Stephen B. Strum & Örn Adalsteinsson & Richard R. Black & Dmitri Segal & Nancy L. Peress & James Waldenfels
S. B. Strum
International Strategic Cancer Alliance, 538 Granite Street, Ashland, OR 97520, USA
e-mail: [email protected]
S. B. Strum American Society of Clinical Cancer Alliance, 538 Granite Street, Ashland, OR 97520, USA. B. Strum
Società Americana di Oncologia Clinica, 538 Granite Street, Ashland, OR 97520, USA
S. B. Strum
American Urological Association, 538 Granite Street, Ashland, OR 97520, USA
Ö. Adalsteinsson
International Strategic Cancer Alliance, 873 E. Baltimore Pike #333, Kennett Square, PA 19348, USA
e-mail: [email protected]
R. R. Black
Nuclear Medicine and PET Imaging, Radisphere Teleradiology Group, Beachwood, OH 44022, USA
e-mail: [email protected]
D. Segal
Valley Radiology Consultants, Poway, CA 92064, USA
e-mail: [email protected]
N. L. Peress
Life Extension Foundation, 5760 S. Scenic Drive, Sault Sainte Marie, MI 49783, USA
e-mail: [email protected]
J. Waldenfels
Life Extension Foundation, 9114 Steeplebush Court, Annandale, VA 22003-4051, USA
e-mail: [email protected]
Parole chiave: Dicloroacetato . DCA . linfoma non-Hodgkin . NHL . PET . PET/CT . Glicolisi . Metabolomica . Warburg
Abbreviazioni:
DCA (dicloroacetato)
NHL (linfoma non-Hodgkin)
PET (tomografia a emissione di positroni)
CT (tomografia computerizzata)
FDG (fluorodesossiglucosio)
SUV (valore di captazione standardizzato)
mg (milligrammi)
kg (chilogrammi)
R-CHOP (rituximab-Citoxan,
Idrossidaunomicina, Oncovin, Prednisone)
Ricevuto: 27 ottobre 2012
Accettato: 23 novembre 2012
Springer Science+Business Media New York 2012
Abstract
L’assorbimento del fluorodesossiglucosio mediante tomografia a emissione di positroni nei tumori di vari tipi di cancro dimostra il ruolo chiave del glucosio nella proliferazione del cancro. Il dicloroacetato è una molecola a 2 carboidrati con un’attività biologica cruciale nell’alterare la scomposizione metabolica del glucosio in acido lattico. Studi su linee cellulari umane dimostrano che il dicloroacetato altera il metabolismo della cellula tumorale da glicolisi a fosforilazione ossidativa, ripristinando così le funzioni mitocondriali che innescano l’apoptosi della cellula tumorale. Le segnalazioni di dicloroacetato in soggetti umani sono rare. Gli autori hanno contattato persone che, nei forum su Internet, avevano riportato eccezionali risposte antitumorali all’automedicazione con dicloroacetato. Con il consenso informato, sono state richieste le cartelle cliniche complete per documentare la risposta al dicloroacetato, sottolineando il contesto della monoterapia con dicloroacetato. Dei dieci pazienti che hanno acconsentito a tale valutazione, solo uno soddisfaceva i criteri di avere cartelle cliniche complete, nonché referti di patologia, diagnostica per immagini e di laboratorio, oltre alla terapia con dicloroacetato come agente singolo. Questo individuo è l’oggetto della presente relazione. In questo caso di un uomo con recidiva documentata dopo una chemioterapia all’avanguardia per il linfoma non-Hodgkin, viene documentata una risposta significativa al dicloroacetato con una remissione completa, che permane dopo 4 anni. Il dicloroacetato sembra essere una terapia innovativa che merita ulteriori indagini nel trattamento del cancro.
Sfondo
Il profilo metabolico delle neoplasie è stato caratterizzato come associato ad adattamenti metabolici diretti a utilizzare preferenzialmente le vie coinvolte nella glicolisi (Warburg et al. 1927), che nella letteratura recente è stato definito fenotipo glicolitico del cancro. (Bui e Thompson 2006; Fang et al. 2008; Gatenby e Gawlinski 2003) In sostanza, questo fenotipo glicolitico è un adattamento darwiniano in quanto la cellula tumorale riduce e mina le vie metaboliche di ossidazione del glucosio utilizzate dalle cellule normali per la produzione di energia e per l’eliminazione delle cellule tumorali (Fang et al. 2008). Una funzione cruciale delle cellule normali compromessa nella lotta contro il cancro è la morte cellulare programmata mitocondriale o apoptosi. Sulla base di queste osservazioni, sono attualmente in fase di sperimentazione clinica agenti che hanno come bersaglio il metabolismo tumorale, e in particolare le vie mitocondriali di produzione di ATP. Il dicloroacetato (DCA) è stato utilizzato negli ultimi 30 anni per trattare l’acidosi lattica congenita, una rara malattia metabolica che si manifesta soprattutto nei bambini e nei giovani adulti. (Berendzen et al. 2006; Kuroda et al. 1986; Stacpoole et al. 1997, 2008, 2006). L’acidosi lattica congenita è associata a vari errori congeniti di disfunzione mitocondriale e quasi un migliaio di pubblicazioni mediche peer-reviewed sono incentrate sull’uso clinico dei DCA per questo disturbo. Sono stati pubblicati anche numerosi altri articoli su vari aspetti della farmacologia, degli effetti metabolici e della tossicologia del DCA, ma solo una decina di articoli sul DCA riguardano la sua attività antitumorale (Bonnet et al. 2007; Bui e Thompson 2006; Cao et al. 2008; Chen et al. 2007; Christofk et al. 2008; Madhok et al. 2010; Michelakis et al. 2010, 2008; Vander Heiden 2010; Wong et al. 2008), molti dei quali si limitano alla valutazione del DCA in linee cellulari tumorali o in modelli animali non umani (Cao et al. 2008; Madhok et al. 2010; Wong et al. 2008; Sun et al. 2010). Solo la pubblicazione di Michelakis et al. (2010) ha valutato il DCA in cinque pazienti umani con glioblastoma multiforme e ha presentato alcune indicazioni cliniche di risposta al DCA.
Il DCA è stato segnalato come efficace agente antitumorale in vitro e come causa di regressione in vivo nel glioblastoma multiforme umano (Michelakis et al. 2010). Questa, tuttavia, è la prima segnalazione di un paziente oncologico sottoposto a monoterapia con DCA con l’induzione di una remissione completa dopo la recidiva di una chemioterapia all’avanguardia con rituximab-CHOP.
I metodi
Popolazione di pazienti
Gli autori SS e OA hanno sollecitato tutte le persone che avevano riportato risposte antitumorali favorevoli al DCA su forum in Internet, chiedendo loro se avrebbero accettato di rendere disponibili le loro cartelle cliniche complete per un’analisi dettagliata, senza alcun costo. Queste persone sono state informate che lo scopo dello studio era quello di verificare se la monoterapia con DCA potesse essere confermata come terapia antitumorale attiva.
Questioni medico-legali
Il consenso informato scritto all’uso di dati medici dettagliati e di immagini radiologiche è stato ottenuto dal soggetto di questo case report. Una copia del consenso scritto è disponibile per la revisione da parte del direttore di questa rivista.
Criteri di esclusione dei pazienti
Sono state richieste copie di tutti i consulti e le visite in ufficio, i referti patologici, gli studi di imaging e di laboratorio, i referti chirurgici, i dati della radioterapia e i dettagli della chemioterapia. Su 10 possibili candidati, 5 hanno fornito cartelle cliniche decisamente insufficienti per consentire una valutazione dell’efficacia del DCA. In altri 3 candidati, un altro trattamento era stato iniziato poco prima o subito dopo l’inizio del DCA. In 1 candidato, il DCA non era mai stato iniziato. A uno dei cinque soggetti con documentazione insufficiente era stato diagnosticato un linfoma non-Hodgkin e sembrava aver avuto una risposta importante alla monoterapia con DCA, ma purtroppo non ha risposto alle nostre comunicazioni per ottenere una documentazione medica completa. Per l’altro individuo (TM), oggetto del presente rapporto, sono state ottenute cartelle cliniche complete. Questo individuo, descritto in questa relazione, non è stato trattato dagli autori.
Onere finanziario per i partecipanti
Per tutti i casi in cui la revisione iniziale delle cartelle cliniche indicava una risposta alla monoterapia con DCA, le cartelle cliniche complete sono state richieste, revisionate ed estratte in una cartella clinica elettronica da SS e OA. In questi casi, i vetrini diagnostici di patologia e/o i blocchi di tessuto sono stati ottenuti e inviati a un esperto di ematopatologia per un secondo parere. I dati di imaging completi, sotto forma di file Dicom, sono stati ottenuti e rivisti dal coautore RB, un radiologo abilitato alla professione, specializzato solo in studi PET e PET/CT. Con questo approccio, è stata ottenuta una valutazione medica altamente dettagliata, senza alcun costo per i partecipanti allo studio.
Presentazione del caso
Il soggetto dello studio TM è attualmente un uomo di 52 anni a cui è stato diagnosticato per la prima volta all’età di 46 anni un linfoma non-Hodgkin (NHL) il 6/1/07. Ha presentato alla fine del 2006 una storia di linfoma non-Hodgkin. Si è presentato alla fine del 2006 con un’anamnesi di herpes zoster, febbri di basso grado e sudorazioni notturne abbondanti. Nei 5 mesi successivi si è verificata una perdita di peso di 15 kg e le diagnosi di infezione sinusale e tubercolosi hanno portato al trattamento con vari farmaci, tra cui ciprofloxacina, clindamicina, INH, rifampicina, pirazimide e altri antibiotici. Una massa di 6,0 cm all’angolo della mandibola inferiore sinistra è stata notata durante la valutazione di medicina interna del 5/1/07, insieme a un livello di proteina C-reattiva di 196, VES 99, ematocrito 33,9 ma un LDH normale di 138. Il 5/7/07, una risonanza magnetica del collo ha mostrato masse multiple all’interno della ghiandola parotidea sinistra e linfonodi cervicali multipli di sinistra fino a 2,0 cm con estensione alla regione sopraclaveare.
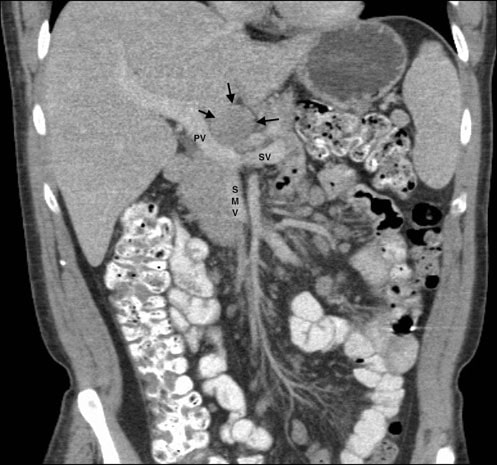
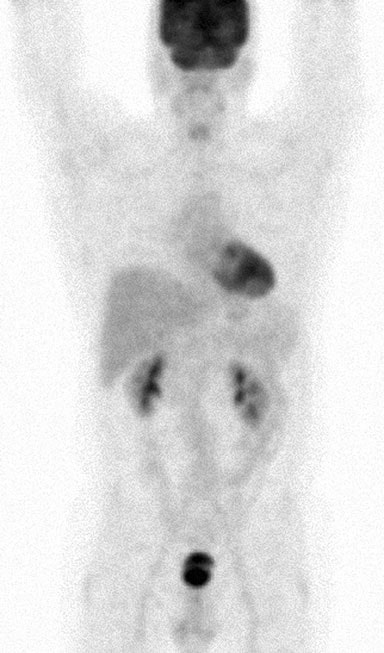
Il 5/11/07 è stato eseguito un agoaspirato di una massa discreta di 3,0 × 3,0 cm nella ghiandola parotidea sinistra. Il referto e i vetrini di questa procedura sono stati richiesti dagli autori numerose volte, ma non sono mai stati ricevuti. La TC del torace, dell’addome e della pelvi del 25 maggio 2007 aveva rivelato splenomegalia, masse renali multiple bilaterali e linfoadenopatia che coinvolgeva i linfonodi mesenterici, della porta epatica, retroperitoneali e dell’asse celiaco (Fig. 1). Questi risultati erano coerenti con una diagnosi di linfoma di stadio IV (> 4 linfonodi coinvolti), con anemia e sintomi sistemici di febbre, sudorazione notturna e perdita di peso, e con un coinvolgimento extra-nodale di almeno la ghiandola parotide.
Il 6/1/07 è stata eseguita una biopsia escissionale della lesione parotidea sinistra. Il referto patologico indicava un linfoma non Hodgkin a cellule B, follicolare e diffuso, con coinvolgimento focale della parotide, e classificato come intermedio sulla base delle dimensioni delle cellule, della morfologia e dell’indice proliferativo (CD71). Gli studi di citometria a flusso dei campioni di tessuto sono risultati coerenti con un linfoma non-Hodgkin a cellule B di origine germinale. L’immunoistochimica è risultata positiva per i marcatori CD3, CD5, CD10, CD20, CD23, CD43, BCL-1 e BCL-2. La colorazione kappa e lambda ha mostrato una popolazione di cellule B monotipiche lambda che esprime il CD10 e che comprende il 38% del tessuto totale.
L’esame fisico effettuato il 26 giugno 2007 da un oncologo medico ha rivelato una massa parotidea sinistra (6,0 × 5,0 cm), un linfonodo giugulodigastrico sinistro (4,0 × 4,0 cm), linfonodi cervicali anteriori superiori di sinistra (3,0 × 3,0 cm), linfonodi di sinistra (3,0 × 3,0 cm) e linfonodi di sinistra (3,0 × 3,0 cm).0 × 3,0 cm), linfonodo giugulodigastrico destro (2,0 × 2,0 cm), linfonodi sopraclaveari destro e sinistro (3,0 × 3,0 cm) e linfonodi ascellari destro e sinistro (2,0 × 3,0 ciascuno). È importante notare che più medici che hanno esaminato il TM hanno riportato misure linfonodali significativamente diverse di siti anatomici specifici nello stesso giorno o nel giro di pochi giorni.
Un consulto ematopatologico di seconda istanza presso un centro medico universitario il 26 giugno 2007 è stato sospetto per un linfoma a cellule B di basso grado, con parziale effusione dell’architettura linfonodale da parte di un’infiltrazione atipica e prevalentemente diffusa di piccole cellule linfoidi; è stata raccomandata una rebiopsia di un linfonodo ingrossato. Un aspirato di midollo osseo eseguito il 29.6.2007 indicava un midollo normocellulare con 1 grande aggregato linfoide non paratrabecolare, contenente piccoli linfociti maturi. La citometria a flusso su quel campione ha mostrato una popolazione clonale di cellule B lambda-limitata, caratterizzata da piccole cellule mature con contorni nucleari irregolari. Insieme alla parziale espressione di CD10 e alla mancanza di espressione di CD 5, è stata posta la diagnosi di linfoma follicolare a cellule centrali e di linfoma a cellule B di basso grado che coinvolge il midollo osseo.
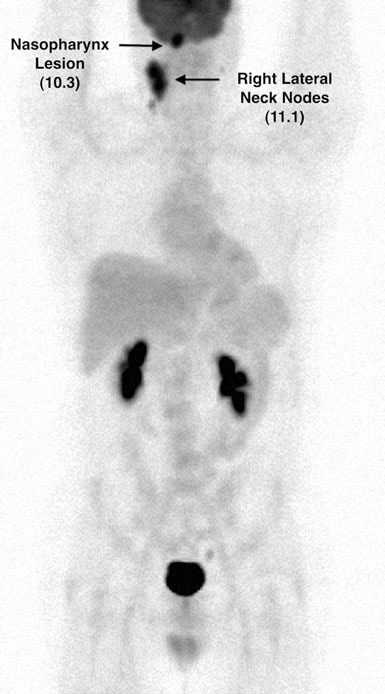
Il 7/12/07 è stata eseguita una PET FDG che ha rivelato anomalie metaboliche in più siti nodali e nell’area parotidea sinistra. Il valore di captazione standardizzato (SUV) è stato corretto mediante standardizzazione con il fegato (Wahl et al. 2009) e i risultati sono stati mostrati nelle Figg. 2 e 3.
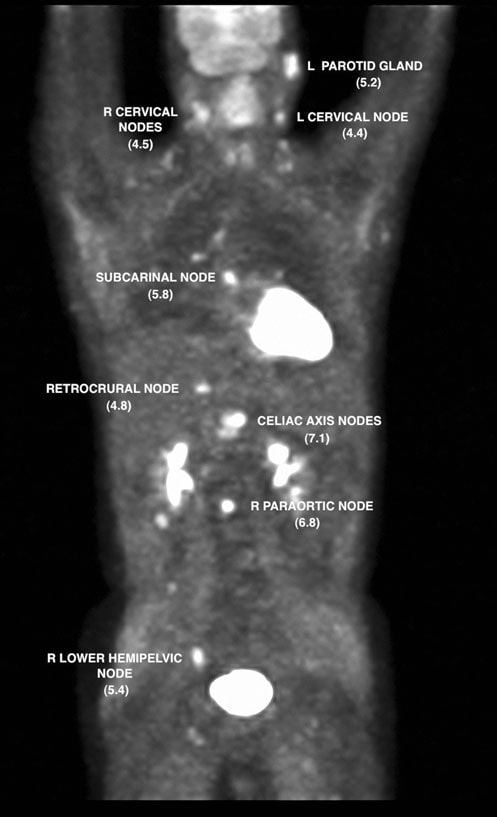
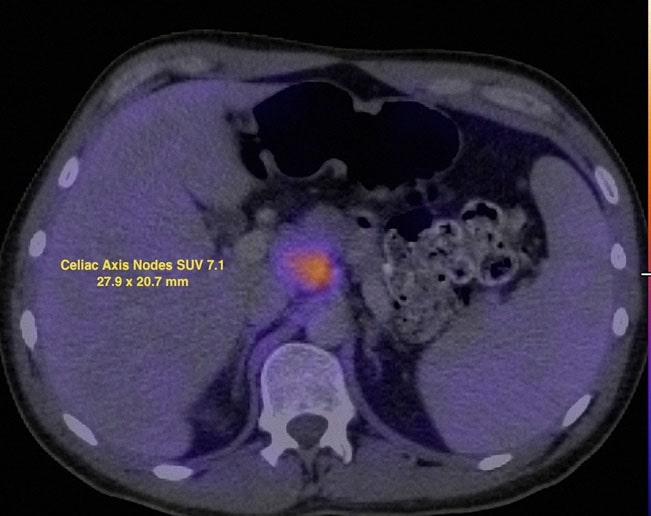
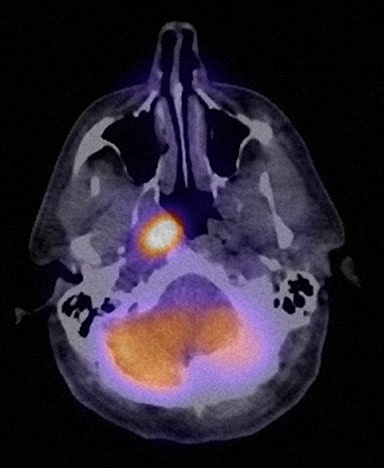
Il 13/7/07 è stato iniziato un ciclo di rituximab-CHOP (R-CHOP) a dose densa, ripetuto ogni 14 giorni. I cicli 2-6 sono stati somministrati il 27/7/07, il 10/8/07, il 24/8/07, il 7/9/07 e il 21/9/07. Il 10/5/07, una TC del collo, del torace, dell’addome e della pelvi ha mostrato una significativa risoluzione di quasi tutte le precedenti aree di adenopatia. Uno studio FDG PET è stato ripetuto il 10/8/07 e ha riportato una captazione focale nella regione sopraciliare R (Fig. 4).
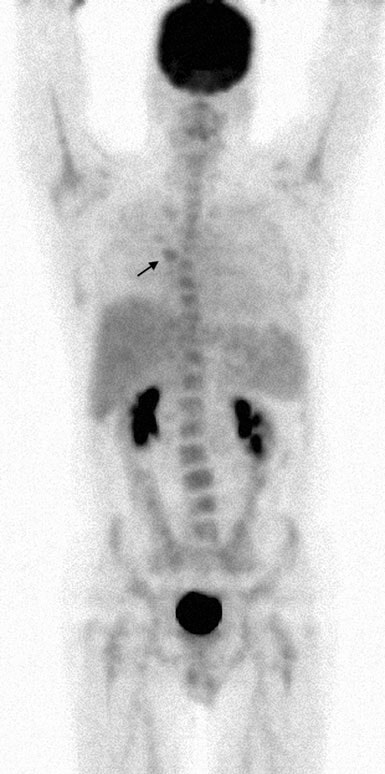
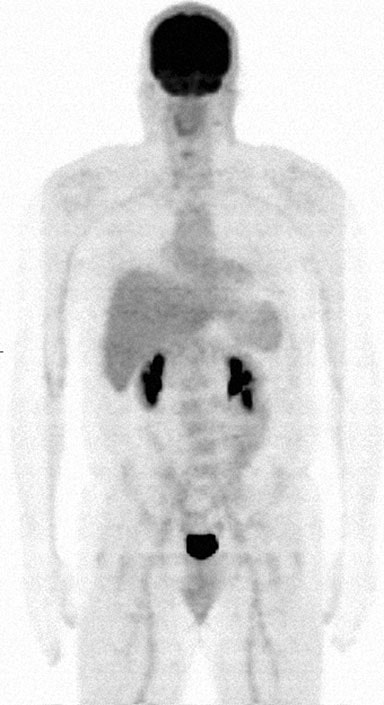
L’esame fisico del 10/9/07 indicava un’area fibrotica residua nella regione cervicale posteriore sinistra di 4,0 per 3,0 cm. La FDG PET n. 3 del 5 agosto 2008 era completamente normale (Fig. 5).
Il paziente è stato considerato in remissione completa a partire dal 1/08. Una PET FDG ripetuta (#4) il 4/11/08 è rimasta normale. Tuttavia, nel 7/08 il TM ha riferito una perdita di peso di 11 libbre nelle precedenti 2 settimane, tosse e sudorazione, nonché febbre di basso grado. Il 7/10/08 è stato riscontrato un nodo cervicale anteriore destro di 2,0 × 2,0 cm all’esame fisico e il 16/08 è stata ottenuta la FDG PET #5. Questa ha mostrato diversi nuovi ipermetabolismi. Questa mostrava diversi nuovi focolai ipermetabolici (Figg. 6 e 7).
Il 4 settembre 2008, TM è stato visitato da un chirurgo della testa e del collo, che ha descritto una grande massa di linfonodi opachi nel collo destro ai livelli I, 2A, 2B e al livello 5 superiore. La cartella clinica descriveva il paziente come estremamente turbato dagli effetti collaterali di nausea e vomito dovuti alla precedente chemioterapia somministrata dal 13/7 al 21/9/07. Il paziente decise di intraprendere una terapia con un’altra persona. In base alle sue ricerche, decise di intraprendere un regime di trattamento che prevedeva l’uso di DCA (dicloroacetato) e di vari integratori, che iniziò il 10 settembre 2008. Il suo “protocollo DCA” consisteva in: DCA 1.000 mg al giorno in un’unica dose giornaliera. Il DCA veniva mescolato con 10 once di Mountain Dew™ contenente 55 mg di caffeina. TM utilizzava anche vitamina B1 a 500 mg/d (fino al 12/10/08), acido alfa lipoico 600 mg bid, tè verde (Jarrow™) 500 mg bid contenente 74 mg di EGCG e 35 mg di caffeina per 500 mg.
Entro 2 settimane dall’inizio di questo regime, il paziente ha riportato una significativa riduzione della sudorazione notturna, della febbre di basso grado, dell’anoressia e dell’affaticamento. Un mese dopo l’inizio del protocollo DCA, i linfonodi del collo erano sensibilmente più piccoli e a 2 mesi non erano più palpabili. A 71 giorni dall’inizio del protocollo DCA, si è verificata la completa risoluzione di tutti i sintomi sistemici. Il paziente ha riferito un buon livello di energia e di appetito, la capacità di dormire bene e nessun effetto collaterale.
La PET FDG n. 6 è stata eseguita il 4 dicembre 2008. Tutte le anomalie riscontrate nell’esame precedente dell’8/16/08 erano scomparse. Grazie agli sforzi degli autori SS e OA, il 2/11/09 è stata ottenuta una consulenza ematopatologica esterna da parte di un centro oncologico internazionale. Le sezioni colorate con ematossilina ed eosina hanno mostrato una ghiandola salivare coinvolta da un denso infiltrato linfoide atipico che mostrava un pattern vagamente nodulare e diffuso. L’infiltrato era composto da piccole cellule linfoidi B monocitoidi. Erano presenti lesioni linfoepiteliali. In base alla morfologia, la diagnosi differenziale includeva un linfoma a cellule B della zona marginale extra-nodale (MALToma) o un linfoma follicolare con differenziazione monocitoide. Gli studi di immunoistochimica (IHC) sono stati eseguiti su vetrini non colorati e sono risultati negativi per BCL-2, CD3, CD43 e cheratina. È stata riscontrata la positività per BCL-6 e CD10 nelle aree vagamente nodulari, CD20, catene kappa e lambda in alcune plasmacellule. L’impressione finale è stata riportata come: “Sebbene non vi siano prove IHC certe di linfoma, la morfologia, il riscontro in citometria a flusso di cellule linfoidi B monotipiche lambda e il riscontro molecolare di riarrangiamento del gene Ig Kappa sono coerenti con un linfoma a cellule B di basso grado, che non può essere ulteriormente classificato”
Il 27/4/09 è stato eseguito lo studio FDG PET/CT n. 7, che non ha mostrato alcuna attività ipermetabolica. Gli stessi risultati sono stati riscontrati nello studio #8 il 9/8/09. Tuttavia, una nota della clinica di medicina interna dell’1/8/10 indicava che il TM aveva notato un ingrossamento dei linfonodi del collo e l’esame fisico confermava un linfonodo cervicale posteriore sinistro di 2 cm di diametro. In concomitanza con ciò, ha iniziato a presentare una lieve sudorazione notturna e affaticamento. La terapia convenzionale è stata offerta ma rifiutata dal paziente, che ha preferito continuare a utilizzare il protocollo DCA. La PET/CT n. 9 è stata eseguita il 2/1/10 e il referto ufficiale indicava un uptake orofaringeo destro che coinvolgeva una lesione di 3,5-4,0 cm con un SUV di 5,5. L’esame dell’esperto RB ha indicato un SUV corretto di 3,0 e la conclusione che questo risultato è “più coerente con la normale distribuzione fisiologica del radiofarmaco” RB ha anche indicato che la misurazione anatomica della lesione orofaringea di cui sopra era sicuramente errata. Gli oncologi locali hanno concluso che il linfoma del paziente si era ripresentato e sono state discusse le opzioni di trattamento locale con radioterapia e lenalidomide da sola o con rituximab. TM ha nuovamente rifiutato un’ulteriore chemioterapia e ha continuato ad assumere DCA. Il controllo ematologico-oncologico dell’1/25/11 ha evidenziato un linfonodo cervicale sinistro di 2,0 cm. Anche in questo caso sono state discusse le opzioni per un ulteriore trattamento, tra cui bendamustina, fludarabina, radioimmunoterapia con Zevalin, ma tutte sono state rifiutate. Il 4/10/11 è stata eseguita una PET/CT FDG #10 che non ha mostrato alcuna evidenza di attività ipermetabolica (Fig. 8).
A novembre 2012, TM riferisce di stare bene e di non avere sintomi sistemici. Nota la comparsa di piccoli linfonodi nel collo che, a suo dire, hanno la dimensione di un pisello. Riferisce di aver continuato a utilizzare il protocollo DCA, ma afferma di aver diminuito la frequenza di somministrazione del DCA a tre volte alla settimana. Dichiara che il suo livello di energia è eccellente, che lavora a tempo pieno, ma che, non potendo ottenere l’assicurazione, non si è sottoposto a ulteriori valutazioni mediche tramite esami fisici, test di laboratorio o studi di imaging. Gli autori SS e OA hanno facilitato l’esecuzione di un recente studio di imaging FDG PET/CT l’11/8/12 e di esami di laboratorio l’11/9/12, senza alcun costo per TM. Lo studio FDG è risultato completamente normale. Tutti gli esami di laboratorio, a parte un aumento dei trigliceridi e delle LDL, sono normali, compresi i biomarcatori come la β-2 μglobulina, i recettori solubili dell’IL-2 e la VES.
Discussione
Le cellule tumorali hanno un metabolismo notevolmente diverso rispetto alle cellule normali da cui derivano. Presentano un metabolismo alterato che consente loro di sostenere tassi proliferativi più elevati e di resistere a vari segnali apoptotici (Bonnet et al. 2007; Bui e Thompson 2006). Questo metabolismo alterato e aumentato significa che le cellule tumorali necessitano di grandi quantità di proteine, lipidi e nucleotidi, oltre che di energia sotto forma di ATP. Quasi 100 anni fa, Otto Warburg pubblicò le sue scoperte, dimostrando che le cellule tumorali presentano un’alterazione dell’energia cellulare che comporta un aumento della glicolisi aerobica. (Warburg 1956a, b; Warburg et al. 1927)
Uno dei meccanismi d’azione proposti per il DCA è quello di spostare il metabolismo delle cellule tumorali dalla via citoplasmatica glicolitica, preferita dalle cellule tumorali, a una via ossidativa del glucosio basata sui mitocondri (fosforilazione ossidativa) (Bonnet et al. 2007). In questo modo, il DCA ripristina la funzione apoptotica dei mitocondri e inverte le alterazioni metaboliche che hanno permesso alle cellule tumorali di sopravvivere, proliferare e metastatizzare. Questa presunta azione del DCA è associata alla sua capacità di invertire l’inibizione della PDH (piruvato deidrogenasi), un enzima cruciale che consente l’ingresso del piruvato nel ciclo di Krebs attraverso la sua ossidazione al substrato chiave acetil CoA. Il DCA agisce inibendo la PDK (piruvato deidrogenasi chinasi) mitocondriale. Pertanto, il DCA inibisce l’inibitore coinvolto nella produzione di PDH. Il DCA ottiene questo risultato impedendo la fosforilazione e l’inattivazione della PDH da parte della PDK. (Constantin-Teodosiu et al. 1999)
L’aggressività delle cellule tumorali è stata associata al grado di iperpolarizzazione del potenziale di membrana mitocondriale (Heerdt et al. 2005). Bonnet et al. hanno definito una serie di effetti mitocondriali del DCA, come segue. Il DCA ha un effetto rapido e dose-dipendente che porta alla depolarizzazione della membrana mitocondriale entro 5 minuti. A causa di questo aspetto dell’attività del DCA, l’AIF (fattore che induce l’apoptosi) viene traslocato nel nucleo della cellula e il citocromo C diventa diffusamente presente nel citoplasma. Il DCA aumenta in modo dose-dipendente la produzione di H2O2 all’interno del Complesso I dell’ETC. La generazione di H2O2 attiva i canali Kv1.5 che a loro volta inibiscono NFAT1, un noto inibitore dell’apoptosi. Altri effetti biochimici del DCA sono l’aumento dell’espressione dell’annexina, l’attivazione delle caspasi 3 e 9, la riduzione della survivin e del PCNA (antigene nucleare delle cellule proliferanti) (Bonnet et al. 2007). Altri studi hanno dimostrato che l’upregulation dei canali Kv1.5 probabilmente riduce il potassio cellulare, attivando le caspasi e l’apoptosi (Remillard e Yuan 2004; Wang 2004; Pan e Mak 2007).
Ci sono stati pochi studi sull’effetto del DCA sulla crescita del cancro umano e la maggior parte di questi sono stati condotti in vitro. Tong et al. hanno studiato il DCA da solo o in combinazione con il 5-FU contro quattro linee cellulari di cancro colorettale umano. Hanno dimostrato che il DCA è sinergico con il 5-FU nell’induzione dell’apoptosi, nell’inibizione della proliferazione delle cellule tumorali, nell’aumento dell’arresto del ciclo cellulare nella fase G1/S, nella diminuzione dell’espressione di Bcl-2 e nell’aumento dell’espressione di Bax e della caspasi-3 (Tong et al. 2011). Lo studio di Michelakis et al. (2010) su cinque pazienti con glioblastoma multiforme descrive la regressione radiologica e fornisce immagini di due pazienti con un’apparente risposta al DCA, anche se il debulking del tumore nel paziente numero 2 sembra confondere l’interpretazione dell’effetto del DCA.
La PET con fluorodesossiglucosio F-18 (F-18 FDG) è ora utilizzata da un numero crescente di oncologi per determinare l’estensione della malignità, documentare la risposta terapeutica e fornire informazioni prognostiche dopo la terapia antineoplastica. Il paziente presentato aveva una PET FDG al basale coerente con i segni e i sintomi clinici di un processo neoplastico altamente attivo. Dopo un trattamento con chemioterapia all’avanguardia con R-CHOP, ha ottenuto una remissione completa di breve durata; la sua ricaduta è stata nuovamente caratterizzata da anomalie della FDG PET. Tuttavia, la risposta alla monoterapia con DCA ha portato a una remissione completa duratura, ora a 4 anni. L’assorbimento di F18 FDG da parte delle neoplasie è considerato un esempio dell’effetto Warburg, cioè dell’utilizzo del glucosio da parte dei tumori in presenza di ossigeno (glicolisi aerobica). Tuttavia, la singolare funzionalità della PET FDG osservata in vari tipi di tumore deriva da molteplici fattori, alcuni dei quali sono:
- Fosforilazione di F18 FDG a F18 FDG-6 fosfato da parte dell’esochinasi (HK)
- Regolazione in aumento dell’HK da parte di fattori di trascrizione inducibili dall’ipossia, ad esempio HIF-1α (Mathupala et al. 1997, 2001)
- Sovraespressione di HK, soprattutto dell’isomero HKII, in molte popolazioni di cellule tumorali
- Ruolo cruciale dell’HK nella bioenergetica delle cellule tumorali, cioè l’effetto Warburg (Bustamante et al. 1981; Bustamante e Pedersen 1977)
- Assenza di glucosio-6-fosfatasi (G6Pasi) nei tumori altamente maligni e a rapida crescita
- Conseguente intrappolamento di F18-FDG-6 fosfato da parte delle cellule tumorali (Higashi et al. 2002)
Nel soggetto dello studio che abbiamo presentato, è stata documentata una remissione completa mediante FDG PET il 12/4/08 e si è protratta per 4 anni senza alcun intervento terapeutico, se non la continuazione dell’uso di DCA, ma con una frequenza di dosaggio inferiore di tre volte a settimana. Al 12/5/12, TM non ha riportato risultati significativi di neuropatia periferica alla dose di ≤ 10 mg/kg/die, il che è coerente con i risultati di Michelakis et al. (2010) che non hanno riportato neuropatia periferica significativa con dosi di DCA inferiori a 6,25 mg/kg due volte al giorno. Inoltre, la TM non ha avuto altri effetti collaterali.
Una spiegazione della risposta drammatica documentata in questo caso è la remissione spontanea di un NHL di grado basso o intermedio. Ciò sembra improbabile alla luce della drastica riduzione e scomparsa dei sintomi sistemici e dell’adenopatia entro poche settimane dall’inizio del DCA. Inoltre, la ricaduta relativamente rapida dopo la chemioterapia iniziale con R-CHOP rende la remissione spontanea un evento improbabile nella malattia di TM.
Il nostro studio, qui presentato, dovrebbe suscitare una discussione approfondita sull’uso della terapia metabolica come il DCA nei pazienti oncologici. Altre questioni che sembrano pertinenti a questa relazione includono il possibile ruolo di una dieta a basso contenuto glicemico nella prevenzione e nel trattamento del cancro e la necessità di concentrarsi sulla metodologia e sul formato di refertazione coinvolti nell’imaging PET. L’utilizzo delle cartelle cliniche di persone che stanno seguendo una terapia antitumorale senza la supervisione di un medico è un approccio unico nel lavoro di investigazione medica. Ottenere le cartelle cliniche dei pazienti, così come i DVD e i referti di imaging, i referti e i vetrini di patologia e i rapporti di laboratorio è impegnativo e richiede molto tempo. Tuttavia, se ulteriori studi su soggetti umani confermeranno che il DCA ha una significativa attività antitumorale, l’approccio utilizzato in questa indagine dovrebbe essere preso in considerazione su scala più ampia.
Conclusioni
L’eccezionale risposta alla monoterapia con DCA del paziente TM, in un contesto di ricaduta dopo la terapia con R-CHOP, giustifica ulteriori studi clinici sul DCA in pazienti umani affetti da varie neoplasie, soprattutto in considerazione del costo trascurabile e della minima tossicità di questa semplice molecola a 2 carboni.
Ringraziamenti
Gli autori desiderano ringraziare la Life Extension Foundation di Fort Lauderdale, Florida, per l’assistenza finanziaria e Robert Vergara della Life Extension per l’aiuto nell’ottimizzazione dei file grafici utilizzati in questa relazione. Jim Tassano è stato fondamentale per allertare gli autori SS e OA sulle segnalazioni di risposte cliniche significative in pazienti oncologici sottoposti a DCA.
Interessi in gioco
Gli autori dichiarano di non avere interessi finanziari o non finanziari in competizione.
Contributi degli autori
SS, OA, NP, JW hanno contribuito alla stesura del manoscritto. SS e OA hanno ottenuto tutte le cartelle cliniche, facilitato la revisione patologica, compilato le cartelle cliniche elettroniche e seguito i partecipanti allo studio. RB ha rivisto tutte le immagini PET e PET/CT, ha fornito SUV e misure corrette. DS ha facilitato l’ottenimento dei DVD chiave degli studi di imaging e ha fornito un parere radiologico.
Informazioni sull’autore
SS è un oncologo medico certificato con oltre 40 anni di pratica clinica e autore di numerosi articoli con revisione paritaria sulla malattia di Hodgkin, il cancro alla prostata e la cura di supporto del paziente oncologico.
RIFERIMENTI
1 Berendzen K, Theriaque DW, Shuster J, Stacpoole PW (2006) Potenziale terapeutico del dicloroacetato per il deficit del complesso della piruvato deidrogenasi. Mitocondrio 6:126-135
2BonnetS, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R et al (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37-51
3BuiT, Thompson CB (2006) Cancer’s sweet tooth. Cancer Cell 9:419- 420
4BustamanteE, Pedersen PL (1977) Elevata glicolisi aerobica di cellule di epatoma di ratto in coltura: ruolo dell’esochinasi mitocondriale. Proc Natl Acad Sci U S A 74:3735-3739
5BustamanteE, Morris HP, Pedersen PL (1981) Metabolismo energetico delle cellule tumorali. Necessità di una forma di esochinasi con una propensione al legame mitocondriale. J Biol Chem 256:8699-8704
6CaoW, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S et al (2008) Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2 in vitro. Prostata 68:1223-1231
7ChenZ, Lu W, Garcia-Prieto C, Huang P (2007) L’effetto Warburg e le sue implicazioni terapeutiche sul cancro. J Bioenerg Biomembr 39:267-274
8ChristofkHR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R et al (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230-233
9Constantin-TeodosiuD, Simpson EJ, Greenhaff PL (1999) L’importanza della disponibilità di piruvato per l’attivazione e l’anaplerosi della PDC nel muscolo scheletrico umano. Am J Physiol 276:E472-E478
10FangJS, Gillies RD, Gatenby RA (2008) Adattamento all’ipossia e all’acidosi nella carcinogenesi e nella progressione tumorale. Semin Cancer Biol 18:330-337
11GatenbyRA, Gawlinski ET (2003) The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res 63:3847-3854
12HeerdtBG, Houston MA, Augenlicht LH (2005) The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res 65:9861-9867
13HigashiT, Saga T, Nakamoto Y, Ishimori T, Mamede MH, Wada M et al (2002) Relazione tra l’indice di ritenzione nella PET a doppia fase (18)F-FDG e l’espressione dell’esochinasi-II e del trasportatore di glucosio-1 nel cancro del pancreas. J Nucl Med 43:173-180
14KurodaY, Ito M, Toshima K, Takeda E, Naito E, Hwang TJ et al (1986) Treatment of chronic congenital lactic acidosis by oral administration of dichloroacetate. J Inherit Metab Dis 9:244-252
15MadhokBM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 102:1746-1752
16MathupalaSP, Rempel A, Pedersen PL (1997) Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J Bioenerg Biomembr 29:339-343
17MathupalaSP, Rempel A, Pedersen PL (2001) Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 276:43407-43412
18MichelakisED, Webster L, Mackey JR (2008) Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer 99:989-994
19MichelakisED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34
20PanJG, Mak TW (2007) Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007:14
21RemillardCV, Yuan JX (2004) Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol 286:L49-L67
22StacpoolePW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 77:535-541
23StacpoolePW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM et al (2006) Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatria 117:1519-1531
24StacpoolePW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW et al (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatria 121:e1223-e1228
25SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2010) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer growth in vitro and in vivo. Breast Cancer Res Treat 120:253-260
26TongJ, Xie G, He J, Li J, Pan F, Liang H (2011) Effetto antitumorale sinergico del dicloroacetato in combinazione con il 5-fluorouracile nel cancro del colon-retto. J Biomed Biotechnol 2011:740564
27VanderHeiden MG (2010) Targeting cell metabolism in cancer patients. Sci Transl Med 2:31ed31
28WahlRL, Jacene H, Kasamon Y, Lodge MA (2009) From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J Nucl Med 50(Suppl 1):122S-150S
29WangZ (2004) Ruolo dei canali K+ nella regolazione della proliferazione e dell’apoptosi delle cellule tumorali. Pflugers Arch 448:274-286
30WarburgO (1956a) Sulla compromissione della respirazione nelle cellule tumorali. Science 124:269-270 Warburg O (1956b) Sull’origine delle cellule cancerose. Scienza 123:309-314
31WarburgO, Wind F, Negelein E (1927) Il metabolismo dei tumori nell’organismo. J Gen Physiol 8:519-530
32WongJY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109:394-402
Contenuti correlati: