Bo Li1,*, Xinzhe Li1,*, Zhenhong Ni1, Yan Zhang1, Yijun Zeng1, Xiaohuan Yan2, Yan Huang3, Jintao He4, Xilin Lyu1, Yaran Wu1, Yuting Wang1, Yingru Zheng2, Fengtian He1
1 Abteilung für Biochemie und Molekularbiologie, College of Basic Medical Sciences, Third Military Medical University,
Chongqing 400038, China2
Abteilung für Geburtshilfe und Gynäkologie, Daping Hospital und Forschungsinstitut für Chirurgie, Third Military MedicalUniversity
, Chongqing 400042, China3
Krebszentrum, Daping Hospital und Forschungsinstitut für Chirurgie, Third Military Medical University, Chongqing 400042,
China4
Studentenbataillon 17, College of Preventive Medicine, Third Military Medical University, Chongqing 400038, China*
Diese Autoren haben gleichermaßen zu dieser Arbeit beigetragen
Korrespondenz:
Yingru Zheng, E-Mail:
[email protected] He, E-Mail: [email protected]
Received: January 24, 2016Accepted
: July 09, 2016Published
: July 19, 2016
Abstrakt
Sowohl Dichloracetat (DCA) als auch Metformin (Met) haben durch die Regulierung des Krebszellstoffwechsels eine vielversprechende Antitumor-Wirksamkeit gezeigt. Allerdings schränken die DCA-vermittelte schützende Autophagie und die Met-induzierte Laktatakkumulation ihr tumortötendes Potenzial ein. Die Überwindung der entsprechenden Defizite wird also ihre therapeutische Wirkung verbessern. In der vorliegenden Studie haben wir festgestellt, dass DCA und Met synergistisch das Wachstum von Eierstockkrebszellen hemmen und deren Apoptose verstärken. Interessanterweise haben wir zum ersten Mal festgestellt, dass Met DCA sensibilisiert, indem es das DCA-induzierte Mcl-1-Protein und die schützende Autophagie drastisch abschwächt, während DCA Met sensibilisiert, indem es die Met-induzierte übermäßige Laktatakkumulation und den Glukoseverbrauch deutlich mindert. Die In-vivo-Experimente an Nacktmäusen zeigten auch, dass DCA und Met synergistisch das Wachstum von Xenotransplantaten von Eierstocktumoren unterdrückten. Diese Ergebnisse könnten den Weg für die Entwicklung neuer Strategien zur Behandlung von Eierstockkrebs auf der Grundlage der kombinierten Verwendung von DCA und Met ebnen.
Schlüsselwörter: Dichloracetat, Metformin, Mcl-1, Krebsstoffwechsel, Eierstockkrebs
EINLEITUNG
Die Sterblichkeitsrate von Eierstockkrebs steht unter den verschiedenen gynäkologischen Krebsarten an erster Stelle. Gegenwärtig sind Chemotherapien auf Platin- und Taxolbasis neben der Operation immer noch das Standardparadigma, doch ihre Nebenwirkungen sind schwerwiegend, und es hat sich auch eine Chemoresistenz entwickelt [1-2]. Daher ist es dringend notwendig, neue Strategien als Alternativen zur herkömmlichen Chemotherapie zu erforschen. In den letzten Jahren wurde immer deutlicher, dass Krebs eine Art Stoffwechselanomalie ist, die durch die Regulierung des Krebsstoffwechsels zur Hemmung des Tumorwachstums in den Vordergrund rückt [3]. Durch gezielte Eingriffe in wichtige Stoffwechselwege können zahlreiche Krebszellen, darunter auch Eierstockkrebszellen, abgetötet werden [4-5]. Unter den verschiedenen Stoffwechselmedikamenten haben Dichloracetat (DCA) und Metformin (Met) aufgrund ihrer positiven Funktionen in der Krebstherapie reizvolle Aussichten gezeigt.
Als Wirkstoff, der auf die Mitochondrien abzielt, kann DCA die Aktivität der Pyruvat-Dehydrogenase-Kinase (PDK) hemmen und anschließend die Aktivität der Pyruvat-Dehydrogenase (PDH) erhöhen, was den Fluss von Kohlenhydraten in die Mitochondrien fördert und dadurch die aerobe Oxidation von Glukose verbessert. Dieser Effekt kehrt die mitochondriale Dysfunktion um und reaktiviert die mitochondrienabhängige Apoptose in verschiedenen Tumorzellen [6-9]. Gleichzeitig hemmt DCA die Glykolyse und verringert die Laktatakkumulation, wodurch die saure Mikroumgebung des Tumors zerstört wird (die saure Mikroumgebung begünstigt im Allgemeinen das Überleben des Tumors) [10]. Obwohl DCA vielversprechende Aussichten bei der Krebsbekämpfung gezeigt hat, wurde berichtet, dass DCA die schützende Autophagie in Dickdarmkrebszellen induziert, was wiederum deren apoptotische Kapazität behindert [11]. Bislang ist noch unklar, ob es noch eine andere Apoptose-assoziierte resistente Determinante gibt, wenn DCA die mitochondriale Apoptose auffrischt.
Met ist ein traditionelles Medikament der Erstlinientherapie bei Typ-2-Diabetes. In den letzten Jahren mehren sich die Hinweise, dass Met in mehreren epidemiologischen Studien auch das Krebsrisiko senken kann [12]. Met unterdrückt das Tumorwachstum, indem es einen Zyklusstillstand bewirkt, die Apoptose fördert und die Autophagie unterdrückt [13-15]. Außerdem kann Met einige Chemotherapeutika wie Paclitaxel, Erlotinib usw. sensibilisieren. [16-17]. Noch bemerkenswerter ist, dass die Anti-Tumor-Wirkung von Met zunehmend mit dem Glukosestoffwechsel von Krebs in Verbindung gebracht wird [18]. Trotz zahlreicher Vorteile in klinischen Studien wird die weitere Anwendung von Met dadurch behindert, dass es zu einer Laktatakkumulation führen kann [19]. Es ist von großem Interesse, ob dieser Nachteil durch die Kombination mit anderen Stoffwechselmedikamenten überwunden werden könnte, damit Met in der Chemotherapie umfassender eingesetzt werden kann.
In Anbetracht ihrer potenziellen gegenseitigen kompensatorischen Wirkungen wollten wir herausfinden, ob DCA und Met sich gegenseitig synergistisch beeinflussen können, um die Zytotoxizität bei Eierstockkrebszellen zu erhöhen. In der vorliegenden Studie konnten wir zeigen, dass DCA und Met gemeinsam die Apoptose in Eierstockkrebszellen auslösen können. Met sensibilisierte DCA durch eine drastische Abschwächung des DCA-induzierten Mcl-1 und der schützenden Autophagie, während DCA Met durch eine deutliche Abschwächung der Met-induzierten übermäßigen Laktatakkumulation und des Glukoseverbrauchs sensibilisierte. Die In-vivo-Experimente an Nacktmäusen zeigten auch, dass DCA und Met synergistisch das Wachstum von Ovarialtumoren unterdrückten. Diese Ergebnisse deuten darauf hin, dass diese therapeutische Strategie eine vielversprechende Wahl für eine künftige metabolismusbasierte gezielte Krebstherapie sein könnte.
ERGEBNISSE
DCA und Met induzieren auf synergistische Weise Apoptose in Eierstockkrebszellen
Um zu untersuchen, ob es einen synergistischen Effekt zwischen DCA und Met bei der Unterdrückung des Wachstums von Eierstockkrebszellen gibt, wurden SKOV3- und OVCAR3-Zellen gemeinsam mit DCA und Met oder jeweils allein behandelt. Wie in Abbildung 1A-1C gezeigt, unterdrückte die gemeinsame Behandlung mit DCA und Met das Wachstum der Eierstockkrebszellen effizienter als die Behandlung mit DCA und Met allein, und die Kombination aus 40mM DCA und 10mM Met konnte die Lebensfähigkeit der Zellen im Vergleich zur Kontrolle auf bis zu 50 % reduzieren. Daher wählten wir in den folgenden Experimenten 40mM DCA und 10mM Met. In ähnlicher Weise wurde die synergistische Hemmwirkung auch bei Gebärmutterhalskrebszellen (HeLa und SiHa), nicht-kleinzelligen Lungenkrebszellen (A549 und GLC-82) und menschlichen hepatozellulären Karzinomzellen (HepG2) beobachtet (Abbildung S1A-1D), was darauf hindeutet, dass der Synergismus zwischen DCA und Met bis zu einem gewissen Grad universell sein könnte. Darüber hinaus induzierten DCA und Met synergistisch die Apoptose in Ovarialkarzinomzellen, was durch eine durchflusszytometrische Analyse der Doppelfärbung von Annexin V-FITC (Fluoresceinisothiocyanat) und PI (Prodiumiodid) (Abbildung 1D), der Hoechst-Färbung von apoptotischen Körpern (Abbildung 1E), der Western-Blot-Analyse von gespaltenem PARP (Poly-ADP-Ribose-Polymerase, ein Marker für Apoptose) (Abbildung 1F) und des Caspase3-Aktivitätstests (Abbildung 1G) nachgewiesen wurde.
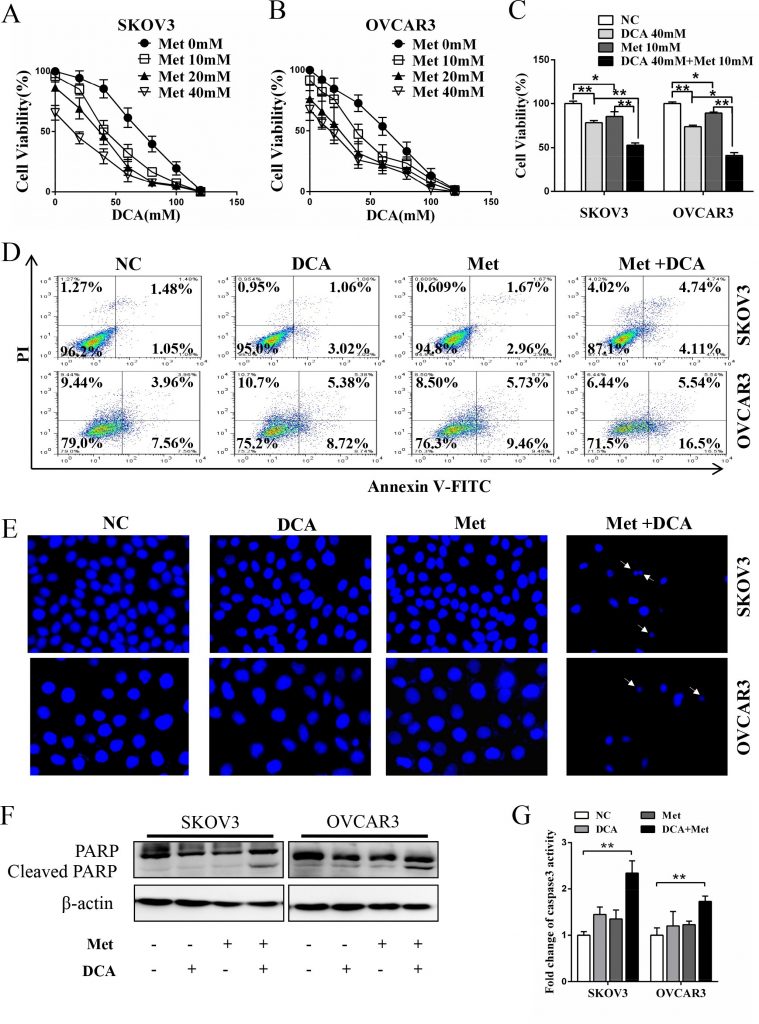
Met sensibilisiert DCA durch Abschwächung von DCA-induziertem Mcl-1
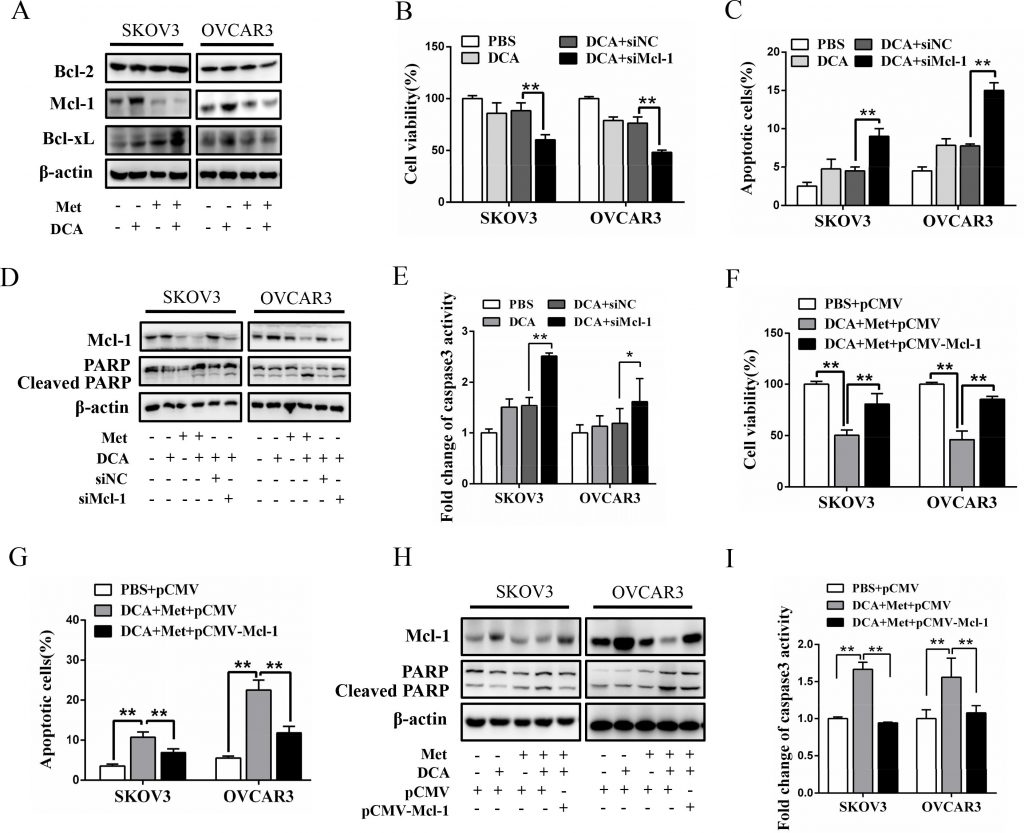
Um den Mechanismus zu untersuchen, durch den Met DCA sensibilisiert, Apoptose zu induzieren, untersuchten wir die Expression der entscheidenden antiapoptotischen Mitglieder der Bcl-2-Familie, einschließlich Mcl-1, Bcl-2 und Bcl-xL [20]. Wie in Abbildung 2A zu sehen ist, erhöhte DCA allein den Spiegel des Mcl-1-Proteins (aber nicht die Bcl-2- und Bcl-xL-Proteine) in Eierstockkrebszellen erheblich, was durch Met deutlich abgeschwächt wurde. Die Ausschaltung von Mcl-1 durch siRNA verstärkte die DCA-vermittelte Hemmung der Zelllebensfähigkeit (Abbildung 2B) und erhöhte die DCA-induzierte Apoptose (Abbildung 2C-2E). Darüber hinaus milderte die ektopische Expression von Mcl-1 die sensibilisierende Wirkung von Met auf DCA auf die Zelllebensfähigkeit und Apoptose drastisch ab (Abbildung 2F-2I). Diese Ergebnisse deuten darauf hin, dass Mcl-1 ein neuartiger resistenter Faktor von DCA ist und Met DCA über die Herunterregulierung von Mcl-1 sensibilisiert.
Met dämpft DCA-induziertes Mcl-1 durch Hemmung der Mcl-1-Translation
Um zu klären, auf welcher Ebene DCA Mcl-1 induziert, wurde zunächst die mRNA von Mcl-1 untersucht. Wie in Abbildung 3A gezeigt, hatte DCA keine offensichtlichen Auswirkungen auf die Mcl-1-mRNA-Expression. Anschließend wurde das Mcl-1-Protein in Gegenwart oder Abwesenheit des Translationshemmers Cycloheximid (CHX) analysiert. Wie in Abbildung 3B und 3C gezeigt, verringerte CHX zeitabhängig das basale (aber nicht DCA-induzierte) Mcl-1-Protein, was darauf hindeutet, dass DCA die Stabilität des Mcl-1-Proteins erhöht. Es wurde berichtet, dass die phosphorylierte ERK (p-ERK) und p-JNK Mcl-1 durch Phosphorylierung von Mcl-1 an Thr163 stabilisieren können [21-22], daher haben wir untersucht, ob p-ERK/p-JNK an der Regulierung von DCA-induziertem Mcl-1 beteiligt ist. Wie in Abbildung 3D zu sehen ist, führte die Behandlung mit DCA zu einer signifikanten Erhöhung von p-ERK (aber nicht p-JNK) und p-Mcl-1Thr163 in Eierstockkrebszellen. Darüber hinaus konnte der MEK1/2-Inhibitor U0126 die DCA-induzierten Mcl-1- und p-Mcl-1Thr163-Werte drastisch abschwächen und das gespaltene PARP deutlich verstärken (Abbildung 3E). Diese Ergebnisse weisen darauf hin, dass p-ERK (aber nicht p-JNK) ein Vermittler von DCA-induziertem Mcl-1 ist. Frühere Studien haben gezeigt, dass DCA reaktive Sauerstoffspezies (ROS) erhöht [23], und ROS ist ein wichtiger Auslöser von p-ERK [24], daher untersuchten wir das ROS-Niveau mit DCFH-DA. Wie in Abbildung S2A gezeigt, erhöhte DCA die Erzeugung von ROS, was darauf hindeutet, dass die Induktion von ROS ein Mechanismus sein könnte, durch den DCA die p-ERK-Aktivierung verstärkt.
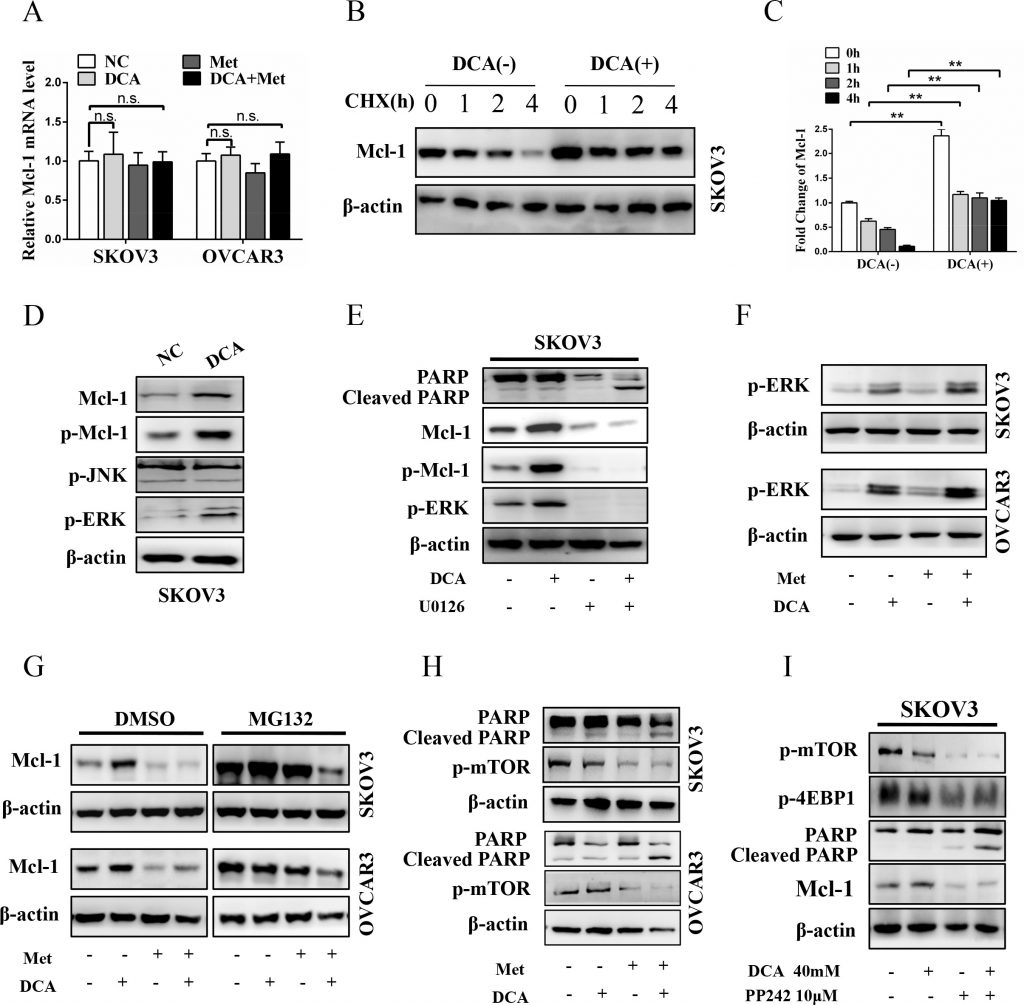
Darüber hinaus steht Ser159 in engem Zusammenhang mit der Stabilität von Mcl-1, und diese Stelle wird hauptsächlich von GSK-3β phosphoryliert [25], daher haben wir getestet, ob GSK-3β auch an der Regulierung der DCA-induzierten Mcl-1-Stabilisierung beteiligt ist. Wie in Abbildung S2B gezeigt, erhöhte DCA die Phosphorylierung von GSK-3β, hatte aber keinen Effekt auf die Gesamt-GSK-3β. Darüber hinaus erhöhte DCA die Phosphorylierung von Akt (ein Upstream-Signalmolekül von GSK-3β), und der Akt-Inhibitor MK-2206 2HCl schwächte die DCA-induzierte GSK-3β-Phosphorylierung, die Mcl-1-Hochregulierung und die Apoptoseresistenz dramatisch ab (Abbildung S2C). Diese Ergebnisse zeigten, dass die p-Akt-vermittelte Phosphorylierung von GSK-3β die DCA-induzierte Mcl-1-Stabilisierung fördert.
Anschließend untersuchten wir, ob Met das DCA-induzierte Mcl-1 durch Hemmung von p-ERK/p-Akt abschwächen kann. Wie in Abbildung 3F und Abbildung S2D gezeigt, konnte Met das DCA-induzierte p-ERK und p-Akt nicht unterdrücken, zusammen mit den Ergebnissen in Abbildung 3A, was darauf hindeutet, dass Met das DCA-induzierte Mcl-1 weder auf transkriptioneller noch auf posttranslationaler Ebene verringert. Anschließend untersuchten wir, ob Met das DCA-induzierte Mcl-1 auf der Translationsebene mit dem Proteasominhibitor MG132 abschwächt. Wie in Abbildung 3G gezeigt, waren die Proteinkonzentrationen von Mcl-1 bei der Behandlung von Zellen mit der Kontrolle, DCA, Met oder einer Kombination davon in Anwesenheit von MG132 im Vergleich zu DMSO gleichermaßen erhöht, was darauf hindeutet, dass Met das DCA-induzierte Mcl-1 durch Hemmung der Mcl-1-Translation abschwächt. Es wurde berichtet, dass die Aktivierung von mTOR die Mcl-1-Translation fördert [26], daher analysierten wir p-mTOR nach der gleichzeitigen Behandlung mit Met und DCA. Wie erwartet, verringerte Met den p-mTOR-Spiegel deutlich (Abbildung 3H), und der mTOR-Inhibitor PP242 hatte eine ähnliche Wirkung wie Met auf die Förderung der Apoptose (Abbildung 3I). Aus den Daten in Abbildung 3 und Abbildung S2 können wir schließen, dass DCA Mcl-1 durch die Steigerung der Phosphorylierung von ERK und GSK-3β hochreguliert und Met die Mcl-1-Translation durch Hemmung von p-mTOR unterdrückt.
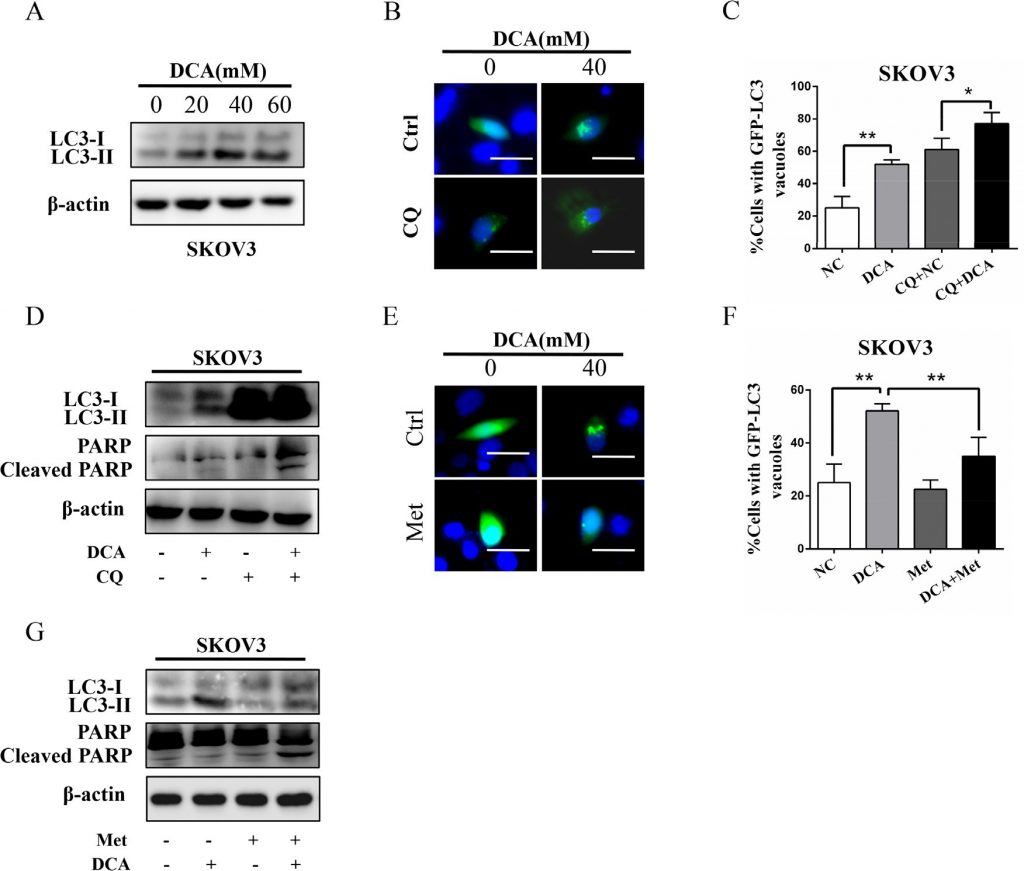
Met vermindert die DCA-induzierte schützende Autophagie
Frühere Studien haben gezeigt, dass die Autophagie eine wichtige Rolle bei der therapeutischen Resistenz von DCA in Dickdarmkrebszellen spielt [11]daher untersuchten wir die Rolle der Autophagie in Eierstockkrebszellen bei Behandlung mit DCA oder/und Met. Wie in Abbildung 4A dargestellt, erhöhte DCA dosisabhängig die Konzentration von MAP1LC3-II (LC3-II), dem Marker der Autophagie. Die Hemmung der Autophagie durch Chloroquin (CQ) oder das Ausschalten von ATG7 verstärkte die DCA-induzierte Apoptose und Zytotoxizität dramatisch (Abbildung 4B-4D, Abbildung S3A-3D), was darauf hindeutet, dass DCA eine schützende Autophagie in Eierstockkrebszellen induziert. Um den Mechanismus der DCA-induzierten Autophagie vorläufig zu untersuchen, untersuchten wir die mRNA-Veränderungen von 7 mit der Autophagie verbundenen Genen in DCA-behandelten Zellen. Wie in Abbildung S3E gezeigt, erhöhte DCA die mRNA-Konzentration von ATG7 in Eierstockkrebszellen dramatisch, was darauf hindeutet, dass ATG7 an der DCA-induzierten schützenden Autophagie beteiligt sein könnte. Anschließend stellten wir fest, dass Met das DCA-induzierte LC3-II deutlich verringerte (Abbildung 4E-4G), was darauf hindeutet, dass Met die DCA-induzierte schützende Autophagie abschwächen könnte. Zusammenfassend könnte man sagen, dass die Schwächung der DCA-induzierten schützenden Autophagie auch für die sensibilisierende Wirkung von Met auf DCA wichtig ist.
DCA mildert den Met-induzierten Glukoseverbrauch und die Laktatproduktion
Um zu klären, ob DCA den Met-Mangel überwinden kann, wurden die Veränderungen von Laktat und Glukose analysiert. Wie in Abbildung 5A-5C dargestellt, erhöhte Met die Laktatproduktion und den Glukoseverbrauch, was durch DCA deutlich gemildert wurde. Außerdem wurden die zelluläre Sauerstoffverbrauchsrate (OCR) und die extrazelluläre Versauerungsrate (ECAR) gemessen. Wie in Abbildung 5D-5F zu sehen ist, verringerte Met das Verhältnis von OCR/ECAR, was durch DCA drastisch abgeschwächt wurde, was zeigt, dass DCA die Met-induzierte Glykolyse durch Wiederherstellung der mitochondrialen Atmung unterdrücken kann. Da DCA ein PDK-Inhibitor ist, der die PDH-Aktivität phosphoryliert und hemmt [9], untersuchten wir das Niveau der p-PDH. Wie in Abbildung 5G zu sehen ist, erhöhte Met den Spiegel von p-PDHE1α (einer Untereinheit von PDH), was durch DCA deutlich rückgängig gemacht wurde. Die Ausschaltung von PDH mit siRNA schwächte die Met-induzierte Laktatproduktion deutlich ab (Abbildung 5H, 5I). Die ektopische Expression von PDK1 und PDK2 verstärkte die Phosphrylierung von PDHE1α und schwächte die durch die gemeinsame Behandlung mit DCA und Met induzierte Apoptose ab (Abbildung 5J, 5K), was zusammen mit den Daten in den Abbildungen 5G-5I darauf hindeutet, dass DCA Met durch die Hemmung des PDK/PDH-Signalwegs bei der Abtötung von Eierstockkrebszellen sensibilisieren kann.
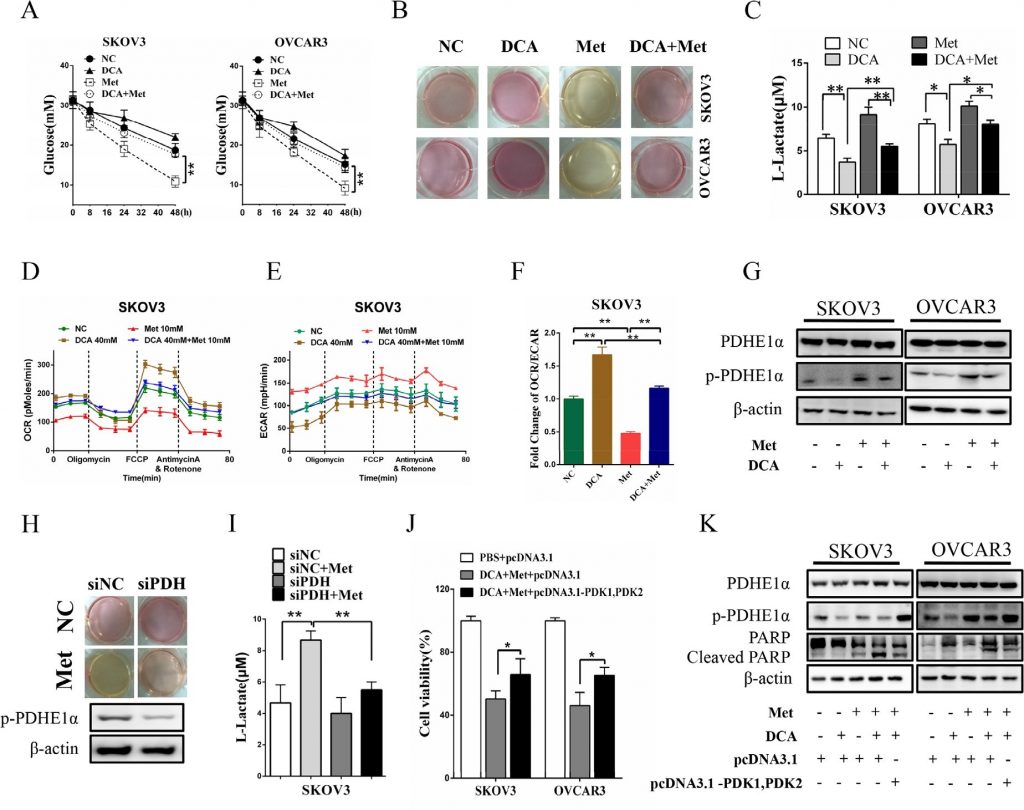
DCA und Met unterdrücken gemeinsam das Wachstum von Eierstockkrebszellen in vivo
Wie in Abbildung 6A und 6B gezeigt, unterdrückte die gemeinsame Behandlung mit DCA und Met das Wachstum von Eierstockkrebs-Xenografts in Nacktmäusen effizienter als die Behandlung mit DCA oder Met allein. Western-Blot-Analysen zeigten, dass DCA und Met synergistisch die Spaltung von PARP erhöhten und Mcl-1 und p-PDHE1α in den Xenotransplantaten herunterregulierten (Abbildung 6C-6D). Diese Ergebnisse deuten darauf hin, dass DCA und Met gemeinsam das Wachstum von Eierstockkrebszellen in vivo hemmen können, indem sie die Defizite des jeweils anderen abschwächen.
DISKUSSION
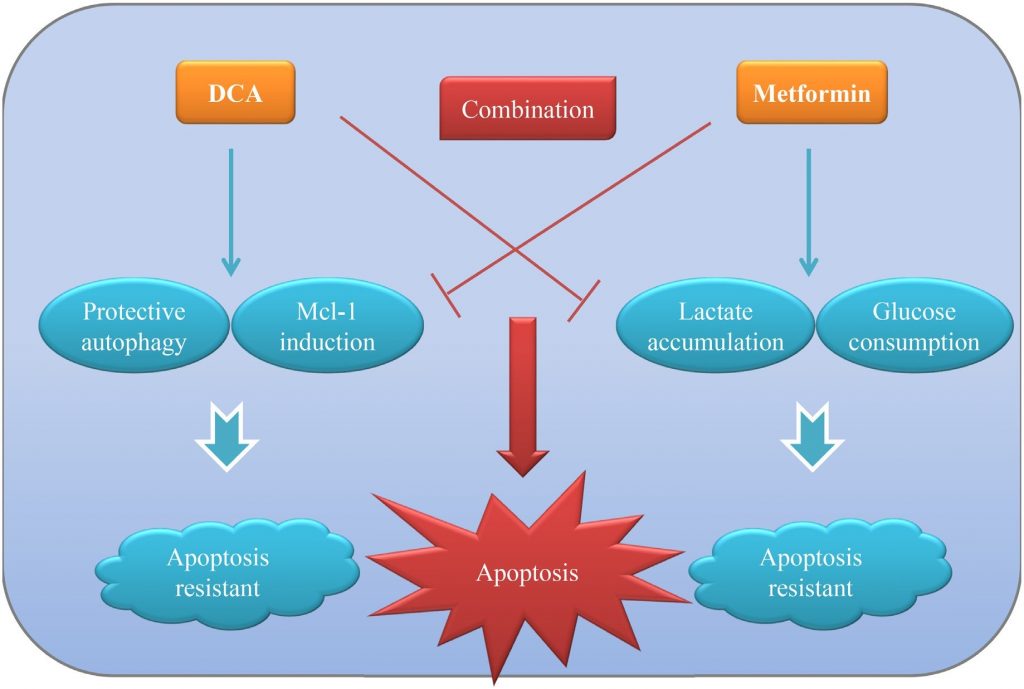
Es hat sich bestätigt, dass die meisten soliden Tumoren durch den „Warburg-Effekt“ gekennzeichnet sind, bei dem sie die Glykolyse zur Energiegewinnung nutzen, obwohl ausreichend Sauerstoff vorhanden ist. Die Beeinflussung dieses abnormen Phänomens hat den Weg für die Entwicklung neuartiger therapeutischer Strategien gegen Krebs zusätzlich zu den herkömmlichen zytotoxischen Medikamenten geebnet. In der vorliegenden Studie konnten wir nachweisen, dass die gemeinsame Behandlung mit DCA und Met (zwei Stoffwechsel-assoziierte Wirkstoffe) das Wachstum von Eierstockkrebszellen in vitro und in vivo effizienter unterdrücken kann als jede Behandlung allein. Met dämpft DCA-induziertes Mcl-1 und schützende Autophagie, während DCA die Met-induzierte übermäßige Laktatakkumulation und den Glukoseverbrauch mindert. Die wechselseitigen Vorteile der beiden Wirkstoffe tragen zu einer intensiven Apoptose bei, die Eierstockkrebszellen effektiver abtötet. Das Funktionsmodell von DCA und Met in Kombination ist in Abbildung dargestellt. 7.
Obwohl seit Jahren Studien durchgeführt werden, sind die Schlüsselfaktoren, die die pro-apoptotische Wirkung von DCA behindern können, noch nicht klar. Unsere Ergebnisse zeigten, dass Mcl-1 ein entscheidender Resistenzfaktor gegen DCA-induzierte Apoptose in Eierstockkrebszellen ist, und die gleichzeitige Behandlung mit Met und DCA verringerte Mcl-1 und verstärkte die Apoptose. Die gleichzeitige Behandlung mit Met und DCA verringerte Mcl-1 und verstärkte die Apoptose. Die gleichzeitige Behandlung führte jedoch zu einem Anstieg von Bcl-xL (Abbildung 2A), was ein Ausgleichsmechanismus sein könnte, um das Überleben der Zellen zu sichern. Über ein ähnliches Phänomen wurde auch in einer früheren Studie berichtet [27] (Die Studie zeigt, dass der Bcl-2/xL-Inhibitor ABT-263 die Apoptose von Krebszellen induziert und gleichzeitig Mcl-1 hochreguliert). Darüber hinaus war der durch die Mitbehandlung induzierte Bcl-xL-Anstieg nur in der SKOV3-Zelllinie (aber nicht in der OVCAR3-Zelllinie) vorhanden, was darauf hindeutet, dass dieser Effekt zellspezifisch sein könnte. Der genaue Mechanismus, durch den die gleichzeitige Behandlung mit Met und DCA die Bcl-xL-Konzentration erhöht, muss natürlich noch ermittelt werden. Weitere Untersuchungen ergaben, dass DCA die Akkumulation von Mcl-1 über die Aktivierung von ERK und Akt induzierte, was Mcl-1 vor dem Proteasom-vermittelten Abbau schützte. Diese Ergebnisse legen nahe, dass die Hemmung von ERK und Akt eine gute Strategie zur Sensibilisierung von DCA bei der Behandlung von Eierstockkrebs sein könnte. Kürzlich wurde jedoch ein widersprüchliches Ergebnis berichtet, wonach DCA die Mcl-1-Konzentration in AML-Zellen [28] und Darmkrebszellen [29] senkt. Die Diskrepanzen deuten darauf hin, dass die Beziehung zwischen DCA und Mcl-1 in verschiedenen Kontexten sehr unterschiedlich sein kann.
Die Autophagie ist ein kataboler Prozess, bei dem essenzielle Stoffwechselprodukte wie Aminosäuren und Lipide recycelt werden, um die bioenergetischen Reserven bei Nährstoffmangel oder anderen dramatischen Stresssituationen wieder aufzufüllen [30-31]. In dieser Studie konnten wir zeigen, dass DCA eine schützende Autophagie in Ovarialkarzinomzellen auslöst, und ATG7 könnte bei diesem Prozess eine Rolle spielen (Abbildung S3E), aber der detaillierte Mechanismus muss noch weiter untersucht werden. Darüber hinaus fanden wir heraus, dass Met DCA sensibilisierte, indem es die DCA-induzierte schützende Autophagie unterdrückte. In Übereinstimmung mit unseren Ergebnissen kann Met die GRP78-abhängige Autophagie unterdrücken, um die Anti-Myelom-Wirkung von Bortezomib zu verstärken [15], und 2DG-induzierte Autophagie hemmen, um 2DG in Prostatakrebszellen zu sensibilisieren [32]. Die genauen Mechanismen, durch die Met die schützende Autophagie unterdrückt, müssen jedoch noch weiter untersucht werden.
Met ist als Einzelwirkstoff oder Sensibilisator in der Krebstherapie gut bekannt, aber ein bemerkenswerter Nachteil ist, dass Met den Glukoseverbrauch fördert und die Laktatakkumulation beschleunigt, was die krebsabhängige aerobe Glykolyse erleichtert. In der vorliegenden Studie haben wir gezeigt, dass DCA diese Nebenwirkung drastisch abschwächen kann. DCA kann die Aktivität der PDKs und des nachgeschalteten p-PDHE1α stark hemmen, was zu einem metabolischen Umbau führt, bei dem die oxidative Phosphorylierung wieder genutzt wird, und eine geringere Laktatakkumulation und einen geringeren Glukoseverbrauch bewirkt. Unter den vier Arten von PDK-Isoenzymen wirkt DCA hauptsächlich durch Hemmung von PDK2 und PDK1 [33]. Wie erwartet, erhöhte die gleichzeitige Überexpression von PDK2 und PDK1 die Phosphorylierung von PDHE1α, hob die sensibilisierende Funktion von DCA auf und hob die tödliche Wirkung von DCA plus Met in Eierstockkrebszellen teilweise auf. Insgesamt deuten unsere Ergebnisse darauf hin, dass DCA die Antitumorfunktion von Met durch die Hemmung der Aktivität von PDKs sensibilisiert. Es ist jedoch zu beachten, dass DCA Met sensibilisieren kann, indem es den Met-induzierten oxidativen Stress in Brustkrebszellen verstärkt [34]. Dies bedeutet, dass der synergistische Mechanismus von DCA auf Met so kompliziert ist, dass er eingehend untersucht werden muss.
Zusammenfassend haben wir gezeigt, dass DCA und Met synergistisch das Wachstum von Eierstockkrebszellen unterdrücken können, was den Weg für die Entwicklung neuer Strategien zur Behandlung von Eierstockkrebs auf der Grundlage der kombinierten Verwendung von DCA und Met ebnen könnte.
MATERIALIEN UND METHODEN
Zelllinien und Reagenzien
Die Zelllinien SKOV3, OVCAR3, HeLa, SiHa, GLC-82, A549 und HepG2 wurden von der American Type Culture Collection (ATCC) erworben und in Dulbecco’s Modified Eagle Medium (DMEM), ergänzt mit 10 % fötalem Rinderserum (FBS), Streptomycin (100 mg/mL) und Penicillin (100 U/mL) bei 37 °C in einem feuchten Inkubator mit 5 %CO2 gezüchtet. DCA und Cycloheximid (CHX) wurden von Sigma-Aldrich (Louis, MO, USA) bezogen. Met, U0126, MG132 und Hoechst 33258 wurden von der Beyotime Company (Shanghai, China) erworben. MK2206 wurde von der Firma Selleck (Shanghai, China) erworben. Caspase-3-Aktivitäts-Assay-Kit und Reactive Oxygen Species Assay-Kit wurden von der Firma Beyotime (Shanghai, China) erworben. Annexin V-FITC und PI wurden von BD Bioscience (BD, NJ, USA) erworben. siRNAs für Mcl-1, ATG7, PDH und Kontroll-siRNA stammen von GenePharma (Shanghai, China). pCMV und pcDNA3.1, pCMV-Mcl-1, pcDNA3.1-PDK1 und pcDNA3.1-PDK2 Expressionsplasmide wurden von Obio Technology (Shanghai, China) erworben.
Western Blot
Ganze Zelllysate wurden vorbereitet und Western Blot wurde wie zuvor beschrieben durchgeführt [35]. Die Antikörper für β-Actin und PDHE1α stammten von der Firma Abcam (San Francisco, CA, USA), die Antikörper für Bcl-2 und Mcl-1 waren von Santa Cruz Biotechnology (Santa Cruz, CA, USA), und die Antikörper für PARP, BCL-xL, GSK-3β, p-ERK (Thr202/Tyr204), p-JNK (Thr183/Tyr185), p-Mcl-1 (Thr163), p-Akt (Ser473), p-4EBP1, p-mTOR und p-GSK-3β (Ser9) waren von Cell Signaling Technology (Boston, MA, USA). Der Antikörper gegen p-PDHE1α (Ser293) stammte von EMD Millipore (Billerica, MA, USA).
Zelltransfektion
SKOV3- und OVCAR3-Zellen wurden in 6-Well-Platten auf 60 bis 70 % Konfluenz gezüchtet. Die siRNA oder das Expressionsplasmid wurde mit 10 μl Lipofectamin 2000 in Opti-MEM (Invitrogen, Carlsbad, CA, USA) für jede Vertiefung gemäß dem Herstellerprotokoll gemischt. Nach 6-stündiger Inkubation mit den Mischungen wurden die Zellen für weitere 6 Stunden in DMEM mit 10 % FBS kultiviert und anschließend entsprechend behandelt.
Zelllebensfähigkeitstest
Der Zelllebensfähigkeitstest wurde mit dem CCK-8-Kit (Dojindo, Shanghai, China) wie zuvor beschrieben durchgeführt [27]. Die Zellen wurden in dreifacher Ausfertigung auf 96-Well-Platten (2×103 Zellen/Well) ausgesät und 12 Stunden lang inkubiert. Dann wurden die Zellen 48 Stunden lang mit verschiedenen Behandlungen oder einer Vehikelkontrolle behandelt, gefolgt von der Zugabe von 10 μl CCK-8-Lösung in jede Vertiefung. Nach der Inkubation bei 37 °C für 1,5 Stunden wurde der OD450nm-Wert mit einem Mikroplattenlesegerät bestimmt.
Durchflusszytometrie
Eierstockkrebszellenwurden mit Annexin V-FITC und PI gemäß den Anweisungen des Herstellers (BD, 561012) inkubiert. Anschließend wurde die Apoptose mit einem Durchflusszytometer analysiert.
Hoechst-Färbung
Nach der 24-stündigen Behandlung wurden die Zellen 10 Minuten lang im Dunkeln mit Hoechst 33258 (Beyotime, Shanghai, China) in einer Konzentration von 10μg/mL gefärbt. Anschließend wurden die Zellen dreimal mit PBS gewaschen und unter dem Fluoreszenzmikroskop fotografiert.
Caspase-3-Aktivitätstest
Die Caspase-3-Aktivität wurde mit dem Caspase3-Aktivitäts-Assay-Kit (Beyotime, C1115) wie zuvor beschrieben untersucht [36]. Kurz gesagt wurden die Kontroll- und behandelten Zellen geerntet, mit eiskaltem PBS gewaschen und in 50 μl gekühltem Zelllysepuffer für 15 Minuten auf Eis resuspendiert. Dann wurden die Lysate zentrifugiert (20.000 g, 10 min, 4°C), und die Überstände wurden sofort für den Caspase3-Aktivitätstest gesammelt.
Messung der gesamten intrazellulären ROS
Die gesamte intrazelluläre ROS wurde mit dem ROS Assay Kit (Beyotime, Shanghai, China) wie zuvor beschrieben getestet [37].
RNA-Isolierung, quantitative Echtzeit-PCR (qPCR)
Die Gesamt-RNA wurde mit TRIzol-Reagenz (ComWin Biotechnology, Beijing, China) aus den Zellen extrahiert und die cDNA des ersten Strangs wurde mit M-MLV-Transkriptase (Invitrogen, Carlsbad, CA, USA) synthetisiert. Die qPCR wurde mit dem QuantiFast SYBR Green PCR Kit (Promega, Shanghai, China) durchgeführt. Die relativen mRNA-Spiegel der Zielgene wurden mit der2-ΔΔCt-Methode berechnet.
Analyse des grün fluoreszierenden Proteins (GFP)-MAP1LC3
Nachdem die Zellen 12 Stunden lang mit dem GFP-MAP1LC3 (GFP-LC3)-Expressionsvektor transfiziert worden waren, wurden sie weitere 24 Stunden lang den angegebenen Behandlungen unterzogen und dann 10 Minuten lang mit 4% Formaldehyd fixiert. Anschließend wurden die Zellen dreimal mit PBS gewaschen und unter einem Fluoreszenzmikroskop beobachtet.
Nachweis von L-Laktat und Glukose
Die Zellen wurden in 6-Wells behandelt, dann wurde das Medium gesammelt und die Konzentrationen von L-Laktat und Glukose wurden mit dem L-Laktat-Assay-Kit (Eton Bioscience, San Diego, CA, USA) und dem kolorimetrischen/fluorometrischen Glukose-Assay-Kit (BioVision, Milpitas, CA, USA) separat bestimmt.
Analyse der zellulären Bioenergetik
Die Zellen wurden in XF96-Platten plattiert und über Nacht wachsen gelassen. Dann wurden die Medien 1 Stunde vor dem Test durch XF96-Medien ersetzt. Rotenon/Antimycin A, FCCP und Oligomycin wurden in XF96-Medien verdünnt und in die zugehörige Kartusche gefüllt, um Endkonzentrationen von 0,5μM, 0,5μM bzw. 1,0μM zu erreichen. Die Injektionen der Medikamente in das Medium erfolgten zu den angegebenen Zeitpunkten. Die OCR (pmol/min) und ECAR (mpH/min) wurden mit dem XF Cell Mito Stress Test Kit (Seahorse Bioscience, North Billerica, MA, USA) unter Verwendung von Seahorse Bioscience XFe und XF Extracellular Flux Analyzers überwacht.
Tierstudie
Sechs Wochen alte weibliche Nacktmäuse wurden von Beijing Huafukang Bioscience (Beijing, China) gekauft und gemäß den Richtlinien des Animal Care and Ethics Committee der Third Military Medical University (Chongqing, China) untergebracht und gepflegt. 5×106 SKOV3-Zellen in 150 μl PBS wurden in die rechte Achselhöhle jeder Nacktmaus implantiert. Wenn sich tastbare Tumore gebildet hatten, wurden die Mäuse nach dem Zufallsprinzip in 4 Gruppen eingeteilt (n = 6 pro Gruppe). Dann wurden den Mäusen 8 Tage lang täglich DCA (50 mg/kg/d) plus Met (100 mg/kg/d) oder beides allein intraperitoneal injiziert, wobei PBS als Kontrolle diente. Die Größe des Xenotransplantats wurde täglich mit einer Schieblehre überwacht, und das Volumen wurde nach folgender Formel geschätzt: Volumen = Breite2×Länge×1/2. Nach der Exzision aus den Mäusen wurden die Xenotransplantat-Tumore fotografiert und die entsprechenden Proteine mittels Western Blot untersucht.
Statistische Analyse
Die Daten wurden als Mittelwert ± SD ausgedrückt. Zur Analyse der Varianz wurden eine einseitige ANOVA und ein t-Test durchgeführt. P< 0,05 wurde als statistisch signifikant angesehen.
DANKSAGUNGEN
Diese Arbeit wurde von der National Natural Science Foundation of China (81472436 und 81272865) und der Natural Science Foundation of Chongqing (cstc2012jjB10025) unterstützt.
INTERESSENKONFLIKTE
Es wurden keine potenziellen Interessenkonflikte offengelegt.
REFERENZEN
1 Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Eierstockkrebs. Lancet. 2014; 384:1376-1388.2 Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489-494.
3 Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297-308.
4 Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism; HNF-1beta confers survival benefits through the Warburg effect and ROS reduction. Oncotarget. 2015; 6:30704-30714. doi: 10.18632/oncotarget.5228.
5 Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. MicroRNA-144 vermittelt metabolische Verschiebung in Eierstockkrebszellen durch direktes Angreifen von Glut1. Tumour Biol. 2015.
6 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolische Modulation des Glioblastoms mit Dichloracetat. Sci Transl Med. 2010; 2:31ra34.
7 Michelakis ED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielle metabolische zielgerichtete Therapie für Krebs. Br J Cancer. 2008; 99:989-994.
8 Papandreou I, Goliasova T, Denko NC. Krebsmedikamente, die auf den Stoffwechsel abzielen: Ist Dichloracetat das neue Paradigma? Int J Cancer. 2011; 128:1001-1008.
9 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle. 2007; 11:37-51.
10 Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Dichloracetat verbessert die durch vom Tumor abgesonderte Milchsäure verursachte Immundysfunktion und erhöht die Antitumor-Immunreaktivität. Int J Cancer. 2013; 133:1107-1118.
11 Gong F, Peng X, Sang Y, Qiu M, Luo C, He Z, Zhao X, Tong A. Dichloracetat induziert schützende Autophagie in LoVo-Zellen: Beteiligung von Cathepsin D/Thioredoxin-ähnlichem Protein 1 und Akt-mTOR-vermittelten Signalen. Cell Death Dis. 2013; 4:e913.
12 Morales DR, Morris AD. Metformin in der Krebsbehandlung und -prävention. Annu Rev Med. 2015; 66:17-29.
13 Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27:3576-3586.
14 Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induziert einzigartige biologische und molekulare Reaktionen in dreifach negativen Brustkrebszellen. Cell Cycle. 2009; 8:2031-2040.
15 Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. Pharmakologische Screens enthüllen Metformin, das die GRP78-abhängige Autophagie unterdrückt und so die Anti-Myelom-Wirkung von Bortezomib verstärkt. Leukemia. 2015; 29:2184-2191.
16 Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. Metformin verstärkt die Chemotherapie-induzierte AMPK-Aktivierung und das antitumorale Wachstum. Clin Cancer Res. 2011; 17:3993-4005.
17 Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE, Cremers S, Szabolcs M, Maurer MA. Metformin und Erlotinib wirken synergistisch bei der Hemmung von basalem Brustkrebs. Oncotarget. 2014; 5:10503-10517. doi: 10.18632/oncotarget.2391.
18 Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, Dumon E, Martineau Y, Danet-Desnoyers G, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia. 2013; 27:2129-2138.
19 Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Targeting metabolic flexibility by simultaneous inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget. 2015; 6:37281-37299. doi: 10.18632/oncotarget.6134.
20 Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008; 7:989-1000.
21 Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 wird in der PEST-Region phosphoryliert und bei ERK-Aktivierung in lebensfähigen Zellen und an zusätzlichen Stellen mit zytotoxischer Okadainsäure oder Taxol stabilisiert. Oncogene. 2004; 23:5301-5315.
22 Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotische Wirkung der c-Jun N-terminalen Kinase-1 durch Mcl-1-Stabilisierung bei TNF-induzierter Hepatozytenapoptose. Gastroenterology. 2009; 136:1423-1434.
23 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016; 371:20-29.
24 Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM, Zhao CQ, Liu PJ, Zhang JM, He JJ. MAPK signaling mediates sinomenine hydrochloride-induced human breast cancer cell death via both reactive oxygen species-dependent and -independent pathways: an in vitro and in vivo study. Cell Death Dis. 2014; 5:e1356.
25 Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glykogensynthase-Kinase-3 reguliert die Permeabilisierung der mitochondrialen Außenmembran und Apoptose durch Destabilisierung von MCL-1. Mol Cell. 2006; 21:749-760.
26 Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, et al. mTORC1 fördert das Überleben durch Translationskontrolle von Mcl-1. Proc Natl Acad Sci U S A. 2008; 105:10853-10858.
27 Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. Der Bcl-2/xL-Inhibitor ABT-263 erhöht die Stabilität von Mcl-1 mRNA und Protein in hepatozellulären Karzinomzellen. Mol Cancer. 2014; 13:98.
28 Emadi A, Sadowska M, Carter-Cooper B, Bhatnagar V, van der Merwe I, Levis MJ, Sausville EA, Lapidus RG. Störung des zellulären oxidativen Zustands durch Dichloracetat und Arsentrioxid zur Behandlung der akuten myeloischen Leukämie. Leuk Res. 2015; 39:719-729.
29 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD, Coomber BL. Dichloracetat beeinflusst die Proliferation, aber nicht das Überleben von menschlichen Darmkrebszellen. Apoptosis. 2015; 20:63-74.
30 Mah LY, Ryan KM. Autophagie und Krebs. Cold Spring Harb Perspect Biol. 2012; 4:a008821.
31 Yang ZJ, Chee CE, Huang S, Sinicrope FA. Die Rolle der Autophagie bei Krebs: therapeutische Implikationen. Mol Cancer Ther. 2011; 10:1533-1541.
32 Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010; 70:2465-2475.
33 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Beweise für die Existenz einer gewebespezifischen Regulierung des Pyruvatdehydrogenase-Komplexes bei Säugetieren. Biochem J. 1998; 329:191-196.
34 Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Dichloracetat verstärkt den apoptotischen Zelltod durch oxidative Schäden und dämpft die Laktatproduktion in mit Metformin behandelten Brustkrebszellen. Breast Cancer Res Treat. 2014; 147:539-550.
35 Lian J, Ni Z, Dai X, Su C, Smith AR, Xu L, He F. Sorafenib sensibilisiert (-)-Gossypol-induzierte Wachstumsunterdrückung in androgenunabhängigen Prostatakrebszellen über Mcl-1-Hemmung und Bak-Aktivierung. Mol Cancer Ther. 2012; 11:416-426.
36 Ou X, Lu Y, Liao L, Li D, Liu L, Liu H, Xu H. Nitidinchlorid induziert Apoptose in menschlichen hepatozellulären Karzinomzellen über einen Weg, an dem p53, p21, Bax und Bcl-2 beteiligt sind. Oncol Rep. 2015; 33:1264-1274.
37 Ni Z, Gong Y, Dai X, Ding W, Wang B, Gong H, Qin L, Cheng P, Li S, Lian J, He F. AU4S: a novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy. 2015; 11:403-415.
Verwandte Inhalte: