiere, Tumorzellen
Die Studie wurde mit 2,0-2,5 Monate alten C57Bl/6-Mäusen mit einem Gewicht von 18-23 g durchgeführt, die in der Tieranlage des R.E. Kavetsky-Instituts für experimentelle Pathologie, Onkologie und Strahlenbiologie der NAS der Ukraine gezüchtet wurden. Die Protokolle der Tierversuche und die Arbeitsverfahren wurden in Übereinstimmung mit den wichtigsten Anforderungen an die Haltung von und die Arbeit mit Labortieren und den Regeln des örtlichen Bioethikausschusses durchgeführt.
In der Studie wurde die LLC-Variante LLC/R9 verwendet, die aus dem Wildtyp-LLC-Stamm durch 9 sequenzielle Chemotherapien in vivo auf der Grundlage von cis-Diamminedichloroplatin (cis-DDP) abgeleitet wurde [13]. LLC/R9-Zellen wurden in RPMI-Kulturmedium (Sigma, USA), ergänzt mit 10 % fetalem Kälberserum (FCS) (Sigma, USA) und 40 mg/ml Gentamycin, bei 37 °C in befeuchteter Atmosphäre mit 5 %CO2 gehalten.
Experimente in vitro
Die Anzahl der Zellen in Suspension und ihre Lebensfähigkeit wurden routinemäßig auf einem Hämozytometer unter Verwendung des Trypanblau-Ausschlusstests analysiert.
Zur Bewertung der Auswirkungen der Laktazidose auf die Lebensfähigkeit von LLC/R9-Zellen wurden 1,5-105 Zellen/Vertiefung in 24-Well-Platten in RPMI 1640-Medium (Sigma, USA) mit Standardglukosegehalt ausgesät. Nach der Inkubation über Nacht wurde das Zellinkubationsmedium durch frisches Medium mit unterschiedlichem Glukose- und Laktatgehalt und mit unterschiedlichem pH-Wert ersetzt, um die Bedingungen des Glukosemangels, der Laktazidose vor dem Hintergrund des Glukosemangels sowie des Standards zu simulieren (Tabelle 1). Glukosemangelmedium wurde auf der Grundlage von RPMI 1640-Medium ohne Glukose (Sigma, USA) hergestellt. Die Laktazidose wurde durch Zugabe von reiner Milchsäure (Sigma, USA) zum Glukosemangelmedium bis zu einer Endkonzentration von 14 ± 0,7 mM und einem pH-Wert von 6,7 erzeugt.
| Medium | Glukosegehalt ,mM | Laktatgehalt, mM | рН |
|---|---|---|---|
| Standard | 9.0 ± 0.5 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Glukose-Mangel | 3.0 ± 0.1 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Laktazidose | 3.0 ± 0.1 | 14.0 ± 0.7 | 6.7 ± 0.01 |
Die Auswirkungen der verschiedenen Inkubationsbedingungen auf das Überleben der Tumorzellen, die ROS-Produktion, den Glukoseverbrauch und die Laktatproduktion wurden amzweiten Tag der Tumorzellinkubation geschätzt.
Der Glukosegehalt in den Kulturmedien und in den Homogenaten des Tumorgewebes wurde mit der Enzym-Glukose-Oxidationsmittel-Methode unter Verwendung des Kits für die Glukoseanalyse in biologischen Flüssigkeiten (Sigma, USA) gemäß den Anweisungen des Herstellers bestimmt. Der Laktatgehalt in den Inkubationsmedien und in den Tumorgewebehomogenaten wurde mit der enzymatischen Spektralphotometriemethode unter Verwendung von Laktatdehydrogenase (Sigma, USA) bestimmt [14]. Die Proben des Mediums und des Tumorgewebes wurden entnommen und bei -20 °С oder in flüssigem Stickstoff aufbewahrt, bis die Messung durchgeführt wurde.
Das Apoptose-Niveau in den Tumorzellen wurde mit Hilfe des Farbstoffs Hoechst 33258 (Sigma, USA) und der Fluoreszenzmikroskopie nach der Standardmethode analysiert.
Die ROS-Produktion in den Tumorzellen wurde mit Hilfe von 2,7-Dichlorfluoresceindiacetat (Sigma, USA) durch Spektrofluorometrie (Anregung bei 495 nm, Emission bei 530 nm) gemäß folgender Formel bestimmt [15].
Alle Messungen wurden wiederholt.
Experimente in vivo
Für In-vivo-Experimente wurden LLC/R9-Zellen in vitro unter Standardbedingungen vermehrt und Mäusen i.m. (1,0-106 Zellen/Tier in 0,1 ml Hanks’scher Lösung) injiziert.
Nach der Inokulation von LLC/R9-Zellen wurden die Tiere in zwei Gruppen aufgeteilt: Gruppe 1 – Mäuse, die mit DCA (Sigma, USA) in einer Gesamtdosis von 1,5 g/kg (LD50/3) behandelt wurden (n = 13); Gruppe 2 – Mäuse, die mit Wasser in der gleichen Dosierung und in der gleichen Menge behandelt wurden (Kontrolle, n = 12).
Die Behandlung wurde am Folgetag nach der Tumorzelltransplantation mit einem metronomischen Schema eingeleitet, fünfmal pro Woche über drei Wochen. DCA wurde ex tempore in Wasser zubereitet und per os in einem Volumen von 0,4 ml/Tier verabreicht.
Das Volumen des Primärtumors wurde auf der Grundlage seines Durchmessers berechnet, der ab dem10. Tag nach der Tumorzellinokulation jeden3:
V = π (d)3/6,
wobei d – Durchmesser des Tumors (mm).
Das Metastasierungsniveau in den tumortragenden Mäusen wurde am 21. Tag nach der Inokulation der Tumorzellen anhand der Anzahl und des Volumens der Lungenmetastasen unter Verwendung eines Binokularmikroskops und einer Millimeterskala bewertet.
Das Gesamtvolumen der Metastasen wurde nach der folgenden Formel berechnet:
V = Σ niπ(di)3/6,
wobei V – Gesamtvolumen der Metastasen (mm3), ni – Anzahl der Metastasen mit einem Durchmesser vondi (mm).
Die Analyse der funktionellen Aktivität der Komponenten der mitochondrialen Atmungskette in den Tumorzellen wurde mit Hilfe der EPR am21. Tag nach der Inokulation der Tumorzellen durchgeführt. Das Tumorgewebe wurde in Proben besonderer Größe(d = 4,0 mm, l = 25-35 mm) geschnitten, eingefroren und bei -70 °C gelagert. Die EPR-Analyse der Proben wurde bei 77 К mit dem Spektralphotometer Е-109 Varian (USA) bei einer Potential-Sweep-Geschwindigkeit von 500 Е/min, einer Modulationsamplitude von 1,25×10 Е, einer SHF-Strahlungsleistung von 10,0 mW und einer konstanten Gerätezeit von 1,0 s durchgeführt. Die Gehalte an Häm-Eisen-Komplexen mit NO (bei gmed = 2,007) in mitochondrialen ETC-Proteinen und Fe-S-Cluster-Proteinen (bei g = 1,94) in Tumorzellen wurden anhand der Daten der EPR-Spektren bestimmt.
Statistische Auswertung die Auswertung der erhaltenen Ergebnisse erfolgte mittels deskriptiver Methoden, nichtlinearer Regressionsanalyse und Student’s t-test unter Verwendung der Programme Microsoft Excel und Microcal Origin.
ERGEBNISSE
Es wurde gezeigt, dass die Laktazidose unter den Bedingungen des Glukosemangels das Überleben von LLC/R9-Zellen signifikant fördert. Tatsächlich unterschied sich die Wachstumskinetik der Tumorzellen, die unter den Bedingungen der Laktazidose bei Glukosemangel inkubiert wurden, nicht signifikant von der der Zellen, die in einem Medium mit Standardglukosegehalt inkubiert wurden, zumindest in der Phase ihres exponentiellen Wachstums. Insbesondere war die Zahl der lebensfähigen Zellen amzweiten Tag der Inkubation unter den Bedingungen der Laktazidose bei Glukosemangel praktisch die gleiche wie bei der Inkubation der Zellen in einem Medium mit Standardglukosegehalt. Gleichzeitig war die Zahl der lebensfähigen Zellen in beiden Fällen (Laktazidose und Standard) um fast 30 % höher (р < 0,05) als bei der Zellbebrütung unter den Bedingungen des Glukosemangels selbst (Abb. 1, a).
Außerdem unterschied sich die Zahl der apoptotischen Zellen unter den Bedingungen der Laktazidose statistisch nicht von dem Index bei der Zellinkubation im Medium mit Standardglukosegehalt und betrug amzweiten Tag 8,5 ± 0,9 %, während die Zahl der apoptotischen Zellen unter den Bedingungen des Glukosemangels fast dreimal so hoch war (p < 0,05) wie unter den Bedingungen der Laktazidose (Abb. 1, b).
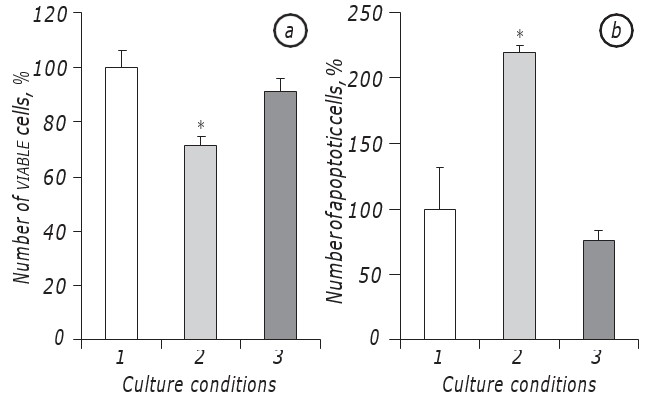
Interessanterweise war der Glukoseverbrauch von LLC/R9-Zellen unter Laktazidose-Bedingungen deutlich geringer. Die niedrige Rate des Glukoseverbrauchs der Tumorzellen bei Laktazidose, die nur amersten Tag ihrer Inkubation registriert wurde, wurde amzweiten Tag wiederhergestellt und war um 70 % niedriger (p < 0,05) als im Fall des Mediums mit Standardglukosegehalt (Tabelle 2). Bei Glukosemangel im Gegensatz zur Laktazidose sank der Glukosespiegel im Inkubationsmedium am2. Tag auf Null, was zusätzlich auf einen verringerten Glukoseverbrauch der LLC/R9-Zellen unter den Bedingungen der Laktazidose hindeutet.
Während die Laktazidose zu einer verringerten Glukoseaufnahme durch LLC/R9-Zellen führte, stieg der Gehalt an intrazellulären ROS in den Zellen, die unter diesen Bedingungen überlebten, deutlich an. Diese Daten sind in Tabelle 2 dargestellt und zeigen, dass der ROS-Gehalt in den Zellen, die unter Laktazidose-Bedingungen bebrütet wurden, um fast 150 % (p < 0,05) bzw. 230 % (p < 0,05) höher war als die entsprechenden Werte für die Zellen, die in Standard- bzw. Glukose-defizienten Medien bebrütet wurden.
Die gewonnenen Daten haben also gezeigt, dass die Laktazidose das Überleben der LLC/R9-Zellen unter den Bedingungen des Glukosemangels in vitro erheblich fördert, was durch die hohe Anzahl der unter diesen ungünstigen Bedingungen überlebenden Zellen und die niedrige Apoptoserate bestätigt wird. Das Überleben der Zellen war mit einem unerwarteten Anstieg des intrazellulären ROS-Niveaus und einem verringerten Glukoseverbrauch in LLC/R9 verbunden.
| Medium | Glukoseverbrauch, % | ROS, % |
|---|---|---|
| Standard | 100.0 ± 5.9 | 100.0 ± 24.8 |
| Glukose-Mangel | 0.0 ± 0.0* | 75.8 ± 10.7 |
| Laktazidose | 29.8 ± 1.5* | 248.7 ± 53.2* |
Anmerkung: *p < 0,05.
Diese Muster des Überlebens von LLC/R9-Zellen unter den Bedingungen der Laktazidose vor dem Hintergrund des Glukosemangels in vitro belegen, dass ein verringerter Laktatgehalt in der Mikroumgebung des Tumors das Überleben der Tumorzellen unter den Bedingungen des metabolischen Stresses verhindern kann und somit eine Antitumorwirkung ausübt. Diese Hypothese wurde von uns mit DCA als einer Verbindung getestet, die in der Lage ist, Laktazidose zu reduzieren.
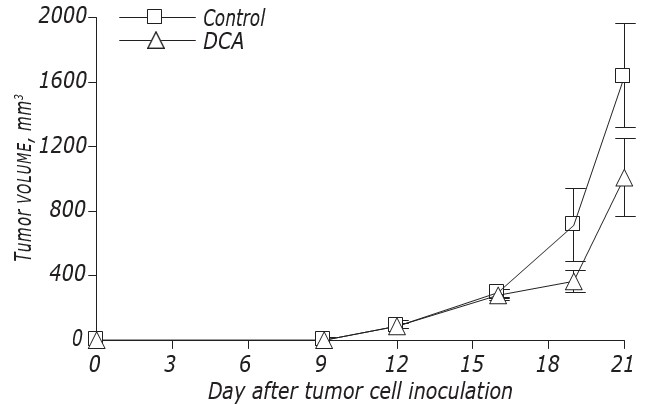
Die Daten zum Einfluss von DCA auf die Wachstumskinetik und Metastasierung von LLC/R9 sind in Abb. 2 und Tabelle 3 dargestellt. Diesen Daten zufolge beeinflusste DCA das Wachstum von Primärtumoren nicht signifikant, bewirkte aber eine deutliche Unterdrückung der Metastasierung. Die Wachstumskinetik der Primärtumore in Mäusen mit LLC/R9, die mit DCA behandelt wurden, unterschied sich praktisch nicht von der der Kontrollmäuse, und am21. Tag nach der Tumortransplantation war das Volumen der Primärtumore in der Versuchsgruppe nur um 39 % geringer als in der Kontrollgruppe (siehe Abb. 2, Tabelle 3). Obwohl DCA keine nennenswerte Unterdrückung des Primärtumorwachstums bewirkte, war seine antimetastatische Aktivität bei LLC/R9 auffällig. Die Anzahl und das Volumen der Lungenmetastasen in den mit DCA behandelten tumortragenden Mäusen waren um 59 % (р < 0,05) bzw. 94 % (р < 0,05) niedriger als diese Indizes in der Kontrollgruppe (siehe Tabelle 3).
| Gruppe von Mäusen | Tumorvolumen,mm3 | Anzahl der Metastasen | Volumen der Metastasen , mm 3 |
|---|---|---|---|
| Kontrolle (n = 13) | 1702.7 ± 333.9 | 10.9 ± 1.2 | 17.9 ± 5.6 |
| DCA (n = 13) | 1046.0 ± 258.3 | 4.5 ± 1.6* | 1.1 ± 0.4* |
Anmerkung: *р < 0,05, Unterschiede sind signifikant im Vergleich zum Wert der Kontrolle.
Eine Analyse des Laktatgehalts in Tumorgewebeproben hat gezeigt, dass DCA unerwartet einen signifikanten Anstieg des Laktatgehalts im Tumorgewebe verursacht, zumindest am21. Tag nach der Tumortransplantation einen signifikanten Anstieg des Laktatgehalts im Tumorgewebe verursachte. Wie aus Tabelle 4 hervorgeht, war der Laktatgehalt im Tumorgewebe der mit DCA behandelten Mäuse um fast 30 % höher (р < 0,05) als der der Kontrollgruppe. In Anbetracht der Fähigkeit von DCA als PDH-Kinase-Inhibitor, den Energiestoffwechsel des malignen Tumors in Richtung oxidative Phosphorylierung zu reorganisieren, haben wir die Laktatproduktion der Tumorzellen als Surrogatmarker für die Hemmung der Glykolyse unter DCA-Einfluss betrachtet. Der durch DCA verursachte Anstieg des Laktatspiegels im Tumor deutet darauf hin, dass seine Verabreichung an Mäuse mit LLC/R9 in einer Gesamtdosis von 1,5 g/kg Körpergewicht des Tieres für die Aktivierung der oxidativen Phosphorylierung in den Tumorzellen unzureichend sein könnte, was zum Teil seine geringe Wirksamkeit gegen Primärtumore erklärt.
| Gruppe von Mäusen | Laktat (µmol/1 g Gewebe) |
|---|---|
| Kontrolle (n = 4) | 11.1 ± 0.6 |
| DCA (n = 5) | 14.4 ± 1.5* |
Anmerkung: р < 0,05, Unterschiede sind signifikant im Vergleich zum Wert der Kontrolle.
Die Analyse der EPR-Spektren von Tumorproben hat gezeigt, dass DCA den Funktionszustand der ETC-Komponenten in den Mitochondrien der Tumorzellen nicht signifikant beeinflusste (Tabelle 5). Zum Beispiel war bei Mäusen mit LLC/R9, die mit DCA behandelt wurden, die Intensität der EPR-Signale, die den Nitrosyl-Häm-Eisen-Protein-Komplexen(gсер= 2,007) in den ETC-Proteinen der Tumorzellmitochondrien entsprechen, nicht signifikant höher als bei Kontrollmäusen. Es ist bekannt, dass die Anhäufung von NO-Komplexen aus Häm-Eisen einerseits auf ein Redox-Ungleichgewicht hinweisen kann, bei dem freie Radikale dominieren, insbesondere auf eine NO-Überproduktion, und andererseits auf eine mögliche Hemmung der Zellatmung durch Nitrosylierung von Häm-Proteinen. DCA, dessen Hauptmechanismus der Antitumorwirkung vermutlich mit der Induktion der ROS-Produktion durch die Mitochondrien zusammenhängt [8, 10, 11]verursachte jedoch keine Erhöhung der Häm-Eisen-Komplexe mit NO im Tumorgewebe. Die jüngste Beobachtung könnte möglicherweise mit den Merkmalen der LLC/R9-Zellen zusammenhängen, nämlich einem extrem hohen Gehalt an diesen Komplexen, der für diesen Tumor charakteristisch ist und dessen fortschreitende Anhäufung während der Tumorentwicklung in vivo von uns auch bei fehlender Behandlung registriert wurde [16].
| Relative EPR-Signalintensität | Relative EPR-Signalintensität | |
| Gruppe von Mäusen | Nitrosyl-Häm-Eisen-Protein-Komplexe (g = 2,007) | Fe-S-Protein (g = 1,94) |
| Kontrolle | 54.3 ± 4.5 | 15.8 ± 0.5 |
| DCA | 97.8 ± 30.1 | 17.8 ± 2.1 |
Das Fehlen einer signifikanten Wirkung von DCA auf die funktionelle Aktivität der mitochondrialen ETC-Komponenten in Tumorzellen wurde auch durch die Daten zur Intensität der EPR-Signale bestätigt, die den Fe-S-Cluster-Proteinen(g = 1,94) entsprechen (Komplexe І, ІІ, ІІІ), die in beiden Tiergruppen praktisch gleich waren (siehe Tabelle 5).
Zusammenfassend haben die Ergebnisse unserer Studie gezeigt, dass die Laktazidose das Überleben der LLC-Variante LLC/R9 unter den Bedingungen des Glukosemangels erheblich fördert. Wenn sich LLC/R9 in vivo entwickelte, übte DCA jedoch keine antitumorale Wirkung auf Primärtumore aus. Die fehlende antitumorale Wirkung von DCA auf das Wachstum von LLC/R9 stand im Einklang mit dem Fehlen einer hemmenden Wirkung von DCA auf den Laktatgehalt im Tumor sowie mit dem Fehlen einer nennenswerten Wirkung von DCA auf die ROS-Produktion der Tumorzellen. Obwohl DCA das Wachstum von LLC/R9 nicht beeinflusste, hemmte es die Metastasierung drastisch; diese Beobachtung konnte nicht durch die DCA-Wirkung innerhalb des Primärtumors erklärt werden, und weitere zusätzliche Studien über seine antimetastatische Wirkung sind erforderlich.
REFERENZEN
1 Feron O. Pyruvat zu Laktat und zurück: vom Warburg-Effekt zum symbiotischen Energieträgeraustausch in Krebszellen. Radiother Oncol 2009; 92: 329-33. doi: 10.1016/j.radonc.2009.06.025.
2 Wu H, Ding Z, Hu D, et al. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J Pathol 2012; 227: 189-99. doi: 10.1002/path.3978.
3 Fiaschi T, Marini A, Giannoni E, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 2012; 72: 5130-40.
4 Solyanik GI, Fedorchuk AG, Pyaskovskaya ON, et al. Anticancer activity of aconitine-containing herbal extract BC1. Exp Oncol 2004; 26: 307-11.
5 Pyaskovskaya ON. Antiangiogene Wirkung von Cyclophosphamid bei experimentellen metastasierenden Tumoren. J Med Chem 2012; 2: 25-9 (auf Ukrainisch).
6 Kolesnik DL, Pyaskovskaya ON, Tregubova NV, Solyanik GI. Lewis-Lungenkarzinom-Variante mit hoher Sensitivität für antiangiogene Antitumortherapie weist eine hohe Kapazität für Autophagie auf. Cytol Genet 2012; 46: 155-60. doi: 10.3103/S009545271203005X.
7 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism 1989; 38: 1124-44.
8 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 2007; 11: 37-51.
9 Wong JY, Huggins GS, Debidda M, et al. Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol 2008; 109: 394-402. doi: 10.1016/j.ygyno.2008.01.038.
10 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2: 31-4. doi: 10.1126/scitranslmed.3000677.
11 Stockwin LH, Yu SX, Borgel S, et al. Natriumdichloracetat zielt selektiv auf Zellen mit Defekten im mitochondrialen ETC Int J Cancer 2010; 127; 2510-19.
12 Kumar A, Kant S, Singh SM. Neue molekulare Mechanismen der antitumoralen Wirkung von Dichloracetat gegen T-Zell-Lymphome: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 2012; 199: 29-37.
13 Pyaskovskaya ON, Dasyukevich OI, Kolesnik DL, et al. Changes in VEGF level and tumor growth characteristics during Lewis lung carcinoma progression towards cis-DDP resistance. Exp Oncol 2007; 29: 197-202.
14 Biochemische Methoden (Lipid- und Energiestoffwechsel). MI Prohorova, ed. L.: Leningrad Univ, 1982. 272 p.
15 Wang H, Joseph JA. Quantifizierung des zellulären oxidativen Stresses mittels Dichlorfluorescein-Assay unter Verwendung eines Mikroplattenlesegeräts. Freie Radikale Biol Med 1999; 27: 612-6.
16 Pyaskovskaya ON, Sorokina LV, Kolesnik DL, et al. Dynamics of changes of antioxidant system indexes during the growth of two Lewis lung carcinoma variants. Exp Oncol 2014; 36: 29-33.