Margaret O James*,1 y Peter W Stacpoole2,3
1 Departamento de Química Médica, Universidad de Florida, Gainesville, FL 32610-0485, EE.UU.
2 Departamento de Medicina, Facultad de Medicina, Universidad de Florida, Gainesville, FL 32610-0485, EE.UU.
3 Departamento de Bioquímica y Biología Molecular, Universidad de Florida, Gainesville, FL 32610-0485, EE.UU
Correspondencia: Tel: +1 352 273 7707 Email: [email protected]
Enviado: 16 de diciembre de 2015
Aceptado: 17 de febrero de 2016
Publicado: 4 de mayo de
Resumen
El fármaco en investigación dicloroacetato (DCA) es un regulador metabólico que se ha utilizado con éxito para tratar enfermedades metabólicas adquiridas y congénitas y, recientemente, tumores sólidos. Su uso clínico ha revelado retos en la selección de las dosis adecuadas. La administración crónica de DCA conduce a la inhibición del metabolismo del DCA y a una posible acumulación a niveles que provocan efectos secundarios. Esto se debe a que la conversión de DCA en glioxilato es catalizada por una enzima, la glutatión transferasa zeta 1 (GSTZ1-1), que es inactivada por el DCA. Los SNP en el gen GSTZ1 dan lugar a la expresión de variantes polimórficas de la enzima que difieren en la actividad y las tasas de inactivación por DCA en condiciones fisiológicas: estas propiedades conducen a una variación considerable entre las personas en la farmacocinética de DCA.
Palabras clave: dicloroacetato; GSTZ1; farmacogenética
Uso clínico del dicloroacetato
Para ser una molécula tan sencilla, el DCA tiene una cartera farmacológica notablemente rica y diversa, que data de casi un siglo [1,2]. Sin embargo, su uso moderno como fármaco de investigación comenzó en 1970, cuando se descubrió su capacidad selectiva para reducir los niveles de glucosa en sangre en animales diabéticos, pero no en los no diabéticos [3 ], y posteriormente se confirmó en humanos [4]. Durante las décadas de 1970 y 1980 se identificaron muchas propiedades metabólicas del DCA relacionadas con el metabolismo de la glucosa y los lípidos. Sin embargo, la mayoría de sus efectos farmacológicos pueden destilarse en unos pocos lugares y mecanismos de acción básicos. En primer lugar, el DCA es un inhibidor no competitivo de la enzima del retículo endoplásmico HMG CoA reductasa, que cataliza el paso que limita la velocidad en la biosíntesis del colesterol. El efecto inhibidor del DCA se observa tanto en el hígado de roedores [5] como en los leucocitos humanos [6]; es probable que explique la reducción del colesterol total y de las lipoproteínas de baja densidad (LDL) asociada al fármaco en pacientes con hipercolesterolemia familiar homocigótica con receptor LDL negativo [7] y su designación como primer producto huérfano para esta enfermedad rara. En segundo lugar, el DCA inhibe la síntesis hepática de triglicéridos de novo en roedores no diabéticos [ 5] y disminuye los niveles circulantes de triglicéridos y de lipoproteínas de muy baja densidad en pacientes con diabetes mellitus de tipo 2 [4]. También disminuye los cuerpos cetónicos sanguíneos en ratas con cetoacidosis diabética inducida experimentalmente [8,9]. Se desconocen los mecanismos precisos que subyacen a estos efectos sobre la síntesis y oxidación de lípidos. En tercer lugar, el DCA estimula el PDC mitocondrial, que oxida irreversiblemente el piruvato a acetil coenzima A (acetil CoA) [10], una propiedad compartida por otros ácidos grasos halogenados de cadena corta [11]. Es la capacidad del DCA para alterar la actividad de la PDC lo que ha generado, con diferencia, la mayor investigación experimental y clínica sobre esta molécula inusual.
La PDC se regula postraduccionalmente principalmente mediante la fosforilación reversible de uno o más de tres residuos de serina en la subunidad E1α de la primera enzima PDH de la PDC que descarboxila el piruvato [12]. Cuatro PDH quinasas (PDK1-4) y dos PDH fosfatasas (PDP 1 y 2) llevan a cabo este aspecto regulador de la PDC en humanos, en los que la enzima fosforilada es catalíticamente inactiva. Las PDK se expresan de forma diferencial en los tejidos, aunque la PDK2 se expresa de forma ubicua [13]. El piruvato, el sustrato de la reacción PDC, inhibe las PDKs uniéndose a un pequeño bolsillo en el N-terminal de la quinasa, lo que conduce a la estimulación de la actividad PDC. Por el contrario, la acumulación de los productos de la reacción, acetil CoA y NADH, provoca la activación de las PDK y la inhibición de la PDC. El DCA, un análogo estructural del piruvato, también se une al sitio de unión del piruvato, resultando en la inhibición de las PDKs con el orden de inhibición siendo PDK2>PDK1˜PDK4>>PDK3 [13]. La exposición crónica al fármaco in vitro [14,15] e in vivo [16] puede estabilizar la PDC y disminuir su recambio, proporcionando un segundo mecanismo por el cual el DCA aumenta la actividad del complejo y una explicación para sus efectos farmacológicos prolongados tras la retirada del fármaco [4].
Aunque el DCA es un inhibidor de PDK relativamente débil (Ki 0.2 mM para PDK2), sus efectos sobre aspectos del metabolismo intermediario son profundos y se relacionan directamente con el papel fundamental que juega la PDC en la regulación de la selección de combustible y la bioenergética celular eucariótica [17,18]. El DCA es todavía un fármaco en investigación no aprobado por la FDA estadounidense. Sin embargo, sigue siendo ampliamente investigado como modulador metabólico para varios trastornos metabólicos congénitos y adquiridos, basándose en su acción estimuladora sobre la PDC (Tabla 1).
| Propiedad | Mecanismo | Ref. |
|---|---|---|
| Aumento de la OXPHOS y de la bioenergética | ↓ PDK ↑ PDC | [2,10] |
| Disminución de la glucosa en sangre en ayunas o diabetes | ↓ PDK ↑ PDC | [4] |
| Disminución del lactato en sangre y LCR | ↓ PDK ↑ PDC | [19] |
| Disminución del colesterol total y LDL en sangre | ↓ HMG CoA reductasa → ↓ síntesis de colesterol | [6,7] |
| Disminución de los triglicéridos en sangre y del colesterol VLDL | ↓ síntesis hepática de TG → ↓ síntesis de VLDL | [4] |
| Reversión del efecto Warburg en cáncer, HAP y otras condiciones proliferativas | ↓ PDK ↑ PDC | [20] |
LCR: Líquido cefalorraquídeo; HMG CoA: Hidroximetilglutaril coenzima A; LDL: lipoproteína de baja densidad; OXPHOS: Fosforilación oxidativa; HAP: Hipertensión arterial pulmonar; CDP: Complejo piruvato deshidrogenasa; PDK: Piruvato deshidrogenasa cinasa; TG: Triglicérido; VLDL: lipoproteína de muy baja densidad.
Farmacocinética y aclaramiento
El DCA oral se absorbe rápidamente y tiene una biodisponibilidad cercana a la unidad tras la dosificación oral y parenteral [21,22]. El fármaco puede detectarse en el plasma humano a los 15 minutos de una dosis oral de 50 mg/kg. En sujetos previamente ingenuos, el DCA oral o intravenoso tiene una semivida plasmática de aproximadamente 1 h [23]. La tasa de metabolismo entre especies sanas es: ratón>rata>humano≥perro [22-25].
Las primeras investigaciones farmacocinéticas de dosis únicas intravenosas de DCA describieron una cinética no lineal a dosis ≥35 mg/kg [26]. Posteriormente se observó que la tasa de aclaramiento plasmático del fármaco disminuía al repetir la dosis intravenosa de 50 mg/kg de peso corporal en adultos gravemente enfermos con acidosis láctica [27,28]. También se observó un retraso en el aclaramiento plasmático del fármaco administrado por vía oral a una dosis de 50 mg/kg en adultos sanos, que necesitaron semanas o meses antes de alcanzar la tasa de aclaramiento original con una segunda dosis [22].
Los principales avances en la comprensión de esta característica inusual de la farmacocinética del DCA surgieron de la investigación probatoria de su toxicología clínica. El DCA crónico puede causar una neuropatía periférica reversible en todas las especies [21,23], un efecto observado clínicamente por primera vez hace 25 años en un chico de 16 años que recibió 50 mg/kg/día durante aproximadamente 4 meses [7]. Por el contrario, en un ensayo controlado aleatorizado (ECA) de 43 niños pequeños con diversos tipos de enfermedades mitocondriales primarias (edad media al ingreso: 5,6 años), el DCA oral administrado a una dosis de 12,5 mg/kg cada 12 h durante 6 meses fue bien tolerado y no causó cambios adversos en la conducción eléctrica del nervio periférico, en comparación con el placebo [29], aunque se observó cierta disminución asintomática de la conducción nerviosa con una exposición más prolongada al fármaco [30]. Por el contrario, un ECA de 30 adolescentes mayores y adultos (edad media de entrada: 30 años) con una enfermedad mitocondrial genética que fueron expuestos a la misma dosis de DCA y a la misma pauta de dosis empleada en el ensayo pediátrico se interrumpió prematuramente debido al empeoramiento o a la nueva aparición de neuropatía periférica [31], una complicación común de las enfermedades mitocondriales [32,33]. Cuando se comparó la cinética del DCA en estas dos poblaciones de pacientes, se descubrió que el aclaramiento plasmático del DCA crónico era notablemente más lento en el grupo de más edad [34], un hallazgo confirmado en estudios con ratas [34,35]. Estos hallazgos dieron lugar a estudios sobre la enzimología de la biotransformación del DCA.
Importancia de la glutatión transferasa zeta 1 en la biotransformación del DCA
Los ácidos carboxílicos pequeños a menudo se glucuronidan [36] o se convierten en sus derivados de la coenzima A y luego en conjugados de aminoácidos [37], sin embargo no hay pruebas de que el DCA forme un glucurónido, un derivado de la coenzima A o un conjugado de aminoácidos [2, James MO,Datos no publicados]. En su lugar, el DCA es un sustrato para una enzima GST inusual, la GSTZ1-1, que lo convierte en glioxilato en una reacción que requiere pero no consume glutatión (GSH) [38,39]. Como otras GSTs, GSTZ1-1 es activa en forma dimérica, y la enzima activa es un homodímero. La decloración del DCA para formar glioxilato, catalizada por la GSTZ1-1, es la única vía de metabolismo primario del DCA observada en el hígado [35]. Además de catalizar el metabolismo del DCA, la GSTZ1-1, también conocida como MAAI, tiene una importante función fisiológica en el catabolismo de la tirosina, donde isomeriza el maleilacetoacetato (MAA) y la maleilacetona (MA) a fumarilacetoacetato y fumarilacetona en reacciones que requieren pero no consumen GSH (Figura 1) [40,41]. La biotransformación del DCA a glioxilato termina sus actividades farmacológica y terapéuticamente importantes que se han discutido anteriormente, por lo que la actividad de GSTZ1-1 controla la duración de la acción del DCA.
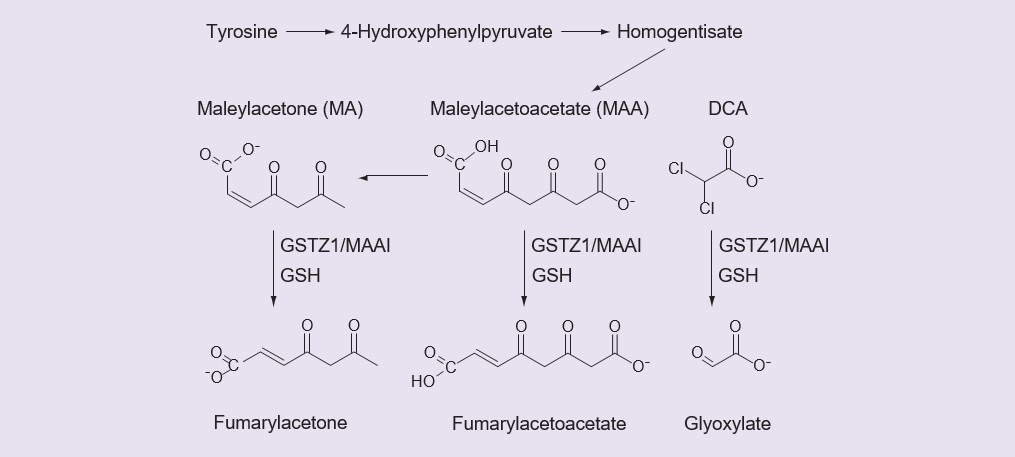
El metabolito inactivo del DCA, el glioxilato, se metaboliza en dióxido de carbono, glicina y oxalato. La fumarilacetona y el fumarilacetoacetato son metabolizados por la fumarilacetoacetato hidrolasa. DCA: Dicloroacetato: GSH: Glutatión; MA: Maleilacetona; MMA: Maleyacetoacetato.
El hígado es el principal lugar de expresión de GSTZ1-1, donde se encuentra tanto en el citosol como en la matriz mitocondrial [38,42]. Estudios en animales han demostrado niveles muy bajos de expresión de GSTZ1-1 en riñón, corazón, tracto gastrointestinal y cerebro [43]. El papel de los tejidos extrahepáticos en el metabolismo del DCA en las personas no se ha explorado en detalle; sin embargo, un estudio demostró que el DCA no se eliminaba de la sangre durante la fase anhepática de los pacientes con trasplante de hígado a los que se administró DCA para contrarrestar la acidosis láctica, lo que indica que no se produjo metabolismo [44]. La fase anhepática fue de corta duración (media: 73 min), por lo que los datos eran limitados, sin embargo este estudio sugiere un papel menor de los tejidos distintos del hígado en la eliminación del DCA, al menos para una dosis única.
Inhibición dela GSTZ1-1 por el DCA
Los estudios con animales y con proteínas GSTZ1-1 humanas recombinantes expresadas han demostrado que la razón por la que múltiples dosis de DCA se eliminan mucho más lentamente que una dosis única es que el DCA es un inhibidor de la GSTZ1-1 basado en un mecanismo [25,34,45-47]. Durante la biotransformación del DCA, se forman aductos a la proteína GSTZ1-1 que inactivan la proteína, y presumiblemente desencadenan su proteólisis [48,49]. La administración de DCA a ratas conduce a la pérdida de actividad y expresión de GSTZ1-1 de forma dependiente de la dosis y del tiempo: la pérdida de función de GSTZ1-1 fue más pronunciada y persistió más tiempo en ratas adultas en comparación con las jóvenes [34,35]. Este efecto de la edad sobre la pérdida de función de la GSTZ1-1 tras la administración de DCA se observó en personas, mostrando los niños un menor aumento de la semivida plasmática y una menor disminución del aclaramiento que los adultos [34]. Además de afectar a la farmacocinética del DCA, la pérdida de GSTZ1-1 (MAAI) provoca la acumulación de los sustratos fisiológicos MA y, presumiblemente, MAA, ambos moléculas reactivas capaces de formar aductos con macromoléculas celulares [34,45]. La inhibición de MAAI también conduce a la desviación del carbono de tirosina hacia la formación de succinilacetona. Esta molécula inhibe un paso proximal en la síntesis del hemo, provocando la acumulación de la molécula reactiva δ-aminolevulinato (δ-ALA). Tanto el MA como el δ-ALA aumentan en la sangre y/o la orina de los seres humanos expuestos crónicamente al DCA [34]: la importancia toxicológica clínica de este efecto aún está por demostrar.
Otros metabolitos del DCA
El único otro metabolito primario conocido del DCA es el ácido monocloroacético (MCA), que se ha encontrado ocasionalmente en cantidades traza en la sangre tras la administración de DCA [34]. El MCA parece formarse a partir del DCA en los glóbulos rojos [34]. No hay evidencia de la formación de MCA a partir de DCA en el hígado. A efectos prácticos, la conversión del DCA en glioxilato determina la farmacocinética de eliminación del DCA, por lo que el conocimiento de los factores que influyen en la tasa y el alcance del metabolismo del DCA catalizado por la GSTZ1-1 en las personas es importante para una dosificación segura y eficaz de los pacientes.
En relación con los efectos del tratamiento con DCA, el glioxilato puede metabolizarse en dióxido de carbono a través de una enzima carboligasa, en glicina por una o más enzimas aminotransferasas o puede oxidarse en oxalato [35]. Se obtuvieron pruebas de las tres vías a partir de estudios en los que se dosificó DCA a ratas [35]. De estas vías, sólo la formación de oxalato podría ser potencialmente deletérea para un paciente, sin embargo en la rata, la formación de oxalato fue una vía menor [35]. Los pacientes adultos en estado crítico con acidosis láctica que fueron tratados con DCA intravenoso o placebo en un ensayo clínico controlado aleatorizado fueron evaluados en busca de cálculos de oxalato y toxicidad relacionada con el oxalato; no se encontró evidencia de diferencias entre los grupos de DCA y placebo [50]. De forma similar, la administración oral de DCA durante varios años en un ensayo de niños con formas congénitas de acidosis láctica no reveló evidencia de cálculos de oxalato o toxicidad [51].
Variantes polimórficas de GSTZ1
Se sospechó que la GSTZ1 humana (NM_145870) presentaba variantes alélicas desde el momento de su descubrimiento [52]. Estudios posteriores han revelado SNPs en las regiones codificantes y no codificantes del gen. Todavía estamos aprendiendo sobre los efectos, si los hay, de la mayoría de estos SNPs en la expresión, estabilidad y actividad de GSTZ1 con DCA. Se trata de un área de investigación activa, ya que estos factores influyen en la farmacocinética y, en última instancia, en el uso seguro del DCA. Los SNPs que conducen a cambios de aminoácidos en la proteína expresada han recibido atención, así como los SNPs que parecen afectar a la traducción de GSTZ1 y, en última instancia, a los niveles de expresión de la proteína enzimática.
Variantes en la secuencia de aminoácidos de la proteína enzimática expresada
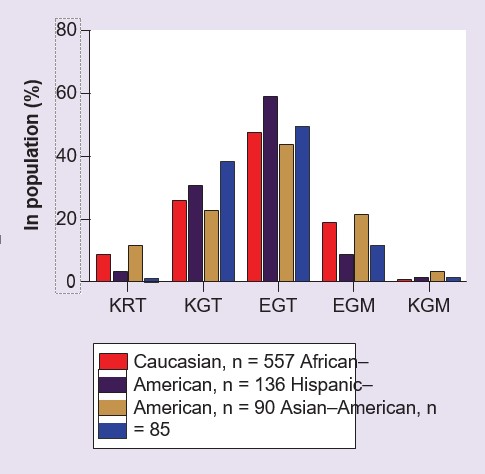
Se identificaron tres SNP comunes en la región codificante de GSTZ1 [53,54], que se ha observado que dan lugar a cinco haplotipos (Tabla 2). Estos cinco haplotipos se encuentran en personas con frecuencias variables, dependiendo de la etnia (Figura 2), sin embargo la variante más frecuentemente encontrada en todas las poblaciones estudiadas hasta la fecha, presente en alrededor del 50% de las personas, es GSTZ1C [54-56]. Este haplotipo también se conoce como EGT, destacando la variante de aminoácidos en las posiciones 32, 42 y 82 de la proteína enzimática [54,55]. En la actualidad, existe relativamente poca información sobre la incidencia de los cinco haplotipos comunes en poblaciones distintas de la caucásica. Se han estudiado las proteínas humanas recombinantes GSTZ1A-1A, 1B-1B, 1C-1C y 1D-1D expresadas con DCA como sustrato, y se descubrió que la GSTZ1A-1A (KRT/KRT) tenía mayor actividad que las otras variantes [54,57]. La GSTZ1F es una variante poco frecuente (Tabla 2) y no se han estudiado las propiedades de la proteína recombinante. Una persona puede ser homocigota o heterocigota para estos haplotipos. En el caso de los heterocigotos, se desconoce si una variante se expresa preferentemente a nivel proteico; sin embargo, las fracciones de citosol hepático preparadas a partir de individuos portadores de la variante GSTZ1A en un alelo presentan una mayor actividad in vitro con DCA que los que carecen de este alelo [56], lo que sugiere la posibilidad de una expresión preferente de la proteína enzimática de la variante KRT.
E: Ácido glutámico; G: Glicina; K: Lisina: M: Metionina; T: Treonina.
| Variante | Posición del nucleótido | Posición de aminoácidos | Porcentaje en la población(%)† | ||||
| 94 | 124 | 245 | 32 | 42 | 82 | ||
| GSTZ1A | A | A | C | Lys (K) | Arg (R) | Thr (T) | 1-10 |
| GSTZ1B | A | G | C | Lys (K) | Gly (G) | Thr (T) | 25-35 |
| GSTZ1C | G | G | C | Glu (E) | Gly (G) | Thr (T) | 45-55 |
| GSTZ1D | G | G | T | Glu (E) | Gly (G) | Met (M) | 10-20 |
| GSTZ1F | A | G | T | Lys (K) | Gly (G) | Met (M) | <1 |
Las variantes polimórficas humanas de GSTZ1-1 y su frecuencia aproximada en la población.
†Laincidencia de cada haplotipo varía con el grupo étnico, ver Figura 2 y [54-56].
Glu (E): Ácido glutámico; Gly (G): Glicina; Lys (K): Lisina: Met (M): Metionina; Thr (T): Treonina.
.
Los estudios con las proteínas humanas recombinantes GSTZ1A-1A, 1B-1B, 1C-1C y 1D-1D expresadas también examinaron la actividad con el sustrato fisiológicamente importante, el MA [54]. Las tasas de metabolismo de MA mostraron un patrón diferente de las actividades medidas con DCA como substrato en que la variante GSTZ1C-1C (EGT/EGT) tuvo la actividad más alta con MA y GSTZ1A-1A la más baja [54].
Además de las variantes comúnmente encontradas descritas anteriormente, existen evidencias de otras dos variantes en la secuencia de codificación. Una de ellas se encontró en la base de datos de etiquetas de secuencia expresada y se denominó GSTZ1E [54], sin embargo esta variante, que tendría prolina en lugar de leucina en la posición 23, no se ha encontrado en personas. Se encontró otra variante en un voluntario para estudios farmacocinéticos de DCA. Esta persona, que era heterocigota para KGT/KGM (1B/1F), eliminó incluso una sola dosis de DCA con extrema lentitud; la secuenciación del ADN de la persona reveló un nuevo SNP en el exón cinco de GSTZ1, que dio lugar a metionina en lugar de valina en la posición 99 de la proteína [55]. Según la estructura cristalina de GSTZ1B [58], la posición 99 está situada en un haz α-helicoidal. A pesar de haber buscado esta variante en muestras de hígado que presentaban una baja actividad con DCA [56], todavía no se ha identificado a ningún otro individuo con este polimorfismo.
Polimorfismos de laregiónpromotora
Se encontraron diez SNPs en muestras de ADN genómico de sujetos africanos y europeos australianos en una región que abarcaba 1500 nucleótidos aguas arriba del sitio de inicio de la transcripción de GSTZ1, que se supone que es la región promotora del gen [59]. Dos de estos SNPs, -1002G>A y -289C>T se asociaron respectivamente con disminuciones y aumentos de la actividad promotora, según se evaluó en células HepG2. Estudios posteriores con ADN de muestras de hígado demostraron que en los estadounidenses de raza blanca, pero no en los afroamericanos, el alelo A del SNP rs 7975, que daba lugar a la expresión de lisina en lugar de ácido glutámico en la posición 32 de la proteína, estaba en desequilibrio de ligamiento con el alelo A del SNP de la región promotora -1002 G>A, rs7160195, y daba lugar a una expresión reducida de la proteína GSTZ1-1 con lisina (K) en la posición 32 [60]. La expresión reducida se observó tanto en heterocigotos como en homocigotos para GSTZ1A (KRT), GSTZ1B (KGT) y GSTZ1F (KGM) en americanos caucásicos [60]. No hubo ningún efecto en la expresión de las variantes GSTZ1-1 que contienen K en las muestras de hígado estudiadas de afroamericanos. Por razones que aún no están claras, no hubo diferencias en los niveles de ARNm de GSTZ1 entre los haplotipos, lo que apunta a una desconexión entre los resultados de la expresión de proteínas y la expresión de ARNm [60]. Encontrar una menor expresión de la proteína GSTZ1-1 para los haplotipos que contienen K en caucásicos sugiere que habrá tasas reducidas de metabolismo inicial del DCA en estos individuos (excepto en aquellos con KRT). Además, esto plantea la posibilidad de que los caucásicos con haplotipos que contienen K que son tratados con DCA experimentarán una re-síntesis menos eficiente de la proteína GSTZ1-1, dando lugar a una eliminación aún más lenta que las personas sin este haplotipo.
Influencia de las variantes de GSTZ1 en la farmacocinética del DCA en personas
Suponiendo que los efectos secundarios del tratamiento crónico con DCA estén relacionados con la acumulación del propio DCA o de los sustratos endógenos reactivos para la GSTZ1, MAA y MA, existe un argumento convincente de que el uso clínico seguro del DCA debería guiarse por una comprensión de la variabilidad en la eliminación del fármaco en las personas durante el tratamiento crónico. La dosis de DCA podría entonces reducirse en aquellos que muestran una eliminación lenta, manteniendo al mismo tiempo niveles sanguíneos efectivos. Si fuera posible predecir quién eliminaría lentamente el DCA, basándose en su composición genética, podría administrarse una prueba diagnóstica para obtener el genotipo para GSTZ1, y la dosis podría basarse en el genotipo. Esto sería menos costoso que medir repetidamente los niveles de DCA en sangre. Con este fin, se ha estudiado la farmacocinética del DCA en voluntarios humanos y en pacientes. En un estudio con voluntarios humanos, se genotipificó a los participantes y se estudió la farmacocinética del DCA tras una dosis única de 25 mg/kg o cinco dosis diarias consecutivas de 25 mg/kg/día [55]. Como se muestra en la Tabla 3, las personas con al menos una variante EGT de GSTZ1 mostraron un mayor aclaramiento de DCA tras cinco dosis que las que no tenían EGT en su genotipo. La Figura 3 ilustra la cinética de eliminación del DCA del plasma en un metabolizador rápido y uno lento del DCA de este estudio [55]. Se encontraron resultados similares en pacientes con enfermedad mitocondrial genética que recibieron 12,5 mg/kg de DCA cada 12 h durante 6, 12 o 30 meses [55 ,61]. Este trabajo también muestra que los niveles de MA excretados en la orina, otra medida de la actividad reducida de GSTZ1/MAAI, fueron mayores en voluntarios y pacientes sin EGT en su genotipo (Tabla 3).
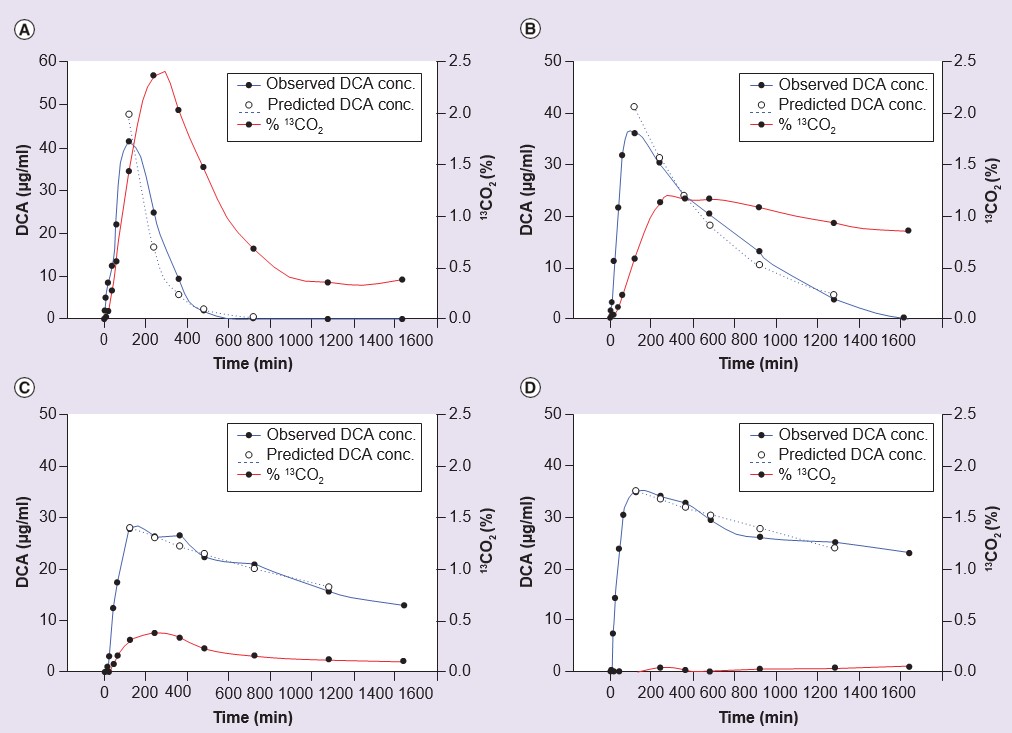
conc.: Concentración; DCA: Dicloroacetato.
| Sunject, duración del tratamiento con DCA | Aclaramiento plasmático, mml/min | Excreción de maleilacetona, μg/g creatinina | Ref. | ||
| Portador EGT | EGT no portador | Portador EGT | EGT no portador | ||
| Voluntarios, 5 días | 2.22 ± 0.72 (7) | 0.73 ± 0.84* (5) | No detectado | 7.2 ± 4.1* | [55] |
| Pacientes, 12 meses | 2.16 ± 0.99 (4) | 0.91, 0.17 | – | – | [55] |
| Pacientes, 6 meses | 1.90 ± 1.13 (11) | 0.53 ± 0.35* (6) | 1.2 ± 0.9 | 6.9 ± 2.6* | [61] |
| Pacientes, 30 meses | 2.08 ± 1.10 (11) | 0.67 ± 0.45* (6) | 1.9 ± 1.1 | 5.5 ± 1.2* | [61] |
Los valores mostrados son la media ± desviación estándar (n) o valores individuales donde n < 3.
*Significativamente diferente de los portadores de EGT, p < 0,05.
El origen étnico de los voluntarios fue el siguiente:
Portadores de EGT: cuatro caucásicos-americanos, dos afroamericanos, un asiático-americano; no portadores de EGT: cinco caucásicos-americanos. Las etnias de los pacientes no estaban disponibles.
DCA: Dicloroacetato.
El análisis de los datos de los estudios de farmacocinética del DCA en voluntarios sugirió que, al igual que se observó in vitro (véase más adelante), los individuos portadores de la variante GSTZ1A, que codifica para KRT, en un alelo mostraron un aclaramiento inicial del DCA más rápido que los demás. Estas personas también mostraron una mayor disminución del aclaramiento después de cinco dosis. La proporción de aclaramiento de la primera a la quinta dosis fue de 3,6 ± 0,8 (media ± error estándar de la media, n = 5) en aquellos con la variante para GSTZ1C (codificación para EGT) pero no GSTZ1A, y fue de 18,2 ± 9,3 (n = 5) en aquellos con la variante GSTZ1A en al menos un alelo. Un individuo homocigoto para GSTZ1A mostró el mayor cambio entre la primera y la quinta dosis, con un descenso del aclaramiento de 16,7 a 0,31 ml/min [55]. La diferencia en el cambio medio en el aclaramiento no fue estadísticamente significativa, debido a la alta variabilidad en el grupo homocigoto o heterocigoto para GSTZ1A, sin embargo señaló una tendencia que estaba de acuerdo con los estudios in vitro de la susceptibilidad de los diferentes haplotipos a la inactivación por DCA.
Estudios invitro del metabolismo del DCA
Los estudios de la expresión y la actividad de GSTZ1 con DCA en el citosol y las mitocondrias del hígado humano han permitido comprender mejor el papel del genotipo en el metabolismo del DCA.
Ontogenia de la actividad
Estudios con citosol de hígado humano preparados a partir de un panel de 230 muestras de hígado de donantes que cubren el rango de edad desde el día 42 de gestación hasta los 84 años han mostrado una expresión y actividad de GSTZ1 muy baja o indetectable prenatalmente así como en las 2 primeras semanas de vida: a partir de entonces, la actividad y la expresión aumentan gradualmente [56]. La expresión y la actividad alcanzaron niveles del rango adulto a la edad de 7 años, aunque se encontró una considerable variabilidad interindividual. La medición de la actividad con DCA y de la expresión de la proteína GSTZ1 en estas muestras reveló que las muestras de individuos con la variante GSTZ1A en un alelo tenían una relación actividad/expresión mayor que todas las demás (Figura 4). Esto concordaba con los hallazgos de la GSTZ1A-1A humana recombinante expresada que mostraba una mayor actividad con el DCA para esta variante [54], y con estudios farmacocinéticos en voluntarios en los que los individuos con un gen GSTZ1A tenían una rápida eliminación inicial del DCA [55]. Aunque el estudio de desarrollo mostró que la actividad en niños menores de 7 años era inferior a la de los adultos, si se tiene en cuenta el mayor tamaño del hígado de los niños en relación con la masa corporal, la actividad con DCA por kilogramo de peso corporal es similar en adultos y niños de entre 2 meses y 7 años [56].
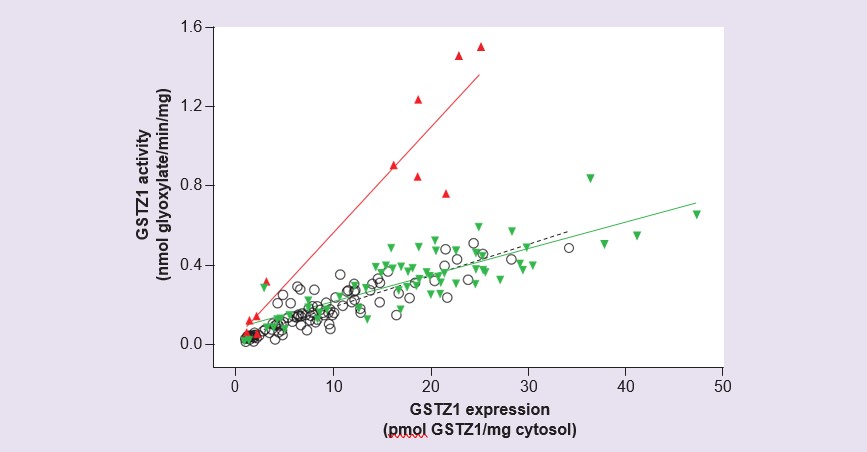
Efecto del cloruro en la inactivación de GSTZ1 por DCA
Aparentemente en desacuerdo con la observación de que la proteína KRT está asociada con un metabolismo más rápido del DCA in vitro [56], y una eliminación más rápida de una dosis única de DCA [55], está el hallazgo de que aquellos con la variante GSTZ1A en un alelo mostraron una mayor reducción en el aclaramiento tras dosis repetidas de DCA que aquellos que eran homocigotos para GSTZ1C. Los recientes descubrimientos sobre el efecto del cloruro en la inactivación de la GSTZ1-1 podrían explicar estas discrepancias. Se encontró que el cloruro protege a GSTZ1-1 de la inactivación por DCA en concentraciones fisiológicamente relevantes en el hígado [62,63], sin embargo la GSTZ1-1 en el citosol hepático de aquellos que eran heterocigotos para GSTZ1A estaba menos protegida de la inactivación por cloruro fisiológico (38 mM) que aquellos con otras variantes alélicas de GSTZ1 (Tabla 4). El efecto del cloruro fue tal que las enzimas GSTZ1-1 en hígados de individuos con la variante GSTZ1A en un alelo se inactivaron dos veces más rápido que los de otros haplotipos. Aún no se ha determinado el mecanismo exacto del efecto del cloruro, pero este fenómeno es una explicación de la mayor pérdida de aclaramiento de DCA tras la dosificación crónica en personas con una o más copias de la variante GSTZ1A. Como ya se ha señalado, es probable que la menor expresión de la enzima en individuos caucásicos con lisina (K) en la posición 32 de la proteína contribuya al muy bajo aclaramiento.
.
| Haplotipo | EC50 mM | Vida media de inactivación, h sin Cl- | Semivida de inactivación, h con 38 mM Cl- |
|---|---|---|---|
| EGT/EGT | 15 ± 3.1 (3) | 0.53, 0.49 | 5.73, 5.02 |
| KRT/EGT | 36 ± 2.2 (3) | 0.38, 0.38 | 2.66, 2.37 |
| EGM/EGM | 16.9 | – | 5.55 |
La EC50 es la concentración de cloruro que protegió a la mitad de la GSTZ1 citosólica de la inactivación tras la incubación durante 2 h con Na DCA, 0,5 mM.
Conclusión
Debido a su capacidad para inhibir la PDH quinasa, el DCA tiene aplicaciones para el tratamiento del cáncer y las enfermedades metabólicas. Tras la administración crónica de DCA a las personas, se reduce su metabolismo y eliminación. La razón de esta reducción en la eliminación tras múltiples dosis de DCA es que la única enzima conocida que metaboliza el DCA, la GSTZ1-1, se inactiva durante el metabolismo del DCA. Esta inhibición de la GSTZ1-1 por el DCA, basada en un mecanismo, da lugar a un aclaramiento más lento no sólo del DCA, sino también de los sustratos endógenos MAA y MA; aún no se ha resuelto si la acumulación de DCA y/o catabolitos de tirosina reactivos es la causa de la neuropatía periférica reversible que se produce en algunos pacientes. Otra posible causa no resuelta de la neuropatía es el modesto aumento de ∂-ALA que sigue al tratamiento crónico con DCA. Para comprender la farmacocinética del DCA es importante saber que el gen GSTZ1 presenta variantes polimórficas en todas las poblaciones humanas estudiadas hasta la fecha. Las propiedades de las proteínas enzimáticas de la variante GSTZ1-1 difieren con respecto a su capacidad para metabolizar el DCA y los sustratos endógenos MAA y MA, especialmente tras la administración crónica. Mientras que todas las variantes de GSTZ1-1 son inactivadas por el DCA, una variante, que tiene lisina en lugar de ácido glutámico en la posición 32 y arginina en lugar de glicina en la posición 42 de la proteína, se inactiva más rápidamente que otras variantes en presencia de concentraciones fisiológicas de cloruro. En caucásicos, pero no en personas de etnia africana, las variantes de GSTZ1 que tienen lisina en la posición 32 están relacionadas con un SNP de la región promotora en el gen GSTZ1 que da lugar a una expresión reducida de la proteína GSTZ1-1; esto también puede presentarse como un fenotipo de metabolizador lento. Es necesario comprender los factores que influyen en la farmacocinética del DCA bajo administración crónica para seleccionar la dosis y el intervalo de dosificación adecuados para administrar este fármaco a los pacientes de forma segura y eficaz.
Perspectivas futuras
De los estudios realizados hasta la fecha se desprende claramente que el haplotipo GSTZ1 es un factor importante que determina el aclaramiento del DCA. Una mayor comprensión de la regulación de la enzima en condiciones fisiológicas y en presencia de DCA conducirá a un uso más seguro y eficaz del fármaco en el futuro. Quedan varias cuestiones por explorar. En los heterocigotos, ¿se expresa preferentemente una variante de la proteína? Los estudios de actividad que muestran una mayor actividad con DCA para fracciones de citosol hepático de individuos heterocigotos para GSTZ1A/GSTZ1C sugieren que la variante 1A (KRT) de la proteína enzimática se expresa preferentemente en heterocigotos, pero esto no se ha demostrado. Esta cuestión tiene implicaciones prácticas, ya que la variante 1A se inactiva más rápidamente que la variante 1C. ¿Debería administrarse una dosis menor de DCA a los individuos que probablemente eliminen el DCA más lentamente que la media en la administración crónica? En caso afirmativo, ¿debería tratarse de todos aquellos que no son homocigotos o heterocigotos para GSTZ1C? ¿Deberían los caucásicos con al menos una variante que contenga K (KRT, KGT, KGM) recibir dosis más bajas de DCA que los de etnia africana porque expresan menos GSTZ1-1? ¿Qué pruebas existen de la baja expresión de las variantes que contienen K en otras poblaciones? ¿Existen diferencias poblacionales en los patrones de distribución de los principales haplotipos? ¿Los efectos de las diferencias farmacogenéticas son los mismos en los niños que en los adultos? Los niños que toman DCA generalmente eliminan el fármaco más rápidamente que los adultos, pero los posibles efectos farmacogenéticos no se han investigado ampliamente. Se necesitan más investigaciones para abordar estas cuestiones, con el fin de garantizar una dosificación segura y eficaz del DCA.
Declaración de intereses financieros y competitivos
El trabajo de los autores discutido en este manuscrito fue financiado en parte por el Servicio de Salud Pública de EE.UU. 1RO1 GM 099871, en parte por el Cerebro y el Banco de Tejidos para los Trastornos del Desarrollo, la Universidad de Maryland, Baltimore y la Universidad de Miami (NO1 HD90011) y en parte por el NIH / NCATS Clínica y Traslacional de Ciencia premio a la Universidad de Florida UL1 TR000064. Los autores no tienen ninguna otra afiliación relevante o participación financiera con ninguna organización o entidad con un interés financiero o conflicto financiero con el tema o los materiales discutidos en el manuscrito aparte de los revelados.
En la elaboración de este manuscrito no se ha contado con la ayuda de ningún redactor.
Resumen
- La selección de una dosis eficaz pero no tóxica de dicloroacetato (DCA) es importante para el uso clínico a largo plazo de DCA en el tratamiento del cáncer y otras enfermedades metabólicas.
- El gen de la enzima única que metaboliza este fármaco, GSTZ1-1, presenta polimorfismos que dan lugar a fenotipos metabolizadores lentos y rápidos tras el tratamiento a largo plazo con DCA.
- Los SNP de la región codificante del gen GSTZ1 dan lugar a cinco haplotipos comunes, con frecuencias variables en las poblaciones. Los individuos homocigotos para el más común, GSTZ1C, son metabolizadores rápidos. Hay indicios de que los haplotipos que codifican proteínas GSTZ1-1 con K en la posición 32 serán metabolizadores lentos, pero no se conocen del todo otros factores que contribuyen al metabolismo lento tras dosis múltiples de DCA.
- Un SNP de la región promotora en personas de etnia caucásica pero no africana está relacionado con un SNP de la región codificante que da lugar a la sustitución de lisina por ácido glutámico en la posición 32 de la proteína GSTZ1, y se asocia con una expresión reducida de la proteína enzimática en el hígado.
- En presencia de DCA, la estabilidad de la enzima GSTZ1-1 varía con la concentración de cloruro. La variante enzimática GSTZ1A (KRT) se inactiva más rápidamente que la variante común GSTZ1C (EGT) bajo concentraciones fisiológicas de cloruro en hígado.
REFERENCIAS
1 Stacpoole PW. Revisión de los efectos farmacológicos y terapéuticos del dicloroacetato de diisopropilamonio (DIPA). J. Clin. Pharmacol. J. New Drugs 9(5), 282-291 (1969).2 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism 38(11), 1124-1144 (1989).
3 Stacpoole PW, Felts JM. Diisopropylammonium dichloroacetate (DIPA) and sodium dichloracetate (DCA): effect on glucose and fat metabolism in normal and diabetic tissue. Metabolism 19(1), 71-78 (1970).
4 Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N. Engl. J. Med. 298(10), 526-530 (1978).
5 Stacpoole PW, Harwood HJ Jr, Varnado CE. Regulation of rat liver hydroxymethylglutaryl coenzyme A reductase by a new class of noncompetitive inhibitors. Effects of dichloroacetate and related carboxylic acids on enzyme activity. J. Clin. Invest. 72(5), 1575-1585 (1983).
6 Harwood HJ Jr, Bridge DM, Stacpoole PW. In vivo regulation of human mononuclear leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase. Estudios en sujetos normales. J. Clin. Invest. 79(4), 1125-1132 (1987).
7 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA, Stacpoole PW. Reducción del colesterol sérico en dos pacientes con hipercolesterolemia familiar homocigota mediante dicloroacetato. Atherosclerosis 33(3), 285-293 (1979).
8 Eichner HL, Stacpoole PW, Forsham PH. Treatment of streptozotocin diabetes with di-isopropylammonium dichloroacetate (DIPA). Diabetes 23(3), 179-182 (1974).
9 Backshear PJ, Holloway PA, Alberti KG. Metabolic interactions of dichloroacetate and insulin in experimental diabetic ketoacidosis. Biochem. J. 146(2), 447-456 (1975).
10 Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (short communication). Biochem. J. 134(2), 651-653 (1973).
11 Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 141(3), 761-774 (1974).
12 Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34(Pt 2), 217-222 (2006).
13 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329(Pt 1), 191-196 (1998).
14 Morten KJ, Caky M, Matthews PM. Stabilization of the pyruvate dehydrogenase E1alpha subunit by dichloroacetate. Neurology 51(5), 1331-1335 (1998).
15 Han Z, Berendzen K, Zhong L et al. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol. Genet. Metab. 93(4), 381-387 (2008).
16 Evans OB, Stacpoole PW. Prolonged hypolactatemia and increased total pyruvate dehydrogenase activity by dichloroacetate. Biochem. Pharmacol. 31(7), 1295-1300 (1982).
17 Denton RM, Randle PJ, Bridges BJ et al. Regulation of mammalian pyruvate dehydrogenase. Mol. Cell. Biochem. 9(1), 27-53 (1975).
18 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284(5), E855-E862 (2003).
19 Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J. Clin. Pharmacol. 43(7), 683-691 (2003).
20 Kankotia S, Stacpoole PW. Dicloroacetato y cáncer: ¿nuevo hogar para un medicamento huérfano? Biochim. Biophys. Acta 1846(2), 617-629 (2014).
21 Stacpoole PW. El dilema del dicloroacetato: peligro ambiental versus mina de oro terapéutica: ¿ambos o ninguno? Environ. Health Perspect. 119(2), 155-158 (2011).
22 Chu PI. Farmacocinética del dicloroacetato de sodio. Ph.D. dissertation, Pharmaceutics Department, University of Florida, FL, USA (1987).
23 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab. Rev. 30(3), 499-539 (1998).
24 Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin. Pharmacol. Ther. 37(1), 89-93 (1985).
25 Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G et al. Pharmacokinetics of oral dichloroacetate in dogs. J. Biochem. Mol. Toxicol. 27(12), 522-525 (2013).
26 Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Efectos metabólicos y farmacocinética del dicloroacetato administrado por vía intravenosa en humanos. Diabetologia 19(2), 109-113 (1980).
27 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Treatment of lactic acidosis with dichloroacetate. N. Engl. J. Med. 309(7), 390-396 (1983).
28 Henderson GN, Curry SH, Derendorf H, Wright EC, Stacpoole PW. Pharmacokinetics of dichloroacetate in adult patients with lactic acidosis. J. Clin. Pharmacol. 37(5), 416-425 (1997).
29 Stacpoole PW, Kerr DS, Barnes C et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117(5), 1519-1531 (2006).
30 Stacpoole PW, Gilbert LR, Neiberger RE et al. Evaluación del tratamiento a largo plazo de niños con acidosis láctica congénita con dicloroacetato. Pediatrics 121(5), e1223-e1228 (2008).
31 Kaufmann P, Engelstad K, Wei Y et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66(3), 324-330 (2006).
32 Stickler DE, Valenstein E, Neiberger RE et al. Peripheral neuropathy in genetic mitochondrial diseases. Pediatr. Neurol. 34(2), 127-131 (2006).r
33 Kaufmann P, Pascual JM, Anziska Y et al. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch. Neurol. 63(5), 746-748 (2006).
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J. Pharmacol. Exp. Ther. 324(3), 1163-1171 (2008).
35 James MO, Yan Z, Cornett R et al. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos. 26(11), 1134-1143 (1998).
36 Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell. Biol. 45(6), 1121-1132 (2013).
37 Vessey DA, Kelley M, Warren RS. Caracterización de las CoA ligasas de mitocondrias de hígado humano que catalizan la activación de ácidos grasos de cadena corta y media y ácidos carboxílicos xenobióticos. Biochim. Biophys. Acta 1428(2-3), 455-462 (1999).
38 James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Glutathione-dependent conversion to glyoxylate, a major pathway of dichloroacetate biotransformation in hepatic cytosol from humans and rats, is reduced in dichloroacetate-treated rats. Drug Metab. Dispos. 25(11), 1223-1227 (1997).
39 Tong Z, Board PG, Anders MW. Glutathione transferase zeta catalyses the oxygenation of the carcinogen dichloroacetic acid to glyoxylic acid. Biochem. J. 331(Pt 2), 371-374 (1998).
49 Fernandez-Canon JM, Penalva MA. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J. Biol. Chem. 273(1), 329-337 (1998).
41 Blackburn AC, Woollatt E, Sutherland GR, Board PG. Characterization and chromosome location of the gene GSTZ1 encoding the human Zeta class glutathione transferase and maleylacetoacetate isomerase. Cytogenet. Cell. Genet. 83(1-2), 109-114 (1998).
42 Li W, James MO, Mckenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrion as a novel site of dichloroacetate biotransformation by glutathione transferase zeta 1. J. Pharmacol. Exp. Ther. 336(1), 87-94 (2011).
43 Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Localización inmunohistoquímica y actividad de la glutatión transferasa zeta (GSTZ1-1) en tejidos de rata. Drug Metab. Dispos. 30(6), 616-625 (2002).
44 Shangraw RE, Fisher DM. Pharmacokinetics of dichloroacetate in patients undergoing liver transplantation. Anesthesiology 84(4), 851-858 (1996).
45 Cornett R, James MO, Henderson GN, Cheung J, Shroads AL, Stacpoole PW. Inhibition of glutathione S-transferase zeta and tyrosine metabolism by dichloroacetate: a potential unifying mechanism for its altered biotransformation and toxicity. Biochem. Biophys. Res. Commun. 262(3), 752-756 (1999).
46 Guo X, Dixit V, Liu H et al. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 34(1), 36-42 (2006).
47 Anderson WB, Board PG, Gargano B, Anders MW. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem. Res. Toxicol. 12(12), 1144-1149 (1999).
48 Anderson WB, Liebler DC, Board PG, Anders MW. Mass spectral characterization of dichloroacetic acid-modified human glutathione transferase zeta. Chem. Res. Toxicol. 15(11), 1387-1397 (2002).
49 Dixit V. Inactivation of glutathione transferase zeta by dichloroacetic acid. PhD dissertation, Medicinal Chemistry Department, University of Florida, FL, USA (2005).
50 Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N. Engl. J. Med. 327(22), 1564-1569 (1992).
51 Abdelmalak M, Lew A, Ramezani R et al. Seguridad a largo plazo del dicloroacetato en la acidosis láctica congénita. Mol. Genet. Metab. 109(2), 139-143 (2013).
52 Board PG, Baker RT, Chelvanayagam G, Jermiin LS. Zeta, a novel class of glutathione transferases in a range of species from plants to humans. Biochem. J. 328(Pt 3), 929-935 (1997).
53 Blackburn AC, Tzeng HF, Anders MW, Board PG. Discovery of a functional polymorphism in human glutathione transferase zeta by expressed sequence tag database ana-lysis. Pharmacogenetics 10(1), 49-57 (2000).
54 Blackburn AC, Coggan M, Tzeng HF et al. GSTZ1d: a new allele of glutathione transferase zeta and maleylacetoacetate isomerase. Pharmacogenetics 11(8), 671-678 (2001).
55 Shroads AL, Langaee T, Coats BS et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J. Clin. Pharmacol. 52(6), 837-849 (2012).
56 Li W, Gu Y, James MO et al. Prenatal and postnatal expression of glutathione transferase zeta 1 in human liver and the roles of haplotype and subject age in determining activity with dichloroacetate. Drug Metab. Dispos. 40(2), 232-239 (2012).Google Scholar
57 Board PG, Anders MW. Glutatión transferasa zeta: descubrimiento, variantes polimórficas, catálisis, inactivación y propiedades de los ratones Gstz1-/-. Drug Metab. Rev 43(2), 215-225 (2011).
58 Polekhina G, Board PG, Blackburn AC, Parker MW. Crystal structure of maleylacetoacetate isomerase/glutathione transferase zeta reveals the molecular basis for its remarkable catalytic promiscuity. Biochemistry 40(6), 1567-1576 (2001).
59 Fang YY, Kashkarov U, Anders MW, Board PG. Polymorphisms in the human glutathione transferase zeta promoter. Pharmacogenet. Genomics 16(5), 307-313 (2006).
60 Langaee TY, Zhong G, Li W et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics 25(5), 239-245 (2015).
61 Shroads AL, Coats BS, Mcdonough CW, Langaee T, Stacpoole PW. Las variaciones del haplotipo en la glutatión transferasa zeta 1 influyen en la cinética y la dinámica del dicloroacetato crónico en niños. J. Clin. Pharmacol. 55(1), 50-55 (2015).
62 Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Chloride and other anions inhibit dichloroacetate-induced inactivation of human liver GSTZ1 in a haplotype-dependent manner. Chem. Biol. Interact. 215C, 33-39 (2014).
63 Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Chloride concentrations in human hepatic cytosol and mitochondria are a function of age. Biochem. Biophys. Res. Commun. 459(3), 463-468 (2015).