Bo Li1,*xinzhe Li1,*, Zhenhong Ni1, Yan Zhang1, Yijun Zeng1, Xiaohuan Yan2, Yan Huang3, Jintao He4, Xilin Lyu1, Yaran Wu1, Yuting Wang1, Yingru Zheng2, Fengtian He1
1 Département de biochimie et de biologie moléculaire, Collège des sciences médicales de base, Troisième université médicale militaire,
Chongqing 400038, Chine
2 Département d’obstétrique et de gynécologie, Hôpital de Daping et Institut de recherche en chirurgie, Troisième université médicale militaire
, Chongqing 400042, Chine
3 Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing 400042,
Chine
4 Battalion 17 of Students, College of Preventive Medicine, Third Military Medical University, Chongqing 400038, Chine
* Ces auteurs ont contribué à parts égales à ce travail
Correspondance :
Yingru Zheng, courriel : [email protected]
Fengtian He, courriel : [email protected]
Reçu : 24 janvier 2016
Accepté : 09 juillet 2016
Publié : 19 juillet 2016
Résumé
Le dichloroacétate (DCA) et la metformine (Met) ont tous deux montré une efficacité antitumorale prometteuse en régulant le métabolisme des cellules cancéreuses. Cependant, l’autophagie protectrice médiée par le DCA et l’accumulation de lactate induite par la Met limitent respectivement leur potentiel de destruction des tumeurs. Ainsi, le fait de surmonter les lacunes correspondantes améliorera leurs effets thérapeutiques. Dans la présente étude, nous avons constaté que le DCA et le Met inhibaient de manière synergique la croissance et augmentaient l’apoptose des cellules cancéreuses ovariennes. De façon intéressante, nous avons révélé pour la première fois que Met sensibilisait le DCA en atténuant considérablement la protéine Mcl-1 induite par le DCA et l’autophagie protectrice, tandis que le DCA sensibilisait Met en atténuant de façon marquée l’accumulation excessive de lactate et la consommation de glucose induites par Met. Les expériences in vivo sur des souris nude ont également montré que le DCA et le Met supprimaient de manière synergique la croissance des tumeurs ovariennes xénogreffées. Ces résultats pourraient ouvrir la voie à l’élaboration de nouvelles stratégies de traitement du cancer de l’ovaire fondées sur l’utilisation combinée du DCA et du Met.
Mots clés : dichloroacétate, metformine, Mcl-1, métabolisme du cancer, cancer de l’ovaire
INTRODUCTION
La mortalité du cancer de l’ovaire est la plus élevée parmi plusieurs types de cancers gynécologiques. À l’heure actuelle, les chimiothérapies à base de platine et de taxol restent le paradigme standard en plus de la chirurgie, mais leurs effets secondaires sont graves et la chimiorésistance est également apparue [1-2]. Il est donc urgent d’explorer de nouvelles stratégies comme alternatives à la chimiothérapie traditionnelle. Ces dernières années, les preuves croissantes ont montré que le cancer est une sorte d’anomalie métabolique, ce qui le place au premier plan en régulant le métabolisme du cancer pour inhiber la croissance tumorale [3]. Le ciblage des voies métaboliques clés permet de tuer de manière significative de nombreuses cellules cancéreuses, y compris celles du cancer de l’ovaire [4-5]. Parmi les différents médicaments métaboliques, le dichloroacétate (DCA) et la metformine (Met) ont montré de charmantes perspectives en raison de leurs fonctions positives dans la thérapie du cancer.
En tant qu’agent ciblant les mitochondries, le DCA peut inhiber l’activité de la pyruvate déshydrogénase kinase (PDK) et augmenter ensuite l’activité de la pyruvate déshydrogénase (PDH), ce qui favorise le flux des glucides vers les mitochondries et améliore ainsi l’oxydation aérobie du glucose. Cet effet inverse le dysfonctionnement mitochondrial et réactive l’apoptose dépendante des mitochondries dans plusieurs cellules tumorales [6-9]. Simultanément, le DCA inhibe la glycolyse et réduit l’accumulation de lactate, ce qui détruit le microenvironnement tumoral acidifié (le microenvironnement acidifié est généralement favorable à la survie des tumeurs) [10]. Bien que le DCA ait montré des perspectives prometteuses dans la lutte contre les cancers, il a été signalé que le DCA induit une autophagie protectrice dans les cellules cancéreuses du côlon, ce qui entrave sa capacité apoptotique [11]. Jusqu’à présent, on ne sait toujours pas s’il existe un autre déterminant de résistance associé à l’apoptose lorsque le DCA relance l’apoptose mitochondriale.
Le Met est un médicament traditionnel de la thérapie de première ligne pour le diabète de type 2. Ces dernières années, des preuves croissantes indiquent que Met peut également réduire le risque de cancer dans plusieurs études épidémiologiques [12]. Met supprime la croissance tumorale en induisant l’arrêt du cycle, en favorisant l’apoptose et en supprimant l’autophagie [13-15]. En outre, Met peut sensibiliser certains médicaments chimiothérapeutiques tels que le paclitaxel, l’erlotinib, etc. [16-17]. Plus intéressant encore, l’effet anti-tumoral de Met est de plus en plus lié au métabolisme du glucose dans le cancer [18]. Malgré plusieurs avantages dans les essais cliniques, l’application de Met est entravée par le fait qu’il pourrait entraîner une accumulation de lactate [19]. Il est très intéressant de savoir si cet inconvénient peut être surmonté en combinant d’autres médicaments métaboliques pour que le Met soit plus largement utilisé en chimiothérapie.
Compte tenu de leurs effets compensatoires mutuels potentiels, nous avons cherché à découvrir si le DCA et le Met peuvent agir en synergie pour améliorer la cytotoxicité des cellules cancéreuses ovariennes. Dans la présente étude, nous avons démontré que le DCA et le Met pouvaient collaborer pour induire l’apoptose dans les cellules cancéreuses ovariennes. Le Met a sensibilisé le DCA en atténuant considérablement le Mcl-1 induit par le DCA et l’autophagie protectrice, tandis que le DCA a sensibilisé le Met en atténuant de façon marquée l’accumulation excessive de lactate et la consommation de glucose induites par le Met. Les expériences in vivo sur des souris nude ont également montré que le DCA et le Met supprimaient de manière synergique la croissance des tumeurs ovariennes xénogreffées. Ces résultats suggèrent que cette stratégie thérapeutique pourrait être un choix prometteur pour une future thérapie ciblée du cancer basée sur le métabolisme.
RÉSULTATS
LeDCA et le Met induisent de façon synergique l’apoptose dans les cellules cancéreuses de l’ovaire
Pour déterminer s’il existe un effet synergique entre le DCA et le Met dans la suppression de la croissance des cellules cancéreuses de l’ovaire, les cellules SKOV3 et OVCAR3 ont été cotraitées avec le DCA et le Met ou avec chacun d’eux séparément. Comme le montrent les figures 1A-1C, le cotraitement avec le DCA et le Met a réprimé plus efficacement la croissance des cellules cancéreuses de l’ovaire par rapport à chacun d’entre eux, et la combinaison de 40mM de DCA et de 10mM de Met a pu inhiber la viabilité des cellules jusqu’à 50 % par rapport au contrôle. Nous avons donc choisi 40mM DCA et 10mM Met dans les expériences suivantes. De même, l’effet d’inhibition synergique a également été observé dans les cellules de cancer du col de l’utérus (HeLa et SiHa), les cellules de cancer du poumon non à petites cellules (A549 et GLC-82) et les cellules de carcinome hépatocellulaire humain (HepG2) (figures S1A-1D), ce qui suggère que la synergie entre DCA et Met peut être universelle dans une certaine mesure. De plus, le DCA et le Met ont induit de façon synergique l’apoptose dans les cellules cancéreuses de l’ovaire, ce qui a été révélé par une analyse par cytométrie en flux de la double coloration d’annexine V-FITC (isothiocyanate de fluorescéine) et de PI (iodure de prodium) (figure 1D), par la coloration de Hoechst des corps apoptotiques (figure 1E), par une analyse par transfert Western de la PARP clivée (poly ADP-ribose polymérase, un marqueur d’apoptose) (figure 1F) et par un test d’activité de la caspase3 (figure 1G).
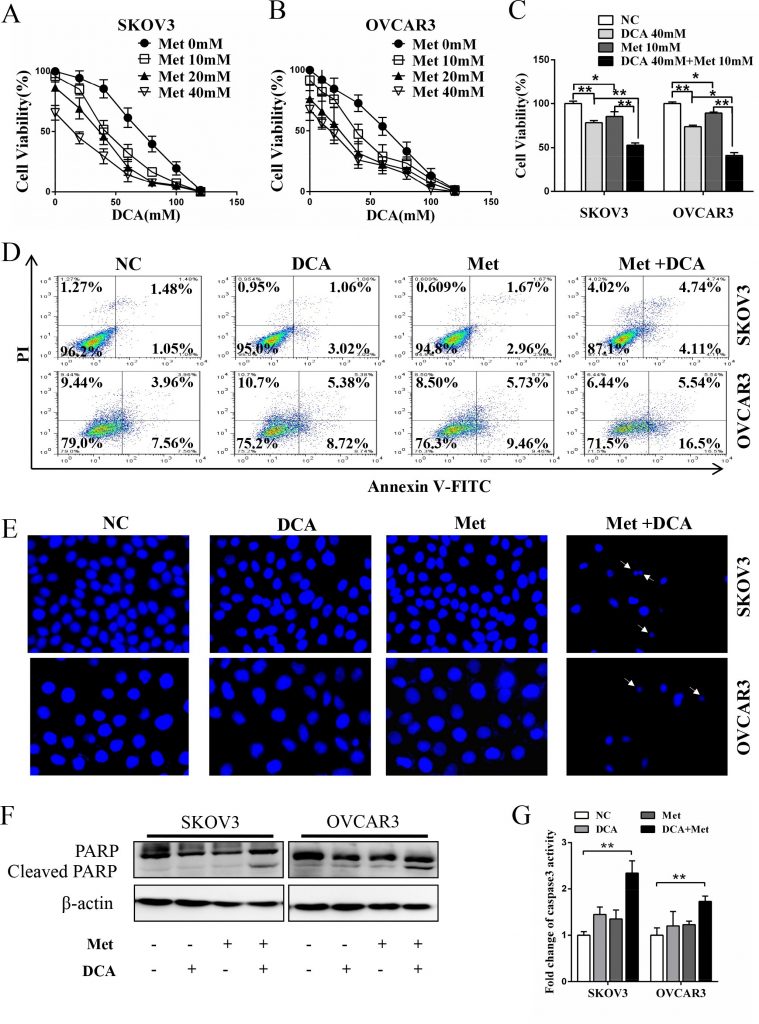
Met sensibilise le DCA en atténuant le Mcl-1 induit par le DCA
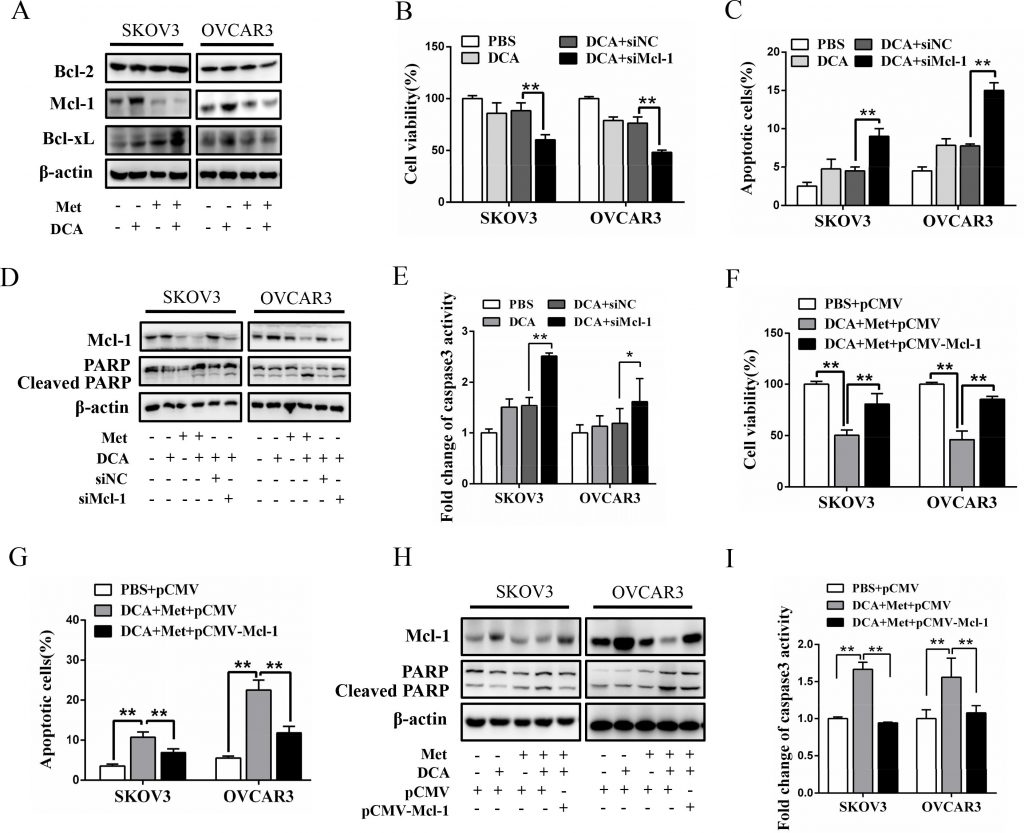
Pour explorer le mécanisme par lequel Met sensibilise le DCA pour induire l’apoptose, nous avons examiné l’expression des membres antiapoptotiques cruciaux de la famille Bcl-2, y compris Mcl-1, Bcl-2 et Bcl-xL [20]. Comme le montre la figure 2A, le DCA seul a augmenté de manière significative le niveau de la protéine Mcl-1 (mais pas les protéines Bcl-2 et Bcl-xL) dans les cellules cancéreuses ovariennes, ce qui a été nettement atténué par Met. L’inhibition de Mcl-1 par un siRNA a renforcé l’inhibition de la viabilité cellulaire induite par le DCA (figure 2B) et augmenté l’apoptose induite par le DCA (figures 2C-2E). En outre, l’expression ectopique de Mcl-1 a considérablement atténué l’effet sensibilisateur de Met au DCA sur la viabilité cellulaire et l’apoptose (figures 2F-2I). Ces résultats indiquent que Mcl-1 est un nouveau facteur de résistance au DCA, et que Met sensibilise au DCA par le biais de la régulation négative de Mcl-1.
Met atténue le Mcl-1 induit par le DCA en inhibant la traduction du Mcl-1
Pour élucider à quel niveau le DCA induit le Mcl-1, on a d’abord examiné l’ARNm du Mcl-1. Comme le montre la figure 3A, le DCA n’a pas eu d’effet évident sur l’expression de l’ARNm de Mcl-1. Ensuite, la protéine Mcl-1 a été analysée en présence ou en l’absence de l’inhibiteur de traduction cycloheximide (CHX). Comme le montrent les figures 3B et 3C, le CHX a diminué de manière dépendante du temps la protéine Mcl-1 basale (mais pas celle induite par le DCA), ce qui indique que le DCA augmente la stabilité de la protéine Mcl-1. Il a été signalé que l’ERK phosphorylé (p-ERK) et le p-JNK peuvent stabiliser Mcl-1 en phosphorylant Mcl-1 sur Thr163 [21-22], nous avons donc cherché à savoir si le p-ERK/p-JNK est impliqué dans la régulation de Mcl-1 induite par le DCA. Comme le montre la figure 3D, le traitement au DCA a augmenté de manière significative p-ERK (mais pas p-JNK) et p-Mcl-1Thr163 dans les cellules cancéreuses ovariennes. De plus, l’inhibiteur de MEK1/2, U0126, a pu atténuer de façon spectaculaire le Mcl-1 et le p-Mcl-1Thr163 induits par le DCA, et a renforcé de façon marquée la PARP clivée (figure 3E). Ces résultats indiquent que p-ERK (mais pas p-JNK) est un médiateur de Mcl-1 induit par le DCA. Des études antérieures ont démontré que le DCA augmente les espèces réactives de l’oxygène (ROS) [23], et les ROS sont un inducteur clé de p-ERK [24], nous avons donc examiné le niveau de ROS avec DCFH-DA. Comme le montre la figure S2A, le DCA a augmenté la production de ROS, ce qui suggère que l’induction de ROS pourrait être un mécanisme par lequel le DCA renforce l’activation de p-ERK.
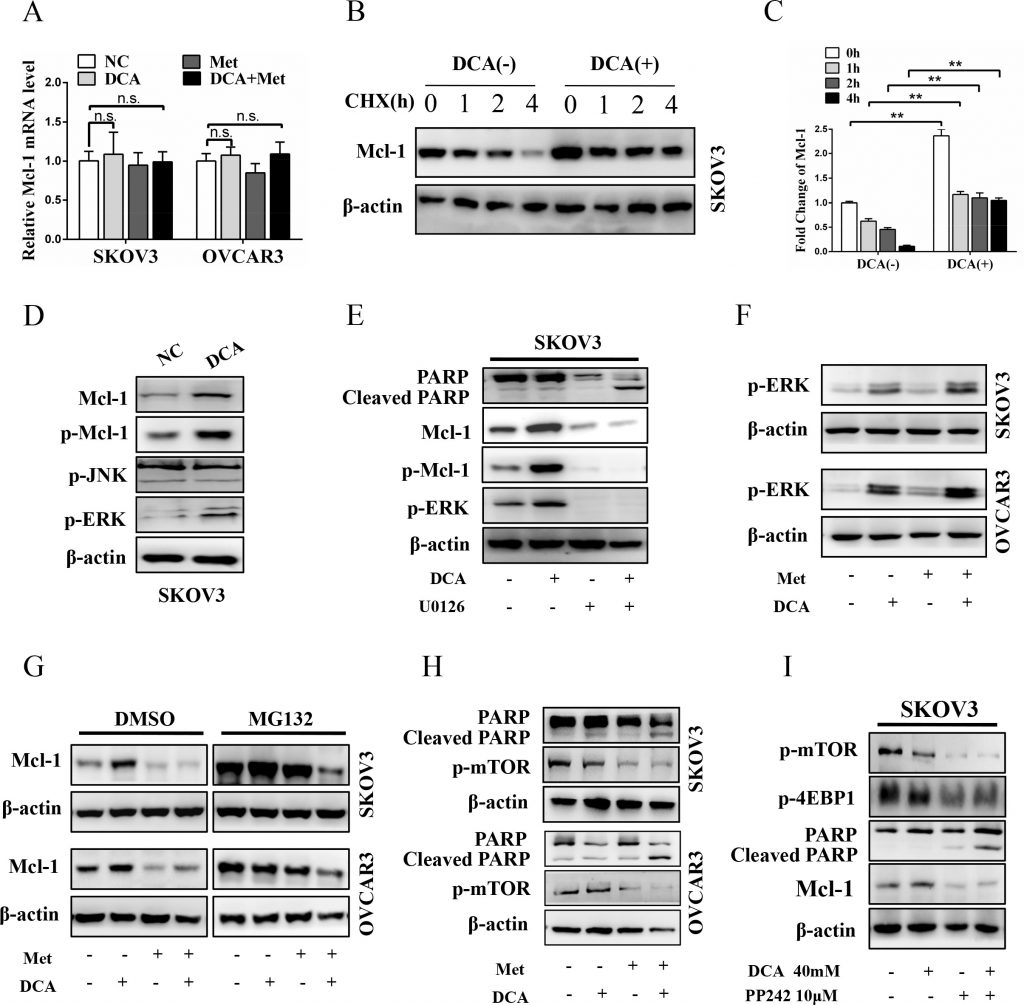
De plus, Ser159 est également étroitement lié à la stabilité de Mcl-1 et ce site est principalement phosphorylé par GSK-3β [25], nous avons donc testé si GSK-3β est également impliqué dans la régulation de la stabilisation de Mcl-1 induite par le DCA. Comme le montre la Figure S2B, le DCA a augmenté la phosphorylation de GSK-3β, mais n’a eu aucun effet sur la GSK-3β totale. De plus, le DCA a augmenté la phosphorylation d’Akt (une molécule de signalisation en amont de GSK-3β), et l’inhibiteur d’Akt MK-2206 2HCl a considérablement atténué la phosphorylation de GSK-3β induite par le DCA, l’upregulation de Mcl-1 et la résistance à l’apoptose (Figure S2C). Ces résultats indiquent que la phosphorylation de GSK-3β médiée par p-Akt favorise la stabilisation de Mcl-1 induite par le DCA.
Par la suite, nous avons examiné si Met peut atténuer Mcl-1 induit par le DCA en inhibant p-ERK/p-Akt. Comme le montrent la figure 3F et la figure S2D, Met ne pouvait pas supprimer p-ERK et p-Akt induits par le DCA, ainsi que les résultats de la figure 3A, ce qui indique que Met ne diminue pas Mcl-1 induit par le DCA au niveau transcriptionnel ou post-traductionnel. Nous avons ensuite analysé si Met atténue le Mcl-1 induit par le DCA au niveau traductionnel avec l’inhibiteur de protéasome MG132. Comme le montre la figure 3G, lorsque les cellules ont été traitées avec le contrôle, le DCA, Met ou une combinaison, les niveaux de protéine de Mcl-1 étaient également élevés en présence de MG132 par rapport au DMSO, ce qui indique que Met atténue Mcl-1 induit par le DCA via l’inhibition de la traduction de Mcl-1. Comme il a été signalé que l’activation de mTOR favorise la traduction de Mcl-1 [26], nous avons analysé p-mTOR après le co-traitement avec Met et DCA. Comme prévu, Met a nettement diminué le niveau de p-mTOR (figure 3H), et l’inhibiteur de mTOR PP242 a eu un effet similaire à celui de Met sur la promotion de l’apoptose (figure 3I). D’après les données de la Figure 3 et de la Figure S2, nous pouvons conclure que le DCA augmente le taux de Mcl-1 en renforçant la phosphorylation de ERK et GSK-3β, et que Met supprime la traduction de Mcl-1 en inhibant p-mTOR.
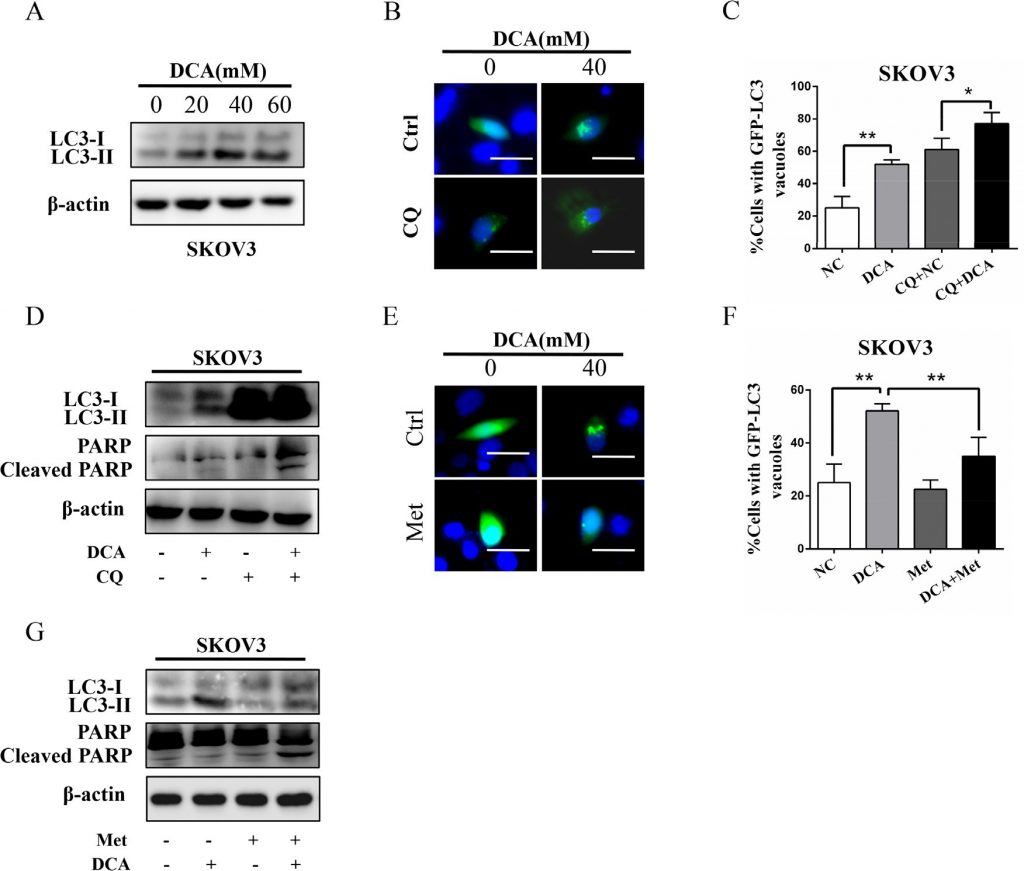
Met diminue l’autophagie protectrice induite par le DCA
Des études précédentes ont montré que l’autophagie joue un rôle important dans la résistance thérapeutique du DCA dans les cellules cancéreuses du côlon [11], nous avons donc examiné le rôle de l’autophagie dans les cellules cancéreuses de l’ovaire lors du traitement avec le DCA ou/et Met. Comme le montre la figure 4A, le DCA a favorisé de manière dose-dépendante le niveau de MAP1LC3-II (LC3-II), le marqueur de l’autophagie. L’inhibition de l’autophagie par la chloroquine (CQ) ou le silence d’ATG7 ont considérablement augmenté l’apoptose et la cytotoxicité induites par le DCA (figures 4B-4D, figures S3A-3D), ce qui indique que le DCA induit une autophagie protectrice dans les cellules cancéreuses ovariennes. Afin d’étudier de manière préliminaire le mécanisme d’autophagie induit par le DCA, nous avons analysé les modifications de l’ARNm de 7 gènes liés à l’autophagie dans les cellules traitées par le DCA. Comme le montre la figure S3E, le DCA a considérablement augmenté le niveau d’ARNm d’ATG7 dans les cellules cancéreuses ovariennes, ce qui suggère que ATG7 peut être impliqué dans l’autophagie protectrice induite par le DCA. Par la suite, nous avons constaté que Met diminuait remarquablement la LC3-II induite par le DCA (Figure 4E-4G), ce qui indique que Met pourrait atténuer l’autophagie protectrice induite par le DCA. En résumé, on peut conclure que l’affaiblissement de l’autophagie protectrice induite par le DCA est également important dans l’effet sensibilisateur de Met au DCA.
Le DCA atténue la consommation de glucose et la production de lactate induites par Met
Pour préciser si le DCA peut surmonter les pénuries de Met, les changements de lactate et de glucose ont été analysés. Comme le montrent les figures 5A-5C, Met a augmenté la production de lactate et la consommation de glucose, ce qui a été remarquablement atténué par le DCA. De plus, le taux de consommation d’oxygène cellulaire (OCR) et le taux d’acidification extracellulaire (ECAR) ont été mesurés. Comme le montrent les figures 5D-5F, Met a diminué le rapport OCR/ECAR, qui a été considérablement atténué par le DCA, ce qui révèle que le DCA peut supprimer la glycolyse induite par Met en rétablissant la respiration mitochondriale. Comme le DCA est un inhibiteur des PDK qui phosphoryle et inhibe l’activité de la PDH [9], nous avons examiné le niveau de p-PDH. Comme le montre la figure 5G, Met a augmenté le niveau de p-PDHE1α (une sous-unité de PDH), ce qui a été nettement inversé par le DCA. La neutralisation de la PDH par un siRNA a atténué de manière significative la production de lactate induite par le Met (Figure 5H, 5I). L’expression ectopique de PDK1 et PDK2 a augmenté la phosphrylation de PDHE1α et a atténué l’apoptose induite par le cotraitement avec le DCA et le Met (Figure 5J, 5K), en combinaison avec les données des Figures 5G-5I, indiquant que le DCA peut sensibiliser le Met par l’inhibition de la voie PDK/PDH en tuant les cellules cancéreuses ovariennes.
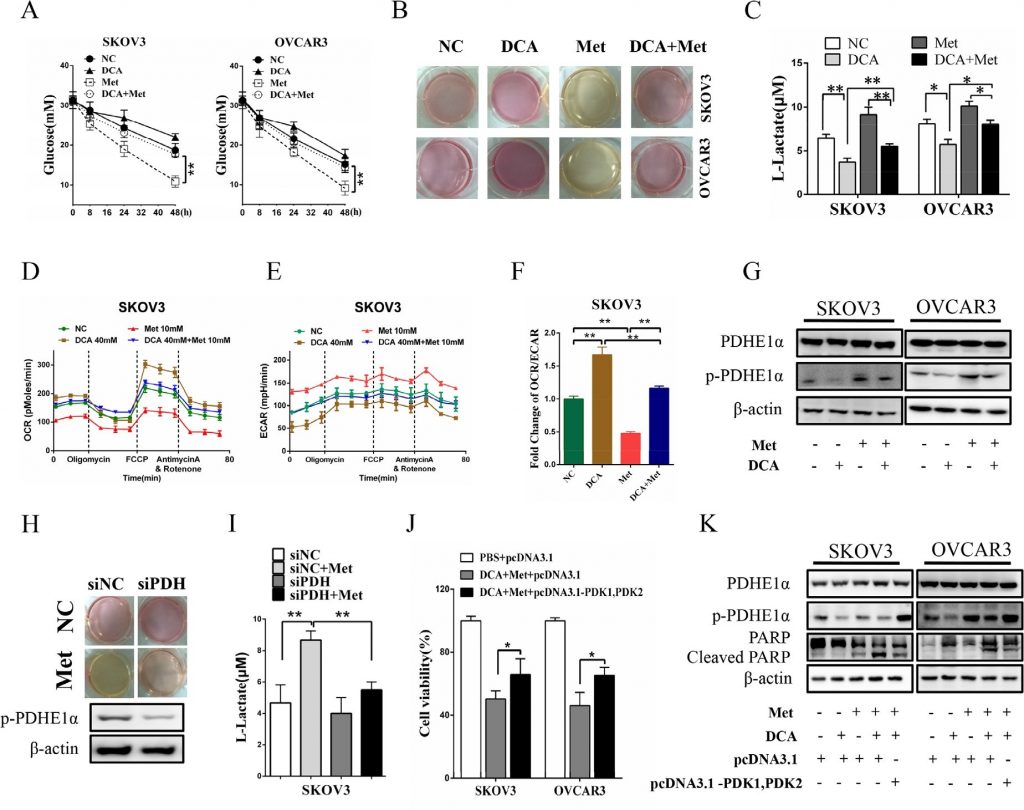
LeDCA et le Met répriment ensemble la croissance des cellules cancéreuses de l’ovaire in vivo
Comme le montrent les figures 6A et 6B, le co-traitement avec le DCA et le Met a supprimé plus efficacement la croissance des xénogreffes de cancer de l’ovaire chez les souris nude par rapport au traitement avec le DCA ou le Met seul. Une analyse par transfert Western a montré que le DCA et le Met augmentaient de manière synergique la PARP clivée et régulaient à la baisse Mcl-1 et p-PDHE1α dans les xénogreffes (figures 6C-6D). Ces résultats suggèrent que le DCA et le Met peuvent inhiber de manière collaborative la croissance des cellules cancéreuses ovariennes in vivo en atténuant les insuffisances de chacun.
DISCUSSION
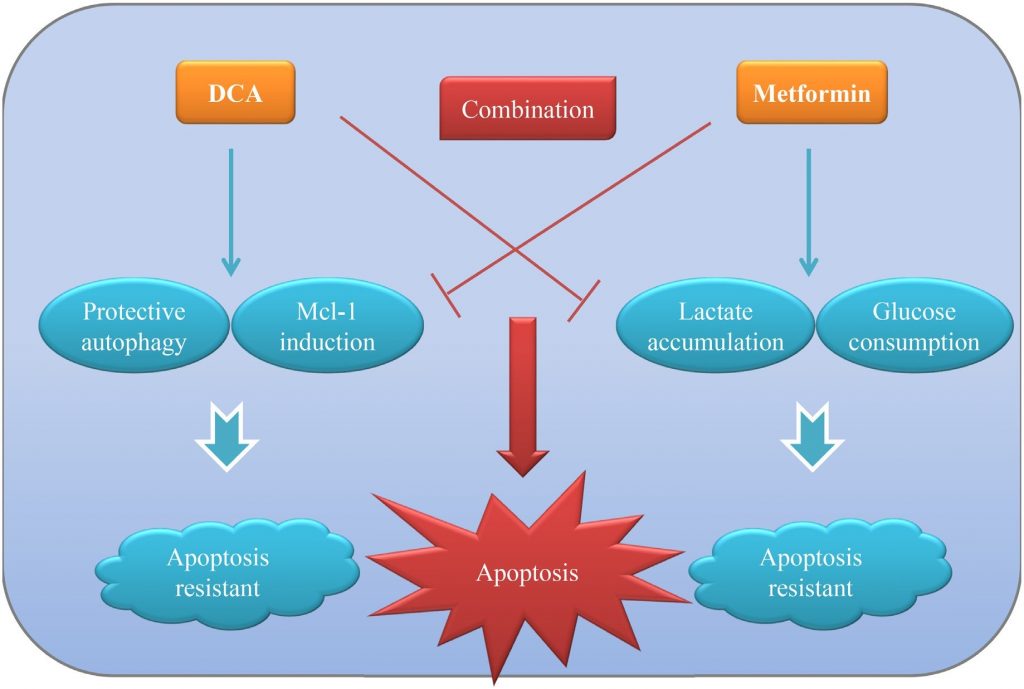
Il a été confirmé que la plupart des tumeurs solides sont caractérisées par » l’effet Warburg « , c’est-à-dire qu’elles utilisent la glycolyse pour la production d’énergie même si l’oxygène est suffisant. Le fait de cibler ce phénomène anormal a ouvert la voie au développement de nouvelles stratégies thérapeutiques contre le cancer, en plus des médicaments cytotoxiques traditionnels. Dans la présente étude, nous avons démontré que le cotretraitement avec le DCA et le Met (deux agents associés au métabolisme) peut réprimer plus efficacement la croissance des cellules cancéreuses ovariennes par rapport à chacun d’eux seul , in vitro et in vivo. Le Met atténue le Mcl-1 et l’autophagie protectrice induits par le DCA, tandis que le DCA atténue l’accumulation excessive de lactate et la consommation de glucose induites par le Met. Les avantages réciproques des deux agents contribuent à une apoptose intense pour tuer plus efficacement les cellules cancéreuses ovariennes. Le modèle de fonctionnement de l’association du DCA et du Met est présenté à la figure 7.
Bien que des études soient en cours depuis des années, les facteurs clés qui peuvent entraver l’effet pro-apoptotique du DCA ne sont pas encore clairs. Nos résultats ont montré que Mcl-1 est un facteur de résistance crucial contre l’apoptose induite par le DCA dans les cellules cancéreuses ovariennes, et que le cotraitement avec Met et DCA a diminué Mcl-1 et augmenté l’apoptose. Toutefois, le cotraitement a entraîné une augmentation de Bcl-xL (figure 2A), qui pourrait être un mécanisme compensatoire pour maintenir la survie des cellules. Un phénomène similaire a également été signalé dans une étude précédente [27] (L’étude montre que l’inhibiteur de Bcl-2/xL ABT-263 induit l’apoptose des cellules cancéreuses tout en régulant à la hausse Mcl-1). De plus, l’augmentation de Bcl-xL induite par le cotraitement n’était présente que dans la lignée cellulaire SKOV3 (mais pas dans OVCAR3), ce qui suggère que cet effet pourrait être spécifique à la cellule. Bien sûr, il reste à déterminer le mécanisme détaillé par lequel le cotraitement avec Met et DCA augmente Bcl-xL. Par une étude plus approfondie, nous avons révélé que le DCA induit l’accumulation de Mcl-1 par l’activation de ERK et d’Akt, ce qui protège Mcl-1 de la dégradation médiée par le protéasome. Ces résultats suggèrent que l’inhibition de ERK et Akt pourrait être une bonne stratégie pour sensibiliser le DCA dans le traitement du cancer des ovaires. Cependant, des résultats contradictoires ont été rapportés récemment : le DCA diminue le niveau de Mcl-1 dans les cellules de LAM [28] et les cellules de cancer colorectal [29]. Ces divergences suggèrent que la relation entre le DCA et Mcl-1 peut être largement différente selon les contextes.
L’autophagie est un processus catabolique qui permet de recycler les métabolites essentiels tels que les acides aminés et les lipides afin de reconstituer les réserves bioénergétiques en présence d’une privation de nutriments ou d’autres stress importants [30-31]. Dans cette étude, nous avons révélé que le DCA induisait une autophagie protectrice dans les cellules cancéreuses ovariennes, et que l’ATG7 pouvait jouer un rôle dans ce processus (figure S3E), mais le mécanisme détaillé doit être étudié plus avant. De plus, nous avons constaté que Met sensibilisait le DCA en supprimant l’autophagie protectrice induite par le DCA. Conformément à nos résultats, Met peut réprimer l’autophagie dépendante de GRP78 pour renforcer l’effet anti-myélome du bortézomib [15], et inhiber l’autophagie induite par le 2DG pour sensibiliser le 2DG dans les cellules cancéreuses de la prostate [32]. Cependant, le(s) mécanisme(s) détaillé(s) par lequel Met supprime l’autophagie protectrice reste(nt) à étudier plus avant.
Le Met a été bien reconnu comme agent unique ou sensibilisateur dans le traitement du cancer, mais un inconvénient notable est que le Met favorise la consommation de glucose et accélère l’accumulation de lactate, ce qui facilite la glycolyse aérobie liée au cancer. Dans la présente étude, nous avons démontré que le DCA pouvait atténuer considérablement cet effet secondaire. Le DCA peut inhiber fortement l’activité des PDK et son aval p-PDHE1α, conduisant à un remodelage métabolique réutilisant la phosphorylation oxydative et entraînant une diminution de l’accumulation de lactate et de la consommation de glucose. Parmi les quatre types d’isoenzymes PDK, le DCA fonctionne principalement en inhibant PDK2 et PDK1 [33]. Comme prévu, la surexpression simultanée de PDK2 et PDK1 a augmenté la phosphorylation de PDHE1α, a aboli la fonction sensibilisante du DCA et a partiellement aboli l’effet létal du DCA plus Met dans les cellules cancéreuses ovariennes. Dans l’ensemble, nos résultats indiquent que le DCA sensibilise la fonction antitumorale de Met en inhibant l’activité des PDK. Cependant, il faut noter que le DCA peut sensibiliser Met en augmentant le stress oxydatif induit par Met dans les cellules du cancer du sein [34]. Cela signifie que le mécanisme de synergie entre le DCA et le Met est très compliqué, ce qui nécessite des études approfondies.
En résumé, nous avons montré que le DCA et le Met peuvent supprimer de façon synergique la croissance des cellules cancéreuses de l’ovaire, ce qui pourrait ouvrir la voie à l’élaboration de nouvelles stratégies de traitement du cancer de l’ovaire fondées sur l’utilisation combinée du DCA et du Met.
MATÉRIEL ET MÉTHODES
Lignées cellulaires et réactifs
Les lignées cellulaires comprenant SKOV3, OVCAR3, HeLa, SiHa, GLC-82, A549 et HepG2 ont été achetées auprès de l’American Type Culture Collection (ATCC) et ont été cultivées dans du Dulbecco’s Modified Eagle Medium (DMEM), complété par 10 % de sérum bovin fœtal (FBS), de la streptomycine (100 mg/mL) et de la pénicilline (100 U/mL) à 37°C dans un incubateur humide à 5 % deCO2. Le DCA et le cycloheximide (CHX) ont été achetés à Sigma-Aldrich (Louis, MO, USA). Met, U0126, MG132 et Hoechst 33258 ont été achetés auprès de Beyotime Company (Shanghai, Chine). Le MK2206 a été acheté auprès de Selleck Company (Shanghai, Chine). Le kit de dosage de l’activité de la caspase-3 et le kit de dosage des espèces réactives de l’oxygène ont été achetés auprès de Beyotime Company (Shanghai, Chine). L’Annexin V-FITC et le PI ont été achetés à BD Bioscience (BD, NJ, USA). Les siRNA de Mcl-1, ATG7, PDH et le siRNA de contrôle ont été fournis par GenePharma (Shanghai, Chine). Les plasmides d’expressionCMV et pcDNA3.1, pCMV-Mcl-1, pcDNA3.1-PDK1 et pcDNA3.1-PDK2 ont été achetés à Obio Technology (Shanghai, Chine).
Western blot
Des lysats de cellules entières ont été préparés et le Western blot a été réalisé comme décrit précédemment [35]. Les anticorps pour la β-actine et PDHE1α provenaient de la société Abcam (San Francisco, CA, USA), les anticorps pour Bcl-2 et Mcl-1 provenaient de Santa Cruz Biotechnology (Santa Cruz, CA, USA), et les anticorps pour PARP, BCL-xL, GSK-3β, p-ERK (Thr202/Tyr204), p-JNK (Thr183/Tyr185), p-Mcl-1 (Thr163), p-Akt (Ser473), p-4EBP1, p-mTOR et p-GSK-3β (Ser9) provenaient de Cell Signaling Technology (Boston, MA, USA). L’anticorps contre p-PDHE1α (Ser293) provient de EMD Millipore (Billerica, MA, USA).
Transfection cellulaire
Les cellules SKOV3 et OVCAR3 ont été cultivées jusqu’à une confluence de 60 à 70 % dans des plaques à 6 puits. Le siRNA ou le plasmide d’expression a été mélangé avec 10 μL de lipofectamine 2000 dans Opti-MEM (Invitrogen, Carlsbad, CA, USA) pour chaque puits, conformément au protocole du fabricant. Après avoir été incubées avec les mélanges pendant 6 h, les cellules ont été cultivées dans du DMEM avec 10 % de FBS pendant 6 h supplémentaires, puis les cellules ont reçu le traitement correspondant.
Test de viabilité cellulaire
Le test de viabilité cellulaire a été réalisé à l’aide du kit CCK-8 (Dojindo, Shanghai, Chine) comme décrit précédemment [27]. Brièvement, les cellules ont été ensemencées sur des plaques à 96 puits (2×103 cellules/puits) en triplicata et incubées pendant 12 h. Ensuite, les cellules ont reçu différents traitements ou un véhicule témoin pendant 48 h, suivis de l’ajout de 10 μl de solution de CCK-8 dans chaque puits. Après incubation à 37°C pendant 1,5 h, la valeur de l’OD450nm a été déterminée avec un lecteur de microplaques.
Cytométrie en flux
Les cellules cancéreusesovariennesont été incubées avec de l’annexine V-FITC et du PI selon les instructions du fabricant (BD, 561012). Puis l’apoptose a été analysée par un cytomètre en flux.
Coloration au Hoechst
Après avoir été traitées pendant 24 h, les cellules ont été colorées avec du Hoechst 33258 (Beyotime, Shanghai, Chine) à 10μg/mL pendant 10 min à l’obscurité. Par la suite, les cellules ont été lavées 3 fois avec du PBS et photographiées au microscope à afluorescence.
Test de l’activité de la Caspase 3
L’activité de la Caspase 3 a été examinée avec le kit de test de l’activité de la Caspase3 (Beyotime, C1115) comme décrit précédemment [36]. Brièvement, les cellules témoins et traitées ont été récoltées, lavées avec du PBS glacé, et remises en suspension dans 50 μl de tampon de lyse cellulaire refroidi pendant 15 min sur la glace. Puis les lysats ont été centrifugés (20 000 g, 10 min, 4°C), et les surnageants ont été collectés pour le dosage de l’activité de la caspase3 immédiatement.
Mesure des ROS intracellulaires totaux
Les ROS intracellulaires totaux ont été testés avec le kit de test ROS (Beyotime, Shanghai, Chine) comme décrit précédemment [37].
Isolation de l’ARN, PCR quantitative en temps réel (qPCR)
L’ARN total a été extrait des cellules avec le réactif TRIzol (ComWin Biotechnology, Beijing, Chine) et l’ADNc premier brin a été synthétisé à l’aide de la transcriptase M-MLV (Invitrogen, Carlsbad, CA, USA). La qPCR a été réalisée avec le kit QuantiFast SYBR Green PCR (Promega, Shanghai, Chine). Les niveaux relatifs d’ARNm des gènes cibles ont été calculés avec la méthode2-ΔΔCt.
Analyse de la protéine fluorescente verte (GFP)-MAP1LC3
Après avoir été transfectées avec le vecteur d’expression GFP-MAP1LC3 (GFP-LC3) pendant 12 heures, les cellules ont reçu les traitements indiqués pendant 24 heures supplémentaires, puis ont été fixées avec du formaldéhyde à 4 % pendant 10 minutes. Ensuite, les cellules ont été lavées 3 fois avec du PBS et observées sous un microscope à fluorescence.
Détection du L-lactate et du glucose
Les cellules ont été traitées dans des puits 6, puis le milieu a été collecté et les concentrations de L-lactate et de glucose ont été déterminées séparément à l’aide du kit de dosage du L-lactate (Eton Bioscience, San Diego, CA, USA) et du kit de dosage colorimétrique/fluorométique du glucose (BioVision, Milpitas, CA, USA).
Analyse bioénergétique cellulaire
Les cellules ont été placées dans des plaques XF96 et laissées à croître pendant une nuit. Puis les milieux ont été remplacés par des milieux XF96 1 h avant le test. La roténone/antimycine A, le FCCP et l’oligomycine ont été dilués dans le milieu XF96 et chargés dans la cartouche d’accompagnement pour obtenir des concentrations finales de 0,5μM, 0,5μM et 1,0μM, respectivement. Les injections des médicaments dans le milieu ont eu lieu aux points de temps spécifiés. L’OCR (pmol/min) et l’ECAR (mpH/min) ont été surveillés avec le kit de test de stress mitotique cellulaire XF (Seahorse Bioscience, North Billerica, MA, USA) en utilisant les analyseurs de flux extracellulaire XFe et XF de Seahorse Bioscience.
Étude sur les animaux
Des souris nues femelles âgées de six semaines ont été achetées auprès de Beijing Huafukang Bioscience (Beijing, Chine). Elles ont été hébergées et soignées conformément aux directives du Comité d’éthique et de protection des animaux de la troisième université médicale militaire (Chongqing, Chine). 5×106 cellules SKOV3 dans 150 μL de PBS ont été implantées dans l’aisselle droite de chaque souris nude. Lorsque des tumeurs palpables se sont formées, les souris ont été randomisées en 4 groupes (n = 6 par groupe). Ensuite, les souris ont reçu une injection intrapéritonéale quotidienne de DCA (50 mg/kg/j) et de Met (100 mg/kg/j) ou de chacun de ces produits seuls pendant 8 jours, avec du PBS comme contrôle. La taille de la tumeur xénogreffe a été contrôlée chaque jour à l’aide d’un pied à coulisse, et le volume a été estimé à l’aide de la formule suivante : volume = largeur2×longueur×1/2. Après excision des souris, les tumeurs xénogreffées ont été photographiées, et les protéines correspondantes ont été examinées par Western blot.
Analyse statistique
Les données ont été exprimées en moyenne ± SD. L’ANOVA à sens unique et le test t ont été utilisés pour analyser la variance. P< 0,05 a été considéré comme statistiquement significatif.
REMERCIEMENTS
Ce travail a été soutenu par la National Natural Science Foundation of China (81472436 et 81272865) et la Natural Science Foundation of Chongqing (cstc2012jjB10025).
CONFLITS D’INTÉRÊTS
Aucun conflit d’intérêt potentiel n’a été révélé.
RÉFÉRENCES
1 Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014 ; 384:1376-1388.2 Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015 ; 521:489-494.
3 Ward PS, Thompson CB. Reprogrammation métabolique : une caractéristique du cancer que même warburg n’a pas anticipée. Cancer Cell. 2012 ; 21:297-308.
4 Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism ; HNF-1beta confers survival benefits through the Warburg effect and ROS reduction. Oncotarget. 2015 ; 6:30704-30714. doi : 10.18632/oncotarget.5228.
5 Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. Le MicroRNA-144 médiateur du changement métabolique dans les cellules cancéreuses ovariennes en ciblant directement Glut1. Tumour Biol. 2015.
6 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Modulation métabolique du glioblastome avec le dichloroacétate. Sci Transl Med. 2010 ; 2:31ra34.
7 Michelakis ED, Webster L, Mackey JR. Le dichloroacétate (DCA) comme thérapie potentielle de ciblage métabolique pour le cancer. Br J Cancer. 2008 ; 99:989-994.
8 Papandreou I, Goliasova T, Denko NC. Les médicaments anticancéreux qui ciblent le métabolisme : le dichloroacétate est-il le nouveau paradigme ? Int J Cancer. 2011 ; 128:1001-1008.
9 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibit cancer growth. Cancer Cell. 2007 ; 11:37-51.
10 Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Le dichloroacétate améliore le dysfonctionnement immunitaire causé par l’acide lactique sécrété par la tumeur et augmente l’immunoréactivité antitumorale. Int J Cancer. 2013 ; 133:1107-1118.
11 Gong F, Peng X, Sang Y, Qiu M, Luo C, He Z, Zhao X, Tong A. Le dichloroacétate induit une autophagie protectrice dans les cellules LoVo : implication de la cathepsine D/thioredoxin-like protein 1 et de la signalisation médiée par Akt-mTOR. Cell Death Dis. 2013 ; 4:e913.
12 Morales DR, Morris AD. La metformine dans le traitement et la prévention du cancer. Annu Rev Med. 2015 ; 66:17-29.
13 Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. La metformine, médicament antidiabétique, exerce un effet antitumoral in vitro et in vivo par une diminution du niveau de cycline D1. Oncogene. 2008 ; 27:3576-3586.
14 Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009 ; 8:2031-2040.
15 Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. Les cribles pharmacologiques révèlent que la metformine supprime l’autophagie dépendante de GRP78 pour renforcer l’effet anti-myélome du bortézomib. Leucémie. 2015 ; 29:2184-2191.
16 Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. La metformine amplifie l’activation de l’AMPK induite par la chimiothérapie et la croissance antitumorale. Clin Cancer Res. 2011 ; 17:3993-4005.
17 Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE, Cremers S, Szabolcs M, Maurer MA. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget. 2014 ; 5:10503-10517. doi : 10.18632/oncotarget.2391.
18 Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, Dumon E, Martineau Y, Danet-Desnoyers G, et al. L’énergie mitochondriale et le statut AKT médient les effets métaboliques et l’apoptose de la metformine dans les cellules leucémiques humaines. Leucémie. 2013 ; 27:2129-2138.
19 Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Cibler la flexibilité métabolique en inhibant simultanément le complexe respiratoire I et la génération de lactate retarde la progression du mélanome. Oncotarget. 2015 ; 6:37281-37299. doi : 10.18632/oncotarget.6134.
20 Lessene G, Czabotar PE, Colman PM. Les antagonistes de la famille BCL-2 pour la thérapie du cancer. Nat Rev Drug Discov. 2008 ; 7:989-1000.
21 Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 est phosphorylé dans la région PEST et stabilisé lors de l’activation de ERK dans les cellules viables, et sur des sites supplémentaires avec l’acide okadaïque cytotoxique ou le taxol. Oncogene. 2004 ; 23:5301-5315.
22 Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology. 2009 ; 136:1423-1434.
23 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. L’activation de l’oxydation mitochondriale par l’inhibition de PDK2 renverse la résistance au cisplatine dans le cancer de la tête et du cou. Cancer Lett. 2016 ; 371:20-29.
24 Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM, Zhao CQ, Liu PJ, Zhang JM, He JJ. La signalisation MAPK médiant la mort cellulaire du cancer du sein humain induite par le chlorhydrate de sinoménine via des voies dépendantes et indépendantes des espèces réactives de l’oxygène : une étude in vitro et in vivo. Cell Death Dis. 2014 ; 5:e1356.
25 Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. La glycogène synthase kinase-3 régule la perméabilisation de la membrane externe mitochondriale et l’apoptose par la déstabilisation de MCL-1. Mol Cell. 2006 ; 21:749-760.
26 Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, et al. mTORC1 favorise la survie par le contrôle translationnel de Mcl-1. Proc Natl Acad Sci U S A. 2008 ; 105:10853-10858.
27 Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. L’inhibiteur de Bcl-2/xL ABT-263 augmente la stabilité de l’ARNm et de la protéine Mcl-1 dans les cellules de carcinome hépatocellulaire. Mol Cancer. 2014 ; 13:98.
28 Emadi A, Sadowska M, Carter-Cooper B, Bhatnagar V, van der Merwe I, Levis MJ, Sausville EA, Lapidus RG. Perturbation de l’état oxydatif cellulaire induit par le dichloroacétate et le trioxyde d’arsenic pour le traitement de la leucémie myéloïde aiguë. Leuk Res. 2015 ; 39:719-729.
29 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD, Coomber BL. Le dichloroacétate affecte la prolifération mais pas la survie des cellules cancéreuses colorectales humaines. Apoptosis. 2015 ; 20:63-74.
30 Mah LY, Ryan KM. L’autophagie et le cancer. Cold Spring Harb Perspect Biol. 2012 ; 4:a008821.
31 Yang ZJ, Chee CE, Huang S, Sinicrope FA. Le rôle de l’autophagie dans le cancer : implications thérapeutiques. Mol Cancer Ther. 2011 ; 10:1533-1541.
32 Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, et al. Targeting cancer cell metabolism : the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010 ; 70:2465-2475.
33 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998 ; 329:191-196.
34 Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Le dichloroacétate augmente la mort cellulaire apoptotique par le biais de dommages oxydatifs et atténue la production de lactate dans les cellules de cancer du sein traitées à la metformine. Breast Cancer Res Treat. 2014 ; 147:539-550.
35 Lian J, Ni Z, Dai X, Su C, Smith AR, Xu L, He F. Le sorafénib sensibilise la suppression de la croissance induite par le (-)-gossypol dans les cellules cancéreuses de la prostate androgéno-indépendantes via l’inhibition de Mcl-1 et l’activation de Bak. Mol Cancer Ther. 2012 ; 11:416-426.
36 Ou X, Lu Y, Liao L, Li D, Liu L, Liu H, Xu H. Le chlorure de nitidine induit l’apoptose dans les cellules de carcinome hépatocellulaire humain par une voie impliquant p53, p21, Bax et Bcl-2. Oncol Rep. 2015 ; 33:1264-1274.
37 Ni Z, Gong Y, Dai X, Ding W, Wang B, Gong H, Qin L, Cheng P, Li S, Lian J, He F. AU4S : un nouveau peptide synthétique pour mesurer l’activité de l’ATG4 dans les cellules vivantes. Autophagy. 2015 ; 11:403-415.
Contenu connexe :