Margaret O James*,1 & Peter W Stacpoole2,3
1 Department of Medicinal Chemistry, University of Florida, Gainesville, FL 32610-0485, USA
2 Department of Medicine, College of Medicine, University of Florida, Gainesville, FL 32610-0485, USA
3 Department of Biochemistry & Molecular Biology, University of Florida, Gainesville, FL 32610-0485, USA
Correspondence: Tel: +1 352 273 7707 Email: [email protected]
Submitted: 16 December 2015
Accepted: 17 February 2016
Published: 4 May 2016
Abstract
The investigational drug dichloroacetate (DCA) is a metabolic regulator that has been successfully used to treat acquired and congenital metabolic diseases and, recently, solid tumors. Its clinical use has revealed challenges in selecting appropriate doses. Chronic administration of DCA leads to inhibition of DCA metabolism and potential accumulation to levels that result in side effects. This is because conversion of DCA to glyoxylate is catalyzed by one enzyme, glutathione transferase zeta 1 (GSTZ1-1), which is inactivated by DCA. SNPs in the GSTZ1 gene result in expression of polymorphic variants of the enzyme that differ in activity and rates of inactivation by DCA under physiological conditions: these properties lead to considerable variation between people in the pharmacokinetics of DCA.
Keywords: dichloroacetate; GSTZ1; pharmacogenetics
Clinical use of dichloroacetate
For being such a simple molecule, DCA has a remarkably rich and diverse pharmacological portfolio, dating almost a century [1,2]. However, its modern use as an investigational drug began in 1970, when its selective ability to lower blood glucose levels in diabetic, but not in nondiabetic, animals was discovered [3] and subsequently confirmed in humans [4]. Many metabolic properties of DCA involving glucose and lipid metabolism were identified during the 1970s and 1980s. However, most of its pharmacological effects can be distilled into a few basic sites and mechanisms of action. First, DCA is a noncompetitive inhibitor of the endoplasmic reticulum enzyme HMG CoA reductase, which catalyzes the rate-limiting step in cholesterol biosynthesis. DCA’s inhibitory effect is observed in both rodent liver [5] and human leukocytes [6]; it likely accounts for the drug-associated reduction in total and low-density lipoprotein (LDL) cholesterol in patients with LDL receptor-negative homozygous familial hypercholesterolemia [7] and for its designation as the first orphan product for this rare disease. Second, DCA inhibits de novo hepatic triglyceride synthesis in nondiabetic rodents [5] and decreases circulating triglyceride and very low density lipoprotein levels in patients with Type 2 diabetes mellitus [4]. It also decreases blood ketone bodies in rats with experimentally induced diabetic ketoacidosis [8,9]. The precise mechanisms underlying these effects on lipid synthesis and oxidation are unknown. Third, DCA stimulates the mitochondrial PDC, which irreversibly oxidizes pyruvate to acetyl coenzyme A (acetyl CoA) [10], a property shared by certain other halogenated short chain fatty acids [11]. It is the ability of DCA to alter PDC activity that has generated by far the greatest experimental and clinical research on this unusual molecule.
PDC is regulated post-translationally mainly by reversible phosphorylation of one or more of three serine residues on the E1α subunit of the first enzyme PDH of PDC that decarboxylates pyruvate [12]. Four PDH kinases (PDK1–4) and two PDH phosphatases (PDP 1 and 2) carry out this regulatory aspect of PDC in humans, in which the phosphorylated enzyme is catalytically inactive. PDKs are differentially expressed in tissues, although PDK2 is expressed ubiquitously [13]. Pyruvate, the substrate for the PDC reaction, inhibits PDKs by binding to a small pocket in the N-terminus of the kinase, leading to stimulation of PDC activity. In contrast, accumulation of the reaction products, acetyl CoA and NADH, results in PDK activation and inhibition of PDC. DCA, a structural analog of pyruvate, also attaches to the pyruvate-binding site, resulting in inhibition of PDKs with the order of inhibition being PDK2>PDK1˜PDK4>>PDK3 [13]. Chronic drug exposure in vitro [14,15] and in vivo [16] may stabilize PDC and decrease its turnover, providing a second mechanism by which DCA increases the activity of the complex and an explanation for its protracted pharmacological effects following drug withdrawal [4].
Although DCA is a relatively weak PDK inhibitor (Ki 0.2 mM for PDK2), its effects on aspects of intermediary metabolism are profound and relate directly to the pivotal role the PDC plays in regulating eukaryotic cellular fuel selection and bioenergetics [17,18]. DCA is as yet a non-US FDA approved investigational drug. However, it continues to be extensively investigated as a metabolic modulator for several congenital and acquired metabolic disorders, based on its stimulatory action on PDC (Table 1).
| Property | Mechanism | Ref. |
|---|---|---|
| Increased OXPHOS and bioenergetics | ↓ PDK ↑ PDC | [2,10] |
| Decreased blood glucose in fasting or diabetes | ↓ PDK ↑ PDC | [4] |
| Decreased blood and CSF lactate | ↓ PDK ↑ PDC | [19] |
| Decreased blood total and LDL cholesterol | ↓ HMG CoA reductase → ↓ cholesterol synthesis | [6,7] |
| Decreased blood triglycerides and VLDL cholesterol | ↓ hepatic TG synthesis → ↓ VLDL syntheis | [4] |
| Reversal of Warburg effect in cancer, PAH and other proliferative conditions | ↓ PDK ↑ PDC | [20] |
CSF: Cerebrospinal fluid; HMG CoA: Hydroxymethylglutaryl coenzyme A; LDL: Low-density lipoprotein; OXPHOS: Oxidative phosphorylation; PAH: Pulmonary arterial hypertension; PDC: Pyruvate dehydrogenase complex; PDK: Pyruvate dehydrogenase kinase; TG: Triglyceride; VLDL: Very low-density lipoprotein.
Pharmacokinetics & clearance
Oral DCA is rapidly absorbed and has a bioavailability approaching unity after both oral and parenteral dosing [21,22]. The drug can be detected in plasma of humans within 15 min of an oral dose of 50 mg/kg. In previously naive subjects, oral or intravenous DCA has a plasma half-life of approximately 1 h [23]. The rate of metabolism among healthy species is: mouse>rat>human≥dog [22–25].
Early pharmacokinetics investigations of single dose intravenous DCA described nonlinear kinetics at doses ≥35 mg/kg [26]. Later it was found that the rate of plasma drug clearance declined upon repeat intravenous dosing of 50 mg/kg body weight in critically ill adults with lactic acidosis [27,28]. Delayed plasma clearance of the drug administered orally at a dose of 50 mg/kg was also observed in healthy adults, who required weeks to months before the original clearance rate was achieved with a second dose [22].
Major advances in understanding this unusual feature of DCA pharmacokinetics arose from probative research of its clinical toxicology. Chronic DCA can cause a reversible peripheral neuropathy across species [21,23], an effect first observed clinically 25 years ago in a 16-year-old boy who received 50 mg/kg/day for approximately 4 months [7]. In contrast, in a randomized, controlled trial (RCT) of 43 young children with various types of primary mitochondrial diseases (mean age at entry: 5.6 years), oral DCA administered at a dose of 12.5 mg/kg every 12 h for 6 months was well tolerated and caused no adverse changes in peripheral nerve electrical conduction, compared with placebo [29], although some asymptomatic decline in nerve conduction was found with longer drug exposure [30]. In contrast, an RCT of 30 older adolescents and adults (mean age of entry: 30 years) with a genetic mitochondrial disease who were exposed to the identical DCA dose and dose schedule employed in the pediatric trial was stopped prematurely because of worsening or new onset of peripheral neuropathy [31], a common complication of mitochondrial diseases [32,33]. When the kinetics of DCA in these two patient populations was compared, it was discovered that the plasma clearance of chronic DCA was markedly slower in the older group [34], a finding confirmed in studies with rats [34,35]. These findings prompted studies of the enzymology of DCA biotransformation.
Importance of glutathione transferase zeta 1 in DCA biotransformation
Small carboxylic acids are often glucuronidated [36] or converted to their coenzyme A derivatives then to amino acid conjugates [37], however there is no evidence that DCA forms a glucuronide, a coenzyme A derivative or an amino acid conjugate [2, James MO,Unpublished data]. Instead DCA is a substrate for an unusual GST enzyme, GSTZ1-1, which converts it to glyoxylate in a reaction that requires but does not consume glutathione (GSH) [38,39]. Like other GSTs, GSTZ1-1 is active in the dimeric form, and the active enzyme is a homodimer. Dechlorination of DCA to form glyoxylate, catalyzed by GSTZ1-1, is the only pathway of primary metabolism of DCA observed in the liver [35]. As well as catalyzing DCA metabolism, GSTZ1-1, also known as MAAI, has an important physiological function in the catabolism of tyrosine, where it isomerizes maleylacetoacetate (MAA) and maleylacetone (MA) to fumarylacetoacetate and fumarylacetone in reactions that require but do not consume GSH (Figure 1) [40,41]. Biotransformation of DCA to glyoxylate terminates its pharmacologically and therapeutically important activities that are discussed above, thus the activity of GSTZ1-1 controls the duration of action of DCA.
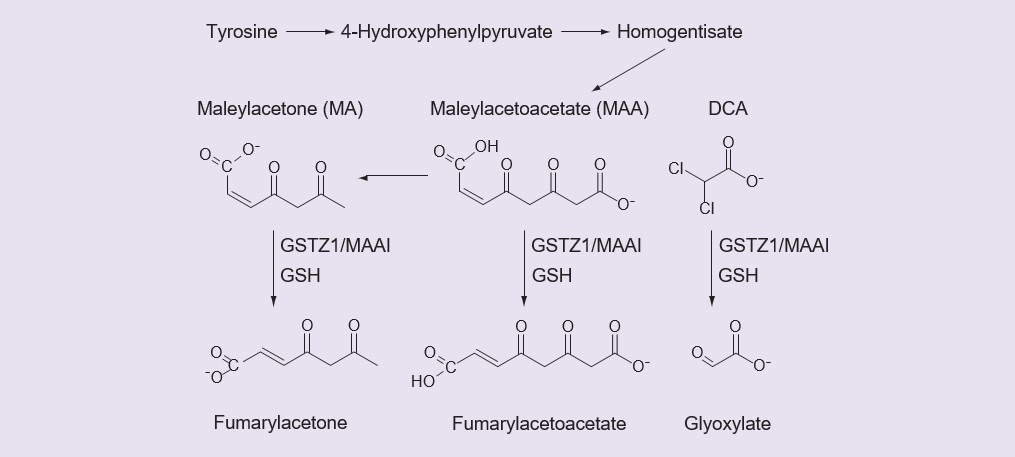
The inactive DCA metabolite, glyoxylate, is further metabolized to carbon dioxide, glycine and oxalate. Fumarylacetone and fumarylacetoacetate are further metabolized by fumarylacetoacetate hydrolase. DCA: Dichloroacetate: GSH: Glutathione; MA: Maleylacetone; MMA: Maleyacetoacetate.
The liver is the main site of GSTZ1-1 expression, where it is found in both the cytosol and the mitochondrial matrix [38,42]. Animal studies have demonstrated very low levels of GSTZ1-1 expression in kidney, heart, GI tract and brain [43]. The role of extrahepatic tissues in DCA metabolism in people has not been explored in detail, however one study showed that DCA was not cleared from the blood during the anhepatic phase of liver transplant patients given DCA to counteract lactic acidosis, indicating no metabolism occurred [44]. The anhepatic phase was of short duration (mean: 73 min), so the data were limited, however this study suggests a minor role of tissues other than liver in DCA elimination, at least for a single dose.
Inhibition of GSTZ1-1 by DCA
Studies with animals and with expressed recombinant human GSTZ1-1 proteins have demonstrated that the reason multiple doses of DCA are eliminated much more slowly than a single dose is that DCA is a mechanism-based inhibitor of GSTZ1-1 [25,34,45–47]. During biotransformation of DCA, adducts to the GSTZ1-1 protein are formed that inactivate the protein, and presumably trigger its proteolysis [48,49]. DCA administration to rats leads to loss of activity and expression of GSTZ1-1 in a dose- and time-dependent manner: the loss of GSTZ1-1 function was more pronounced and persisted longer in adult rats compared with juveniles [34,35]. This effect of age on loss of GSTZ1-1 function following DCA administration was observed in people, with children showing a smaller increase in plasma half-life and lower decrease in clearance than adults [34]. As well as affecting DCA pharmacokinetics, loss of GSTZ1-1 (MAAI) results in accumulation of the physiological substrates MA and, presumably, MAA, both of which are reactive molecules capable of adduct formation with cellular macromolecules [34,45]. MAAI inhibition also leads to diversion of tyrosine carbon to succinylacetone formation. This molecule inhibits a proximal step in heme synthesis, causing accumulation of the reactive molecule δ-aminolevulinate (δ-ALA). Both MA and δ-ALA increase in the blood and/or urine of humans chronically exposed to DCA [34]: the clinical toxicological significance of this effect has yet to be demonstrated.
Other metabolites of DCA
The only other known primary metabolite of DCA is monochloroacetic acid (MCA), which has been occasionally found in trace amounts in the blood following administration of DCA [34]. The MCA appears to be formed from DCA in the red blood cells [34]. There is no evidence for formation of MCA from DCA in the liver. For practical purposes, the conversion of DCA to glyoxylate determines the pharmacokinetics of elimination of DCA, thus knowledge of the factors that influence the rate and extent of GSTZ1-1-catalyzed DCA metabolism in people is important for safe and effective dosing of patients.
Relevant to the effects of DCA treatment, glyoxylate can be further metabolized to carbon dioxide via a carboligase enzyme, to glycine by one or more aminotransferase enzymes or can be oxidized to oxalate [35]. Evidence for all three pathways was gained from studies in which rats were dosed with DCA [35]. Of these pathways, only the formation of oxalate could potentially be deleterious to a patient, however in the rat, formation of oxalate was a minor pathway [35]. Critically ill adult patients with lactic acidosis who were treated with intravenous DCA or placebo in a randomized controlled clinical trial were assessed for oxalate stones and oxalate-related toxicity; no evidence was found for differences between the DCA and placebo groups [50]. Similarly, oral administration of DCA for several years in a trial of children with congenital forms of lactic acidosis did not reveal evidence of oxalate stones or toxicity [51].
Polymorphic variants of GSTZ1
Human GSTZ1 (NM_145870) was suspected to exhibit allelic variants from the time it was discovered [52]. Subsequent studies have revealed SNPs in the coding and noncoding regions of the gene. We are still learning about the effects, if any, of most of these SNPs on GSTZ1 expression, stability and activity with DCA. This is an active area of investigation, as these factors impact the pharmacokinetics and ultimately the safe use of DCA. SNPs that lead to amino acid changes in the expressed protein have received attention, as well as SNPs that appear to affect translation of GSTZ1 and ultimately expression levels of the enzyme protein.
Variants in the amino acid sequence of the expressed enzyme protein
Three common SNPs were identified in the coding region of GSTZ1 [53,54], which have been observed to give rise to five haplotypes (Table 2). These five haplotypes are found in people with varying frequencies, depending on ethnicity (Figure 2), however the most frequently found variant in all populations studied to date, present in about 50% of people, is GSTZ1C [54–56]. This haplotype is also referred to as EGT, highlighting the variant amino acids at positions 32, 42 and 82 in the enzyme protein [54,55]. At present, there is relatively little information on the incidence of the five common haplotypes in populations other than Caucasians. Expressed recombinant human GSTZ1A-1A, 1B-1B, 1C-1C and 1D-1D proteins have been studied with DCA as substrate, and it was found that GSTZ1A-1A (KRT/KRT) had higher activity than the other variants [54,57]. GSTZ1F is a rare variant (Table 2) and the properties of the recombinant protein have not been studied. An individual person may be homo- or heterozygous for these haplotypes. For heterozygotes, it is not known if one variant is preferentially expressed at the protein level, however liver cytosol fractions prepared from individuals carrying the GSTZ1A variant on one allele have higher in vitro activity with DCA than those without this allele [56], suggesting the possibility of preferential expression of the KRT variant enzyme protein.
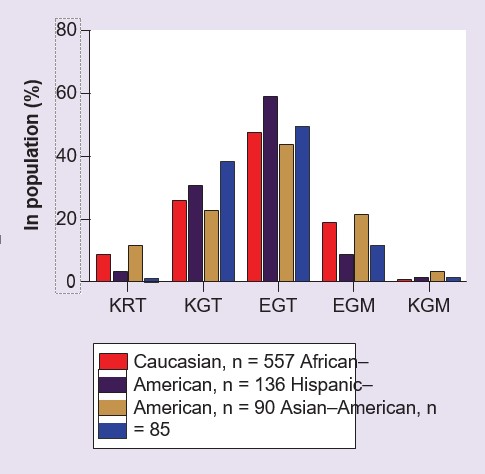
E: Glutamic acid; G: Glycine; K: Lysine: M: Metionine; T: Threonine.
| Variant | Nucleotide position | Amino acid position | Percentage in population(%)† | ||||
| 94 | 124 | 245 | 32 | 42 | 82 | ||
| GSTZ1A | A | A | C | Lys (K) | Arg (R) | Thr (T) | 1–10 |
| GSTZ1B | A | G | C | Lys (K) | Gly (G) | Thr (T) | 25–35 |
| GSTZ1C | G | G | C | Glu (E) | Gly (G) | Thr (T) | 45–55 |
| GSTZ1D | G | G | T | Glu (E) | Gly (G) | Met (M) | 10–20 |
| GSTZ1F | A | G | T | Lys (K) | Gly (G) | Met (M) | <1 |
The GSTZ1-1 human polymorphic variants and their approximate frequency in the population.
†The incidence of each haplotype varies with ethnic group, see Figure 2 and [54–56].
Glu (E): Glutamic acid; Gly (G): Glycine; Lys (K): Lysine: Met (M): Methionine; Thr (T): Threonine.
.
Studies with the expressed recombinant human GSTZ1A-1A, 1B-1B, 1C-1C and 1D-1D proteins also examined activity with the physiologically important substrate, MA [54]. The rates of MA metabolism showed a different pattern from activities measured with DCA as substrate in that the GSTZ1C-1C (EGT/EGT) variant had the highest activity with MA and GSTZ1A-1A the lowest [54].
As well as the commonly found variants described above, there is evidence for two other coding sequence variants. One of them was found in the expressed sequence tag database and was named GSTZ1E [54], however this variant, which would have proline in place of leucine at position 23, has not been found in people. Another variant was found in a volunteer for pharmacokinetic studies of DCA. This person, who was heterozygous for KGT/KGM (1B/1F), eliminated even a single dose of DCA extremely slowly; sequencing of the person’s DNA revealed a novel SNP in exon five of GSTZ1, which resulted in methionine instead of valine at position 99 of the protein [55]. Based on the crystal structure of GSTZ1B [58], position 99 is located in an α-helical bundle. Despite seeking this variant in liver samples that exhibited low activity with DCA [56], no other individuals with this polymorphism have yet been identified.
Promoter region polymorphisms
Ten SNPs in genomic DNA samples from African and Australian European subjects were found in a region that spanned 1500 nucleotides upstream of the transcription start site of GSTZ1, assumed to be the promoter region of the gene [59]. Two of these SNPs, -1002G>A and -289C>T were associated respectively with decreases and increases in promoter activity, as assessed in HepG2 cells. Subsequent studies with DNA from liver samples showed that in Caucasian–Americans but not African–Americans the A allele of SNP rs 7975, which resulted in expression of lysine instead of glutamic acid at position 32 of the protein, was in linkage disequilibrium with the A allele of the promoter region SNP -1002 G>A, rs7160195, and resulted in reduced expression of GSTZ1-1 protein with lysine (K) at position 32 [60]. The reduced expression was observed in heterozygotes as well as homozygotes for GSTZ1A (KRT), GSTZ1B (KGT) and GSTZ1F (KGM) in Caucasian–Americans [60]. There was no effect on expression of K-containing GSTZ1-1 variants in liver samples studied from African–Americans. For reasons that are not yet clear, there were no differences in the mRNA levels of GSTZ1 between haplotypes, pointing out a disconnect between results for protein expression and mRNA expression [60]. Finding lower expression of the GSTZ1-1 protein for K-containing haplotypes in Caucasians suggests that there will be reduced rates of initial metabolism of DCA in these individuals (except those with KRT). Furthermore, this raises the possibility that Caucasians with K-containing haplotypes who are treated with DCA will experience less efficient re-synthesis of the GSTZ1-1 protein, leading to even slower elimination than people without this haplotype.
Influence of GSTZ1 variants on DCA pharmacokinetics in people
Assuming that the side effects of chronic DCA treatment are related to accumulation of either DCA itself or the reactive endogenous substrates for GSTZ1, MAA and MA, there is a convincing argument that the safe clinical use of DCA should be guided by an understanding of the variability in elimination of the drug in people during chronic treatment. The dosage of DCA could then be reduced in those who exhibit slow elimination, while maintaining effective blood levels. If it were possible to predict who would eliminate DCA slowly, based on their genetic make-up, a diagnostic test could be administered to obtain the genotype for GSTZ1, and dose could be based on genotype. This would be less expensive than repeatedly measuring DCA levels in blood. To this end, the pharmacokinetics of DCA have been studied in human volunteers and in patients. In one human volunteer study, participants were genotyped and the pharmacokinetics of DCA were studied after a single dose of 25 mg/kg or five consecutive daily doses of 25 mg/kg/day [55]. As shown in Table 3, persons with at least one EGT variant of GSTZ1 showed greater clearance of DCA after five doses than those who had no EGT in their genotype. Figure 3 illustrates the kinetics of DCA elimination from plasma in one rapid and one slow metabolizer of DCA from this study [55]. Similar results were found in patients with genetic mitochondrial disease who received 12.5 mg/kg DCA every 12 h for 6, 12 or 30 months [55,61]. This work also shows that the levels of MA excreted in the urine, another measure of reduced activity of GSTZ1/MAAI, were higher in volunteers and patients with no EGT in their genotype (Table 3).
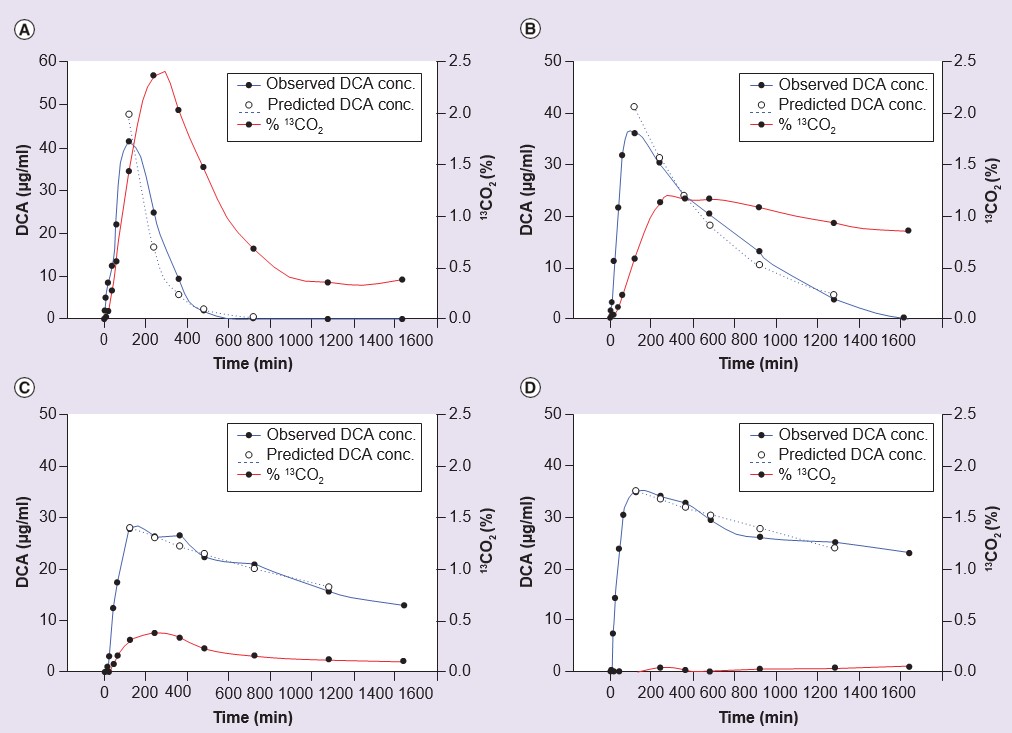
conc.: Concentration; DCA: Dichloroacetate.
| Sunject, duration of DCA treatment | Plasma clearance, mml/min | Maleylacetone excretion, μg/g creatinine | Ref. | ||
| EGT carrier | EGT noncarrier | EGT carrier | EGT noncarrier | ||
| Volunteers, 5 days | 2.22 ± 0.72 (7) | 0.73 ± 0.84* (5) | Not detected | 7.2 ± 4.1* | [55] |
| Patients, 12 months | 2.16 ± 0.99 (4) | 0.91, 0.17 | – | – | [55] |
| Patients, 6 months | 1.90 ± 1.13 (11) | 0.53 ± 0.35* (6) | 1.2 ± 0.9 | 6.9 ± 2.6* | [61] |
| Patients, 30 months | 2.08 ± 1.10 (11) | 0.67 ± 0.45* (6) | 1.9 ± 1.1 | 5.5 ± 1.2* | [61] |
Values shown are mean ± standard deviation (n) or individual values where n < 3.
*Significantly different from EGT carriers, p < 0.05.
The ethnic origin of the volunteers was as follows:
EGT carriers: four Caucasian–American, two African–American, one Asian–American; EGT noncarriers: five Caucasian–American. The ethnicities of the patients were not available.
DCA: Dichloroacetate.
Analysis of the data from the studies of DCA pharmacokinetics in volunteers suggested that, as found in vitro (see below), individuals carrying the GSTZ1A variant, coding for KRT, on one allele exhibited more rapid initial clearance of DCA than others. These persons also showed a greater decline in clearance after five doses. The ratio of clearance from the first to fifth dose was 3.6 ± 0.8 (mean ± standard error of the mean, n = 5) in those with the variant for GSTZ1C (coding for EGT) but not GSTZ1A, and was 18.2 ± 9.3 (n = 5) in those with the GSTZ1A variant on at least one allele. An individual who was homozygous for GSTZ1A exhibited the greatest change from the first to fifth dose, with clearance dropping from 16.7 to 0.31 ml/min [55]. The difference in mean change in clearance was not statistically significant, because of high variability in the group that was homo- or hetero-zygous for GSTZ1A, however it pointed out a trend that was in agreement with in vitro studies of the susceptibility of the different haplotypes to inactivation by DCA.
In vitro studies of DCA metabolism
Further insight into the role of genotype in DCA metabolism has been gained from studies of GSTZ1 expression and activity with DCA in human liver cytosol and mitochondria.
Ontogeny of activity
Studies with human liver cytosol prepared from a panel of 230 liver samples from donors covering the age range of gestation day 42 up to 84 years have shown very low or undetectable expression and activity of GSTZ1 prenatally as well as in the first 2 weeks of life: thereafter, activity and expression rise gradually [56]. Expression and activity attained levels in the adult range by age 7 years, however considerable interindividual variability was found. Measurement of activity with DCA and expression of GSTZ1 protein in these samples revealed that samples from individuals with the GSTZ1A variant on one allele had a higher ratio of activity/expression than all others (Figure 4). This agreed with findings from expressed recombinant human GSTZ1A-1A that showed higher activity with DCA for this variant [54], and with pharmacokinetic studies in volunteers where individuals with one GSTZ1A gene had rapid initial clearance of DCA [55]. Although the developmental study showed that activity in children under age 7 years was lower than adults, consideration of the larger size of children’s livers relative to body mass means that per kilogram body weight, activity with DCA is similar in adults and children aged 2 months to 7 years [56].
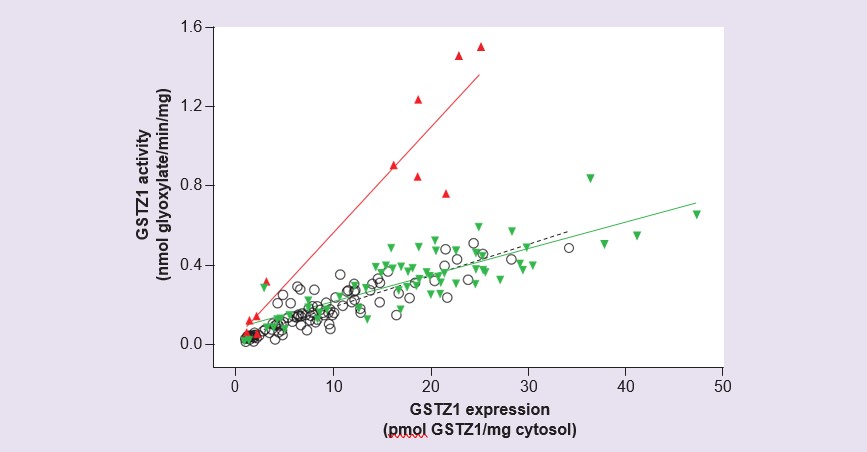
Effect of chloride on inactivation of GSTZ1 by DCA
Apparently at odds with the observation that the KRT protein is associated with more rapid metabolism of DCA in vitro [56], and more rapid elimination of a single dose of DCA [55], is the finding that those with the GSTZ1A variant on one allele showed a greater reduction in clearance after repeated DCA doses than those who were homozygous for GSTZ1C. Recent findings of the effect of chloride on GSTZ1-1 inactivation could explain these discrepancies. It was found that chloride protects GSTZ1-1 from inactivation by DCA at physiologically relevant concentrations in the liver [62,63], however the GSTZ1-1 in liver cytosol of those who were heterozygous for GSTZ1A was less protected from inactivation by physiological (38 mM) chloride than those with other GSTZ1 allelic variants (Table 4). The effect of chloride was such that the GSTZ1-1 enzymes in livers from individuals with the GSTZ1A variant on one allele were inactivated twice as quickly as those with other haplotypes. The exact mechanism of the chloride effect has yet to be determined, but this phenomenon is one explanation for the greater loss in clearance of DCA following chronic dosing in persons with one or more copies of the GSTZ1A variant. As noted above, the lower expression of enzyme in Caucasian individuals with lysine (K) at position 32 of the protein is likely to contribute to the very low clearance.
.
| Haplotype | EC50 mM | Inactivation half-life, h without Cl– | Inactivation half-life, h with 38 mM Cl– |
|---|---|---|---|
| EGT/EGT | 15 ± 3.1 (3) | 0.53, 0.49 | 5.73, 5.02 |
| KRT/EGT | 36 ± 2.2 (3) | 0.38, 0.38 | 2.66, 2.37 |
| EGM/EGM | 16.9 | – | 5.55 |
EC50 is the concentration of chloride that protected half the cytosolic GSTZ1 from inactivation following incubation for 2 h with Na DCA, 0.5 mM.
Conclusion
Because of its ability to inhibit PDH kinase, DCA has applications for treatment of cancer and metabolic diseases. Upon chronic administration of DCA to people, its metabolism and elimination is reduced. The reason for this reduction in clearance after multiple DCA doses is that the only enzyme known to metabolize DCA, GSTZ1-1, is inactivated during DCA metabolism. This mechanism-based inhibition of GSTZ1-1 by DCA results in the slowed clearance not only of DCA, but also of the endogenous substrates MAA and MA; whether accumulation of DCA and/or reactive tyrosine catabolites mediates the reversible peripheral neuropathy that occurs in some patients remains unresolved. Another unresolved possible cause of the neuropathy is the modest increase in ∂-ALA that follows chronic DCA treatment. Pertinent to an understanding of DCA pharmacokinetics is that the GSTZ1 gene exhibits polymorphic variants in all human populations studied to date. The properties of the GSTZ1-1 variant enzyme proteins differ with respect to their ability to metabolize DCA and the endogenous substrates MAA and MA, especially upon chronic administration. While all GSTZ1-1 variants are inactivated by DCA, one variant, which has lysine instead of glutamic acid at position 32 and arginine instead of glycine at position 42 of the protein, is more rapidly inactivated than other variants in the presence of physiological concentrations of chloride. In Caucasians, but not in persons of African ethnicity, GSTZ1 variants that have lysine at position 32 are linked with a promoter-region SNP in the GSTZ1 gene that results in reduced expression of GSTZ1-1 protein; this may also present as a slow metabolizer phenotype. An understanding of the factors that influence DCA pharmacokinetics under chronic administration is required to select the right dose and dosing interval to safely and effectively administer this drug to patients.
Future perspective
It is clear from studies to date that GSTZ1 haplotype is an important factor that determines the clearance of DCA. A greater understanding of the regulation of the enzyme under physiological conditions and in the presence of DCA will lead to safer and more effective use of the drug in the future. Several questions remain to be explored. In heterozygotes, is one variant of the protein preferentially expressed? Activity studies that show higher activity with DCA for liver cytosol fractions from individuals who are heterozygous for GSTZ1A/GSTZ1C suggest the 1A (KRT) enzyme protein variant is preferentially expressed in heterozygotes, but this has not been demonstrated. This question has practical implications, since the 1A variant is inactivated more rapidly than the 1C variant. Should a lower dose of DCA be administered to individuals who are likely to clear DCA more slowly than average on chronic administration? If so, should this be all those who are not homozygous or heterozygous for GSTZ1C? Should Caucasians with at least one K-containing variant (KRT, KGT, KGM) be given lower doses of DCA than those of African ethnicity because they express less GSTZ1-1? What is the evidence for low expression of K-containing variants in other populations? Are there population differences in the distribution patterns of the major haplotypes? Are the effects of pharmacogenetic differences the same in children as in adults? Children taking DCA generally clear the drug more rapidly than adults, but possible pharmacogenetic effects have not been extensively investigated. Further research is needed to address these questions, so as to ensure safe and effective dosing of DCA.
Financial & competing interests disclosure
The authors’ work discussed in this manuscript was funded in part by the US Public Health Service 1RO1 GM 099871, in part by the Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore and University of Miami (NO1 HD90011) and in part by the NIH/NCATS Clinical and Translational Science award to the University of Florida UL1 TR000064. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Executive summary
- Selection of an effective but nontoxic dose of dichloroacetate (DCA) is important for the long-term clinical use of DCA in treatment of cancer and other metabolic diseases.
- The gene for the single enzyme that metabolizes this drug, GSTZ1-1, exhibits polymorphisms that result in slow and rapid metabolizer phenotypes following long-term treatment with DCA.
- Coding region SNPs in the GSTZ1 gene result in five common haplotypes, with varying frequencies in populations. Individuals who are homozygous for the most common, GSTZ1C, are rapid metabolizers. There are indications that haplotypes coding for GSTZ1-1 proteins with K at position 32 will be slow metabolizers, but other factors contributing to slow metabolism following multiple DCA doses are not fully understood.
- A promoter region SNP in people with Caucasian but not African ethnicity is linked with a coding region SNP that results in substitution of lysine for glutamic acid at position 32 of the GSTZ1 protein, and is associated with reduced expression of the enzyme protein in liver.
- In the presence of DCA, the stability of the GSTZ1-1 enzyme varies with the concentration of chloride. The GSTZ1A variant enzyme (KRT) is inactivated more rapidly than the common GSTZ1C variant (EGT) under physiological chloride concentrations in liver.
REFERENCES
1 Stacpoole PW. Review of the pharmacologic and therapeutic effects of diisopropylammonium dichloroacetate (DIPA). J. Clin. Pharmacol. J. New Drugs 9(5), 282–291 (1969).2 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism 38(11), 1124–1144 (1989).
3 Stacpoole PW, Felts JM. Diisopropylammonium dichloroacetate (DIPA) and sodium dichloracetate (DCA): effect on glucose and fat metabolism in normal and diabetic tissue. Metabolism 19(1), 71–78 (1970).
4 Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N. Engl. J. Med. 298(10), 526–530 (1978).
5 Stacpoole PW, Harwood HJ Jr, Varnado CE. Regulation of rat liver hydroxymethylglutaryl coenzyme A reductase by a new class of noncompetitive inhibitors. Effects of dichloroacetate and related carboxylic acids on enzyme activity. J. Clin. Invest. 72(5), 1575–1585 (1983).
6 Harwood HJ Jr, Bridge DM, Stacpoole PW. In vivo regulation of human mononuclear leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase. Studies in normal subjects. J. Clin. Invest. 79(4), 1125–1132 (1987).
7 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA, Stacpoole PW. Reduction of serum cholesterol in two patients with homozygous familial hypercholesterolemia by dichloroacetate. Atherosclerosis 33(3), 285–293 (1979).
8 Eichner HL, Stacpoole PW, Forsham PH. Treatment of streptozotocin diabetes with di-isopropylammonium dichloroacetate (DIPA). Diabetes 23(3), 179–182 (1974).
9 Backshear PJ, Holloway PA, Alberti KG. Metabolic interactions of dichloroacetate and insulin in experimental diabetic ketoacidosis. Biochem. J. 146(2), 447–456 (1975).
10 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (short communication). Biochem. J. 134(2), 651–653 (1973).
11 Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 141(3), 761–774 (1974).
12 Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34(Pt 2), 217–222 (2006).
13 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329(Pt 1), 191–196 (1998).
14 Morten KJ, Caky M, Matthews PM. Stabilization of the pyruvate dehydrogenase E1alpha subunit by dichloroacetate. Neurology 51(5), 1331–1335 (1998).
15 Han Z, Berendzen K, Zhong L et al. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol. Genet. Metab. 93(4), 381–387 (2008).
16 Evans OB, Stacpoole PW. Prolonged hypolactatemia and increased total pyruvate dehydrogenase activity by dichloroacetate. Biochem. Pharmacol. 31(7), 1295–1300 (1982).
17 Denton RM, Randle PJ, Bridges BJ et al. Regulation of mammalian pyruvate dehydrogenase. Mol. Cell. Biochem. 9(1), 27–53 (1975).
18 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284(5), E855–E862 (2003).
19 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J. Clin. Pharmacol. 43(7), 683–691 (2003).
20 Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim. Biophys. Acta 1846(2), 617–629 (2014).
21 Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine-both or neither? Environ. Health Perspect. 119(2), 155–158 (2011).
22 Chu PI. Pharmacokinetics of sodium dichloroacetate. Ph.D. dissertation, Pharmaceutics Department, University of Florida, FL, USA (1987).
23 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab. Rev. 30(3), 499–539 (1998).
24 Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin. Pharmacol. Ther. 37(1), 89–93 (1985).
25 Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G et al. Pharmacokinetics of oral dichloroacetate in dogs. J. Biochem. Mol. Toxicol. 27(12), 522–525 (2013).
26 Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 19(2), 109–113 (1980).
27 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N. Engl. J. Med. 309(7), 390–396 (1983).
28 Henderson GN, Curry SH, Derendorf H, Wright EC, Stacpoole PW. Pharmacokinetics of dichloroacetate in adult patients with lactic acidosis. J. Clin. Pharmacol. 37(5), 416–425 (1997).
29 Stacpoole PW, Kerr DS, Barnes C et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117(5), 1519–1531 (2006).
30 Stacpoole PW, Gilbert LR, Neiberger RE et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121(5), e1223–e1228 (2008).
31 Kaufmann P, Engelstad K, Wei Y et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66(3), 324–330 (2006).
32 Stickler DE, Valenstein E, Neiberger RE et al. Peripheral neuropathy in genetic mitochondrial diseases. Pediatr. Neurol. 34(2), 127–131 (2006).r
33 Kaufmann P, Pascual JM, Anziska Y et al. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch. Neurol. 63(5), 746–748 (2006).
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J. Pharmacol. Exp. Ther. 324(3), 1163–1171 (2008).
35 James MO, Yan Z, Cornett R et al. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos. 26(11), 1134–1143 (1998).
36 Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell. Biol. 45(6), 1121–1132 (2013).
37 Vessey DA, Kelley M, Warren RS. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim. Biophys. Acta 1428(2–3), 455–462 (1999).
38 James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Glutathione-dependent conversion to glyoxylate, a major pathway of dichloroacetate biotransformation in hepatic cytosol from humans and rats, is reduced in dichloroacetate-treated rats. Drug Metab. Dispos. 25(11), 1223–1227 (1997).
39 Tong Z, Board PG, Anders MW. Glutathione transferase zeta catalyses the oxygenation of the carcinogen dichloroacetic acid to glyoxylic acid. Biochem. J. 331(Pt 2), 371–374 (1998).
49 Fernandez-Canon JM, Penalva MA. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J. Biol. Chem. 273(1), 329–337 (1998).
41 Blackburn AC, Woollatt E, Sutherland GR, Board PG. Characterization and chromosome location of the gene GSTZ1 encoding the human Zeta class glutathione transferase and maleylacetoacetate isomerase. Cytogenet. Cell. Genet. 83(1–2), 109–114 (1998).
42 Li W, James MO, Mckenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrion as a novel site of dichloroacetate biotransformation by glutathione transferase zeta 1. J. Pharmacol. Exp. Ther. 336(1), 87–94 (2011).
43 Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Immunohistochemical localization and activity of glutathione transferase zeta (GSTZ1-1) in rat tissues. Drug Metab. Dispos. 30(6), 616–625 (2002).
44 Shangraw RE, Fisher DM. Pharmacokinetics of dichloroacetate in patients undergoing liver transplantation. Anesthesiology 84(4), 851–858 (1996).
45 Cornett R, James MO, Henderson GN, Cheung J, Shroads AL, Stacpoole PW. Inhibition of glutathione S-transferase zeta and tyrosine metabolism by dichloroacetate: a potential unifying mechanism for its altered biotransformation and toxicity. Biochem. Biophys. Res. Commun. 262(3), 752–756 (1999).
46 Guo X, Dixit V, Liu H et al. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 34(1), 36–42 (2006).
47 Anderson WB, Board PG, Gargano B, Anders MW. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem. Res. Toxicol. 12(12), 1144–1149 (1999).
48 Anderson WB, Liebler DC, Board PG, Anders MW. Mass spectral characterization of dichloroacetic acid-modified human glutathione transferase zeta. Chem. Res. Toxicol. 15(11), 1387–1397 (2002).
49 Dixit V. Inactivation of glutathione transferase zeta by dichloroacetic acid. PhD dissertation, Medicinal Chemistry Department, University of Florida, FL, USA (2005).
50 Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N. Engl. J. Med. 327(22), 1564–1569 (1992).
51 Abdelmalak M, Lew A, Ramezani R et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol. Genet. Metab. 109(2), 139–143 (2013).
52 Board PG, Baker RT, Chelvanayagam G, Jermiin LS. Zeta, a novel class of glutathione transferases in a range of species from plants to humans. Biochem. J. 328(Pt 3), 929–935 (1997).
53 Blackburn AC, Tzeng HF, Anders MW, Board PG. Discovery of a functional polymorphism in human glutathione transferase zeta by expressed sequence tag database ana-lysis. Pharmacogenetics 10(1), 49–57 (2000).
54 Blackburn AC, Coggan M, Tzeng HF et al. GSTZ1d: a new allele of glutathione transferase zeta and maleylacetoacetate isomerase. Pharmacogenetics 11(8), 671–678 (2001).
55 Shroads AL, Langaee T, Coats BS et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J. Clin. Pharmacol. 52(6), 837–849 (2012).
56 Li W, Gu Y, James MO et al. Prenatal and postnatal expression of glutathione transferase zeta 1 in human liver and the roles of haplotype and subject age in determining activity with dichloroacetate. Drug Metab. Dispos. 40(2), 232–239 (2012).Google Scholar
57 Board PG, Anders MW. Glutathione transferase zeta: discovery, polymorphic variants, catalysis, inactivation, and properties of Gstz1-/- mice. Drug Metab. Rev 43(2), 215–225 (2011).
58 Polekhina G, Board PG, Blackburn AC, Parker MW. Crystal structure of maleylacetoacetate isomerase/glutathione transferase zeta reveals the molecular basis for its remarkable catalytic promiscuity. Biochemistry 40(6), 1567–1576 (2001).
59 Fang YY, Kashkarov U, Anders MW, Board PG. Polymorphisms in the human glutathione transferase zeta promoter. Pharmacogenet. Genomics 16(5), 307–313 (2006).
60 Langaee TY, Zhong G, Li W et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics 25(5), 239–245 (2015).
61 Shroads AL, Coats BS, Mcdonough CW, Langaee T, Stacpoole PW. Haplotype variations in glutathione transferase zeta 1 influence the kinetics and dynamics of chronic dichloroacetate in children. J. Clin. Pharmacol. 55(1), 50–55 (2015).
62 Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Chloride and other anions inhibit dichloroacetate-induced inactivation of human liver GSTZ1 in a haplotype-dependent manner. Chem. Biol. Interact. 215C, 33–39 (2014).
63 Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Chloride concentrations in human hepatic cytosol and mitochondria are a function of age. Biochem. Biophys. Res. Commun. 459(3), 463–468 (2015).