Бо Ли1,*, Синьчжэ Ли1,*, Zhenhong Ni1, Yan Zhang1, Yijun Zeng1, Xiaohuan Yan2, Yan Huang3, Jintao He4, Xilin Lyu1, Yaran Wu1, Yuting Wang1, Yingru Zheng2, Fengtian He1
1 Кафедра биохимии и молекулярной биологии, Колледж фундаментальных медицинских наук, Третий военный медицинский университет,
Чунцин 400038, Китай
2 Кафедра акушерства и гинекологии, госпиталь Дапин и Научно-исследовательский институт хирургии, Третий военный медицинский
университет, Чунцин 400042, Китай
3 Онкологический центр, госпиталь Дапин и научно-исследовательский институт хирургии, Третий военно-медицинский университет, Чунцин 400042,
Китай
4 Батальон 17 студентов, колледж профилактической медицины, Третий военно-медицинский университет, Чунцин 400038, Китай
Эти авторы внесли равный вклад в данную работу
Корреспонденция:
Yingru Zheng, e-mail: [email protected]
Fengtian He, e-mail: [email protected]
Принято: 24 января 2016 г.
Принято: 09 июля 2016 г.
Опубликовано: 19 июля 2016 г
Аннотация
Дихлорацетат (ДХА) и метформин (Мет) показали многообещающую противоопухолевую эффективность, регулируя метаболизм раковых клеток. Однако опосредованная DCA защитная аутофагия и индуцированное Met накопление лактата ограничивают их потенциал уничтожения опухолей соответственно. Поэтому преодоление соответствующих недостатков позволит улучшить их терапевтический эффект. В настоящем исследовании мы обнаружили, что DCA и Met синергически подавляют рост и усиливают апоптоз клеток рака яичников. Интересно, что мы впервые обнаружили, что Met сенсибилизировал DCA через резкое ослабление индуцированного DCA белка Mcl-1 и защитной аутофагии, а DCA сенсибилизировал Met через заметное ослабление индуцированного Met чрезмерного накопления лактата и потребления глюкозы. Эксперименты in vivo на голых мышах также показали, что DCA и Met синергически подавляли рост ксенотрансплантатов опухолей яичников. Эти результаты могут проложить путь к разработке новых стратегий лечения рака яичников на основе комбинированного применения DCA и Met.
Ключевые слова: дихлорацетат, метформин, Mcl-1, метаболизм рака, рак яичников
ВВЕДЕНИЕ
Смертность от рака яичников занимает первое место среди нескольких видов гинекологических онкологических заболеваний. В настоящее время химиотерапия на основе платины и таксола остается стандартной парадигмой в дополнение к хирургическому вмешательству, однако ее побочные эффекты тяжелы, кроме того, возникла химиорезистентность [1-2]. Поэтому необходимо срочно изучить новые стратегии в качестве альтернативы традиционной химиотерапии. В последние годы все больше доказательств показывают, что рак является своего рода метаболическим нарушением, что выдвигает на передний план регулирование метаболизма рака для подавления роста опухоли [3]. Нацеливание на ключевые метаболические пути значительно убивает многочисленные раковые клетки, включая клетки рака яичников [4-5]. Среди различных метаболических препаратов, дихлорацетат (DCA) и метформин (Met) показали блестящие перспективы из-за их положительных функций в терапии рака.
Как препарат, нацеленный на митохондрии, DCA может подавлять активность киназы пируватдегидрогеназы (PDK) и впоследствии увеличивать активность пируватдегидрогеназы (PDH), что способствует притоку углеводов в митохондрии и тем самым усиливает аэробное окисление глюкозы. Этот эффект обращает вспять митохондриальную дисфункцию и реактивирует митохондрий-зависимый апоптоз в некоторых опухолевых клетках [6-9]. Одновременно DCA ингибирует гликолиз и снижает накопление лактата, что разрушает закисленное микроокружение опухоли (закисленное микроокружение в целом благоприятствует выживанию опухоли) [10]. Хотя DCA показал многообещающие перспективы в борьбе с раком, сообщалось, что DCA индуцирует защитную аутофагию в клетках рака толстой кишки, что, в свою очередь, препятствует их апоптотической способности [11]. До сих пор остается неясным, существует ли какой-либо другой апоптоз-ассоциированный устойчивый детерминант, когда DCA обновляет митохондриальный апоптоз.
Мет — традиционный препарат первой линии терапии сахарного диабета 2 типа. В последние годы появляется все больше доказательств того, что Met может также снижать риск развития рака в нескольких эпидемиологических исследованиях [12]. Met подавляет рост опухоли, вызывая остановку цикла, способствуя апоптозу и подавляя аутофагию [13-15]. Кроме того, Met может сенсибилизировать некоторые химиотерапевтические препараты, такие как паклитаксел, эрлотиниб и т.д. [16-17]. Более того, противоопухолевый эффект Met все чаще связывают с метаболизмом глюкозы при раке [18]. Несмотря на ряд преимуществ в клинических испытаниях, дальнейшее применение Met затруднено, поскольку он может приводить к накоплению лактата [19]. Представляет большой интерес возможность преодоления этого недостатка путем комбинирования других метаболических препаратов для более широкого применения Met в химиотерапии.
Учитывая их потенциальные взаимные компенсаторные эффекты, мы попытались выяснить, могут ли DCA и Met синергизировать друг друга для усиления цитотоксичности клеток рака яичников. В настоящем исследовании мы продемонстрировали, что DCA и Met могут совместно вызывать апоптоз в клетках рака яичников. Met сенсибилизировал DCA через резкое ослабление вызываемого DCA Mcl-1 и защитной аутофагии, в то время как DCA сенсибилизировал Met через заметное ослабление вызываемого Met чрезмерного накопления лактата и потребления глюкозы. Эксперименты in vivo на голых мышах также показали, что DCA и Met синергически подавляли рост ксенотрансплантатов опухолей яичников. Эти результаты позволяют предположить, что данная терапевтическая стратегия может стать многообещающим выбором для будущей целенаправленной терапии рака на основе метаболизма.
РЕЗУЛЬТАТЫ
DCA и Met синергично индуцируют апоптоз в клетках рака яичников
Чтобы выяснить, существует ли синергичный эффект между DCA и Met в подавлении роста клеток рака яичников, клетки SKOV3 и OVCAR3 подвергали совместной обработке DCA и Met или каждым из них в отдельности. Как показано на рис. 1A-1C, совместная обработка DCA и Met более эффективно подавляла рост клеток рака яичников по сравнению с каждой из них в отдельности, а комбинация 40 мМ DCA и 10 мМ Met подавляла жизнеспособность клеток до 50% по сравнению с контролем. Поэтому в последующих экспериментах мы выбрали 40 мМ DCA и 10 мМ Met. Аналогичным образом, синергический эффект ингибирования также наблюдался в клетках рака шейки матки (HeLa и SiHa), немелкоклеточного рака легких (A549 и GLC-82) и клетках гепатоцеллюлярной карциномы человека (HepG2) (рис. S1A-1D), предполагая, что синергизм между DCA и Met может быть в определенной степени универсальным. Более того, DCA и Met синергично индуцировали апоптоз в клетках рака яичников, что было выявлено с помощью анализа проточной цитометрии двойного окрашивания аннексина V-FITC (изотиоцианат флуоресцеина) и PI (йодид продия) (Рисунок 1D), окрашивания апоптотических тел Hoechst (Рисунок 1E), западного блот-анализа расщепленной PARP (поли-АДФ-рибоза полимераза, маркер апоптоза) (Рисунок 1F) и анализа активности каспазы3 (Рисунок 1G).
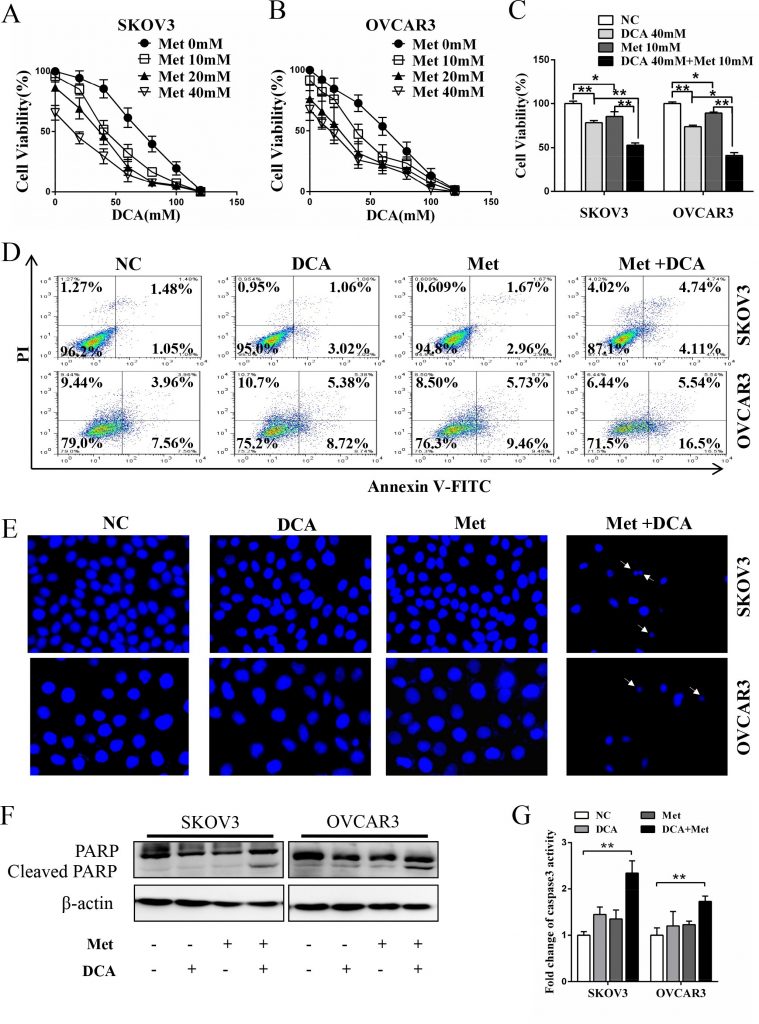
Met сенсибилизирует ДКА через ослабление ДКА-индуцированного Mcl-1
Чтобы изучить механизм, посредством которого Met сенсибилизирует ДКА к индуцированию апоптоза, мы исследовали экспрессию важнейших антиапоптотических членов семейства Bcl-2, включая Mcl-1, Bcl-2 и Bcl-xL [20]. Как показано на рисунке 2А, только DCA значительно повышал уровень белка Mcl-1 (но не белков Bcl-2 и Bcl-xL) в клетках рака яичников, который заметно снижался под действием Met. Глушение Mcl-1 с помощью siRNA усиливало DCA-опосредованное ингибирование жизнеспособности клеток (Рисунок 2B) и усиливало DCA-индуцированный апоптоз (Рисунок 2C-2E). Более того, эктопическая экспрессия Mcl-1 значительно ослабляла сенсибилизирующее действие Met на жизнеспособность и апоптоз клеток (Рисунок 2F-2I). Эти результаты показали, что Mcl-1 является новым фактором устойчивости к DCA, а Met сенсибилизирует DCA через снижение уровня Mcl-1.
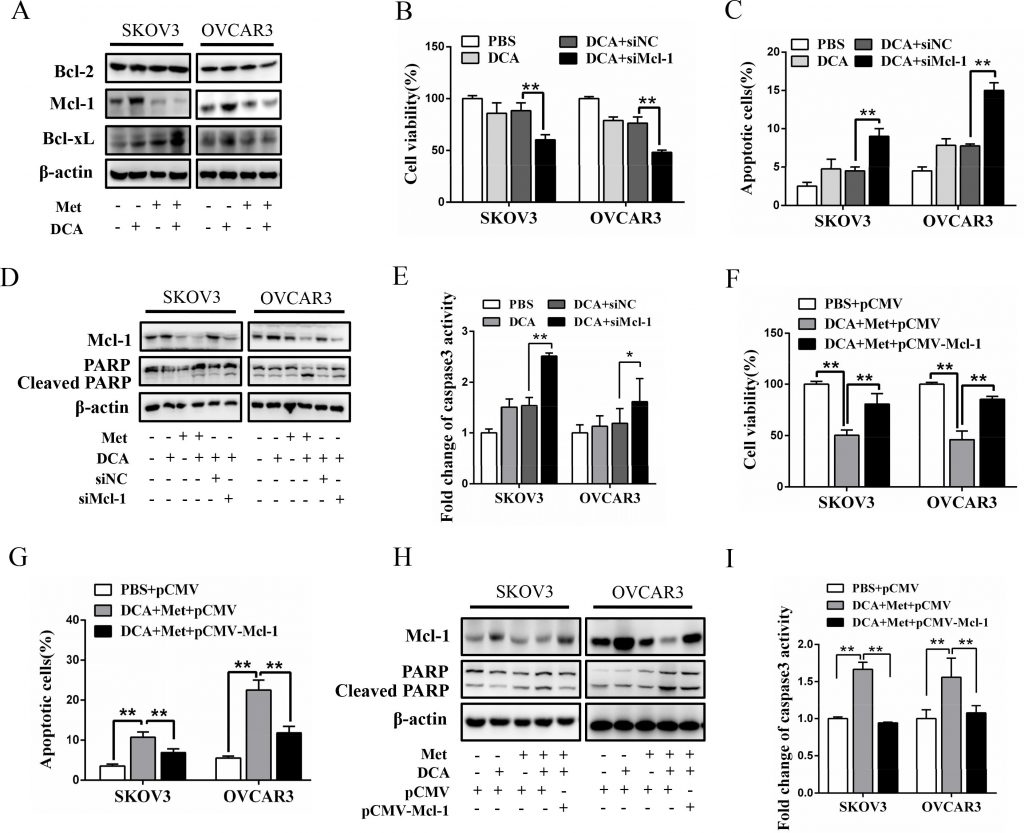
Met ослабляет DCA-индуцированный Mcl-1 через ингибирование трансляции Mcl-1
Чтобы выяснить, на каком уровне DCA индуцирует Mcl-1, сначала была исследована мРНК Mcl-1. Как показано на рисунке 3А, ДКА не оказывал явного влияния на экспрессию мРНК Mcl-1. Затем был проведен анализ белка Mcl-1 в присутствии или отсутствии ингибитора трансляции циклогексимида (CHX). Как показано на рисунках 3B и 3C, CHX в зависимости от времени снижал базальный (но не индуцированный DCA) белок Mcl-1, что указывает на то, что DCA повышает стабильность белка Mcl-1. Сообщалось, что фосфорилированный ERK (p-ERK) и p-JNK могут стабилизировать Mcl-1 путем фосфорилирования Mcl-1 на Thr163 [21-22], поэтому мы исследовали, участвует ли p-ERK/p-JNK в регуляции Mcl-1, индуцированной DCA. Как показано на рисунке 3D, лечение DCA значительно повышало p-ERK (но не p-JNK) и p-Mcl-1Thr163 в клетках рака яичников. Более того, ингибитор MEK1/2 U0126 значительно снижал уровень Mcl-1 и p-Mcl-1Thr163, вызванный DCA, и заметно повышал уровень расщепленного PARP (рис. 3E). Эти результаты показали, что p-ERK (но не p-JNK) является медиатором Mcl-1, индуцированного DCA. Предыдущие исследования показали, что DCA повышает уровень реактивных форм кислорода (ROS) [23], а ROS является ключевым индуктором p-ERK [24], поэтому мы исследовали уровень ROS с помощью DCFH-DA. Как показано на рисунке S2A, DCA увеличивал генерацию ROS, что позволяет предположить, что индукция ROS может быть механизмом, с помощью которого DCA усиливает активацию p-ERK.
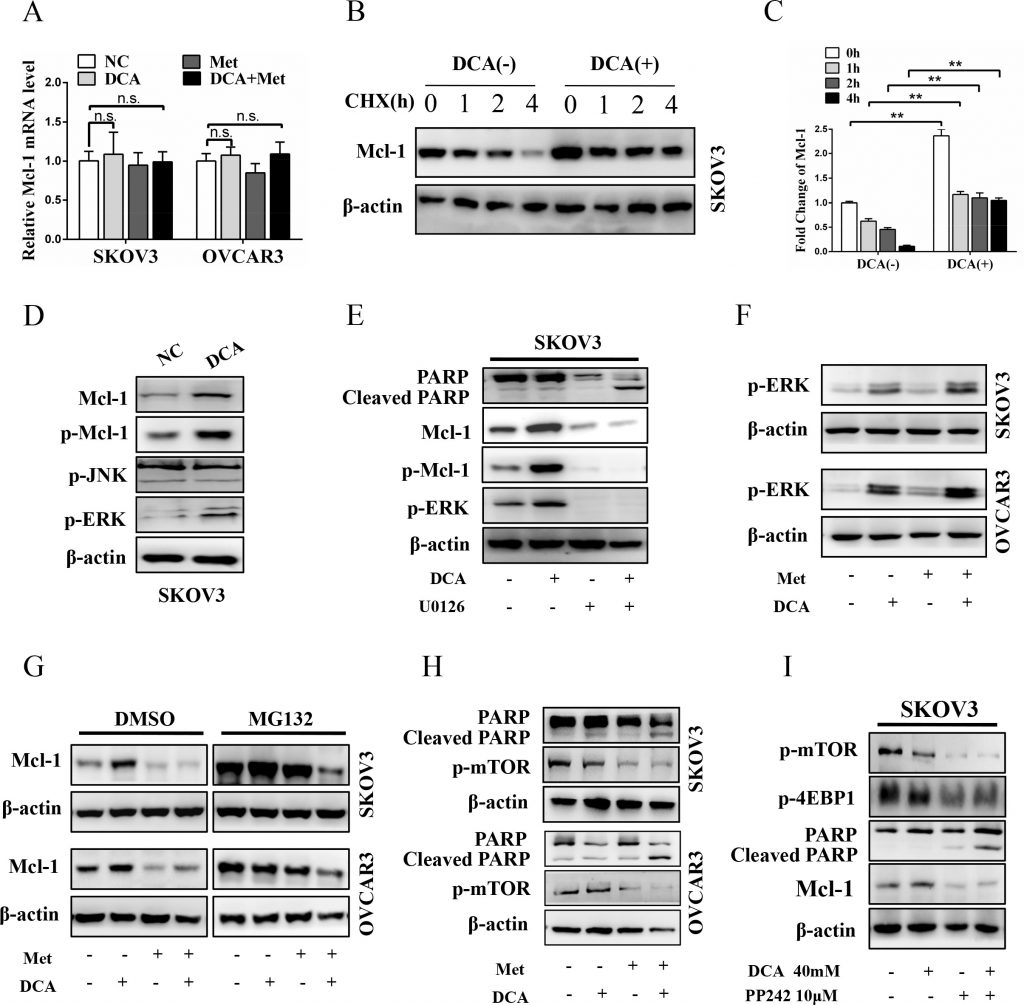
Кроме того, Ser159 также тесно связан со стабильностью Mcl-1, и этот сайт в основном фосфорилируется GSK-3β [25], поэтому мы проверили, участвует ли GSK-3β в регуляции DCA-индуцированной стабилизации Mcl-1. Как показано на рисунке S2B, DCA увеличивал фосфорилирование GSK-3β, но не влиял на общее количество GSK-3β. Более того, DCA усиливал фосфорилирование Akt (сигнальной молекулы GSK-3β), а ингибитор Akt MK-2206 2HCl значительно ослаблял DCA-индуцированное фосфорилирование GSK-3β, повышение уровня Mcl-1 и устойчивость к апоптозу (Рисунок S2C). Эти результаты показали, что p-Akt-опосредованное фосфорилирование GSK-3β способствует DCA-индуцированной стабилизации Mcl-1.
Затем мы изучили, может ли Met ослабить DCA-индуцированную стабилизацию Mcl-1 путем ингибирования p-ERK/p-Akt. Как показано на рисунке 3F и рисунке S2D, Met не смог подавить DCA-индуцированные p-ERK и p-Akt, как и результаты на рисунке 3A, что указывает на то, что Met снижает DCA-индуцированный Mcl-1 ни на транскрипционном, ни на посттрансляционном уровне. Затем мы проанализировали, ослабляет ли Met индуцированный DCA Mcl-1 на трансляционном уровне с помощью ингибитора протеасомы MG132. Как показано на рисунке 3G, когда клетки обрабатывали контролем, DCA, Met или комбинацией, уровни белка Mcl-1 были одинаково повышены в присутствии MG132 по сравнению с DMSO, что указывает на то, что Met ослабляет DCA-индуцированный Mcl-1 через ингибирование трансляции Mcl-1. Сообщалось, что активация mTOR способствует трансляции Mcl-1 [26], поэтому мы проанализировали p-mTOR после совместной обработки Met и DCA. Как и ожидалось, Met заметно снижал уровень p-mTOR (Рисунок 3H), а ингибитор mTOR PP242 оказывал сходное с Met действие на стимулирование апоптоза (Рисунок 3I). Согласно данным, представленным на рисунке 3 и рисунке S2, можно сделать вывод, что DCA повышает уровень Mcl-1 через усиление фосфорилирования ERK и GSK-3β, а Met подавляет трансляцию Mcl-1 через ингибирование p-mTOR.
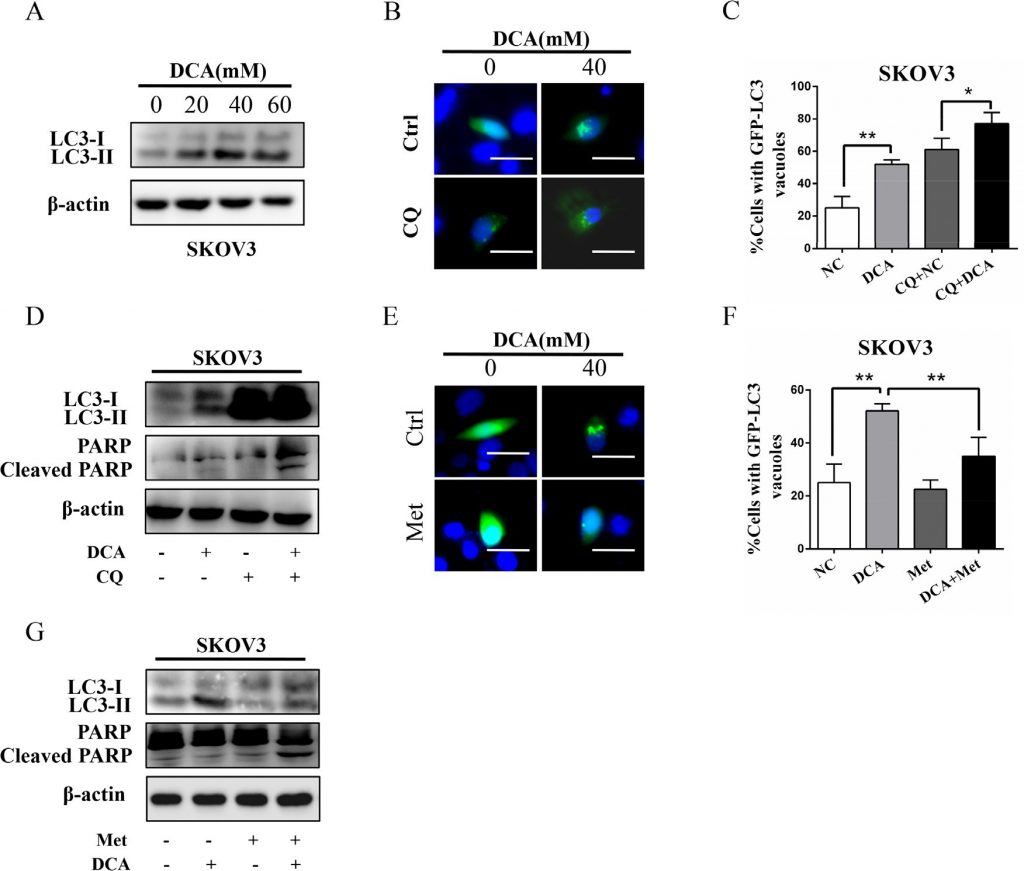
Met снижает DCA-индуцированную защитную аутофагию
Предыдущие исследования показали, что аутофагия играет важную роль в терапевтической устойчивости DCA в клетках рака толстой кишки [11], поэтому мы изучили роль аутофагии в клетках рака яичников при лечении DCA и/или Met. Как показано на рисунке 4A, DCA дозозависимо повышал уровень MAP1LC3-II (LC3-II), маркера аутофагии. Ингибирование аутофагии хлорохином (CQ) или молчание ATG7 резко усиливало апоптоз и цитотоксичность, вызванные DCA (Рисунок 4B-4D, Рисунок S3A-3D), что указывает на то, что DCA индуцирует защитную аутофагию в клетках рака яичников. Для предварительного изучения механизма DCA-индуцированной аутофагии мы проанализировали изменения мРНК 7 генов, связанных с аутофагией, в клетках, обработанных DCA. Как показано на рисунке S3E, DCA резко повысил уровень мРНК ATG7 в клетках рака яичников, предполагая, что ATG7 может быть вовлечен в вызванную DCA защитную аутофагию. Впоследствии мы обнаружили, что Met значительно снизил уровень LC3-II, вызванный DCA (рис. 4E-4G), что указывает на то, что Met может ослаблять защитную аутофагию, вызванную DCA. Таким образом, можно сделать вывод, что ослабление DCA-индуцированной защитной аутофагии также играет важную роль в сенсибилизирующем эффекте Met на DCA.
DCA облегчает Met-индуцированное потребление глюкозы и производство лактата
Чтобы выяснить, может ли DCA преодолеть дефицит Met, были проанализированы изменения лактата и глюкозы. Как показано на рисунке 5A-5C, Met увеличивал выработку лактата и потребление глюкозы, что значительно снижалось под действием DCA. Кроме того, были измерены скорость потребления кислорода клетками (OCR) и скорость внеклеточного закисления (ECAR). Как показано на рисунке 5D-5F, Met снижал соотношение OCR/ECAR, которое резко снижалось под действием DCA, что свидетельствует о том, что DCA может подавлять гликолиз, вызванный Met, через восстановление митохондриального дыхания. Поскольку DCA является ингибитором PDKs, который фосфорилирует и подавляет активность PDH [9], мы исследовали уровень p-PDH. Как показано на рисунке 5G, Met повышал уровень p-PDHE1α (субъединица PDH), что заметно снижалось под действием DCA. Выключение PDH с помощью siRNA значительно ослабило индуцированную Met продукцию лактата (Рисунок 5H, 5I). Эктопическая экспрессия PDK1 и PDK2 усилила фосфрилирование PDHE1α и ослабила апоптоз, индуцированный совместной обработкой DCA и Met (рис. 5J, 5K), что в совокупности с данными на рисунках 5G-5I указывает на то, что DCA может сенсибилизировать Met через ингибирование пути PDK/PDH в уничтожении клеток рака яичников.
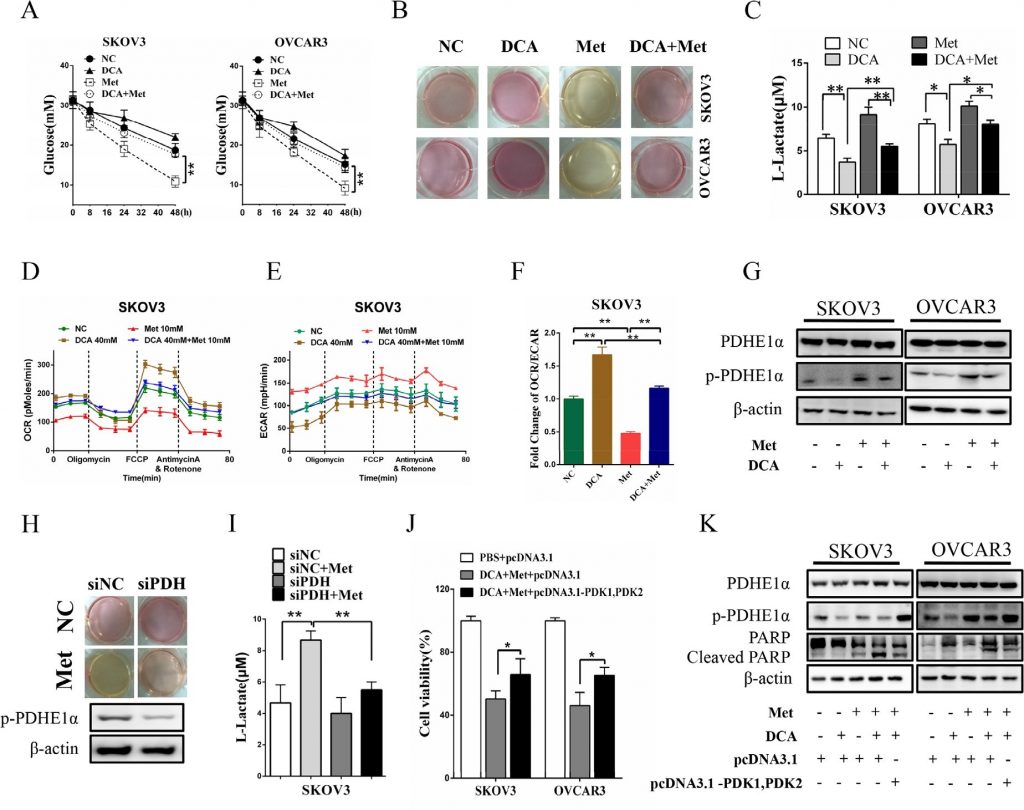
DCA и Met совместно подавляют рост клеток рака яичников in vivo
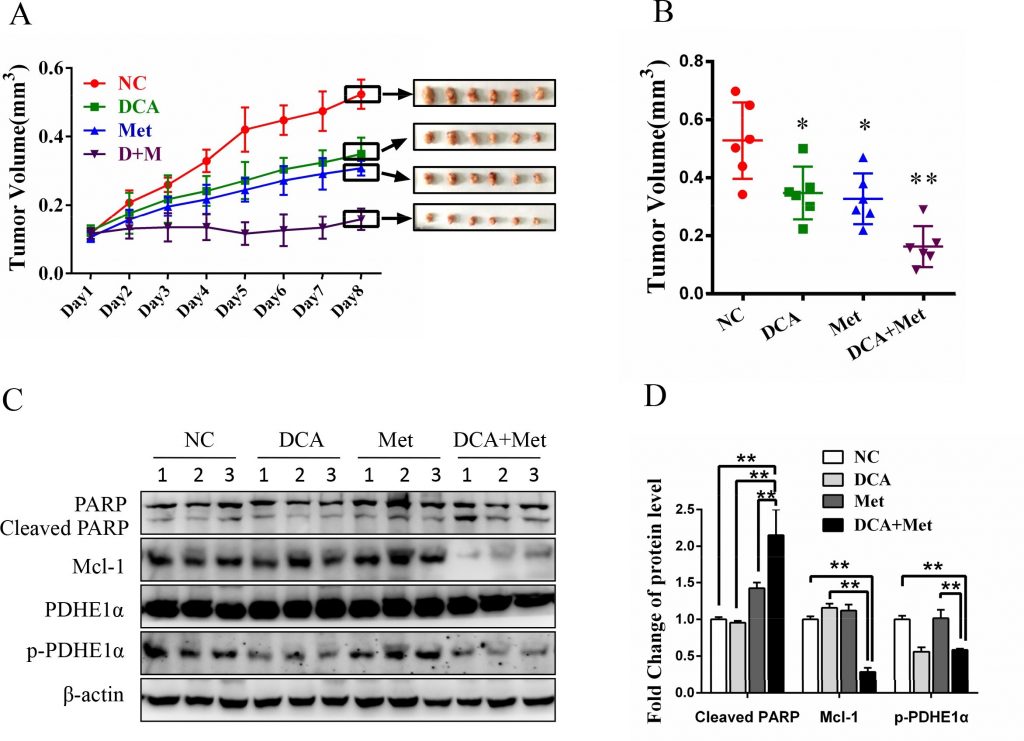
Как показано на рисунках 6A и 6B, совместное лечение DCA и Met более эффективно подавляло рост ксенотрансплантатов рака яичников у обнаженных мышей по сравнению с лечением только DCA или только Met. Вестерн-блот анализ показал, что DCA и Met синергично увеличивали расщепленный PARP, и снижали Mcl-1 и p-PDHE1α в ксенотрансплантатах (Рисунок 6C-6D). Эти результаты свидетельствуют о том, что DCA и Met могут совместно подавлять рост клеток рака яичников in vivo за счет ослабления недостатка друг друга.
ДИСКУССИЯ
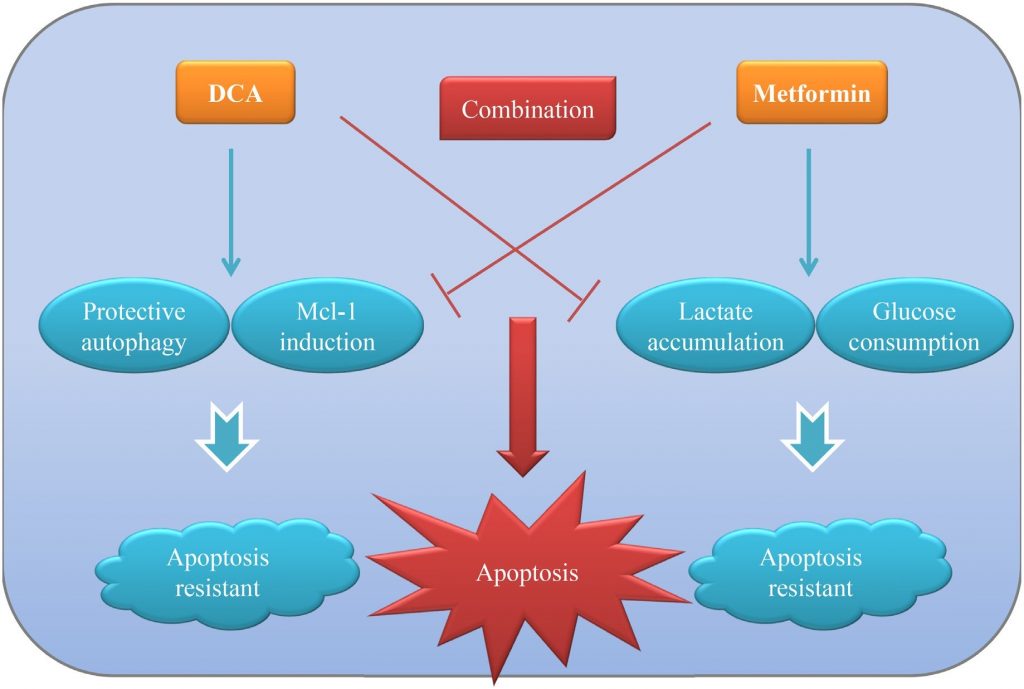
Подтверждено, что для большинства солидных опухолей характерен «эффект Варбурга», при котором они используют гликолиз для производства энергии даже при достаточном количестве кислорода. Борьба с этим аномальным явлением открыла путь для разработки новых терапевтических стратегий лечения рака в дополнение к традиционным цитотоксическим препаратам. В настоящем исследовании мы продемонстрировали, что совместное воздействие DCA и Met (двух метаболически связанных агентов) может более эффективно подавлять рост клеток рака яичников по сравнению с каждым из них в отдельности in vitro и in vivo. Met ослабляет индуцированную DCA Mcl-1 и защитную аутофагию, а DCA смягчает индуцированное Met чрезмерное накопление лактата и потребление глюкозы. Взаимное действие двух агентов способствует интенсивному апоптозу для более эффективного уничтожения клеток рака яичников. Рабочая модель комбинации DCA и Met показана на рисунке
7.
Хотя исследования ведутся уже много лет, ключевые факторы, которые могут препятствовать про-апоптотическому эффекту DCA, пока не ясны. Наши результаты показали, что Mcl-1 является важнейшим фактором устойчивости против DCA-индуцированного апоптоза в клетках рака яичников, а совместная обработка Met и DCA снижала Mcl-1 и усиливала апоптоз. Однако ко-обработка привела к увеличению Bcl-xL (рис. 2A), что может быть компенсаторным механизмом для поддержания выживания клеток. Подобное явление также было отмечено в предыдущем исследовании [27] (Исследование показывает, что ингибитор Bcl-2/xL ABT-263 индуцирует апоптоз раковых клеток, одновременно повышая Mcl-1). Более того, вызванное котерапии увеличение Bcl-xL наблюдалось только в клеточной линии SKOV3 (но не в OVCAR3), что позволяет предположить, что этот эффект может быть клеточно-специфичным. Конечно, еще предстоит выяснить подробный механизм, с помощью которого совместная обработка Met и DCA повышает уровень Bcl-xL. Дальнейшие исследования показали, что DCA индуцирует накопление Mcl-1 через активацию ERK и Akt, которые защищают Mcl-1 от деградации, опосредованной протеасомой. Эти результаты позволяют предположить, что ингибирование ERK и Akt может быть хорошей стратегией для сенсибилизации DCA в лечении рака яичников. Однако недавно были получены противоречивые данные о том, что DCA снижает уровень Mcl-1 в клетках AML [28] и клетках колоректального рака [29]. Эти расхождения позволяют предположить, что взаимосвязь между DCA и Mcl-1 может быть в значительной степени различной в разных контекстах.
Аутофагия — это катаболический процесс, направленный на утилизацию необходимых метаболитов, таких как аминокислоты и липиды, для пополнения биоэнергетического резерва в условиях дефицита питательных веществ или других резких стрессов [30-31]. В данном исследовании мы обнаружили, что DCA индуцирует защитную аутофагию в клетках рака яичников, и ATG7 может играть определенную роль в этом процессе (Рисунок S3E), но подробный механизм требует дальнейшего изучения. Более того, мы обнаружили, что Met сенсибилизирует DCA через подавление DCA-индуцированной защитной аутофагии. В соответствии с нашими результатами, Met может подавлять GRP78-зависимую аутофагию для усиления антимиеломного эффекта бортезомиба [15] и ингибировать 2DG-индуцированную аутофагию для сенсибилизации 2DG в клетках рака простаты [32]. Однако детальный механизм (механизмы), посредством которого Met подавляет защитную аутофагию, еще предстоит изучить.
Met получил широкое признание как единственный агент или сенсибилизатор в терапии рака, однако заметным недостатком является то, что Met способствует потреблению глюкозы и ускоряет накопление лактата, что облегчает аэробный гликолиз, зависимый от рака. В настоящем исследовании мы продемонстрировали, что DCA может значительно ослабить этот побочный эффект. DCA может сильно ингибировать активность PDKs и их нисходящего потока p-PDHE1α, что приводит к метаболической перестройке, переиспользующей окислительное фосфорилирование и вызывающей снижение накопления лактата и потребления глюкозы. Среди четырех типов изоферментов PDK, DCA в основном функционирует через ингибирование PDK2 и PDK1 [33]. Как и ожидалось, одновременная сверхэкспрессия PDK2 и PDK1 усилила фосфорилирование PDHE1α, отменила сенсибилизирующую функцию DCA и частично аннулировала летальный эффект DCA плюс Met в клетках рака яичников. В целом, наши результаты показывают, что DCA сенсибилизирует противоопухолевую функцию Met через ингибирование активности PDKs. Однако следует отметить, что DCA может сенсибилизировать Met путем усиления Met-индуцированного окислительного стресса в клетках рака молочной железы [34]. Это означает, что механизм синергизма DCA с Met настолько сложен, что требует глубокого изучения.
В итоге мы показали, что DCA и Met могут синергически подавлять рост клеток рака яичников, что может проложить путь к разработке новых стратегий лечения рака яичников на основе комбинированного использования DCA и Met.
МАТЕРИАЛЫ И МЕТОДЫ
Клеточные линии и реактивы
Клеточные линии, включая SKOV3, OVCAR3, HeLa, SiHa, GLC-82, A549 и HepG2, были приобретены в American Type Culture Collection (ATCC) и культивировались в среде Dulbecco’s Modified Eagle Medium (DMEM), дополненной 10% фетальной бычьей сывороткой (FBS), стрептомицином (100 мг/мл) и пенициллином (100 Ед/мл) при 37°C в 5%CO2 влажном инкубаторе. DCA и циклогексимид (CHX) были приобретены у Sigma-Aldrich (Луис, МО, США). Met, U0126, MG132 и Hoechst 33258 были приобретены у компании Beyotime (Шанхай, Китай). MK2206 был приобретен у компании Selleck (Шанхай, Китай). Набор для определения активности каспазы-3 и набор для определения реактивных форм кислорода были приобретены у компании Beyotime (Шанхай, Китай). Аннексин V-FITC и PI были приобретены у BD Bioscience (BD, NJ, США). siRNAs к Mcl-1, ATG7, PDH и контрольная siRNA были приобретены у GenePharma (Шанхай, Китай). pCMV и pcDNA3.1, pCMV-Mcl-1, pcDNA3.1-PDK1 и pcDNA3.1-PDK2 экспрессионные плазмиды были приобретены у Obio Technology (Шанхай, Китай).
Вестерн-блот
Готовили лизаты целых клеток и проводили Вестерн-блот, как описано ранее [35]. Антитела к β-актину и PDHE1α были получены от компании Abcam (San Francisco, CA, USA), антитела к Bcl-2 и Mcl-1 — от Santa Cruz Biotechnology (Santa Cruz, CA, USA), антитела к PARP, BCL-xL, GSK-3β, p-ERK (Thr202/Tyr204), p-JNK (Thr183/Tyr185), p-Mcl-1 (Thr163), p-Akt (Ser473), p-4EBP1, p-mTOR и p-GSK-3β (Ser9) были получены от Cell Signaling Technology (Boston, MA, USA). Антитело против p-PDHE1α (Ser293) было получено от EMD Millipore (Billerica, MA, USA).
Трансфекция клеток
Клетки SKOV3 и OVCAR3 выращивали до 60%-70% конфлюентности в 6-луночных планшетах. SiRNA или экспрессионную плазмиду смешивали с 10 мкл lipofectamine 2000 в Opti-MEM (Invitrogen, Carlsbad, CA, USA) для каждой лунки в соответствии с протоколом производителя. После инкубации со смесями в течение 6 ч, клетки культивировали в DMEM с 10% FBS еще 6 ч. Затем клетки получали соответствующую обработку.
Анализ жизнеспособности клеток
Анализ жизнеспособности клеток проводили с использованием набора CCK-8 (Dojindo, Шанхай, Китай), как описано ранее [27]. Клетки высевали на 96-луночные планшеты (2×103 клеток/лунку) в трех экземплярах и инкубировали в течение 12 ч. Затем клетки подвергали различным видам лечения или контролю с использованием транспортного средства в течение 48 ч, после чего в каждую лунку добавляли 10 мкл раствора CCK-8. После инкубации при 37°C в течение 1,5 ч значение OD450 нм определяли с помощью микропланшетного ридера.
Проточная цитометрия
Клетки рака яичников инкубировали с аннексином V-FITC и PI в соответствии с инструкциями производителя (BD, 561012). Затем апоптоз анализировали с помощью проточного цитометра.
Окрашивание Hoechst
После обработки в течение 24 ч клетки окрашивали Hoechst 33258 (Beyotime, Шанхай, Китай) в концентрации 10 мкг/мл в течение 10 мин в темноте. Затем клетки промывали 3 раза PBS и фотографировали под микроскопом афлуоресценции.
Анализ активности каспазы 3
Активность каспазы 3 исследовали с помощью набора Caspase3 Activity Assay kit (Beyotime, C1115), как описано ранее [36]. Вкратце, контрольные и обработанные клетки собирали, промывали ледяным PBS и ресуспендировали в 50 мкл охлажденного буфера для лизиса клеток в течение 15 минут на льду. Затем лизаты центрифугировали (20 000 g, 10 мин, 4°C), и супернатанты сразу же собирали для анализа активности каспазы3.
Измерение общего внутриклеточного ROS
Общий внутриклеточный ROS тестировали с помощью набора ROS Assay Kit (Beyotime, Шанхай, Китай), как описано ранее [37].
Выделение РНК, количественная ПЦР в реальном времени (qPCR)
Общую РНК выделяли из клеток с помощью реагента TRIzol (ComWin Biotechnology, Beijing, China) и синтезировали кДНК первой нити с использованием транскриптазы M-MLV (Invitrogen, Carlsbad, CA, USA). QPCR проводили с помощью набора QuantiFast SYBR Green PCR Kit (Promega, Шанхай, Китай). Относительные уровни мРНК целевых генов рассчитывали методом2-ΔΔCt.
Анализ зеленого флуоресцентного белка (GFP)-MAP1LC3
После трансфекции экспрессионным вектором GFP-MAP1LC3 (GFP-LC3) в течение 12 ч, клетки подвергали указанной обработке в течение еще 24 ч, а затем фиксировали 4% формальдегидом в течение 10 мин. Затем клетки промывали 3 раза PBS и наблюдали под флуоресцентным микроскопом.
Определение L-лактата и глюкозы
Клетки обрабатывали в 6 лунках, затем собирали среду и определяли концентрацию L-лактата и глюкозы отдельно с помощью L-Lactate Assay Kit (Eton Bioscience, San Diego, CA, USA) и Glucose Colorimetric/Fluorometic Assay Kit (BioVision, Milpitas, CA, USA).
Анализ биоэнергетики клеток
Клетки высевали в планшеты XF96 и оставляли расти на ночь. Затем среду заменяли на среду XF96 за 1 ч до начала анализа. Ротенон/антимицин А, FCCP и олигомицин разводили в среде XF96 и загружали в прилагаемый картридж для достижения конечных концентраций 0,5 мкМ, 0,5 мкМ и 1,0 мкМ, соответственно. Инъекции препаратов в среду проводились в указанные временные точки. OCR (пмоль/мин) и ECAR (мпч/мин) отслеживались с помощью набора XF Cell Mito Stress Test Kit (Seahorse Bioscience, North Billerica, MA, USA) с использованием анализаторов внеклеточного потока Seahorse Bioscience XFe и XF.
Исследование на животных
Шестинедельные самки голых мышей были куплены у Beijing Huafukang Bioscience (Пекин, Китай), содержались и ухаживались в соответствии с правилами Комитета по уходу за животными и этике Третьего военно-медицинского университета (Чунцин, Китай). 5×106 клеток SKOV3 в 150 мкл PBS имплантировали в правую подмышечную впадину каждой обнаженной мыши. После образования пальпируемых опухолей мышей рандомизировали на 4 группы (n = 6 на группу). Затем мышам ежедневно внутрибрюшинно вводили DCA (50 мг/кг/день) плюс Met (100 мг/кг/день) или каждый из них в отдельности в течение 8 дней, в качестве контроля брали PBS. Размер опухоли ксенотрансплантата отслеживали каждый день с помощью штангенциркуля, а объем оценивали по следующей формуле: объем = ширина2×длина×1/2. После вырезания у мышей ксенотрансплантаты опухолей фотографировали, а соответствующие белки исследовали методом Вестерн-блоттинга.
Статистический анализ
Данные были выражены как среднее ± SD. Для анализа дисперсии использовали односторонний ANOVA и t-тест. P<0,05 считалось статистически значимым.
БЛАГОДАРНОСТИ
Эта работа была поддержана Национальным фондом естественных наук Китая (81472436 и 81272865) и Фондом естественных наук Чунцина (cstc2012jjB10025).
КОНФЛИКТЫ ИНТЕРЕСОВ
Потенциальные конфликты интересов не были раскрыты.
ССЫЛКИ
1 Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Рак яичников. Lancet. 2014; 384:1376-1388.2 Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489-494.
3 Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipated. Cancer Cell. 2012; 21:297-308.
4 Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ясноклеточная карцинома яичников встречает метаболизм; HNF-1beta обеспечивает преимущества выживания благодаря эффекту Варбурга и снижению ROS. Oncotarget. 2015; 6:30704-30714. doi: 10.18632/oncotarget.5228.
5 Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. МикроРНК-144 опосредует метаболический сдвиг в клетках рака яичников путем прямого нацеливания на Glut1. Tumour Biol. 2015.
6 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Метаболическая модуляция глиобластомы с помощью дихлорацетата. Sci Transl Med. 2010; 2:31ra34.
7 Michelakis ED, Webster L, Mackey JR. Дихлорацетат (ДХА) как потенциальная метаболически-таргетная терапия рака. Br J Cancer. 2008; 99:989-994.
8 Papandreou I, Goliasova T, Denko NC. Противораковые препараты, направленные на метаболизм: является ли дихлорацетат новой парадигмой? Int J Cancer. 2011; 128:1001-1008.
9 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37-51.
10 Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013; 133:1107-1118.
11 Gong F, Peng X, Sang Y, Qiu M, Luo C, He Z, Zhao X, Tong A. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013; 4:e913.
12 Morales DR, Morris AD. Метформин в лечении и профилактике рака. Annu Rev Med. 2015; 66:17-29.
13 Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. Противодиабетический препарат метформин оказывает противоопухолевое действие in vitro и in vivo через снижение уровня циклина D1. Oncogene. 2008; 27:3576-3586.
14 Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Метформин вызывает уникальные биологические и молекулярные реакции в клетках тройного негативного рака молочной железы. Cell Cycle. 2009; 8:2031-2040.
15 Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. Фармакологические экраны выявляют метформин, который подавляет GRP78-зависимую аутофагию для усиления антимиеломного эффекта бортезомиба. Leukemia. 2015; 29:2184-2191.
16 Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. Метформин усиливает индуцированную химиотерапией активацию AMPK и противоопухолевый рост. Clin Cancer Res. 2011; 17:3993-4005.
17 Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE, Cremers S, Szabolcs M, Maurer MA. Метформин и эрлотиниб синергично ингибируют базальный рак молочной железы. Oncotarget. 2014; 5:10503-10517. doi: 10.18632/oncotarget.2391.
18 Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, Dumon E, Martineau Y, Danet-Desnoyers G, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia. 2013; 27:2129-2138.
19 Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Нацеливание на метаболическую гибкость путем одновременного ингибирования дыхательного комплекса I и выработки лактата замедляет прогрессию меланомы. Oncotarget. 2015; 6:37281-37299. doi: 10.18632/oncotarget.6134.
20 Lessene G, Czabotar PE, Colman PM. Антагонисты семейства BCL-2 для терапии рака. Nat Rev Drug Discov. 2008; 7:989-1000.
21 Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 фосфорилируется в области PEST и стабилизируется при активации ERK в жизнеспособных клетках, а также в дополнительных участках при воздействии цитотоксической окадаиновой кислоты или таксола. Oncogene. 2004; 23:5301-5315.
22 Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Антиапоптотический эффект c-Jun N-terminal Kinase-1 через стабилизацию Mcl-1 в TNF-индуцированном апоптозе гепатоцитов. Гастроэнтерология. 2009; 136:1423-1434.
23 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016; 371:20-29.
24 Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM, Zhao CQ, Liu PJ, Zhang JM, He JJ. MAPK-сигнализация опосредует индуцированную синоменином гидрохлоридом гибель клеток рака молочной железы человека как через реактивные виды кислорода, так и через независимые пути: исследование in vitro и in vivo. Cell Death Dis. 2014; 5:e1356.
25 Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Гликогенсинтаза киназа-3 регулирует проницаемость внешней мембраны митохондрий и апоптоз путем дестабилизации MCL-1. Mol Cell. 2006; 21:749-760.
26 Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, et al. mTORC1 способствует выживанию через трансляционный контроль Mcl-1. Proc Natl Acad Sci U S A. 2008; 105:10853-10858.
27 Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. Ингибитор Bcl-2/xL ABT-263 повышает стабильность мРНК и белка Mcl-1 в клетках гепатоцеллюлярной карциномы. Mol Cancer. 2014; 13:98.
28 Emadi A, Sadowska M, Carter-Cooper B, Bhatnagar V, van der Merwe I, Levis MJ, Sausville EA, Lapidus RG. Возмущение клеточного окислительного состояния, индуцированное дихлорацетатом и триоксидом мышьяка при лечении острого миелоидного лейкоза. Leuk Res. 2015; 39:719-729.
29 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD, Coomber BL. Дихлорацетат влияет на пролиферацию, но не на выживание клеток колоректального рака человека. Apoptosis. 2015; 20:63-74.
30 Mah LY, Ryan KM. Аутофагия и рак. Cold Spring Harb Perspect Biol. 2012; 4:a008821.
31 Yang ZJ, Chee CE, Huang S, Sinicrope FA. Роль аутофагии в раке: терапевтические последствия. Mol Cancer Ther. 2011; 10:1533-1541.
32 Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010; 70:2465-2475.
33 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Доказательства существования тканеспецифической регуляции комплекса пируватдегидрогеназы млекопитающих. Biochem J. 1998; 329:191-196.
34 Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Дихлорацетат усиливает апоптотическую гибель клеток через окислительное повреждение и ослабляет выработку лактата в клетках рака молочной железы, обработанных метформином. Breast Cancer Res Treat. 2014; 147:539-550.
35 Lian J, Ni Z, Dai X, Su C, Smith AR, Xu L, He F. Sorafenib sensitizes (-)-gossypol-induced growth suppression in androgen-independent prostate cancer cells through Mcl-1 inhibition and Bak activation. Mol Cancer Ther. 2012; 11:416-426.
36 Ou X, Lu Y, Liao L, Li D, Liu L, Liu H, Xu H. Нитидинхлорид индуцирует апоптоз в клетках гепатоцеллюлярной карциномы человека через путь с участием p53, p21, Bax и Bcl-2. Oncol Rep. 2015; 33:1264-1274.
37 Ni Z, Gong Y, Dai X, Ding W, Wang B, Gong H, Qin L, Cheng P, Li S, Lian J, He F. AU4S: новый синтетический пептид для измерения активности ATG4 в живых клетках. Autophagy. 2015; 11:403-415.
Связанный контент: