Bo Li1,*, Xinzhe Li1,*, Zhenhong Ni1, Yan Zhang1, Yijun Zeng1, Xiaohuan Yan2, Yan Huang3, Jintao He4, Xilin Lyu1, Yaran Wu1, Yuting Wang1, Yingru Zheng2, Fengtian He1
1 Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias Médicas Básicas, Tercera Universidad Médica Militar,
Chongqing 400038, China
2 Departamento de Obstetricia y Ginecología, Hospital de Daping e Instituto de Investigación de Cirugía, Tercera Universidad Médica Militar
, Chongqing 400042, China
3 Centro Oncológico, Hospital de Daping e Instituto de Investigación Quirúrgica, Tercera Universidad Médica Militar, Chongqing 400042,
China
4 Batallón 17 de Estudiantes, Facultad de Medicina Preventiva, Tercera Universidad Médica Militar, Chongqing 400038, China
* Estos autores han contribuido a partes iguales a este trabajo
Correspondencia:
Yingru Zheng, correo electrónico: [email protected]
Fengtian He, correo electrónico: [email protected]
Recibido: 24 de enero de 2016
Aceptado: 09 de julio de 2016
Publicado: 19 de julio de 2016
Resumen
Tanto el dicloroacetato (DCA) como la metformina (Met) han mostrado una prometedora eficacia antitumoral al regular el metabolismo de las células cancerosas. Sin embargo, la autofagia protectora mediada por DCA y la acumulación de lactato inducida por Met limitan su potencial de eliminación de tumores, respectivamente. Por tanto, la superación de las carencias correspondientes mejorará sus efectos terapéuticos. En el presente estudio, descubrimos que el DCA y la Met inhibían sinérgicamente el crecimiento y potenciaban la apoptosis de las células de cáncer de ovario. Curiosamente, por primera vez revelamos que Met sensibilizaba a DCA a través de la atenuación dramática de la proteína Mcl-1 inducida por DCA y la autofagia protectora, mientras que DCA sensibilizaba a Met a través del marcado alivio de la acumulación excesiva de lactato y el consumo de glucosa inducidos por Met. Los experimentos in vivo en ratones desnudos también demostraron que el DCA y la Met suprimían sinérgicamente el crecimiento de los tumores de ovario xenoinjertados. Estos resultados pueden allanar el camino para el desarrollo de nuevas estrategias para el tratamiento del cáncer de ovario basadas en el uso combinado de DCA y Met.
Palabras clave: dicloroacetato, metformina, Mcl-1, metabolismo del cáncer, cáncer de ovario
INTRODUCCIÓN
La mortalidad del cáncer de ovario ocupa el primer lugar entre varios tipos de cánceres ginecológicos. En la actualidad, las quimioterapias basadas en platino y taxol siguen siendo el paradigma estándar, además de la cirugía; sin embargo, sus efectos secundarios son graves y también ha surgido la quimiorresistencia [1-2]. Por lo tanto, es urgente explorar nuevas estrategias como alternativas a la quimioterapia tradicional. En los últimos años, las crecientes evidencias han demostrado que el cáncer es un tipo de anomalías metabólicas, lo que lo empuja a la vanguardia mediante la regulación del metabolismo del cáncer para inhibir el crecimiento tumoral [3]. Dirigirse a vías metabólicas clave mata significativamente numerosas células cancerosas, incluidas las de cáncer de ovario [4-5]. Entre los diversos fármacos metabólicos, el dicloroacetato (DCA) y la metformina (Met) han mostrado unas perspectivas encantadoras debido a sus funciones positivas en la terapia del cáncer.
Como agente dirigido a las mitocondrias, el DCA puede inhibir la actividad de la piruvato deshidrogenasa cinasa (PDK) y, posteriormente, aumentar la actividad de la piruvato deshidrogenasa (PDH), lo que promueve el flujo de carbohidratos a las mitocondrias y, por lo tanto, mejora la oxidación aeróbica de la glucosa. Este efecto revierte la disfunción mitocondrial y reactiva la apoptosis dependiente de mitocondrias en varias células tumorales [6-9]. Simultáneamente, el DCA inhibe la glucólisis y reduce la acumulación de lactato, lo que destruye el microambiente tumoral acidificado (El microambiente acidificado generalmente favorece la supervivencia del tumor) [10]. Aunque el DCA ha mostrado perspectivas prometedoras en la lucha contra el cáncer, se ha informado de que el DCA induce una autofagia protectora en las células de cáncer de colon, lo que a su vez dificulta su capacidad apoptótica [11]. Hasta ahora, todavía no está claro si existe algún otro determinante resistente asociado a la apoptosis cuando el DCA refresca la apoptosis mitocondrial.
La Met es un fármaco tradicional de la terapia de primera línea para la diabetes tipo 2. En los últimos años, cada vez más evidencias indican que Met también puede reducir el riesgo de cáncer en varios estudios epidemiológicos [12]. Met suprime el crecimiento tumoral mediante la inducción de la detención del ciclo, la promoción de la apoptosis y la supresión de la autofagia [13-15]. Además, Met puede sensibilizar algunos fármacos quimioterapéuticos como paclitaxel, erlotinib, etc. [ 16-17]. Y lo que es más llamativo, el efecto antitumoral de la Met está cada vez más relacionado con el metabolismo de la glucosa en el cáncer [18]. A pesar de varias ventajas en los ensayos clínicos, la Met se ve obstaculizada para su aplicación posterior porque podría conducir a la acumulación de lactato [19]. Es de gran interés saber si esta desventaja podría superarse mediante la combinación de otros fármacos metabólicos para que el Met se utilice más ampliamente en quimioterapia.
Dados sus potenciales efectos compensatorios mutuos, intentamos descubrir si el DCA y el Met pueden sinergizar entre sí para potenciar la citotoxicidad en células de cáncer de ovario. En el presente estudio, demostramos que el DCA y la Met podían inducir conjuntamente la apoptosis en células de cáncer de ovario. La Met sensibilizó al DCA atenuando drásticamente la Mcl-1 inducida por el DCA y la autofagia protectora, mientras que el DCA sensibilizó a la Met aliviando notablemente la acumulación excesiva de lactato y el consumo de glucosa inducidos por la Met. Los experimentos in vivo en ratones desnudos también demostraron que el DCA y la Met suprimían sinérgicamente el crecimiento de los tumores de ovario xenoinjertados. Estos resultados sugieren que esta estrategia terapéutica puede ser una opción prometedora para futuras terapias dirigidas contra el cáncer basadas en el metabolismo.
RESULTADOS
El DCAy el Met inducen sinérgicamente la apoptosis en células de cáncer de ovario
Para investigar si existe un efecto sinérgico entre el DCA y el Met en la supresión del crecimiento de células de cáncer de ovario, se cotrataron células SKOV3 y OVCAR3 con DCA y Met o con cada uno de ellos por separado. Como se muestra en la Figura 1A-1C, el cotratamiento con DCA y Met reprimió más eficazmente el crecimiento de las células de cáncer de ovario en comparación con cada uno por separado, y la combinación de 40mM DCA y 10mM Met podría inhibir la viabilidad celular a tan bajo como 50% en comparación con el control. Así que elegimos 40mM DCA y 10mM Met en los experimentos posteriores. De forma similar, el efecto de inhibición sinérgica también se observó en células de cáncer de cuello de útero (HeLa y SiHa), células de cáncer de pulmón no microcítico (A549 y GLC-82) y células de carcinoma hepatocelular humano (HepG2) (Figura S1A-1D), lo que sugiere que el sinergismo entre DCA y Met puede ser universal hasta cierto punto. Además, el DCA y la Met indujeron sinérgicamente la apoptosis en las células de cáncer de ovario, según revelaron los análisis de citometría de flujo de la tinción doble de anexina V-FITC (isotiocianato de fluoresceína) y PI (yoduro de predio) (Figura 1D), la tinción Hoechst de los cuerpos apoptóticos (Figura 1E), el análisis Western blot de la PARP (poli ADP-ribosa polimerasa, un marcador de apoptosis) escindida (Figura 1F) y el ensayo de la actividad de la caspasa3 (Figura 1G).
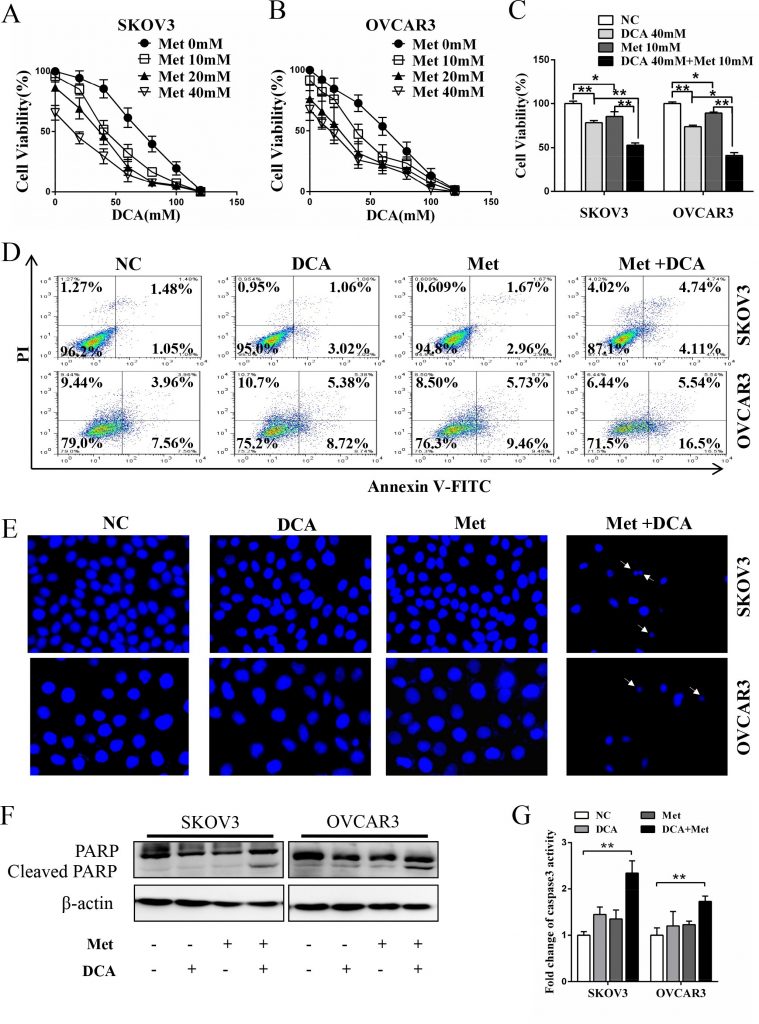
Met sensibiliza al DCA mediante la atenuación de la Mcl-1 inducida por el DCA
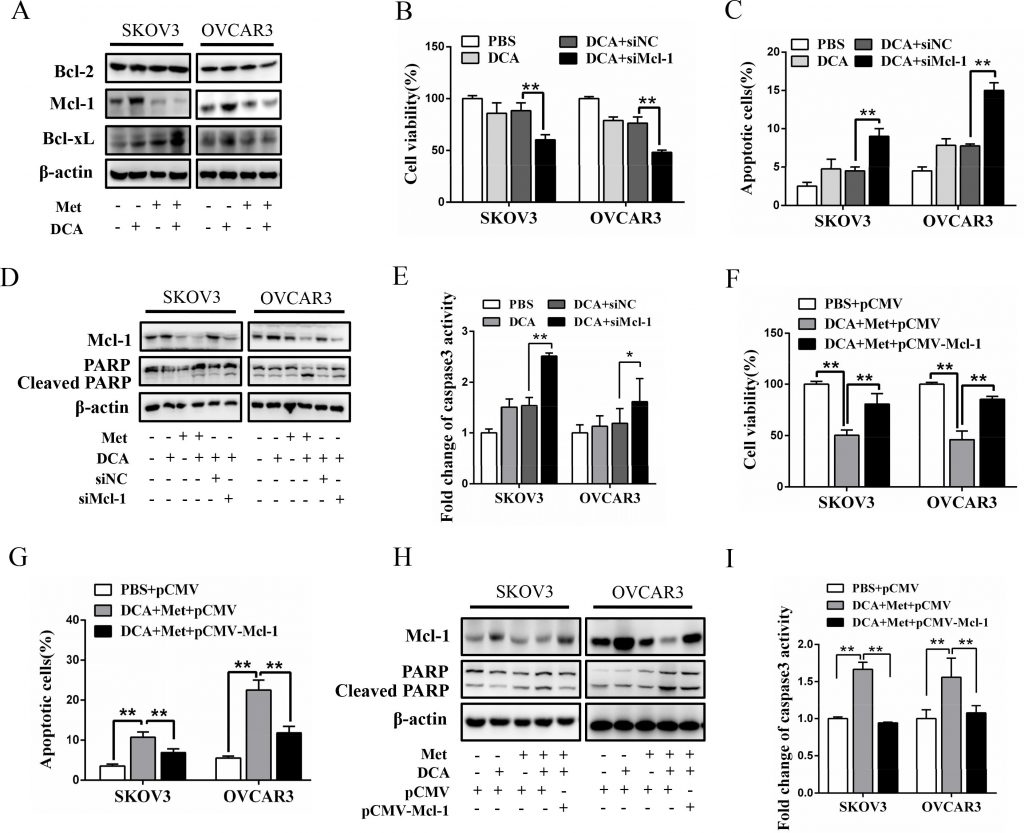
Para explorar el mecanismo por el cual Met sensibiliza al DCA para inducir la apoptosis, examinamos la expresión de los miembros cruciales de la familia antiapoptótica Bcl-2, incluyendo Mcl-1, Bcl-2 y Bcl-xL [20]. Como se muestra en la Figura 2A, el DCA por sí solo aumentó significativamente el nivel de la proteína Mcl-1 (pero no las proteínas Bcl-2 y Bcl-xL) en las células de cáncer de ovario, lo que fue notablemente atenuado por Met. El silenciamiento de Mcl-1 mediante siRNA mejoró la inhibición de la viabilidad celular mediada por DCA (Figura 2B) y aumentó la apoptosis inducida por DCA (Figura 2C-2E). Además, la expresión ectópica de Mcl-1 alivió drásticamente el efecto sensibilizador de Met al DCA sobre la viabilidad celular y la apoptosis (Figura 2F-2I). Estos resultados indican que Mcl-1 es un nuevo factor de resistencia al DCA, y que Met sensibiliza al DCA a través de la regulación a la baja de Mcl-1.
Met atenúa la Mcl-1 inducida por DCA mediante la inhibición de la traducción de Mcl-1
Para dilucidar en qué nivel el DCA induce Mcl-1, se examinó en primer lugar el ARNm de Mcl-1. Como se muestra en la Figura 3A, el DCA no tuvo un efecto obvio sobre la expresión del ARNm de Mcl-1. Posteriormente, se examinó la proteína Mcl-1 en el ARNm. Posteriormente, se analizó la proteína Mcl-1 en presencia o ausencia del inhibidor traslacional cicloheximida (CHX). Como se muestra en las Figuras 3B y 3C, la CHX disminuyó de forma dependiente del tiempo la proteína Mcl-1 basal (pero no la inducida por DCA), lo que indica que el DCA aumenta la estabilidad de la proteína Mcl-1. Se ha reportado que la ERK fosforilada (p-ERK) y p-JNK pueden estabilizar Mcl-1 a través de la fosforilación de Mcl-1 en Thr163 [21-22], por lo que investigamos si p-ERK/p-JNK está involucrada en la regulación de Mcl-1 inducida por DCA. Como se muestra en la Figura 3D, el tratamiento con DCA elevó significativamente p-ERK (pero no p-JNK) y p-Mcl-1Thr163 en células de cáncer de ovario. Además, el inhibidor de MEK1/2 U0126 pudo atenuar drásticamente la Mcl-1 y la p-Mcl-1Thr163 inducidas por el DCA, y reforzó notablemente la PARP escindida (Figura 3E). Estos resultados indicaron que p-ERK (pero no p-JNK) es un mediador de la Mcl-1 inducida por el DCA. Estudios previos han demostrado que el DCA eleva las especies reactivas de oxígeno (ROS) [23], y ROS es un inductor clave de p-ERK [24], por lo que examinamos el nivel de ROS con DCFH-DA. Como se muestra en la Figura S2A, el DCA aumentó la generación de ROS, lo que sugiere que la inducción de ROS puede ser un mecanismo por el cual el DCA aumenta la activación de p-ERK.
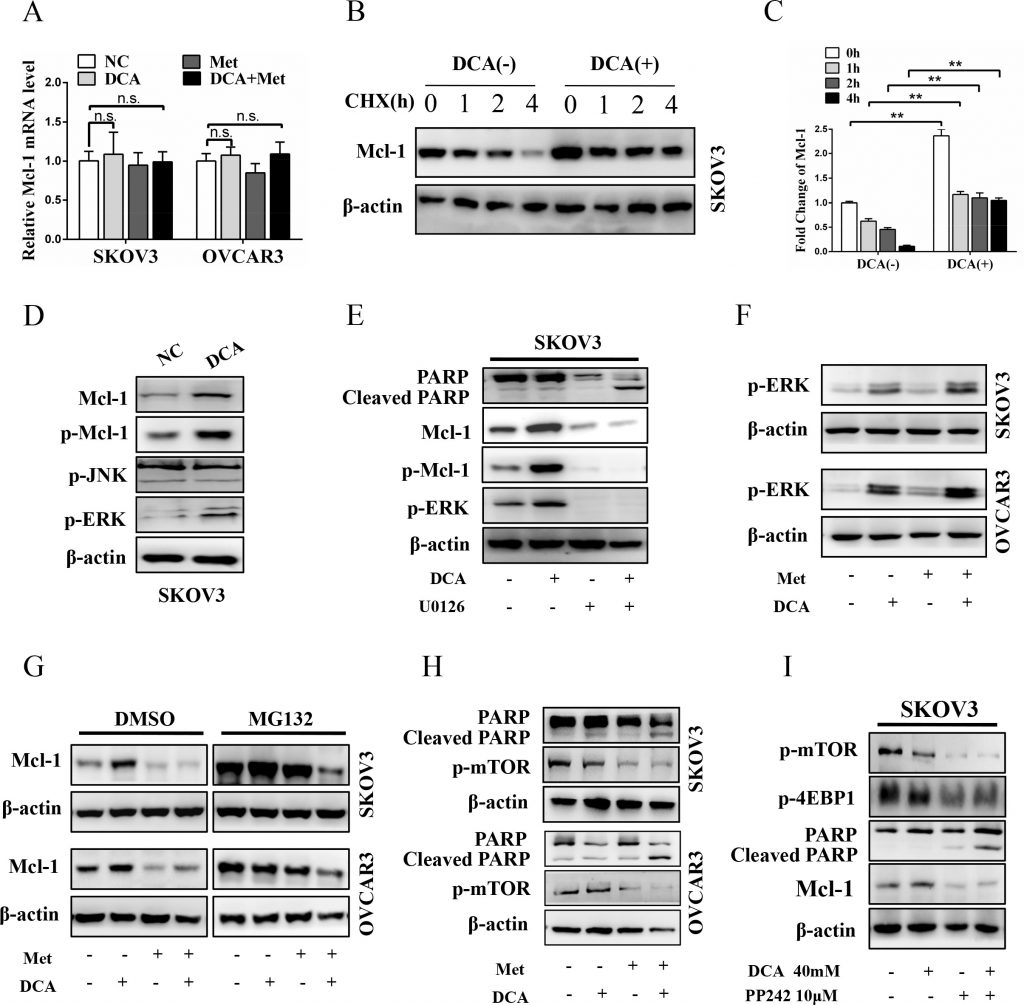
Además, Ser159 también está estrechamente relacionado con la estabilidad de Mcl-1 y este sitio es principalmente fosforilado por GSK-3β [25], por lo que probamos si GSK-3β también está implicada en la regulación de la estabilización de Mcl-1 inducida por DCA. Como se muestra en la Figura S2B, el DCA aumentó la fosforilación de GSK-3β, pero no tuvo efecto sobre GSK-3β total. Además, el DCA aumentó la fosforilación de Akt (una molécula de señal ascendente de GSK-3β), y el inhibidor de Akt MK-2206 2HCl atenuó drásticamente la fosforilación de GSK-3β inducida por el DCA, la regulación al alza de Mcl-1 y la resistencia a la apoptosis (Figura S2C). Estos resultados indicaron que la fosforilación de GSK-3β mediada por p-Akt promueve la estabilización de Mcl-1 inducida por DCA.
Posteriormente, examinamos si Met puede atenuar la Mcl-1 inducida por DCA mediante la inhibición de p-ERK/p-Akt. Como se muestra en la Figura 3F y en la Figura S2D, Met no pudo suprimir la p-ERK y la p-Akt inducidas por DCA, junto con los resultados de la Figura 3A, lo que indica que Met disminuye la Mcl-1 inducida por DCA ni a nivel transcripcional ni a nivel postraduccional. A continuación, analizamos si Met atenúa la Mcl-1 inducida por DCA a nivel transcripcional con el inhibidor del proteasoma MG132. Como se muestra en la Figura 3G, cuando las células fueron tratadas con control, DCA, Met o la combinación, los niveles proteicos de Mcl-1 fueron igualmente elevados en presencia de MG132 en comparación con DMSO, lo que indica que Met atenúa la Mcl-1 inducida por DCA a través de la inhibición de la traducción de Mcl-1. Se ha informado de la activación de mcl-1 a nivel transcripcional con el inhibidor del proteasoma MG132. Se ha descrito que la activación de mTOR promueve la traducción de Mcl-1 [26], por lo que analizamos p-mTOR tras el cotratamiento con Met y DCA. Como era de esperar, Met disminuyó notablemente el nivel de p-mTOR (Figura 3H), y el inhibidor de mTOR PP242 tuvo un efecto similar al de Met en la promoción de la apoptosis (Figura 3I). De acuerdo con los datos de la Figura 3 y la Figura S2, podemos concluir que el DCA regula al alza Mcl-1 a través del aumento de la fosforilación de ERK y GSK-3β, y Met suprime la traducción de Mcl-1 a través de la inhibición de p-mTOR.
Metdisminuye la autofagia protectora inducida por DCA
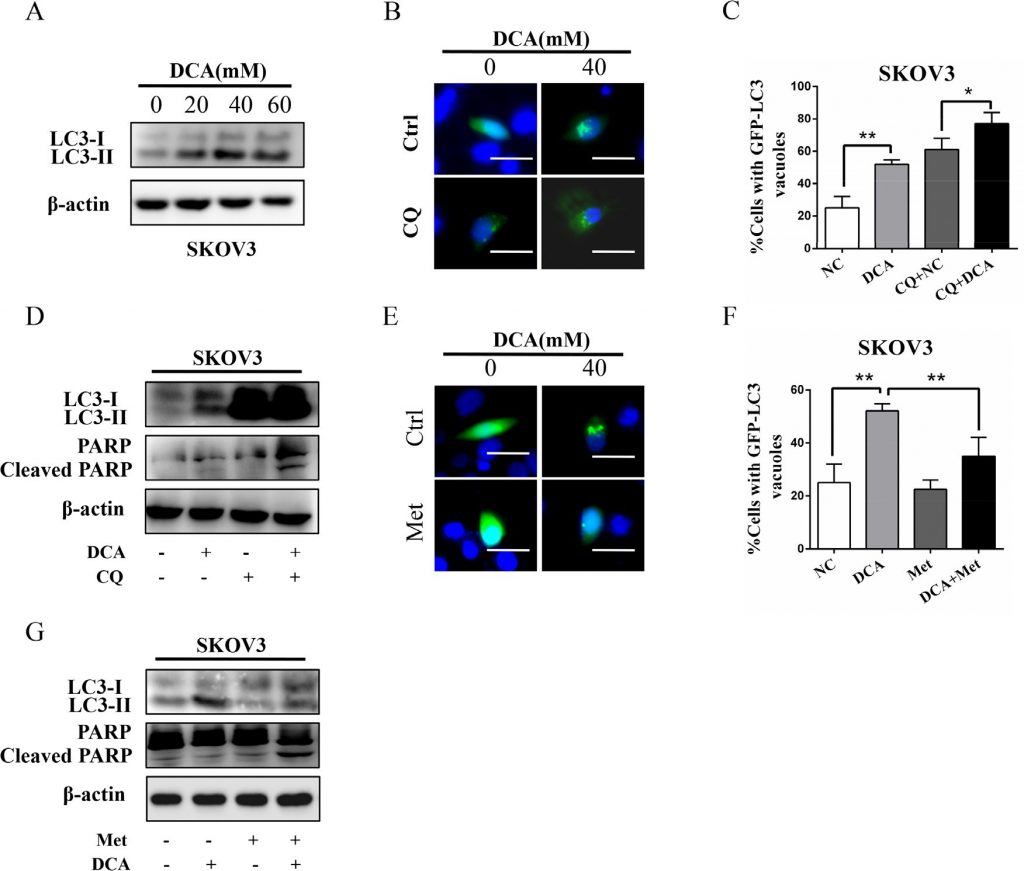
Estudios previos han demostrado que la autofagia juega un papel importante en la resistencia terapéutica del DCA en células de cáncer de colon [11], por lo que examinamos el papel de la autofagia en células de cáncer de ovario tras el tratamiento con DCA y/o Met. Como se muestra en la Figura 4A, el DCA promovió de forma dosis-dependiente el nivel de MAP1LC3-II (LC3-II), el marcador de autofagia. La inhibición de la autofagia mediante cloroquina (CQ) o el silenciamiento de ATG7 mejoraron drásticamente la apoptosis y la citotoxicidad inducidas por el DCA (Figura 4B-4D, Figura S3A-3D), lo que indica que el DCA induce una autofagia protectora en las células de cáncer de ovario. Para investigar preliminarmente el mecanismo de la autofagia inducida por el DCA, analizamos los cambios del ARNm de 7 genes relacionados con la autofagia en las células tratadas con DCA. Como se muestra en la Figura S3E, el DCA aumentó drásticamente el nivel de ARNm de ATG7 en las células de cáncer de ovario, lo que sugiere que ATG7 puede estar implicado en la autofagia protectora inducida por el DCA. Posteriormente, observamos que Met disminuía notablemente la LC3-II inducida por DCA (Figura 4E-4G), lo que indica que Met podría atenuar la autofagia protectora inducida por DCA. En resumen, podría deducirse que el debilitamiento de la autofagia protectora inducida por el DCA también es importante en el efecto sensibilizador de Met frente al DCA.
ElDCA alivia el consumo de glucosa y la producción de lactato inducidos porMet
Para aclarar si el DCA puede superar la escasez de Met, se analizaron los cambios de lactato y glucosa. Como se muestra en la Figura 5A-5C, la Met aumentó la producción de lactato y el consumo de glucosa, que se vieron notablemente aliviados por el DCA. Además, se midieron la tasa de consumo de oxígeno celular (OCR) y la tasa de acidificación extracelular (ECAR). Como se muestra en la Figura 5D-5F, Met disminuyó la relación de OCR/ECAR, que fue dramáticamente atenuada por el DCA, revelando que el DCA puede suprimir la glucólisis inducida por Met a través de la recuperación de la respiración mitocondrial. Como el DCA es un inhibidor de PDKs que fosforila e inhibe la actividad de PDH [9], examinamos el nivel de p-PDH. Como se muestra en la Figura 5G, Met elevó el nivel de p-PDHE1α (una subunidad de PDH), que fue marcadamente revertido por DCA. El silenciamiento de la PDH con siRNA atenuó significativamente la producción de lactato inducida por Met (Figura 5H, 5I). La expresión ectópica de PDK1 y PDK2 aumentó la fosforilación de PDHE1α y atenuó la apoptosis inducida por el tratamiento conjunto con DCA y Met (figuras 5J y 5K), lo que, junto con los datos de las figuras 5G-5I, indica que el DCA puede sensibilizar a Met mediante la inhibición de la vía PDK/PDH en la destrucción de células de cáncer de ovario.
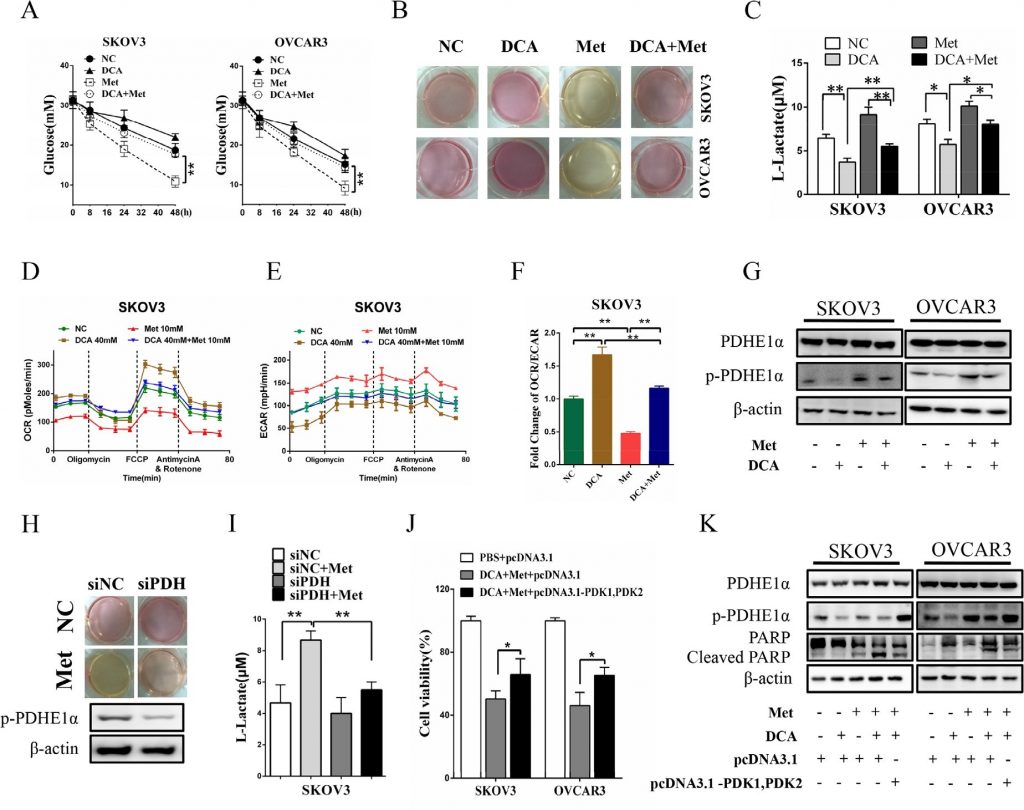
El DCAy la Met reprimen en colaboración el crecimiento de célulasde cáncer de ovario in vivo
Como se muestra en las figuras 6A y 6B, el tratamiento conjunto con DCA y Met suprimió de forma más eficaz el crecimiento de xenoinjertos de cáncer de ovario en ratones desnudos en comparación con el tratamiento con DCA o Met por separado. El análisis de Western blot mostró que el DCA y la Met aumentaban sinérgicamente la PARP escindida y reducían la Mcl-1 y la p-PDHE1α en los xenoinjertos (figuras 6C-6D). Estos resultados sugieren que el DCA y la Met pueden inhibir de forma colaborativa el crecimiento de células de cáncer de ovario in vivo mediante la atenuación de las carencias de cada uno.
DISCUSIÓN
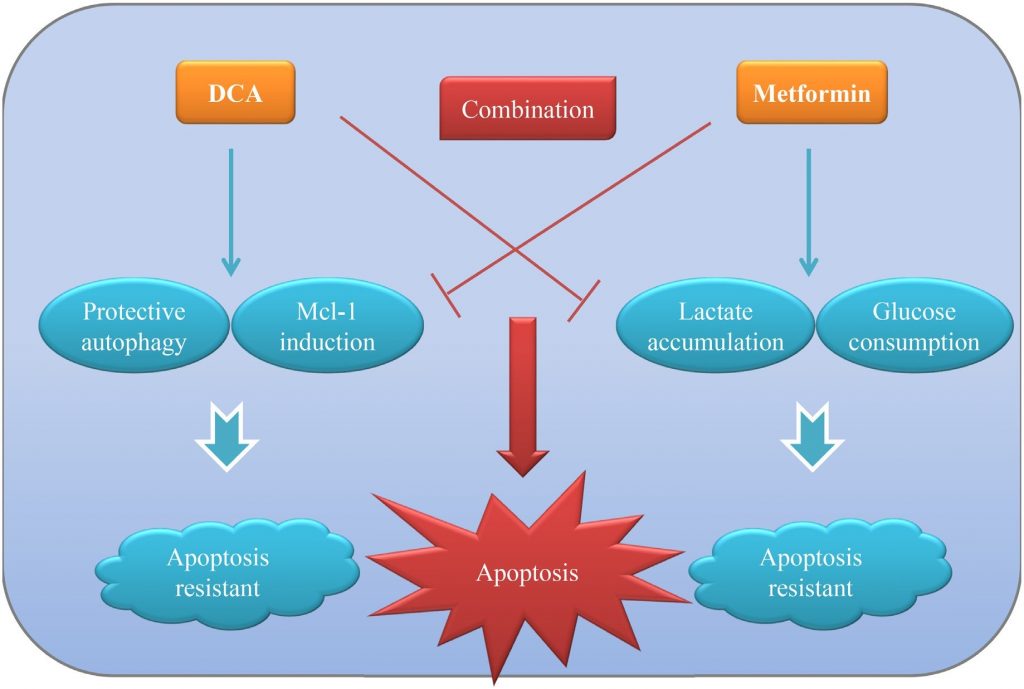
Se ha confirmado que la mayoría de los tumores sólidos se caracterizan por el «efecto Warburg», según el cual utilizan la glucólisis para la producción de energía aunque el oxígeno sea suficiente. Dirigirse a este fenómeno anómalo ha allanado el camino para el desarrollo de nuevas estrategias terapéuticas contra el cáncer, además de los fármacos citotóxicos tradicionales. En el presente estudio, demostramos que el tratamiento conjunto con DCA y Met (dos agentes metabólicamente asociados) puede reprimir más eficazmente el crecimiento de las células de cáncer de ovario en comparación con cada uno por separado in vitro e in vivo. Met atenúa la Mcl-1 inducida por DCA y la autofagia protectora, mientras que DCA alivia la acumulación excesiva de lactato y el consumo de glucosa inducidos por Met. Los beneficios recíprocos de los dos agentes contribuyen a una intensa apoptosis para eliminar las células de cáncer de ovario con mayor eficacia. El modelo de funcionamiento de la combinación de DCA y Met se muestra en la figura 7.
Aunque se vienen realizando estudios desde hace años, aún no están claros los factores clave que pueden impedir el efecto proapoptótico del DCA. Nuestros resultados mostraron que Mcl-1 es un factor resistente crucial contra la apoptosis inducida por DCA en células de cáncer de ovario, y el cotratamiento con Met y DCA disminuyó Mcl-1 y aumentó la apoptosis. Sin embargo, el cotratamiento provocó un aumento de Bcl-xL (Figura 2A), que puede ser un mecanismo compensatorio para mantener la supervivencia celular. En un estudio anterior [27] también se había descrito un fenómeno similar (el estudio muestra que el inhibidor de Bcl-2/xL ABT-263 induce la apoptosis de las células cancerosas al tiempo que aumenta Mcl-1). Además, el aumento de Bcl-xL inducido por el cotratamiento sólo se produjo en la línea celular SKOV3 (pero no en la OVCAR3), lo que sugiere que este efecto puede ser específico de la célula. Por supuesto, queda por determinar el mecanismo detallado por el que el cotratamiento con Met y DCA aumenta la Bcl-xL. Mediante estudios adicionales, revelamos que el DCA inducía la acumulación de Mcl-1 a través de la activación de ERK y Akt, que protegían a Mcl-1 de la degradación mediada por el proteasoma. Estos resultados sugieren que la inhibición de ERK y Akt puede ser una buena estrategia para sensibilizar al DCA en el tratamiento del cáncer de ovario. Sin embargo, recientemente se ha informado de un resultado contradictorio según el cual el DCA disminuye el nivel de Mcl-1 en células de LMA [28] y de cáncer colorrectal [29]. Las discrepancias sugieren que la relación entre el DCA y Mcl-1 puede ser muy diferente en distintos contextos.
La autofagia es un proceso catabólico para reciclar metabolitos esenciales como aminoácidos y lípidos para reponer su reserva bioenergética en presencia de privación de nutrientes u otros estreses dramáticos [30-31]. En este estudio, revelamos que el DCA induce una autofagia protectora en las células de cáncer de ovario, y que ATG7 puede desempeñar un papel en este proceso (Figura S3E), pero el mecanismo detallado necesita ser estudiado más a fondo. Además, descubrimos que Met sensibilizaba al DCA mediante la supresión de la autofagia protectora inducida por el DCA. En consonancia con nuestros hallazgos, Met puede reprimir la autofagia dependiente de GRP78 para potenciar el efecto antimieloma de bortezomib [15], e inhibir la autofagia inducida por 2DG para sensibilizar a 2DG en células de cáncer de próstata [32]. Sin embargo, queda por investigar en detalle el mecanismo o mecanismos por los que Met suprime la autofagia protectora.
Met ha sido bien reconocido como agente único o sensibilizador en la terapia del cáncer, pero una desventaja notable es que Met promueve el consumo de glucosa y acelera la acumulación de lactato, lo que facilita la glucólisis aeróbica adicta al cáncer. En el presente estudio, demostramos que el DCA podía atenuar drásticamente este efecto secundario. El DCA puede inhibir fuertemente la actividad de las PDKs y su corriente descendente p-PDHE1α, conduciendo a una remodelación metabólica reutilizando la fosforilación oxidativa y causando una menor acumulación de lactato y consumo de glucosa. Entre los cuatro tipos de isoenzimas PDK, DCA funciona principalmente a través de la inhibición de PDK2 y PDK1 [33]. Como era de esperar, la sobreexpresión simultánea de PDK2 y PDK1 aumentó la fosforilación de PDHE1α, abolió la función sensibilizadora del DCA y anuló parcialmente el efecto letal del DCA más Met en células de cáncer de ovario. En conjunto, nuestros resultados indican que el DCA sensibiliza la función antitumoral de Met a través de la inhibición de la actividad de las PDKs. Sin embargo, debe tenerse en cuenta que el DCA puede sensibilizar a Met potenciando el estrés oxidativo inducido por Met en células de cáncer de mama [34]. Esto significa que el mecanismo sinérgico del DCA a la Met es tan complicado, que necesita estudios profundos.
En resumen, demostramos que DCA y Met pueden suprimir sinérgicamente el crecimiento de células de cáncer de ovario, lo que puede allanar el camino para desarrollar nuevas estrategias para el tratamiento del cáncer de ovario basadas en el uso combinado de DCA y Met.
MATERIALES Y MÉTODOS
Líneas celulares y reactivos
Las líneas celulares SKOV3, OVCAR3, HeLa, SiHa, GLC-82, A549 y HepG2 se adquirieron a la American Type Culture Collection (ATCC) y se cultivaron en Dulbecco’s Modified Eagle Medium (DMEM), suplementado con un 10% de suero bovino fetal (FBS), estreptomicina (100 mg/mL) y penicilina (100 U/mL) a 37°C en un incubador húmedocon un 5% deCO2. El DCA y la cicloheximida (CHX) se adquirieron a Sigma-Aldrich (Louis, MO, EE.UU.). Met, U0126, MG132 y Hoechst 33258 se adquirieron a Beyotime Company (Shanghai, China). MK2206 se adquirió a Selleck Company (Shanghai, China). El kit de ensayo de la actividad de la caspasa 3 y el kit de ensayo de especies reactivas de oxígeno se adquirieron a Beyotime Company (Shanghai, China). Annexin V-FITC y PI se adquirieron a BD Bioscience (BD, NJ, EE.UU.). Los siRNA para Mcl-1, ATG7, PDH y el siRNA de control se adquirieron a GenePharma (Shanghai, China). Los plásmidos de expresión pCMV y pcDNA3.1, pCMV-Mcl-1, pcDNA3.1-PDK1 y pcDNA3.1-PDK2 se adquirieron a Obio Technology (Shanghai, China).
Western blot
Se prepararon lisados de células enteras y se realizó Western blot como se ha descrito previamente [35]. Los anticuerpos para β-actina y PDHE1α eran de la empresa Abcam (San Francisco, CA, EE.UU.), los anticuerpos para Bcl-2 y Mcl-1 eran de Santa Cruz Biotechnology (Santa Cruz, CA, EE.UU.), y los anticuerpos para PARP BCL-xL, GSK-3β, p-ERK (Thr202/Tyr204), p-JNK (Thr183/Tyr185), p-Mcl-1 (Thr163), p-Akt (Ser473), p-4EBP1, p-mTOR y p-GSK-3β (Ser9) eran de Cell Signaling Technology (Boston, MA, EE.UU.). El anticuerpo contra p-PDHE1α (Ser293) era de EMD Millipore (Billerica, MA, EE.UU.).
Transfección celular
Las células SKOV3 y OVCAR3 se cultivaron hasta una confluencia del 60% al 70% en placas de 6 pocillos. El ARNsi o el plásmido de expresión se mezclaron con 10 μL de lipofectamina 2000 en Opti-MEM (Invitrogen, Carlsbad, CA, EE.UU.) para cada pocillo según el protocolo del fabricante. Tras incubarlas con las mezclas durante 6 h, las células se cultivaron en DMEM con FBS al 10% durante otras 6 h. A continuación, se administró a las células el tratamiento correspondiente.
Ensayo de viabilidad celular
El ensayo de viabilidad celular se realizó utilizando el kit CCK-8 (Dojindo, Shanghai, China) tal y como se ha descrito previamente [27]. Brevemente, se sembraron células en placas de 96 pocillos (2×103 células/pocillo) por triplicado y se incubaron durante 12 h. A continuación, se administraron a las células diferentes tratamientos o el vehículo de control durante 48 h, seguido de la adición de 10 μl de solución de CCK-8 a cada pocillo. Tras incubar a 37 °C durante 1,5 h, se determinó el valor de OD450nm con un lector de microplacas.
Citometría de flujo
Las células de cáncer deovariose incubaron con anexina V-FITC y PI siguiendo las instrucciones del fabricante (BD, 561012). A continuación, se analizó la apoptosis mediante un citómetro de flujo.
Tinción Hoechst
Tras 24 h de tratamiento, las células se tiñeron con Hoechst 33258 (Beyotime, Shanghai, China) a 10μg/mL durante 10 min en la oscuridad. Posteriormente, las células se lavaron 3 veces con PBS y se fotografiaron al microscopio de afluorescencia.
Ensayo de la actividad de la caspasa 3
La actividad de la caspasa 3 se examinó con el kit de ensayo de la actividad de la caspasa 3 (Beyotime, C1115) como se describió anteriormente [36]. Brevemente, se cosecharon las células de control y las tratadas, se lavaron con PBS helado y se resuspendieron en 50 μl de tampón de lisis celular refrigerado durante 15 min en hielo. A continuación, se centrifugaron los lisados (20.000 g, 10 min, 4 °C) y se recogieron inmediatamente los sobrenadantes para el ensayo de la actividad de la caspasa3.
Medición de la ROS intracelular total
La ROS intracelular total se analizó con ROS Assay Kit (Beyotime, Shanghai, China) como se describió previamente [37].
Aislamiento de ARN, PCR cuantitativa en tiempo real (qPCR)
El ARN total se extrajo de las células con el reactivo TRIzol (ComWin Biotechnology, Pekín, China) y el ADNc de primera cadena se sintetizó utilizando la transcriptasa M-MLV (Invitrogen, Carlsbad, CA, EE.UU.). La qPCR se realizó con QuantiFast SYBR Green PCR Kit (Promega, Shanghai, China). Los niveles relativos de ARNm de los genes diana se calcularon con el método2-ΔΔCt.
Análisis de la proteína verde fluorescente (GFP)-MAP1LC3
Tras transfectar con el vector de expresión GFP-MAP1LC3 (GFP-LC3) durante 12 h, las células recibieron los tratamientos indicados durante otras 24 h y, a continuación, se fijaron con formaldehído al 4% durante 10 min. Posteriormente, las células se lavaron 3 veces con PBS y se observaron al microscopio de fluorescencia.
Detección de L-lactato y glucosa
Las células se trataron en pocillos de 6 y, a continuación, se recogió el medio y se determinaron las concentraciones de L-lactato y glucosa por separado utilizando el L-Lactate Assay Kit (Eton Bioscience, San Diego, CA, EE.UU.) y el Glucose Colorimetric/Fluorometic Assay Kit (BioVision, Milpitas, CA, EE.UU.).
Análisis de bioenergética celular
Las células se sembraron en placas XF96 y se dejaron crecer durante toda la noche. A continuación, el medio se sustituyó por medio XF96 1 h antes del ensayo. La rotenona/antimicina A, la FCCP y la oligomicina se diluyeron en medio XF96 y se cargaron en el cartucho adjunto para alcanzar concentraciones finales de 0,5μM, 0,5μM y 1,0μM, respectivamente. Las inyecciones de los fármacos en el medio se produjeron en los puntos temporales especificados. El OCR (pmol/min) y el ECAR (mpH/min) se monitorizaron con el XF Cell Mito Stress Test Kit (Seahorse Bioscience, North Billerica, MA, EE.UU.) utilizando los analizadores de flujo extracelular XFe y XF de Seahorse Bioscience.
Estudio con animales
Se compraron ratones desnudos hembra de seis semanas de edad a Beijing Huafukang Bioscience (Pekín, China), y se alojaron y cuidaron según las normas del Comité de Ética y Cuidado de los Animales de la Tercera Universidad Médica Militar (Chongqing, China). se implantaron 5×106 células SKOV3 en 150 μL de PBS en la axila derecha de cada ratón desnudo. Cuando se formaron tumores palpables, los ratones se distribuyeron aleatoriamente en 4 grupos (n = 6 por grupo). A continuación, los ratones fueron inyectados por vía intraperitoneal todos los días con DCA (50 mg/kg/d) más Met (100 mg/kg/d) o cada uno por separado durante 8 días, tomando PBS como control. El tamaño del tumor xenoinjertado se controló cada día con un calibrador deslizante, y el volumen se estimó mediante la siguiente fórmula: volumen = anchura2×longitud×1/2. Una vez extirpados de los ratones, se fotografiaron los tumores xenoinjertados y se examinaron las proteínas correspondientes mediante Western blot.
Análisis estadístico
Los datos se expresaron como media ± DE. Se utilizaron ANOVA unidireccional y la prueba t para analizar la varianza. P< 0,05 se consideró estadísticamente significativo.
AGRADECIMIENTOS
Este trabajo fue financiado por la Fundación Nacional de Ciencias Naturales de China (81472436 y 81272865) y la Fundación de Ciencias Naturales de Chongqing (cstc2012jjB10025).
CONFLICTOS DE INTERESES
No se declararon posibles conflictos de intereses.
REFERENCIAS
1 Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014; 384:1376-1388.2 Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Caracterización del genoma completo del cáncer de ovario quimiorresistente. Nature. 2015; 521:489-494.
3 Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297-308.
4 Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism; HNF-1beta confers survival benefits through the Warburg effect and ROS reduction. Oncotarget. 2015; 6:30704-30714. doi: 10.18632/oncotarget.5228.
5 Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumor Biol. 2015.
6 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Modulación metabólica del glioblastoma con dicloroacetato. Sci Transl Med. 2010; 2:31ra34.
7 Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008; 99:989-994.
8 Papandreou I, Goliasova T, Denko NC. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer. 2011; 128:1001-1008.
9 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37-51.
10 Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013; 133:1107-1118.
11 Gong F, Peng X, Sang Y, Qiu M, Luo C, He Z, Zhao X, Tong A. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013; 4:e913.
12 Morales DR, Morris AD. Metformina en el tratamiento y prevención del cáncer. Annu Rev Med. 2015; 66:17-29.
13 Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27:3576-3586.
14 Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009; 8:2031-2040.
15 Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia. 2015; 29:2184-2191.
16 Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. La metformina amplifica la activación AMPK inducida por la quimioterapia y el crecimiento antitumoral. Clin Cancer Res. 2011; 17:3993-4005.
17 Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE, Cremers S, Szabolcs M, Maurer MA. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget. 2014; 5:10503-10517. doi: 10.18632/oncotarget.2391.
18 Escocia S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, Dumon E, Martineau Y, Danet-Desnoyers G, et al. mitocondrial energética y AKT estado mediar efectos metabólicos y la apoptosis de la metformina en células leucémicas humanas. Leukemia. 2013; 27:2129-2138.
19 Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget. 2015; 6:37281-37299. doi: 10.18632/oncotarget.6134.
20 Lessene G, Czabotar PE, Colman PM. Antagonistas de la familia BCL-2 para la terapia del cáncer. Nat Rev Drug Discov. 2008; 7:989-1000.
21 Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004; 23:5301-5315.
22 Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology. 2009; 136:1423-1434.
23 Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016; 371:20-29.
24 Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM, Zhao CQ, Liu PJ, Zhang JM, He JJ. MAPK signaling mediates sinomenine hydrochloride-induced human breast cancer cell death via both reactive oxygen species-dependent and -independent pathways: an in vitro and in vivo study. Cell Death Dis. 2014; 5:e1356.
25 Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. La glucógeno sintasa quinasa-3 regula la permeabilización de la membrana externa mitocondrial y la apoptosis mediante la desestabilización de MCL-1. Mol Cell. 2006; 21:749-760.
26 Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, et al. mTORC1 promueve la supervivencia a través del control traslacional de Mcl-1. Proc Natl Acad Sci U S A. 2008; 105:10853-10858.
27 Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer. 2014; 13:98.
28 Emadi A, Sadowska M, Carter-Cooper B, Bhatnagar V, van der Merwe I, Levis MJ, Sausville EA, Lapidus RG. Perturbación del estado oxidativo celular inducida por dicloroacetato y trióxido de arsénico para el tratamiento de la leucemia mieloide aguda. Leuk Res. 2015; 39:719-729.
29 Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD, Coomber BL. El dicloroacetato afecta a la proliferación pero no a la supervivencia de las células de cáncer colorrectal humano. Apoptosis. 2015; 20:63-74.
30 Mah LY, Ryan KM. Autofagia y cáncer. Cold Spring Harb Perspect Biol. 2012; 4:a008821.
31 Yang ZJ, Chee CE, Huang S, Sinicrope FA. El papel de la autofagia en el cáncer: implicaciones terapéuticas. Mol Cancer Ther. 2011; 10:1533-1541.
32 Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010; 70:2465-2475.
33 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998; 329:191-196.
34 Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Cáncer de mama Res Treat. 2014; 147:539-550.
35 Lian J, Ni Z, Dai X, Su C, Smith AR, Xu L, He F. Sorafenib sensitizes (-)-gossypol-induced growth suppression in androgen-independent prostate cancer cells via Mcl-1 inhibition and Bak activation. Mol Cancer Ther. 2012; 11:416-426.
36 Ou X, Lu Y, Liao L, Li D, Liu L, Liu H, Xu H. El cloruro de nitina induce la apoptosis en células de carcinoma hepatocelular humano a través de una vía que implica p53, p21, Bax y Bcl-2. Oncol Rep. 2015; 33:1264-1274.
37 Ni Z, Gong Y, Dai X, Ding W, Wang B, Gong H, Qin L, Cheng P, Li S, Lian J, He F. AU4S: a novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy. 2015; 11:403-415.
Contenido relacionado: