Margaret O James*,1 & Peter W Stacpoole2,3
1 Département de chimie médicinale, Université de Floride, Gainesville, FL 32610-0485, USA
2 Département de médecine, Collège de médecine, Université de Floride, Gainesville, FL 32610-0485, USA
3 Département de biochimie et de biologie moléculaire, Université de Floride, Gainesville, FL 32610-0485, USA
Correspondance : Tél : +1 352 273 7707 Courriel : [email protected]
Soumis: 16 décembre 2015
Accepté : 17 février 2016
Publié : 4 mai 2016
Résumé
Le médicament expérimental dichloroacétate (DCA) est un régulateur métabolique qui a été utilisé avec succès pour traiter les maladies métaboliques acquises et congénitales et, récemment, les tumeurs solides. Son utilisation clinique a révélé des difficultés dans le choix des doses appropriées. L’administration chronique de DCA entraîne une inhibition du métabolisme du DCA et une accumulation potentielle à des niveaux qui entraînent des effets secondaires. Cela est dû au fait que la conversion du DCA en glyoxylate est catalysée par une enzyme, la glutathion transférase zeta 1 (GSTZ1-1), qui est inactivée par le DCA. Les SNP dans le gène GSTZ1 entraînent l’expression de variantes polymorphes de l’enzyme qui diffèrent en termes d’activité et de taux d’inactivation par le DCA dans des conditions physiologiques : ces propriétés entraînent une variation considérable entre les personnes dans la pharmacocinétique du DCA.
Mots clés : dichloroacétate ; GSTZ1 ; pharmacogénétique
Utilisation clinique du dichloroacétate
Pour une molécule aussi simple, le DCA possède un portefeuille pharmacologique remarquablement riche et diversifié, datant de près d’un siècle [1,2]. Cependant, son utilisation moderne en tant que médicament expérimental a débuté en 1970, lorsque sa capacité sélective à réduire la glycémie chez les animaux diabétiques, mais pas chez les animaux non diabétiques, a été découverte [3], puis confirmée chez l’homme [4]. De nombreuses propriétés métaboliques du DCA impliquant le métabolisme du glucose et des lipides ont été identifiées au cours des années 1970 et 1980. Cependant, la plupart de ses effets pharmacologiques peuvent se résumer à quelques sites et mécanismes d’action fondamentaux. Premièrement, le DCA est un inhibiteur non compétitif de l’enzyme HMG CoA réductase du réticulum endoplasmique, qui catalyse l’étape limitant la vitesse de la biosynthèse du cholestérol. L’effet inhibiteur du DCA est observé à la fois dans le foie des rongeurs [5] et dans les leucocytes humains [6]; il explique probablement la réduction du cholestérol total et du cholestérol des lipoprotéines de basse densité (LDL) associée au médicament chez les patients atteints d’hypercholestérolémie familiale homozygote négative pour les récepteurs LDL [7] et sa désignation comme premier produit orphelin pour cette maladie rare. Deuxièmement, le DCA inhibe la synthèse de novo des triglycérides hépatiques chez les rongeurs non diabétiques [5] et diminue les taux de triglycérides circulants et de lipoprotéines de très basse densité chez les patients atteints de diabète sucré de type 2 [4]. Il diminue également les corps cétoniques sanguins chez les rats atteints d’acidocétose diabétique induite expérimentalement [8,9]. Les mécanismes précis qui sous-tendent ces effets sur la synthèse et l’oxydation des lipides sont inconnus. Troisièmement, le DCA stimule la PDC mitochondriale, qui oxyde de manière irréversible le pyruvate en acétyl coenzyme A (acétyl CoA) [10], une propriété partagée par certains autres acides gras halogénés à chaîne courte [11]. C’est la capacité du DCA à modifier l’activité de la PDC qui a suscité de loin la plus grande recherche expérimentale et clinique sur cette molécule inhabituelle.
La PDC est régulée de manière post-traductionnelle principalement par la phosphorylation réversible d’un ou plusieurs des trois résidus de sérine sur la sous-unité E1α de la première enzyme PDH de la PDC qui décarboxyle le pyruvate [12]. Quatre PDH kinases (PDK1-4) et deux PDH phosphatases (PDP 1 et 2) réalisent cet aspect régulateur de la PDC chez l’homme, dans lequel l’enzyme phosphorylée est catalytiquement inactive. Les PDK sont exprimées de manière différentielle dans les tissus, bien que la PDK2 soit exprimée de manière ubiquitaire [13]. Le pyruvate, le substrat de la réaction PDC, inhibe les PDK en se liant à une petite poche dans l’extrémité N-terminale de la kinase, ce qui entraîne une stimulation de l’activité PDC. En revanche, l’accumulation des produits de la réaction, l’acétyl CoA et le NADH, entraîne l’activation des PDK et l’inhibition de la PDC. Le DCA, un analogue structurel du pyruvate, se fixe également au site de liaison du pyruvate, ce qui entraîne une inhibition des PDK, l’ordre d’inhibition étant PDK2>PDK1˜PDK4>>PDK3 [13]. L’exposition chronique à un médicament in vitro [14,15] et in vivo [16] peut stabiliser le PDC et diminuer son renouvellement, ce qui constitue un deuxième mécanisme par lequel le DCA augmente l’activité du complexe et une explication de ses effets pharmacologiques prolongés après le retrait du médicament [4].
Bien que le DCA soit un inhibiteur de la PDK relativement faible (Ki 0,2 mM pour la PDK2), ses effets sur certains aspects du métabolisme intermédiaire sont profonds et sont directement liés au rôle central que joue la PDC dans la régulation de la sélection des combustibles et de la bioénergétique des cellules eucaryotes [17,18]. Le DCA n’est pas encore un médicament expérimental approuvé par la FDA américaine. Cependant, il continue à faire l’objet de recherches approfondies en tant que modulateur métabolique pour plusieurs troubles métaboliques congénitaux et acquis, sur la base de son action stimulante sur la PDC (tableau 1).
| Propriété | Mécanisme | Réf. |
|---|---|---|
| Augmentation de l’OXPHOS et de la bioénergétique | ↓ PDK ↑ PDC | [2,10] |
| Diminution de la glycémie en cas de jeûne ou de diabète | ↓ PDK ↑ PDC | [4] |
| Diminution du lactate sanguin et du LCR | ↓ PDK ↑ PDC | [19] |
| Diminution du cholestérol total et du LDL dans le sang | ↓ HMG CoA réductase → ↓ synthèse du cholestérol | [6,7] |
| Diminution des triglycérides sanguins et du cholestérol VLDL | ↓ synthèse hépatique des TG → ↓ synthèse du VLDL | [4] |
| Inversion de l’effet Warburg dans le cancer, l’HTAP et d’autres conditions prolifératives | ↓ PDK ↑ PDC | [20] |
LCR : liquide céphalorachidien ; HMG CoA : Hydroxyméthylglutaryl coenzyme A ; LDL : lipoprotéine de basse densité ; OXPHOS : Phosphorylation oxydative ; HTAP : hypertension artérielle pulmonaire ; PDC : complexe pyruvate déshydrogénase ; PDK : pyruvate déshydrogénase kinase ; TG : Triglycérides ; VLDL : lipoprotéines de très basse densité.
Pharmacocinétique et clairance
Le DCA oral est rapidement absorbé et a une biodisponibilité proche de l’unité après administration orale et parentérale [21,22]. Le médicament peut être détecté dans le plasma de l’homme dans les 15 minutes qui suivent une dose orale de 50 mg/kg. Chez des sujets préalablement naïfs, le DCA administré par voie orale ou intraveineuse a une demi-vie plasmatique d’environ 1 h [23]. Le taux de métabolisme chez les espèces saines est le suivant : souris>rat>humain≥chien [22-25].
Les premières études pharmacocinétiques du DCA intraveineux en dose unique ont décrit une cinétique non linéaire à des doses ≥35 mg/kg [26]. Plus tard, il a été constaté que le taux de clairance plasmatique du médicament diminuait lors de l’administration intraveineuse répétée de 50 mg/kg de poids corporel chez des adultes gravement malades souffrant d’acidose lactique [27,28]. Un retard de la clairance plasmatique du médicament administré par voie orale à une dose de 50 mg/kg a également été observé chez des adultes en bonne santé, qui ont eu besoin de semaines à plusieurs mois avant d’atteindre le taux de clairance initial avec une deuxième dose [22].
Des progrès importants dans la compréhension de cette caractéristique inhabituelle de la pharmacocinétique du DCA ont été réalisés grâce à des recherches probantes sur sa toxicologie clinique. Le DCA chronique peut provoquer une neuropathie périphérique réversible chez toutes les espèces [21,23], un effet observé pour la première fois en clinique il y a 25 ans chez un garçon de 16 ans qui avait reçu 50 mg/kg/jour pendant environ 4 mois [7]. En revanche, dans un essai contrôlé randomisé (ECR) portant sur 43 jeunes enfants atteints de divers types de maladies mitochondriales primaires (âge moyen au moment de l’admission : 5,6 ans), le DCA administré par voie orale à la dose de 12,5 mg/kg toutes les 12 heures pendant 6 mois a été bien toléré et n’a provoqué aucun changement indésirable dans la conduction électrique des nerfs périphériques, par rapport au placebo [29], bien qu’un certain déclin asymptomatique de la conduction nerveuse ait été constaté avec une exposition plus longue au médicament [30]. En revanche, un ECR mené auprès de 30 adolescents et adultes plus âgés (âge moyen d’admission : 30 ans) atteints d’une maladie mitochondriale génétique et exposés à la même dose de DCA et au même schéma posologique que ceux utilisés dans l’essai pédiatrique a été interrompu prématurément en raison de l’aggravation ou de l’apparition d’une neuropathie périphérique [31], une complication fréquente des maladies mitochondriales [32,33]. Lorsqu’on a comparé la cinétique du DCA dans ces deux populations de patients, on a découvert que la clairance plasmatique du DCA chronique était nettement plus lente dans le groupe le plus âgé [34], ce qui a été confirmé par des études sur des rats [34,35]. Ces résultats ont incité à étudier l’enzymologie de la biotransformation du DCA.
Importance de la glutathion transférase zeta 1 dans la biotransformation du DCA
Les petits acides carboxyliques sont souvent glucuronidés [36] ou convertis en leurs dérivés de coenzyme A puis en conjugués d’acides aminés [37], mais rien ne prouve que le DCA forme un glucuronide, un dérivé de coenzyme A ou un conjugué d’acide aminé [2, James MO, données non publiées]. Le DCA est plutôt un substrat pour une enzyme GST inhabituelle, la GSTZ1-1, qui le convertit en glyoxylate dans une réaction qui nécessite mais ne consomme pas de glutathion (GSH) [38,39]. Comme les autres GST, la GSTZ1-1 est active sous la forme dimérique, et l’enzyme active est un homodimère. La déchloration du DCA pour former du glyoxylate, catalysée par la GSTZ1-1, est la seule voie du métabolisme primaire du DCA observée dans le foie [35]. En plus de catalyser le métabolisme du DCA, la GSTZ1-1, également connue sous le nom de MAAI, a une fonction physiologique importante dans le catabolisme de la tyrosine, où elle isomérise le maléylacétoacétate (MAA) et la maléylacétone (MA) en fumarylacétoacétate et fumarylacétone dans des réactions qui nécessitent mais ne consomment pas de GSH (Figure 1) [40,41]. La biotransformation du DCA en glyoxylate met fin à ses activités pharmacologiques et thérapeutiques importantes qui sont discutées ci-dessus, ainsi l’activité de GSTZ1-1 contrôle la durée d’action du DCA.
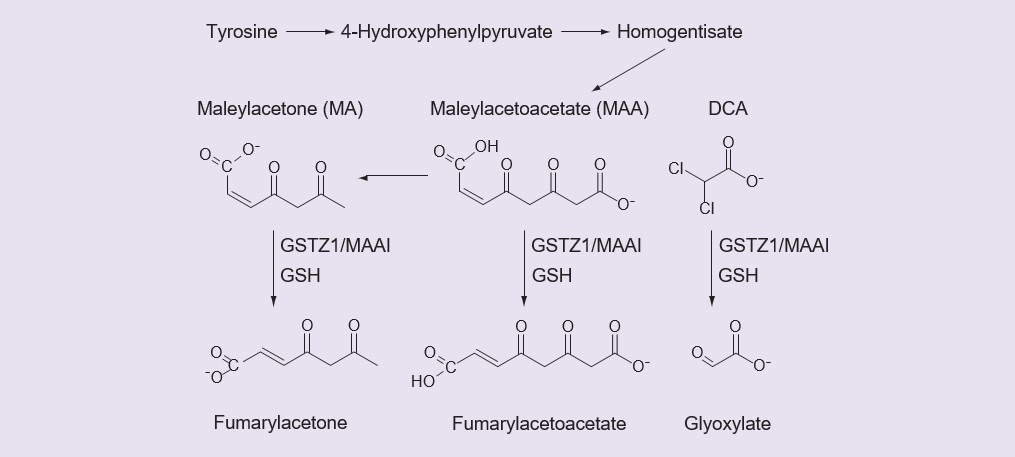
Le métabolite inactif du DCA, le glyoxylate, est ensuite métabolisé en dioxyde de carbone, glycine et oxalate. La fumarylacétone et le fumarylacétoacétate sont ensuite métabolisés par la fumarylacétoacétate hydrolase. DCA : Dichloroacétate : GSH : Glutathion ; MA : Maléylacétone ; MMA : Maléacétoacétate.
Le foie est le principal site d’expression de GSTZ1-1, où on le trouve à la fois dans le cytosol et dans la matrice mitochondriale [38,42]. Des études animales ont démontré des niveaux très faibles d’expression de GSTZ1-1 dans les reins, le cœur, le tube digestif et le cerveau [43]. Le rôle des tissus extra-hépatiques dans le métabolisme du DCA chez l’homme n’a pas été exploré en détail, mais une étude a montré que le DCA n’était pas éliminé du sang pendant la phase anhépatique de patients ayant subi une transplantation hépatique et à qui l’on avait administré du DCA pour contrer l’acidose lactique, ce qui indique qu’aucun métabolisme n’a eu lieu [44]. La phase anhépatique étant de courte durée (moyenne : 73 min), les données sont limitées, mais cette étude suggère un rôle mineur des tissus autres que le foie dans l’élimination du DCA, du moins pour une dose unique.
Inhibition de la GSTZ1-1 par le DCA
Des études sur des animaux et sur des protéines humaines recombinantes exprimées de la GSTZ1-1 ont démontré que la raison pour laquelle des doses multiples de DCA sont éliminées beaucoup plus lentement qu’une dose unique est que le DCA est un inhibiteur de la GSTZ1-1 basé sur un mécanisme [25,34,45-47]. Au cours de la biotransformation du DCA, des produits d’addition à la protéine GSTZ1-1 sont formés, ce qui inactive la protéine et déclenche vraisemblablement sa protéolyse [48,49]. L’administration de DCA à des rats entraîne une perte d’activité et d’expression de GSTZ1-1 en fonction de la dose et du temps : la perte de fonction de GSTZ1-1 était plus prononcée et persistait plus longtemps chez les rats adultes que chez les jeunes [34,35]. Cet effet de l’âge sur la perte de la fonction GSTZ1-1 après l’administration de DCA a été observé chez l’homme, les enfants présentant une augmentation plus faible de la demi-vie plasmatique et une diminution plus faible de la clairance que les adultes [34]. En plus d’affecter la pharmacocinétique du DCA, la perte de la fonction GSTZ1-1 (MAAI) entraîne une accumulation des substrats physiologiques MA et, vraisemblablement, MAA, qui sont tous deux des molécules réactives capables de former des adduits avec les macromolécules cellulaires [34,45]. L’inhibition de la MAAI entraîne également le détournement du carbone de la tyrosine vers la formation de succinylacétone. Cette molécule inhibe une étape proximale de la synthèse de l’hème, provoquant l’accumulation de la molécule réactive δ-aminolevulinate (δ-ALA). Le MA et le δ-ALA augmentent tous deux dans le sang et/ou l’urine des humains exposés de manière chronique au DCA [34]: la signification toxicologique clinique de cet effet reste à démontrer.
Autres métabolites du DCA
Le seul autre métabolite primaire connu du DCA est l’acide monochloroacétique (MCA), qui a été occasionnellement retrouvé à l’état de traces dans le sang après administration de DCA [34]. Le MCA semble être formé à partir du DCA dans les globules rouges [34]. Il n’existe aucune preuve de la formation du MCA à partir du DCA dans le foie. À des fins pratiques, la conversion du DCA en glyoxylate détermine la pharmacocinétique de l’élimination du DCA. Il est donc important de connaître les facteurs qui influencent la vitesse et l’étendue du métabolisme du DCA catalysé par la GSTZ1-1 chez l’humain afin d’établir une posologie sûre et efficace pour les patients.
En ce qui concerne les effets du traitement par DCA, le glyoxylate peut être métabolisé en dioxyde de carbone par une enzyme carboligase, en glycine par une ou plusieurs enzymes aminotransférases ou être oxydé en oxalate [35]. Des études au cours desquelles des rats ont reçu des doses de DCA ont mis en évidence ces trois voies [35]. Parmi ces voies, seule la formation d’oxalate peut potentiellement être délétère pour un patient, mais chez le rat, la formation d’oxalate est une voie mineure [35]. Des patients adultes gravement malades souffrant d’acidose lactique et traités par DCA intraveineux ou par placebo dans le cadre d’un essai clinique contrôlé et randomisé ont été évalués pour détecter la présence de calculs d’oxalate et la toxicité liée à l’oxalate ; aucune preuve de différences entre les groupes DCA et placebo n’a été trouvée [50]. De même, l’administration orale de DCA pendant plusieurs années dans un essai portant sur des enfants atteints de formes congénitales d’acidose lactique n’a pas révélé de signes de calculs d’oxalate ou de toxicité [51].
Variants polymorphes de GSTZ1
La GSTZ1 humaine (NM_145870) a été suspectée de présenter des variants alléliques dès sa découverte [52]. Des études ultérieures ont révélé des SNP dans les régions codantes et non codantes du gène. Nous en sommes encore à découvrir les effets, s’il y en a, de la plupart de ces SNP sur l’expression, la stabilité et l’activité de GSTZ1 avec le DCA. Il s’agit d’un domaine d’investigation actif, car ces facteurs ont un impact sur la pharmacocinétique et, en fin de compte, sur l’utilisation sûre du DCA. Les SNP qui entraînent des modifications des acides aminés dans la protéine exprimée ont retenu l’attention, de même que les SNP qui semblent affecter la traduction de la GSTZ1 et, en fin de compte, les niveaux d’expression de la protéine enzymatique.
Variants dans la séquence d’acides aminés de la protéine enzymatique exprimée
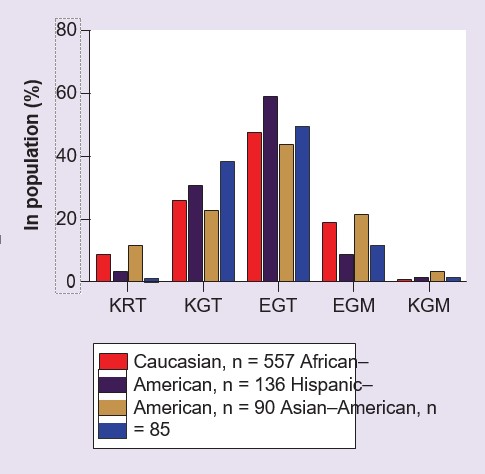
Trois SNP communs ont été identifiés dans la région codante de GSTZ1 [53,54], qui ont été observés pour donner lieu à cinq haplotypes (tableau 2). Ces cinq haplotypes sont présents chez les personnes avec des fréquences variables, en fonction de l’ethnie (figure 2), mais la variante la plus fréquemment trouvée dans toutes les populations étudiées à ce jour, présente chez environ 50 % des personnes, est GSTZ1C [54-56]. Cet haplotype est également appelé EGT, ce qui met en évidence la variante des acides aminés aux positions 32, 42 et 82 dans la protéine enzymatique [54,55]. À l’heure actuelle, il existe relativement peu d’informations sur l’incidence des cinq haplotypes communs dans des populations autres que les Caucasiens. Les protéines humaines recombinantes exprimées GSTZ1A-1A, 1B-1B, 1C-1C et 1D-1D ont été étudiées avec du DCA comme substrat, et il a été constaté que la GSTZ1A-1A (KRT/KRT) avait une activité plus élevée que les autres variantes [54,57]. La GSTZ1F est une variante rare (tableau 2) et les propriétés de la protéine recombinante n’ont pas été étudiées. Un individu peut être homo- ou hétérozygote pour ces haplotypes. Pour les hétérozygotes, on ne sait pas si une variante est exprimée préférentiellement au niveau de la protéine, cependant les fractions du cytosol du foie préparées à partir d’individus portant la variante GSTZ1A sur un allèle ont une activité in vitro avec le DCA plus élevée que ceux qui ne portent pas cet allèle [56], ce qui suggère la possibilité d’une expression préférentielle de la protéine enzymatique de la variante KRT.
E : acide glutamique ; G : glycine ; K : lysine : M : Métionine ; T : Thréonine.
| Variante | Position du nucléotide | Position des acides aminés | Pourcentage dans la population(%)† | ||||
| 94 | 124 | 245 | 32 | 42 | 82 | ||
| GSTZ1A | A | A | C | Lys (K) | Arg (R) | Thr (T) | 1-10 |
| GSTZ1B | A | G | C | Lys (K) | Gly (G) | Thr (T) | 25-35 |
| GSTZ1C | G | G | C | Glu (E) | Gly (G) | Thr (T) | 45-55 |
| GSTZ1D | G | G | T | Glu (E) | Gly (G) | Met (M) | 10-20 |
| GSTZ1F | A | G | T | Lys (K) | Gly (G) | Met (M) | <1 |
Les variants polymorphes humains de GSTZ1-1 et leur fréquence approximative dans la population.
†L’incidence de chaque haplotype varie selon le groupe ethnique, voir la figure 2 et [54-56].
Glu (E) : acide glutamique ; Gly (G) : Glycine ; Lys (K) : Lysine : Met (M) : Méthionine ; Thr (T) : Thréonine.
.
Des études réalisées avec les protéines humaines recombinantes exprimées GSTZ1A-1A, 1B-1B, 1C-1C et 1D-1D ont également examiné l’activité avec le substrat physiologiquement important, le MA [54]. Les taux de métabolisme de la MA ont montré un schéma différent des activités mesurées avec le DCA comme substrat, en ce sens que la variante GSTZ1C-1C (EGT/EGT) avait la plus forte activité avec la MA et la GSTZ1A-1A la plus faible [54].
En plus des variantes communément trouvées décrites ci-dessus, il existe des preuves pour deux autres variantes de la séquence codante. L’un d’eux a été trouvé dans la base de données des étiquettes de séquences exprimées et a été nommé GSTZ1E [54], cependant cette variante, qui aurait une proline à la place de la leucine en position 23, n’a pas été trouvée chez l’homme. Une autre variante a été trouvée chez un volontaire pour des études pharmacocinétiques du DCA. Cette personne, qui était hétérozygote pour KGT/KGM (1B/1F), éliminait même une dose unique de DCA extrêmement lentement ; le séquençage de l’ADN de cette personne a révélé un nouveau SNP dans l’exon cinq de GSTZ1, qui se traduisait par une méthionine au lieu d’une valine en position 99 de la protéine [55]. D’après la structure cristalline de GSTZ1B [58], la position 99 est située dans un faisceau α-hélicoïdal. Malgré la recherche de ce variant dans des échantillons de foie qui présentaient une faible activité avec le DCA [56], aucun autre individu présentant ce polymorphisme n’a encore été identifié.
Polymorphismes de la région promotrice
Dix SNP dans des échantillons d’ADN génomique de sujets africains et australiens ont été trouvés dans une région qui s’étend sur 1500 nucléotides en amont du site de début de transcription de GSTZ1, supposée être la région promotrice du gène [59]. Deux de ces SNP, -1002G>A et -289C>T, ont été associés respectivement à une diminution et à une augmentation de l’activité du promoteur, évaluée sur des cellules HepG2. Des études ultérieures réalisées avec de l’ADN provenant d’échantillons de foie ont montré que chez les Américains de type caucasien, mais pas chez les Afro-Américains, l’allèle A du SNP rs 7975, qui entraîne l’expression de la lysine au lieu de l’acide glutamique en position 32 de la protéine, était en déséquilibre de liaison avec l’allèle A du SNP -1002 G>A, rs7160195, de la région promotrice et entraînait une expression réduite de la protéine GSTZ1-1 avec la lysine (K) en position 32 [60]. L’expression réduite a été observée chez les hétérozygotes ainsi que chez les homozygotes pour GSTZ1A (KRT), GSTZ1B (KGT) et GSTZ1F (KGM) chez les Américains de race blanche [60]. Il n’y avait aucun effet sur l’expression des variants K de la GSTZ1-1 dans les échantillons de foie étudiés chez les Afro-Américains. Pour des raisons qui ne sont pas encore claires, il n’y avait pas de différences dans les niveaux d’ARNm de GSTZ1 entre les haplotypes, ce qui indique une déconnexion entre les résultats de l’expression des protéines et de l’expression de l’ARNm [60]. Le fait de trouver une expression plus faible de la protéine GSTZ1-1 pour les haplotypes contenant K chez les Caucasiens suggère que les taux de métabolisme initial du DCA seront réduits chez ces individus (à l’exception de ceux qui présentent un TK). En outre, cela soulève la possibilité que les Caucasiens présentant des haplotypes contenant K qui sont traités au DCA connaissent une re-synthèse moins efficace de la protéine GSTZ1-1, ce qui entraîne une élimination encore plus lente que les personnes ne présentant pas cet haplotype.
Influence des variantes de GSTZ1 sur la pharmacocinétique du DCA chez les personnes
En supposant que les effets secondaires d’un traitement chronique au DCA sont liés à l’accumulation soit du DCA lui-même, soit des substrats endogènes réactifs de la GSTZ1, la MAA et la MA, il existe un argument convaincant pour que l’utilisation clinique sûre du DCA soit guidée par une compréhension de la variabilité de l’élimination du médicament chez les personnes au cours d’un traitement chronique. La dose de DCA pourrait alors être réduite chez les personnes qui présentent une élimination lente, tout en maintenant des taux sanguins efficaces. S’il était possible de prédire quelles personnes élimineraient lentement le DCA, en fonction de leur constitution génétique, un test diagnostique pourrait être administré pour obtenir le génotype de GSTZ1, et la dose pourrait être basée sur le génotype. Cette méthode serait moins coûteuse que la mesure répétée des taux de DCA dans le sang. À cette fin, la pharmacocinétique du DCA a été étudiée chez des volontaires humains et chez des patients. Dans une étude sur des volontaires humains, les participants ont été génotypés et la pharmacocinétique du DCA a été étudiée après une dose unique de 25 mg/kg ou cinq doses quotidiennes consécutives de 25 mg/kg/jour [55]. Comme le montre le tableau 3, les personnes présentant au moins une variante EGT de GSTZ1 ont montré une plus grande clairance du DCA après cinq doses que celles qui n’avaient pas d’EGT dans leur génotype. La figure 3 illustre la cinétique d’élimination du DCA du plasma chez un métaboliseur rapide et un métaboliseur lent du DCA de cette étude [55]. Des résultats similaires ont été obtenus chez des patients atteints d’une maladie mitochondriale génétique qui ont reçu 12,5 mg/kg de DCA toutes les 12 heures pendant 6, 12 ou 30 mois [55,61]. Ce travail montre également que les niveaux de MA excrétés dans l’urine, une autre mesure de l’activité réduite de la GSTZ1/MAAI, étaient plus élevés chez les volontaires et les patients sans EGT dans leur génotype (Tableau 3).
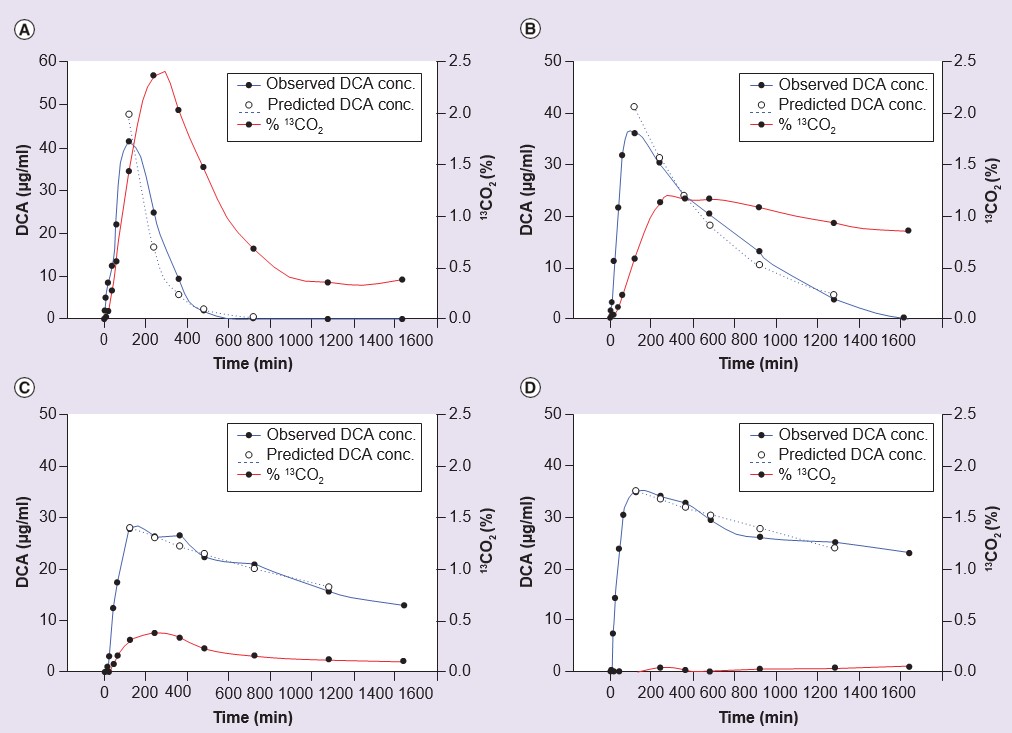
conc : Concentration ; DCA : Dichloroacétate.
| Sunject, durée du traitement par DCA | Clairance plasmatique, mml/min | Excrétion de maléylacétone, μg/g de créatinine | Réf. | ||
| Porteur d’EGT | Non-porteur d’EGT | EGT porteur | EGT non porteur | ||
| Volontaires, 5 jours | 2.22 ± 0.72 (7) | 0.73 ± 0.84* (5) | Non détecté | 7.2 ± 4.1* | [55] |
| Patients, 12 mois | 2.16 ± 0.99 (4) | 0.91, 0.17 | – | – | [55] |
| Patients, 6 mois | 1.90 ± 1.13 (11) | 0.53 ± 0.35* (6) | 1.2 ± 0.9 | 6.9 ± 2.6* | [61] |
| Patients, 30 mois | 2.08 ± 1.10 (11) | 0.67 ± 0.45* (6) | 1.9 ± 1.1 | 5.5 ± 1.2* | [61] |
Les valeurs indiquées sont la moyenne ± l’écart-type (n) ou les valeurs individuelles lorsque n < 3.
*Significativement différent des porteurs d’EGT, p < 0,05.
L’origine ethnique des volontaires était la suivante :
porteurs d’EGT : quatre caucasiens-américains, deux afro-américains, un asiatique-américain ; non porteurs d’EGT : cinq caucasiens-américains. Les origines ethniques des patients n’étaient pas disponibles.
DCA : Dichloroacétate.
L’analyse des données provenant des études de la pharmacocinétique du DCA chez des volontaires a suggéré que, comme cela a été constaté in vitro (voir ci-dessous), les personnes portant la variante GSTZ1A, codant pour l’EGT, sur un allèle présentaient une clairance initiale plus rapide du DCA que les autres. Ces personnes ont également présenté un déclin plus important de la clairance après cinq doses. Le rapport de la clairance de la première à la cinquième dose était de 3,6 ± 0,8 (moyenne ± erreur standard de la moyenne, n = 5) chez les personnes présentant le variant pour GSTZ1C (codant pour EGT) mais pas pour GSTZ1A, et était de 18,2 ± 9,3 (n = 5) chez les personnes présentant le variant GSTZ1A sur au moins un allèle. Un individu homozygote pour GSTZ1A a présenté le changement le plus important entre la première et la cinquième dose, la clairance passant de 16,7 à 0,31 ml/min [55]. La différence dans le changement moyen de la clairance n’était pas statistiquement significative, en raison de la grande variabilité dans le groupe homo- ou hétérozygote pour GSTZ1A, mais elle soulignait une tendance qui était en accord avec les études in vitro de la susceptibilité des différents haplotypes à l’inactivation par le DCA.
Étudesin vitro du métabolisme du DCA
Des études sur l’expression et l’activité de GSTZ1 avec le DCA dans le cytosol et les mitochondries du foie humain ont permis de mieux comprendre le rôle du génotype dans le métabolisme du DCA.
Ontogenèse de l’activité
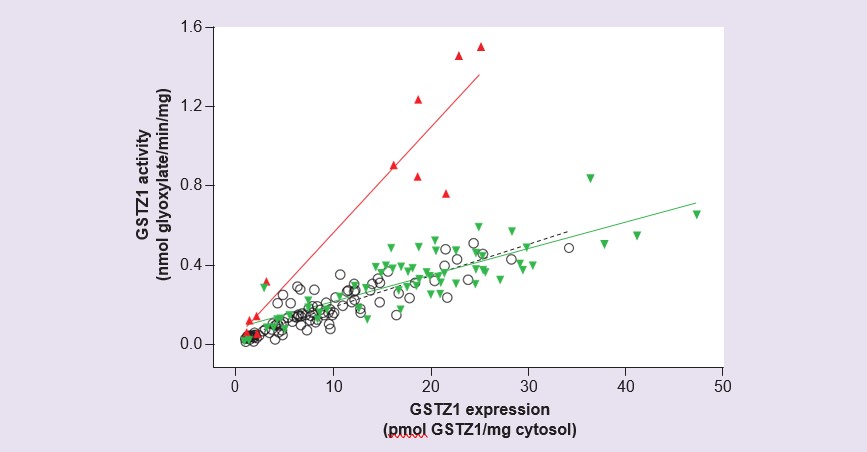
Des études réalisées avec du cytosol de foie humain préparé à partir d’un panel de 230 échantillons de foie provenant de donneurs couvrant la tranche d’âge du 42e jour de gestation jusqu’à 84 ans ont montré une expression et une activité de GSTZ1 très faibles ou indétectables avant la naissance ainsi que pendant les deux premières semaines de vie : par la suite, l’activité et l’expression augmentent progressivement [56]. L’expression et l’activité atteignent les niveaux de l’adulte à l’âge de 7 ans, mais une variabilité interindividuelle considérable a été constatée. La mesure de l’activité avec le DCA et de l’expression de la protéine GSTZ1 dans ces échantillons a révélé que les échantillons des individus présentant la variante GSTZ1A sur un allèle présentaient un rapport activité/expression plus élevé que tous les autres (figure 4). Ces résultats concordent avec ceux obtenus à partir de l’expression de la GSTZ1A-1A humaine recombinante qui a montré une activité plus élevée du DCA pour cette variante [54], et avec les études pharmacocinétiques menées chez des volontaires où les personnes possédant un gène GSTZ1A présentaient une clairance initiale rapide du DCA [55]. Bien que l’étude sur le développement ait montré que l’activité chez les enfants de moins de 7 ans était inférieure à celle des adultes, la prise en compte de la taille plus importante du foie des enfants par rapport à la masse corporelle signifie que par kilogramme de poids corporel, l’activité avec le DCA est similaire chez les adultes et les enfants âgés de 2 mois à 7 ans [56].
Effet du chlorure sur l’inactivation de la GSTZ1 par le DCA
En contradiction apparente avec l’observation selon laquelle la protéine KRT est associée à un métabolisme plus rapide du DCA in vitro [56] et à une élimination plus rapide d’une dose unique de DCA [55], on a constaté que les personnes présentant la variante GSTZ1A sur un allèle présentaient une réduction plus importante de la clairance après des doses répétées de DCA que celles qui étaient homozygotes pour GSTZ1C. Des découvertes récentes concernant l’effet du chlorure sur l’inactivation du GSTZ1-1 pourraient expliquer ces divergences. On a constaté que le chlorure protège la GSTZ1-1 de l’inactivation par le DCA à des concentrations physiologiquement pertinentes dans le foie [62,63], mais la GSTZ1-1 dans le cytosol du foie des personnes hétérozygotes pour la GSTZ1A était moins protégée de l’inactivation par le chlorure physiologique (38 mM) que celle des personnes ayant d’autres variantes alléliques de la GSTZ1 (tableau 4). L’effet du chlorure était tel que les enzymes GSTZ1-1 dans les foies des individus présentant la variante GSTZ1A sur un allèle étaient inactivées deux fois plus rapidement que ceux présentant d’autres haplotypes. Le mécanisme exact de l’effet du chlorure n’a pas encore été déterminé, mais ce phénomène est une des explications de la plus grande perte de clairance du DCA à la suite d’une dose chronique chez les personnes ayant une ou plusieurs copies de la variante GSTZ1A. Comme on l’a mentionné plus haut, l’expression plus faible de l’enzyme chez les personnes de race blanche ayant une lysine (K) en position 32 de la protéine est susceptible de contribuer à la très faible clairance.
.
| Haplotype | EC50 mM | Demi-vie d’inactivation, h sans Cl- | Demi-vie d’inactivation, h avec 38 mM Cl- |
|---|---|---|---|
| EGT/EGT | 15 ± 3.1 (3) | 0.53, 0.49 | 5.73, 5.02 |
| KRT/EGT | 36 ± 2.2 (3) | 0.38, 0.38 | 2.66, 2.37 |
| EGM/EGM | 16.9 | – | 5.55 |
La CE50 est la concentration de chlorure qui a protégé la moitié de la GSTZ1 cytosolique de l’inactivation après une incubation de 2 h avec du Na DCA, 0,5 mM.
Conclusion
En raison de sa capacité à inhiber la PDH kinase, le DCA a des applications pour le traitement du cancer et des maladies métaboliques. Lors de l’administration chronique de DCA à des personnes, son métabolisme et son élimination sont réduits. La raison de cette réduction de la clairance après de multiples doses de DCA est que la seule enzyme connue pour métaboliser le DCA, la GSTZ1-1, est inactivée au cours du métabolisme du DCA. Cette inhibition de la GSTZ1-1 par le DCA, fondée sur un mécanisme, entraîne un ralentissement de la clairance non seulement du DCA, mais aussi des substrats endogènes MAA et MA ; la question de savoir si l’accumulation de DCA et/ou de catabolites réactifs de la tyrosine est à l’origine de la neuropathie périphérique réversible qui se produit chez certains patients n’est pas encore résolue. Une autre cause possible non résolue de la neuropathie est l’augmentation modeste de ∂-ALA qui suit le traitement chronique au DCA. Pour comprendre la pharmacocinétique du DCA, il faut savoir que le gène GSTZ1 présente des variantes polymorphes dans toutes les populations humaines étudiées à ce jour. Les propriétés des protéines enzymatiques du variant GSTZ1-1 diffèrent quant à leur capacité à métaboliser le DCA et les substrats endogènes MAA et MA, en particulier lors d’une administration chronique. Alors que toutes les variantes de la GSTZ1-1 sont inactivées par le DCA, une variante, dont la lysine remplace l’acide glutamique en position 32 et l’arginine remplace la glycine en position 42 de la protéine, est plus rapidement inactivée que les autres variantes en présence de concentrations physiologiques de chlorure. Chez les Caucasiens, mais pas chez les personnes d’origine africaine, les variants de GSTZ1 qui ont une lysine en position 32 sont liés à un SNP de la région promotrice du gène GSTZ1 qui entraîne une expression réduite de la protéine GSTZ1-1 ; ceci peut également se présenter comme un phénotype de métaboliseur lent. Une compréhension des facteurs qui influencent la pharmacocinétique du DCA en cas d’administration chronique est nécessaire pour sélectionner la bonne dose et le bon intervalle d’administration afin d’administrer ce médicament aux patients de manière sûre et efficace.
Perspectives d’avenir
Les études menées à ce jour montrent clairement que l’haplotype GSTZ1 est un facteur important qui détermine la clairance du DCA. Une meilleure compréhension de la régulation de l’enzyme dans des conditions physiologiques et en présence de DCA permettra à l’avenir une utilisation plus sûre et plus efficace du médicament. Plusieurs questions restent à explorer. Chez les hétérozygotes, une variante de la protéine est-elle exprimée de manière préférentielle ? Les études d’activité qui montrent une activité plus élevée avec le DCA pour les fractions du cytosol du foie provenant d’individus hétérozygotes pour la GSTZ1A/GSTZ1C suggèrent que la variante de la protéine enzymatique 1A (KRT) est exprimée préférentiellement chez les hétérozygotes, mais cela n’a pas été démontré. Cette question a des implications pratiques, puisque la variante 1A est inactivée plus rapidement que la variante 1C. Faut-il administrer une dose plus faible de DCA aux personnes qui sont susceptibles d’éliminer le DCA plus lentement que la moyenne lors d’une administration chronique ? Dans l’affirmative, devrait-il s’agir de toutes les personnes qui ne sont pas homozygotes ou hétérozygotes pour le GSTZ1C? Les Caucasiens présentant au moins une variante contenant K (KRT, KGT, KGM) devraient-ils recevoir des doses de DCA plus faibles que les personnes d’origine africaine parce qu’ils expriment moins de GSTZ1-1 ? Quelles sont les preuves de la faible expression des variants contenant K dans d’autres populations ? Existe-t-il des différences de population dans les schémas de distribution des principaux haplotypes ? Les effets des différences pharmacogénétiques sont-ils les mêmes chez les enfants que chez les adultes ? Les enfants prenant du DCA éliminent généralement le médicament plus rapidement que les adultes, mais les effets pharmacogénétiques possibles n’ont pas été étudiés de manière approfondie. Des recherches supplémentaires sont nécessaires pour répondre à ces questions, afin de garantir un dosage sûr et efficace du DCA.
Divulgation d’intérêts financiers et concurrents
Le travail des auteurs discuté dans ce manuscrit a été financé en partie par le US Public Health Service 1RO1 GM 099871, en partie par la Brain and Tissue Bank for Developmental Disorders, Université du Maryland, Baltimore et Université de Miami (NO1 HD90011) et en partie par le NIH/NCATS Clinical and Translational Science award à l’Université de Floride UL1 TR000064. Les auteurs n’ont pas d’autres affiliations pertinentes ou d’implication financière avec une organisation ou une entité ayant un intérêt financier ou un conflit financier avec le sujet ou les matériaux discutés dans le manuscrit en dehors de ceux divulgués.
Aucune aide à la rédaction n’a été utilisée pour la production de ce manuscrit.
Résumé
- La sélection d’une dose efficace mais non toxique de dichloroacétate (DCA) est importante pour l’utilisation clinique à long terme du DCA dans le traitement du cancer et d’autres maladies métaboliques.
- Le gène de l’enzyme unique qui métabolise ce médicament, GSTZ1-1, présente des polymorphismes qui entraînent des phénotypes de métaboliseurs lents et rapides après un traitement à long terme au DCA.
- Les SNP de la région codante du gène GSTZ1 donnent lieu à cinq haplotypes communs, dont la fréquence varie selon les populations. Les personnes homozygotes pour le plus courant, GSTZ1C, sont des métaboliseurs rapides. Il semble que les haplotypes codant pour les protéines GSTZ1-1 avec K en position 32 soient des métaboliseurs lents, mais les autres facteurs contribuant au métabolisme lent après l’administration de doses multiples de DCA ne sont pas entièrement compris.
- Un SNP de la région promotrice chez les personnes d’origine caucasienne mais non africaine est lié à un SNP de la région codante qui entraîne la substitution de la lysine par l’acide glutamique en position 32 de la protéine GSTZ1, et est associé à une expression réduite de la protéine enzymatique dans le foie.
- En présence de DCA, la stabilité de l’enzyme GSTZ1-1 varie en fonction de la concentration de chlorure. L’enzyme variante GSTZ1A (KRT) est inactivée plus rapidement que la variante commune GSTZ1C (EGT) sous des concentrations physiologiques de chlorure dans le foie.
RÉFÉRENCES
1 Stacpoole PW. Revue des effets pharmacologiques et thérapeutiques du dichloroacétate de diisopropylammonium (DIPA). J. Clin. Pharmacol. J. New Drugs 9(5), 282-291 (1969).2 Stacpoole PW. La pharmacologie du dichloroacétate. Metabolism 38(11), 1124-1144 (1989).
3 Stacpoole PW, Felts JM. Le dichloroacétate de diisopropylammonium (DIPA) et le dichloroacétate de sodium (DCA) : effet sur le métabolisme du glucose et des graisses dans les tissus normaux et diabétiques. Metabolism 19(1), 71-78 (1970).
4 Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N. Engl. J. Med. 298(10), 526-530 (1978).
5 Stacpoole PW, Harwood HJ Jr, Varnado CE. Regulation of rat liver hydroxymethylglutaryl coenzyme A reductase by a new class of noncompetitive inhibitors. Effets du dichloroacétate et des acides carboxyliques apparentés sur l’activité enzymatique. J. Clin. Invest. 72(5), 1575-1585 (1983).
6 Harwood HJ Jr, Bridge DM, Stacpoole PW. In vivo regulation of human mononuclear leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase. Studies in normal subjects. J. Clin. Invest. 79(4), 1125-1132 (1987).
7 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA, Stacpoole PW. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis 33(3), 285-293 (1979).
8 Eichner HL, Stacpoole PW, Forsham PH. Treatment of streptozotocin diabetes with di-isopropylammonium dichloroacetate (DIPA). Diabetes 23(3), 179-182 (1974).
9 Backshear PJ, Holloway PA, Alberti KG. Interactions métaboliques du dichloroacétate et de l’insuline dans l’acidocétose diabétique expérimentale. Biochem. J. 146(2), 447-456 (1975).
10 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (short communication). Biochem. J. 134(2), 651-653 (1973).
11 Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 141(3), 761-774 (1974).
12 Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34(Pt 2), 217-222 (2006).
13 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329(Pt 1), 191-196 (1998).
14 Morten KJ, Caky M, Matthews PM. Stabilisation de la sous-unité E1alpha de la pyruvate déshydrogénase par le dichloroacétate. Neurology 51(5), 1331-1335 (1998).
15 Han Z, Berendzen K, Zhong L et al. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol. Genet. Metab. 93(4), 381-387 (2008).
16 Evans OB, Stacpoole PW. Prolonged hypolactatemia and increased total pyruvate dehydrogenase activity by dichloroacetate. Biochem. Pharmacol. 31(7), 1295-1300 (1982).
17 Denton RM, Randle PJ, Bridges BJ et al. Regulation of mammalian pyruvate dehydrogenase. Mol. Cell. Biochem. 9(1), 27-53 (1975).
18 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284(5), E855-E862 (2003).
19 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacité du dichloroacétate comme médicament pour abaisser le taux de lactate. J. Clin. Pharmacol. 43(7), 683-691 (2003).
20 Kankotia S, Stacpoole PW. Le dichloroacétate et le cancer : un nouveau foyer pour un médicament orphelin ? Biochim. Biophys. Acta 1846(2), 617-629 (2014).
21 Stacpoole PW. Le dilemme du dichloroacétate : danger environnemental ou mine d’or thérapeutique – les deux ou aucun ? Environ. Health Perspect. 119(2), 155-158 (2011).
22 Chu PI. Pharmacocinétique du dichloroacétate de sodium. Thèse de doctorat, Pharmaceutics Department, University of Florida, FL, USA (1987).
23 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacocinétique, métabolisme et toxicologie du dichloroacétate. Drug Metab. Rev. 30(3), 499-539 (1998).
24 Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. Concentrations plasmatiques et effets métaboliques du dichloroacétate de sodium intraveineux. Clin. Pharmacol. Ther. 37(1), 89-93 (1985).
25 Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G et al. Pharmacokinetics of oral dichloroacetate in dogs. J. Biochem. Mol. Toxicol. 27(12), 522-525 (2013).
26 Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Effets métaboliques et pharmacocinétique du dichloroacétate administré par voie intraveineuse chez l’homme. Diabetologia 19(2), 109-113 (1980).
27 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N. Engl. J. Med. 309(7), 390-396 (1983).
28 Henderson GN, Curry SH, Derendorf H, Wright EC, Stacpoole PW. Pharmacocinétique du dichloroacétate chez les patients adultes atteints d’acidose lactique. J. Clin. Pharmacol. 37(5), 416-425 (1997).
29 Stacpoole PW, Kerr DS, Barnes C et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117(5), 1519-1531 (2006).
30 Stacpoole PW, Gilbert LR, Neiberger RE et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121(5), e1223-e1228 (2008).
31 Kaufmann P, Engelstad K, Wei Y et al. Dichloroacetate causes toxic neuropathy in MELAS : a randomized, controlled clinical trial. Neurology 66(3), 324-330 (2006).
32 Stickler DE, Valenstein E, Neiberger RE et al. Peripheral neuropathy in genetic mitochondrial diseases. Pediatr. Neurol. 34(2), 127-131 (2006).r
33 Kaufmann P, Pascual JM, Anziska Y et al. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch. Neurol. 63(5), 746-748 (2006).
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate : possible relevance to toxicity. J. Pharmacol. Exp. Ther. 324(3), 1163-1171 (2008).
35 James MO, Yan Z, Cornett R et al. Pharmacocinétique et métabolisme du [14C]dichloroacétate chez les rats Sprague-Dawley mâles. Identification des conjugués de la glycine, y compris l’hippurate, comme métabolites urinaires du dichloroacétate. Drug Metab. Dispos. 26(11), 1134-1143 (1998).
36 Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases : their role in drug metabolism and detoxification. Int. J. Biochem. Cell. Biol. 45(6), 1121-1132 (2013).
37 Vessey DA, Kelley M, Warren RS. Caractérisation des CoA ligases des mitochondries du foie humain catalysant l’activation des acides gras à chaîne courte et moyenne et des acides carboxyliques xénobiotiques. Biochim. Biophys. Acta 1428(2-3), 455-462 (1999).
38 James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Glutathione-dependent conversion to glyoxylate, a major pathway of dichloroacetate biotransformation in hepatic cytosol from humans and rats, is reduced in dichloroacetate-treated rats. Drug Metab. Dispos. 25(11), 1223-1227 (1997).
39 Tong Z, Board PG, Anders MW. Glutathione transferase zeta catalyses the oxygenation of the carcinogen dichloroacetic acid to glyoxylic acid. Biochem. J. 331(Pt 2), 371-374 (1998).
49 Fernandez-Canon JM, Penalva MA. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J. Biol. Chem. 273(1), 329-337 (1998).
41 Blackburn AC, Woollatt E, Sutherland GR, Board PG. Characterization and chromosome location of the gene GSTZ1 encoding the human Zeta class glutathione transferase and maleylacetoacetate isomerase. Cytogenet. Cell. Genet. 83(1-2), 109-114 (1998).
42 Li W, James MO, Mckenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrion as a novel site of dichloroacetate biotransformation by glutathione transferase zeta 1. J. Pharmacol. Exp. Ther. 336(1), 87-94 (2011).
43 Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Localisation immunohistochimique et activité de la glutathion transférase zeta (GSTZ1-1) dans les tissus du rat. Drug Metab. Dispos. 30(6), 616-625 (2002).
44 Shangraw RE, Fisher DM. Pharmacocinétique du dichloroacétate chez les patients subissant une transplantation hépatique. Anesthesiology 84(4), 851-858 (1996).
45 Cornett R, James MO, Henderson GN, Cheung J, Shroads AL, Stacpoole PW. Inhibition de la glutathione S-transférase zeta et du métabolisme de la tyrosine par le dichloroacétate : un mécanisme unificateur potentiel pour sa biotransformation altérée et sa toxicité. Biochem. Biophys. Res. Commun. 262(3), 752-756 (1999).
46 Guo X, Dixit V, Liu H et al. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 34(1), 36-42 (2006).
47 Anderson WB, Board PG, Gargano B, Anders MW. Inactivation de la glutathion transférase zeta par l’acide dichloroacétique et d’autres acides alpha-haloalcanoïques sans fluor. Chem. Res. Toxicol. 12(12), 1144-1149 (1999).
48 Anderson WB, Liebler DC, Board PG, Anders MW. Mass spectral characterization of dichloroacetic acid-modified human glutathione transferase zeta. Chem. Res. Toxicol. 15(11), 1387-1397 (2002).
49 Dixit V. Inactivation of glutathione transferase zeta by dichloroacetic acid. Thèse de doctorat, Medicinal Chemistry Department, University of Florida, FL, USA (2005).
50 Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N. Engl. J. Med. 327(22), 1564-1569 (1992).
51 Abdelmalak M, Lew A, Ramezani R et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol. Genet. Metab. 109(2), 139-143 (2013).
52 Conseil PG, Baker RT, Chelvanayagam G, Jermiin LS. Zeta, une nouvelle classe de glutathion transférases dans une gamme d’espèces allant des plantes aux humains. Biochem. J. 328(Pt 3), 929-935 (1997).
53 Blackburn AC, Tzeng HF, Anders MW, Board PG. Discovery of a functional polymorphism in human glutathione transferase zeta by expressed sequence tag database ana-lysis. Pharmacogenetics 10(1), 49-57 (2000).
54 Blackburn AC, Coggan M, Tzeng HF et al. GSTZ1d : a new allele of glutathione transferase zeta and maleylacetoacetate isomerase. Pharmacogenetics 11(8), 671-678 (2001).
55 Shroads AL, Langaee T, Coats BS et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J. Clin. Pharmacol. 52(6), 837-849 (2012).
56 Li W, Gu Y, James MO et al. Expression prénatale et postnatale de la glutathion transférase zeta 1 dans le foie humain et rôles de l’haplotype et de l’âge du sujet dans la détermination de l’activité avec le dichloroacétate. Drug Metab. Dispos. 40(2), 232-239 (2012).Google Scholar
57 Board PG, Anders MW. Glutathion transférase zeta : découverte, variantes polymorphes, catalyse, inactivation et propriétés des souris Gstz1-/-. Drug Metab. Rev 43(2), 215-225 (2011).
58 Polekhina G, Board PG, Blackburn AC, Parker MW. La structure cristalline de la maleylacetoacetate isomerase/glutathione transferase zeta révèle la base moléculaire de sa remarquable promiscuité catalytique. Biochemistry 40(6), 1567-1576 (2001).
59 Fang YY, Kashkarov U, Anders MW, Board PG. Polymorphismes dans le promoteur de la glutathion transférase zeta humaine. Pharmacogenet. Genomics 16(5), 307-313 (2006).
60 Langaee TY, Zhong G, Li W et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics 25(5), 239-245 (2015).
61 Shroads AL, Coats BS, Mcdonough CW, Langaee T, Stacpoole PW. Les variations d’haplotype de la glutathion transférase zeta 1 influencent la cinétique et la dynamique du dichloroacétate chronique chez les enfants. J. Clin. Pharmacol. 55(1), 50-55 (2015).
62 Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Le chlorure et d’autres anions inhibent l’inactivation induite par le dichloroacétate de la GSTZ1 du foie humain d’une manière dépendante de l’haplotype. Chem. Biol. Interact. 215C, 33-39 (2014).
63 Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Les concentrations de chlorure dans le cytosol et les mitochondries hépatiques humaines sont une fonction de l’âge. Biochem. Biophys. Res. Commun. 459(3), 463-468 (2015).