Margaret O James*,1 & Peter W Stacpoole2,3
1 Abteilung für Medizinische Chemie, Universität von Florida, Gainesville, FL 32610-0485, USA
2 Abteilung für Medizin, College of Medicine, Universität von Florida, Gainesville, FL 32610-0485, USA
3 Abteilung für Biochemie und Molekularbiologie, Universität von Florida, Gainesville, FL 32610-0485, USA
Korrespondenz: Tel: +1 352 273 7707 E-Mail: [email protected]
Eingereicht: 16. Dezember 2015Akzeptiert
: 17 February 2016Published
: 4 May 2016
Zusammenfassung
Das Prüfpräparat Dichloracetat (DCA) ist ein Stoffwechselregulator, der erfolgreich zur Behandlung erworbener und angeborener Stoffwechselkrankheiten und seit kurzem auch solider Tumore eingesetzt wird. Bei seiner klinischen Anwendung hat sich gezeigt, dass die Auswahl geeigneter Dosen schwierig ist. Die chronische Verabreichung von DCA führt zu einer Hemmung des DCA-Stoffwechsels und zu einer potenziellen Anreicherung auf Werte, die zu Nebenwirkungen führen. Dies liegt daran, dass die Umwandlung von DCA in Glyoxylat von einem Enzym, der Glutathiontransferase zeta 1 (GSTZ1-1), katalysiert wird, das durch DCA inaktiviert wird. SNPs im GSTZ1-Gen führen zur Expression polymorpher Varianten des Enzyms, die sich unter physiologischen Bedingungen in der Aktivität und der Geschwindigkeit der Inaktivierung durch DCA unterscheiden: Diese Eigenschaften führen zu erheblichen Unterschieden in der Pharmakokinetik von DCA zwischen den Menschen.
Schlüsselwörter: Dichloracetat; GSTZ1; Pharmakogenetik
Klinische Anwendung von Dichloracetat
Dafür, dass DCA ein so einfaches Molekül ist, verfügt es über ein bemerkenswert reichhaltiges und vielfältiges pharmakologisches Portfolio, das fast ein Jahrhundert alt ist [1,2]. Seine moderne Verwendung als Prüfpräparat begann jedoch 1970, als seine selektive Fähigkeit zur Senkung des Blutzuckerspiegels bei diabetischen, aber nicht bei nicht-diabetischen Tieren entdeckt [3 ] und später beim Menschen bestätigt wurde [4]. In den 1970er und 1980er Jahren wurden zahlreiche metabolische Eigenschaften von DCA im Zusammenhang mit dem Glukose- und Lipidstoffwechsel festgestellt. Die meisten seiner pharmakologischen Wirkungen lassen sich jedoch auf einige grundlegende Wirkorte und -mechanismen zurückführen. Erstens ist DCA ein nichtkompetitiver Inhibitor des Enzyms HMG-CoA-Reduktase des endoplasmatischen Retikulums, das den ratenbegrenzenden Schritt der Cholesterinbiosynthese katalysiert. Die hemmende Wirkung von DCA wird sowohl in der Leber von Nagetieren [5] als auch in menschlichen Leukozyten [6] beobachtet; sie ist wahrscheinlich der Grund für die arzneimittelassoziierte Senkung des Gesamtcholesterins und des Cholesterins aus Lipoproteinen niedriger Dichte (LDL) bei Patienten mit LDL-Rezeptor-negativer homozygoter familiärer Hypercholesterinämie [7 ] und für seine Ausweisung als erstes Arzneimittel für seltene Krankheiten. Zweitens hemmt DCA die hepatische De-novo-Synthese von Triglyceriden bei nicht-diabetischen Nagetieren [5] und senkt die zirkulierenden Triglycerid- und Lipoproteinspiegel sehr niedriger Dichte bei Patienten mit Diabetes mellitus Typ 2 [4]. Es verringert auch die Ketonkörper im Blut von Ratten mit experimentell induzierter diabetischer Ketoazidose [8,9]. Die genauen Mechanismen, die diesen Wirkungen auf die Lipidsynthese und -oxidation zugrunde liegen, sind nicht bekannt. Drittens stimuliert DCA die mitochondriale PDC, die Pyruvat irreversibel zu Acetyl-Coenzym A (Acetyl-CoA) oxidiert [10], eine Eigenschaft, die auch bestimmte andere kurzkettige halogenierte Fettsäuren aufweisen [11]. Die Fähigkeit von DCA, die PDC-Aktivität zu verändern, hat die bei weitem größte experimentelle und klinische Forschung zu diesem ungewöhnlichen Molekül ausgelöst.
Die PDC wird posttranslational hauptsächlich durch die reversible Phosphorylierung eines oder mehrerer von drei Serinresten auf der E1α-Untereinheit des ersten Enzyms PDH der PDC reguliert, das Pyruvat decarboxyliert [12]. Beim Menschen übernehmen vier PDH-Kinasen (PDK1-4) und zwei PDH-Phosphatasen (PDP 1 und 2) diesen regulatorischen Aspekt der PDC, bei dem das phosphorylierte Enzym katalytisch inaktiv ist. Die PDKs werden in den Geweben unterschiedlich exprimiert, wobei PDK2 ubiquitär exprimiert wird [13]. Pyruvat, das Substrat für die PDC-Reaktion, hemmt PDKs durch Bindung an eine kleine Tasche im N-Terminus der Kinase, was zu einer Stimulation der PDC-Aktivität führt. Im Gegensatz dazu führt die Anhäufung der Reaktionsprodukte Acetyl-CoA und NADH zu einer PDK-Aktivierung und Hemmung der PDC. DCA, ein strukturelles Analogon von Pyruvat, bindet ebenfalls an die Pyruvat-Bindungsstelle, was zu einer Hemmung der PDKs führt, wobei die Reihenfolge der Hemmung PDK2>PDK1˜PDK4>>PDK3 ist [13]. Eine chronische Arzneimittelexposition in vitro [14,15] und in vivo [16] kann PDC stabilisieren und seinen Umsatz verringern, was einen zweiten Mechanismus darstellt, durch den DCA die Aktivität des Komplexes erhöht, und eine Erklärung für seine lang anhaltenden pharmakologischen Wirkungen nach dem Absetzen der Droge liefert [4].
Obwohl DCA ein relativ schwacher PDK-Inhibitor ist (Ki 0,2 mM für PDK2), sind seine Auswirkungen auf Aspekte des Intermediärstoffwechsels tiefgreifend und stehen in direktem Zusammenhang mit der zentralen Rolle, die der PDC bei der Regulierung der eukaryotischen zellulären Brennstoffauswahl und Bioenergetik spielt [17,18]. DCA ist ein noch nicht von der FDA zugelassenes Prüfpräparat. Es wird jedoch weiterhin intensiv als Stoffwechselmodulator für verschiedene angeborene und erworbene Stoffwechselstörungen untersucht, die auf seiner stimulierenden Wirkung auf die PDC beruhen (Tabelle 1).
| Eigenschaft | Mechanismus | Ref. |
|---|---|---|
| Erhöhte OXPHOS und Bioenergetik | ↓ PDK ↑ PDC | [2,10] |
| Verminderte Blutglukose bei Nüchternheit oder Diabetes | ↓ PDK ↑ PDC | [4] |
| Verringertes Blut- und Liquorlaktat | ↓ PDK ↑ PDC | [19] |
| Verringertes Gesamt- und LDL-Cholesterin im Blut | ↓ HMG CoA-Reduktase → ↓ Cholesterin-Synthese | [6,7] |
| Verringerung der Triglyceride und des VLDL-Cholesterins im Blut | ↓ hepatische TG-Synthese → ↓ VLDL-Syntheis | [4] |
| Umkehrung des Warburg-Effekts bei Krebs, PAH und anderen proliferativen Zuständen | ↓ PDK ↑ PDC | [20] |
Liquor: Liquor cerebrospinalis; HMG CoA: Hydroxymethylglutaryl-Coenzym A; LDL: Low-Density-Lipoprotein; OXPHOS: Oxidative Phosphorylierung; PAH: Pulmonale arterielle Hypertonie; PDC: Pyruvatdehydrogenase-Komplex; PDK: Pyruvatdehydrogenase-Kinase; TG: Triglycerid; VLDL: Lipoprotein sehr niedriger Dichte.
Pharmakokinetik und Clearance
Oral eingenommenes DCA wird rasch resorbiert und hat sowohl nach oraler als auch nach parenteraler Verabreichung eine Bioverfügbarkeit von annähernd eins [21,22]. Der Wirkstoff kann im Plasma von Menschen innerhalb von 15 Minuten nach einer oralen Dosis von 50 mg/kg nachgewiesen werden. Bei zuvor naiven Probanden hat oral oder intravenös verabreichtes DCA eine Plasmahalbwertszeit von etwa 1 Stunde [23]. Die Metabolisierungsrate bei gesunden Tierarten ist: Maus>Ratte>Mensch≥Hund [22-25].
Frühe pharmakokinetische Untersuchungen der intravenösen DCA-Einzeldosis beschrieben eine nichtlineare Kinetik bei Dosen ≥35 mg/kg [26]. Später wurde festgestellt, dass die Rate der Plasma-Clearance bei wiederholter intravenöser Verabreichung von 50 mg/kg Körpergewicht bei kritisch kranken Erwachsenen mit Laktatazidose abnahm [27,28]. Eine verzögerte Plasmaclearance des oral verabreichten Medikaments in einer Dosis von 50 mg/kg wurde auch bei gesunden Erwachsenen beobachtet, die Wochen bis Monate benötigten, bevor die ursprüngliche Clearance-Rate mit einer zweiten Dosis erreicht wurde [22].
Wesentliche Fortschritte beim Verständnis dieser ungewöhnlichen Eigenschaft der Pharmakokinetik von DCA ergaben sich aus der Erforschung der klinischen Toxikologie des Wirkstoffs. Chronisches DCA kann bei verschiedenen Spezies eine reversible periphere Neuropathie verursachen [21,23], eine Wirkung, die erstmals vor 25 Jahren bei einem 16-jährigen Jungen klinisch beobachtet wurde, der etwa vier Monate lang 50 mg/kg/Tag erhielt [7]. Im Gegensatz dazu wurde in einer randomisierten, kontrollierten Studie (RCT) mit 43 Kleinkindern mit verschiedenen Arten primärer mitochondrialer Erkrankungen (Durchschnittsalter bei Studienbeginn: 5,6 Jahre) oral verabreichtes DCA in einer Dosis von 12,5 mg/kg alle 12 Stunden über einen Zeitraum von 6 Monaten gut vertragen und verursachte im Vergleich zu Placebo keine nachteiligen Veränderungen der elektrischen Nervenleitung [29], obwohl bei längerer Medikamentenexposition eine gewisse asymptomatische Abnahme der Nervenleitung festgestellt wurde [30]. Im Gegensatz dazu wurde eine RCT mit 30 älteren Jugendlichen und Erwachsenen (mittleres Eintrittsalter: 30 Jahre) mit einer genetischen mitochondrialen Erkrankung, die mit der gleichen DCA-Dosis und dem gleichen Dosierungsschema wie in der pädiatrischen Studie behandelt wurden, vorzeitig abgebrochen, weil sich die periphere Neuropathie verschlimmerte oder neu auftrat [31], eine häufige Komplikation mitochondrialer Erkrankungen [32,33]. Beim Vergleich der Kinetik von DCA in diesen beiden Patientengruppen wurde festgestellt, dass die Plasma-Clearance von chronischem DCA in der älteren Gruppe deutlich langsamer war [34], ein Ergebnis, das in Studien mit Ratten bestätigt wurde [34,35]. Diese Ergebnisse gaben Anlass zu Studien über die Enzymologie der DCA-Biotransformation.
Bedeutung der Glutathiontransferase zeta 1 für die Biotransformation von DCA
Kleine Carbonsäuren werden häufig glucuronidiert [36] oder in ihre Coenzym-A-Derivate und dann in Aminosäurekonjugate umgewandelt [37]. Es gibt jedoch keine Hinweise darauf, dass DCA ein Glucuronid, ein Coenzym-A-Derivat oder ein Aminosäurekonjugat bildet [2, James MO, unveröffentlichte Daten]. Stattdessen ist DCA ein Substrat für ein ungewöhnliches GST-Enzym, GSTZ1-1, das es in einer Reaktion, die Glutathion (GSH) erfordert, aber nicht verbraucht, in Glyoxylat umwandelt [38,39]. Wie andere GSTs ist auch GSTZ1-1 in dimerer Form aktiv, und das aktive Enzym ist ein Homodimer. Die Dechlorierung von DCA zu Glyoxylat, die von GSTZ1-1 katalysiert wird, ist der einzige Weg des Primärstoffwechsels von DCA, der in der Leber beobachtet wurde [35]. Neben der Katalyse des DCA-Stoffwechsels hat GSTZ1-1, auch bekannt als MAAI, eine wichtige physiologische Funktion beim Tyrosinkatabolismus, wo es Maleylacetoacetat (MAA) und Maleylaceton (MA) in Reaktionen, die GSH erfordern, aber nicht verbrauchen, zu Fumarylacetoacetat und Fumarylaceton isomerisiert (Abbildung 1) [40,41]. Die Biotransformation von DCA zu Glyoxylat beendet seine pharmakologisch und therapeutisch wichtigen Aktivitäten, die oben erörtert wurden, so dass die Aktivität von GSTZ1-1 die Wirkungsdauer von DCA kontrolliert.
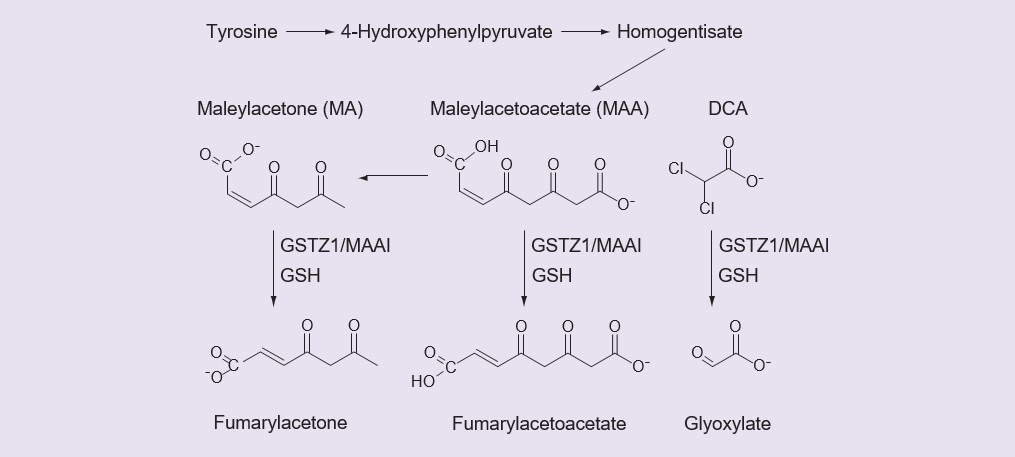
Der inaktive DCA-Metabolit, Glyoxylat, wird weiter zu Kohlendioxid, Glycin und Oxalat verstoffwechselt. Fumarylaceton und Fumarylacetoacetat werden durch Fumarylacetoacetat-Hydrolase weiter metabolisiert. DCA: Dichloracetat: GSH: Glutathion; MA: Maleylaceton; MMA: Maleyacetoacetat.
GSTZ1-1 wird hauptsächlich in der Leber exprimiert, wo es sowohl im Zytosol als auch in der mitochondrialen Matrix vorkommt [38,42]. In Tierstudien wurde eine sehr geringe Expression von GSTZ1-1 in Niere, Herz, GI-Trakt und Gehirn nachgewiesen [43]. Die Rolle des extrahepatischen Gewebes beim DCA-Stoffwechsel beim Menschen wurde nicht im Detail untersucht. Eine Studie zeigte jedoch, dass DCA während der anhepatischen Phase von Lebertransplantationspatienten, denen DCA zur Bekämpfung der Laktatazidose verabreicht wurde, nicht aus dem Blut ausgeschieden wurde, was darauf hindeutet, dass kein Stoffwechsel stattfand [44]. Die anhepatische Phase war von kurzer Dauer (Mittelwert: 73 Minuten), so dass die Daten begrenzt waren, jedoch deutet diese Studie darauf hin, dass andere Gewebe als die Leber bei der DCA-Elimination eine untergeordnete Rolle spielen, zumindest bei einer Einzeldosis.
Hemmung von GSTZ1-1 durch DCA
Studien mit Tieren und mit exprimierten rekombinanten menschlichen GSTZ1-1-Proteinen haben gezeigt, dass der Grund dafür, dass mehrere DCA-Dosen viel langsamer ausgeschieden werden als eine Einzeldosis, darin liegt, dass DCA ein auf einem Mechanismus beruhender Inhibitor von GSTZ1-1 ist [25,34,45-47]. Während der Biotransformation von DCA werden Addukte an das GSTZ1-1-Protein gebildet, die das Protein inaktivieren und vermutlich seine Proteolyse auslösen [48, 49]. Die Verabreichung von DCA an Ratten führt zu einem dosis- und zeitabhängigen Verlust der Aktivität und Expression von GSTZ1-1: Der Verlust der GSTZ1-1-Funktion war bei erwachsenen Ratten ausgeprägter und hielt länger an als bei Jungtieren [34,35]. Diese Auswirkung des Alters auf den Verlust der GSTZ1-1-Funktion nach DCA-Verabreichung wurde auch bei Menschen beobachtet, wobei Kinder einen geringeren Anstieg der Plasmahalbwertszeit und eine geringere Abnahme der Clearance zeigten als Erwachsene [34]. Der Verlust von GSTZ1-1 (MAAI) wirkt sich nicht nur auf die Pharmakokinetik von DCA aus, sondern führt auch zur Akkumulation der physiologischen Substrate MA und vermutlich MAA, beides reaktive Moleküle, die zur Adduktbildung mit zellulären Makromolekülen fähig sind [34,45]. Die Hemmung von MAAI führt auch zu einer Umleitung von Tyrosin-Kohlenstoff zur Succinylacetonbildung. Dieses Molekül hemmt einen proximalen Schritt in der Häm-Synthese und verursacht die Anhäufung des reaktiven Moleküls δ-Aminolävulinat (δ-ALA). Sowohl MA als auch δ-ALA nehmen im Blut und/oder Urin von Menschen zu, die chronisch DCA ausgesetzt sind [34]: die klinische toxikologische Bedeutung dieses Effekts muss noch nachgewiesen werden.
Andere Metaboliten von DCA
Der einzige andere bekannte Primärmetabolit von DCA ist Monochloressigsäure (MCA), die gelegentlich in Spuren im Blut nach Verabreichung von DCA gefunden wurde [34]. MCA scheint in den roten Blutkörperchen aus DCA gebildet zu werden [34]. Es gibt keine Hinweise auf die Bildung von MCA aus DCA in der Leber. Für praktische Zwecke bestimmt die Umwandlung von DCA in Glyoxylat die Pharmakokinetik der Ausscheidung von DCA, so dass die Kenntnis der Faktoren, die die Geschwindigkeit und das Ausmaß des GSTZ1-1-katalysierten DCA-Stoffwechsels bei Menschen beeinflussen, für eine sichere und wirksame Dosierung bei Patienten wichtig ist.
Was die Auswirkungen der DCA-Behandlung betrifft, so kann Glyoxylat über ein Carboligase-Enzym weiter zu Kohlendioxid verstoffwechselt werden, durch ein oder mehrere Aminotransferase-Enzyme zu Glycin, oder es kann zu Oxalat oxidiert werden [35]. Alle drei Wege wurden in Studien nachgewiesen, in denen Ratten mit DCA behandelt wurden [35]. Von diesen Wegen könnte nur die Bildung von Oxalat potenziell schädlich für einen Patienten sein, bei der Ratte war die Bildung von Oxalat jedoch ein untergeordneter Weg [35]. Kritisch kranke erwachsene Patienten mit Laktatazidose, die in einer randomisierten, kontrollierten klinischen Studie mit intravenösem DCA oder Placebo behandelt wurden, wurden auf Oxalatsteine und oxalatbedingte Toxizität untersucht; es gab keine Hinweise auf Unterschiede zwischen der DCA- und der Placebogruppe [50]. Auch die orale Verabreichung von DCA über mehrere Jahre in einer Studie an Kindern mit kongenitalen Formen der Laktatazidose ergab keine Hinweise auf Oxalatsteine oder Toxizität [51].
Polymorphe Varianten von GSTZ1
Beim menschlichen GSTZ1 (NM_145870) wurde seit seiner Entdeckung vermutet, dass es allele Varianten aufweist [52]. In nachfolgenden Studien wurden SNPs in den kodierenden und nicht kodierenden Regionen des Gens entdeckt. Wir sind noch dabei, die Auswirkungen der meisten dieser SNPs auf die Expression, Stabilität und Aktivität von GSTZ1 mit DCA zu erforschen. Dies ist ein aktiver Forschungsbereich, da diese Faktoren die Pharmakokinetik und letztlich die sichere Anwendung von DCA beeinflussen. SNPs, die zu Aminosäureveränderungen im exprimierten Protein führen, haben Aufmerksamkeit erhalten, ebenso wie SNPs, die die Translation von GSTZ1 und letztlich die Expressionsmengen des Enzymproteins zu beeinflussen scheinen.
Varianten in der Aminosäuresequenz des exprimierten Enzymproteins
In der kodierenden Region von GSTZ1 wurden drei häufige SNPs identifiziert [53,54], die zu fünf Haplotypen geführt haben (Tabelle 2). Diese fünf Haplotypen kommen bei Menschen je nach ethnischer Zugehörigkeit mit unterschiedlicher Häufigkeit vor (Abbildung 2). Die in allen bisher untersuchten Populationen am häufigsten gefundene Variante, die bei etwa 50 % der Menschen auftritt, ist jedoch GSTZ1C [54-56]. Dieser Haplotyp wird auch als EGT bezeichnet, was auf die variierenden Aminosäuren an den Positionen 32, 42 und 82 im Enzymprotein hinweist [54,55]. Gegenwärtig gibt es relativ wenig Informationen über die Häufigkeit der fünf häufigen Haplotypen in anderen Populationen als Kaukasiern. Exprimierte rekombinante menschliche GSTZ1A-1A-, 1B-1B-, 1C-1C- und 1D-1D-Proteine wurden mit DCA als Substrat untersucht, und es wurde festgestellt, dass GSTZ1A-1A (KRT/KRT) eine höhere Aktivität als die anderen Varianten aufweist [54,57]. GSTZ1F ist eine seltene Variante (Tabelle 2), und die Eigenschaften des rekombinanten Proteins sind nicht untersucht worden. Eine einzelne Person kann homo- oder heterozygot für diese Haplotypen sein. Bei Heterozygoten ist nicht bekannt, ob eine Variante auf Proteinebene bevorzugt exprimiert wird, jedoch weisen Leberzytosolfraktionen von Personen, die die GSTZ1A-Variante auf einem Allel tragen, eine höhere In-vitro-Aktivität mit DCA auf als solche ohne dieses Allel [56], was auf die Möglichkeit einer bevorzugten Expression des Enzymproteins der KRT-Variante hinweist.
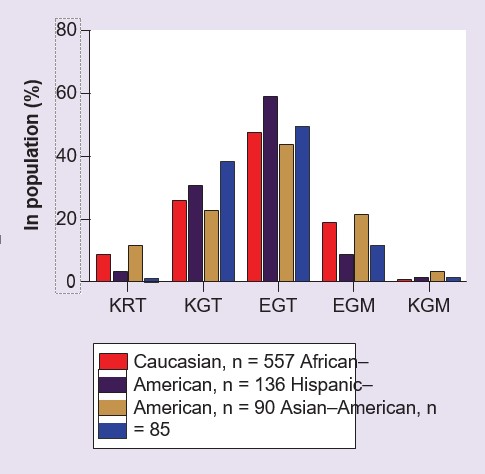
E: Glutaminsäure; G: Glycin; K: Lysin: M: Metionin; T: Threonin.
| Variante | Nukleotid-Position | Position der Aminosäure | Prozentsatz in der Bevölkerung (%)† | ||||
| 94 | 124 | 245 | 32 | 42 | 82 | ||
| GSTZ1A | A | A | C | Lys (K) | Arg (R) | Thr (T) | 1-10 |
| GSTZ1B | A | G | C | Lys (K) | Gly (G) | Thr (T) | 25-35 |
| GSTZ1C | G | G | C | Glu (E) | Gly (G) | Thr (T) | 45-55 |
| GSTZ1D | G | G | T | Glu (E) | Gly (G) | Met (M) | 10-20 |
| GSTZ1F | A | G | T | Lys (K) | Gly (G) | Met (M) | <1 |
Die polymorphen Varianten von GSTZ1-1 beim Menschen und ihre ungefähre Häufigkeit in der Bevölkerung.
†DieHäufigkeit der einzelnen Haplotypen variiert je nach ethnischer Gruppe, siehe Abbildung 2 und [54-56].
Glu (E): Glutaminsäure; Gly (G): Glycin; Lys (K): Lysin: Met (M): Methionin; Thr (T): Threonin.
.
In Studien mit den exprimierten rekombinanten humanen GSTZ1A-1A, 1B-1B, 1C-1C und 1D-1D-Proteinen wurde auch die Aktivität mit dem physiologisch wichtigen Substrat MA untersucht [54]. Die Raten des MA-Stoffwechsels zeigten ein anderes Muster als die mit DCA als Substrat gemessenen Aktivitäten, da die GSTZ1C-1C (EGT/EGT)-Variante die höchste Aktivität mit MA und GSTZ1A-1A die niedrigste hatte [54].
Neben den oben beschriebenen, häufig vorkommenden Varianten gibt es Hinweise auf zwei weitere kodierende Sequenzvarianten. Eine davon wurde in der Expressed-Sequence-Tag-Datenbank gefunden und als GSTZ1E bezeichnet [54]. Diese Variante, bei der anstelle von Leucin an Position 23 Prolin vorkommt, wurde jedoch nicht bei Menschen gefunden. Eine weitere Variante wurde bei einem Probanden gefunden, der an pharmakokinetischen Studien mit DCA teilnahm. Diese Person, die heterozygot für KGT/KGM (1B/1F) war, eliminierte selbst eine Einzeldosis DCA extrem langsam; die Sequenzierung der DNA der Person ergab einen neuartigen SNP im Exon 5 von GSTZ1, der zu Methionin anstelle von Valin an Position 99 des Proteins führte [55]. Ausgehend von der Kristallstruktur von GSTZ1B [58] befindet sich Position 99 in einem α-helicalen Bündel. Trotz der Suche nach dieser Variante in Leberproben, die eine geringe Aktivität mit DCA aufwiesen [56], wurden bisher keine weiteren Personen mit diesem Polymorphismus identifiziert.
Polymorphismen in der Promotorregion
Zehn SNPs in genomischen DNA-Proben von afrikanischen und australischen europäischen Probanden wurden in einer Region gefunden, die sich 1500 Nukleotide stromaufwärts der Transkriptionsstartstelle von GSTZ1 erstreckt und als Promotorregion des Gens angenommen wird [59]. Zwei dieser SNPs, -1002G>A und -289C>T, wurden mit einer Abnahme bzw. Zunahme der Promotoraktivität assoziiert, die in HepG2-Zellen gemessen wurde. Nachfolgende Studien mit DNA aus Leberproben zeigten, dass bei kaukasischen Amerikanern, nicht aber bei Afroamerikanern, das A-Allel des SNP rs 7975, das zur Expression von Lysin anstelle von Glutaminsäure an Position 32 des Proteins führte, in einem Kopplungsungleichgewicht mit dem A-Allel des SNP -1002 G>A, rs7160195, in der Promotorregion stand und zu einer reduzierten Expression des GSTZ1-1-Proteins mit Lysin (K) an Position 32 führte [60]. Die verminderte Expression wurde sowohl bei Heterozygoten als auch bei Homozygoten für GSTZ1A (KRT), GSTZ1B (KGT) und GSTZ1F (KGM) bei kaukasischen Amerikanern beobachtet [60 ]. Die Expression von K-haltigen GSTZ1-1-Varianten in untersuchten Leberproben von Afroamerikanern zeigte keine Auswirkungen. Aus noch nicht geklärten Gründen gab es keine Unterschiede in den mRNA-Spiegeln von GSTZ1 zwischen den Haplotypen, was auf eine Diskrepanz zwischen den Ergebnissen für die Proteinexpression und die mRNA-Expression hindeutet [60]. Die Feststellung einer geringeren Expression des GSTZ1-1-Proteins bei K-enthaltenden Haplotypen bei Kaukasiern deutet darauf hin, dass der anfängliche Metabolismus von DCA bei diesen Personen (mit Ausnahme derjenigen mit KRT) geringer ist. Darüber hinaus besteht die Möglichkeit, dass Kaukasier mit K-haltigen Haplotypen, die mit DCA behandelt werden, eine weniger effiziente Re-Synthese des GSTZ1-1-Proteins erfahren, was zu einer noch langsameren Eliminierung führt als Personen ohne diesen Haplotyp.
Einfluss von GSTZ1-Varianten auf die Pharmakokinetik von DCA bei Menschen
Wenn man davon ausgeht, dass die Nebenwirkungen einer chronischen DCA-Behandlung mit der Akkumulation entweder von DCA selbst oder der reaktiven endogenen Substrate für GSTZ1, MAA und MA, zusammenhängen, gibt es ein überzeugendes Argument dafür, dass die sichere klinische Anwendung von DCA durch ein Verständnis der Variabilität der Eliminierung des Arzneimittels bei Menschen während einer chronischen Behandlung geleitet werden sollte. Die DCA-Dosierung könnte dann bei denjenigen, die eine langsame Ausscheidung aufweisen, reduziert werden, wobei die wirksamen Blutspiegel beibehalten würden. Wenn es möglich wäre vorherzusagen, wer DCA aufgrund seiner genetischen Veranlagung langsam ausscheidet, könnte ein diagnostischer Test durchgeführt werden, um den Genotyp für GSTZ1 zu ermitteln, und die Dosis könnte sich nach dem Genotyp richten. Dies wäre weniger kostspielig als die wiederholte Messung des DCA-Spiegels im Blut. Zu diesem Zweck wurde die Pharmakokinetik von DCA an freiwilligen Probanden und an Patienten untersucht. In einer Studie an menschlichen Freiwilligen wurden die Teilnehmer genotypisiert und die Pharmakokinetik von DCA nach einer Einzeldosis von 25 mg/kg oder fünf aufeinanderfolgenden Tagesdosen von 25 mg/kg/Tag untersucht [55]. Wie aus Tabelle 3 hervorgeht, wiesen Personen mit mindestens einer EGT-Variante von GSTZ1 nach fünf Dosen eine größere Clearance von DCA auf als diejenigen, die kein EGT in ihrem Genotyp hatten. Abbildung 3 veranschaulicht die Kinetik der DCA-Elimination aus dem Plasma bei einem schnellen und einem langsamen Metabolisierer von DCA aus dieser Studie [55]. Ähnliche Ergebnisse wurden bei Patienten mit genetisch bedingter mitochondrialer Erkrankung gefunden, die 6, 12 oder 30 Monate lang alle 12 Stunden 12,5 mg/kg DCA erhielten [55,61]. Diese Arbeit zeigt auch, dass die im Urin ausgeschiedenen MA-Konzentrationen, ein weiteres Maß für die verminderte Aktivität von GSTZ1/MAAI, bei Probanden und Patienten ohne EGT in ihrem Genotyp höher waren (Tabelle 3).
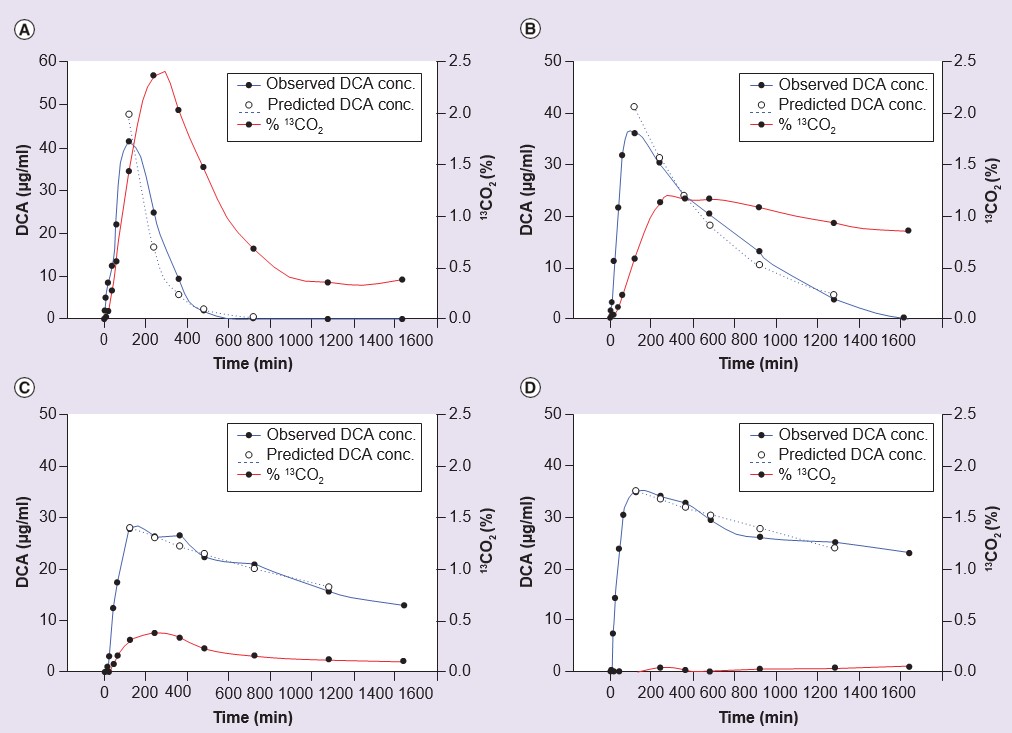
konz: Konzentration; DCA: Dichloroacetat.
| Sunject, Dauer der DCA-Behandlung | Plasma-Clearance, mml/min | Maleylaceton-Ausscheidung, μg/g Kreatinin | Ref. | ||
| EGT-Träger | EGT Nicht-Träger | EGT-Träger | EGT Nicht-Träger | ||
| Freiwillige, 5 Tage | 2.22 ± 0.72 (7) | 0.73 ± 0.84* (5) | Nicht erkannt | 7.2 ± 4.1* | [55] |
| Patienten, 12 Monate | 2.16 ± 0.99 (4) | 0.91, 0.17 | – | – | [55] |
| Patienten, 6 Monate | 1.90 ± 1.13 (11) | 0.53 ± 0.35* (6) | 1.2 ± 0.9 | 6.9 ± 2.6* | [61] |
| Patienten, 30 Monate | 2.08 ± 1.10 (11) | 0.67 ± 0.45* (6) | 1.9 ± 1.1 | 5.5 ± 1.2* | [61] |
Die angegebenen Werte sind Mittelwerte ± Standardabweichung (n) oder Einzelwerte, wenn n < 3.
*Signifikant verschieden von EGT-Trägern, p < 0,05.
Die ethnische Herkunft der Probanden war wie folgt:
EGT-Träger: vier kaukasische Amerikaner, zwei Afroamerikaner, ein Asiate; EGT-Nicht-Träger: fünf kaukasische Amerikaner. Die Ethnien der Patienten waren nicht verfügbar.
DCA: Dichloroacetat.
Die Analyse der Daten aus den Studien zur Pharmakokinetik von DCA bei freiwilligen Probanden deutet darauf hin, dass Personen, die die GSTZ1A-Variante, die für KRT kodiert, auf einem Allel tragen, eine schnellere anfängliche Clearance von DCA aufweisen als andere. Diese Personen zeigten auch eine stärkere Abnahme der Clearance nach fünf Dosen. Das Verhältnis der Clearance von der ersten bis zur fünften Dosis betrug 3,6 ± 0,8 (Mittelwert ± Standardfehler des Mittelwerts, n = 5) bei Personen mit der Variante für GSTZ1C (kodierend für EGT), aber nicht für GSTZ1A, und 18,2 ± 9,3 (n = 5) bei Personen mit der GSTZ1A-Variante auf mindestens einem Allel. Eine Person, die homozygot für GSTZ1A war, wies die größte Veränderung von der ersten bis zur fünften Dosis auf, wobei die Clearance von 16,7 auf 0,31 ml/min sank [55]. Der Unterschied in der mittleren Veränderung der Clearance war aufgrund der hohen Variabilität in der Gruppe, die homo- oder heterozygot für GSTZ1A war, statistisch nicht signifikant, wies jedoch auf einen Trend hin, der mit In-vitro-Studien über die Anfälligkeit der verschiedenen Haplotypen für die Inaktivierung durch DCA übereinstimmte.
In-vitro-Untersuchungen des DCA-Stoffwechsels
Weitere Einblicke in die Rolle des Genotyps beim DCA-Stoffwechsel ergaben sich aus Untersuchungen der GSTZ1-Expression und -Aktivität mit DCA im Zytosol und in den Mitochondrien der menschlichen Leber.
Ontogenese der Aktivität
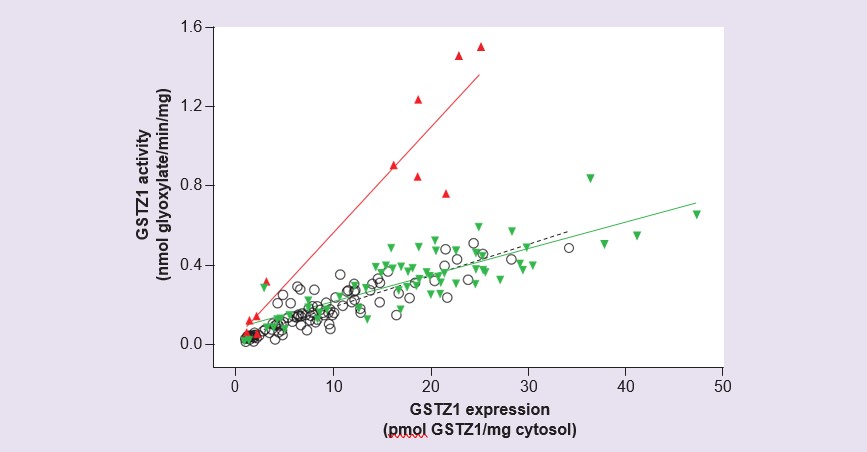
Studien mit menschlichem Leberzytosol, das aus einer Gruppe von 230 Leberproben von Spendern im Alter von 42 bis 84 Jahren hergestellt wurde, haben gezeigt, dass die Expression und Aktivität von GSTZ1 pränatal und in den ersten zwei Lebenswochen sehr gering oder nicht nachweisbar ist: Danach steigen Aktivität und Expression allmählich an [56]. Expression und Aktivität erreichten im Alter von 7 Jahren Werte im Erwachsenenbereich, wobei jedoch eine erhebliche interindividuelle Variabilität festgestellt wurde. Die Messung der Aktivität mit DCA und der Expression des GSTZ1-Proteins in diesen Proben ergab, dass die Proben von Personen mit der GSTZ1A-Variante auf einem Allel ein höheres Verhältnis von Aktivität/Expression aufwiesen als alle anderen (Abbildung 4). Dies stimmt mit den Ergebnissen von exprimiertem rekombinantem humanem GSTZ1A-1A überein, das für diese Variante eine höhere Aktivität mit DCA zeigte [54], und mit pharmakokinetischen Studien an Freiwilligen, bei denen Personen mit einem GSTZ1A-Gen eine schnelle anfängliche Clearance von DCA aufwiesen [55]. Obwohl die Entwicklungsstudie zeigte, dass die Aktivität bei Kindern unter 7 Jahren geringer war als bei Erwachsenen, bedeutet die Berücksichtigung der größeren Größe der Kinderleber im Verhältnis zur Körpermasse, dass die Aktivität mit DCA pro Kilogramm Körpergewicht bei Erwachsenen und Kindern im Alter von 2 Monaten bis 7 Jahren ähnlich ist [56].
Wirkung von Chlorid auf die Inaktivierung von GSTZ1 durch DCA
In scheinbarem Widerspruch zu der Beobachtung, dass das KRT-Protein mit einem schnelleren Metabolismus von DCA in vitro [56] und einer schnelleren Ausscheidung einer einzelnen DCA-Dosis [55] verbunden ist, steht die Feststellung, dass Personen mit der GSTZ1A-Variante auf einem Allel eine stärkere Verringerung der Clearance nach wiederholten DCA-Dosen aufwiesen als Personen, die homozygot für GSTZ1C waren. Neuere Erkenntnisse über die Wirkung von Chlorid auf die GSTZ1-1-Inaktivierung könnten diese Diskrepanzen erklären. Es wurde festgestellt, dass Chlorid GSTZ1-1 vor der Inaktivierung durch DCA in physiologisch relevanten Konzentrationen in der Leber schützt [62,63], jedoch war das GSTZ1-1 im Leberzytosol von Personen, die heterozygot für GSTZ1A waren, weniger vor der Inaktivierung durch physiologisches (38 mM) Chlorid geschützt als bei Personen mit anderen GSTZ1-Allelvarianten (Tabelle 4). Die Wirkung von Chlorid war so, dass die GSTZ1-1-Enzyme in den Lebern von Personen mit der GSTZ1A-Variante auf einem Allel doppelt so schnell inaktiviert wurden wie die mit anderen Haplotypen. Der genaue Mechanismus des Chlorid-Effekts muss noch bestimmt werden, aber dieses Phänomen ist eine Erklärung für den größeren Verlust der Clearance von DCA nach chronischer Verabreichung bei Personen mit einer oder mehreren Kopien der GSTZ1A-Variante. Wie bereits erwähnt, trägt die geringere Expression des Enzyms bei kaukasischen Personen mit Lysin (K) an Position 32 des Proteins wahrscheinlich zu der sehr geringen Clearance bei.
.
| Haplotyp | EC50 mM | Inaktivierungshalbwertszeit, h ohne Cl- | Inaktivierungshalbwertszeit, h mit 38 mM Cl- |
|---|---|---|---|
| EGT/EGT | 15 ± 3.1 (3) | 0.53, 0.49 | 5.73, 5.02 |
| KRT/EGT | 36 ± 2.2 (3) | 0.38, 0.38 | 2.66, 2.37 |
| EGM/EGM | 16.9 | – | 5.55 |
EC50 ist die Chloridkonzentration, die die Hälfte des zytosolischen GSTZ1 nach 2-stündiger Inkubation mit Na DCA, 0,5 mM, vor Inaktivierung schützt.
Schlussfolgerung
Aufgrund seiner Fähigkeit, die PDH-Kinase zu hemmen, wird DCA für die Behandlung von Krebs und Stoffwechselkrankheiten eingesetzt. Bei chronischer Verabreichung von DCA an den Menschen sind sein Stoffwechsel und seine Ausscheidung verringert. Der Grund für diese Verringerung der Ausscheidung nach mehrfacher DCA-Gabe ist, dass das einzige Enzym, von dem bekannt ist, dass es DCA verstoffwechselt, GSTZ1-1, während des DCA-Stoffwechsels inaktiviert wird. Diese auf einem Mechanismus beruhende Hemmung von GSTZ1-1 durch DCA führt zu einer verlangsamten Clearance nicht nur von DCA, sondern auch von den endogenen Substraten MAA und MA; ob die Akkumulation von DCA und/oder reaktiven Tyrosinkataboliten die reversible periphere Neuropathie verursacht, die bei einigen Patienten auftritt, ist noch nicht geklärt. Eine weitere ungelöste mögliche Ursache der Neuropathie ist der bescheidene Anstieg von ∂-ALA, der auf eine chronische DCA-Behandlung folgt. Für das Verständnis der Pharmakokinetik von DCA ist von Bedeutung, dass das GSTZ1-Gen in allen bisher untersuchten menschlichen Populationen polymorphe Varianten aufweist. Die Eigenschaften der Enzymproteine der GSTZ1-1-Variante unterscheiden sich hinsichtlich ihrer Fähigkeit, DCA und die endogenen Substrate MAA und MA zu metabolisieren, insbesondere bei chronischer Verabreichung. Während alle GSTZ1-1-Varianten durch DCA inaktiviert werden, wird eine Variante, die Lysin anstelle von Glutaminsäure an Position 32 und Arginin anstelle von Glycin an Position 42 des Proteins aufweist, in Gegenwart von physiologischen Chloridkonzentrationen schneller inaktiviert als andere Varianten. Bei Kaukasiern, nicht aber bei Personen afrikanischer Ethnizität, sind GSTZ1-Varianten mit Lysin an Position 32 mit einem SNP in der Promotorregion des GSTZ1-Gens verbunden, der zu einer verminderten Expression des GSTZ1-1-Proteins führt; dies kann auch als Phänotyp eines langsamen Metabolisierers auftreten. Ein Verständnis der Faktoren, die die Pharmakokinetik von DCA bei chronischer Verabreichung beeinflussen, ist erforderlich, um die richtige Dosis und das richtige Dosierungsintervall für eine sichere und wirksame Verabreichung dieses Medikaments an Patienten auszuwählen.
Ausblick auf die Zukunft
Aus den bisherigen Studien geht hervor, dass der GSTZ1-Haplotyp ein wichtiger Faktor ist, der die Clearance von DCA bestimmt. Ein besseres Verständnis der Regulierung des Enzyms unter physiologischen Bedingungen und in Gegenwart von DCA wird in Zukunft zu einem sichereren und wirksameren Einsatz des Medikaments führen. Mehrere Fragen müssen noch erforscht werden. Wird bei Heterozygoten eine Variante des Proteins bevorzugt exprimiert? Aktivitätsstudien, die eine höhere Aktivität mit DCA für Leberzytosolfraktionen von Personen zeigen, die heterozygot für GSTZ1A/GSTZ1C sind, lassen vermuten, dass die Enzymproteinvariante 1A (KRT) bei Heterozygoten bevorzugt exprimiert wird, was jedoch nicht nachgewiesen wurde. Diese Frage hat praktische Auswirkungen, da die 1A-Variante schneller inaktiviert wird als die 1C-Variante. Sollte eine niedrigere DCA-Dosis an Personen verabreicht werden, die DCA bei chronischer Verabreichung wahrscheinlich langsamer als der Durchschnitt abbauen? Wenn ja, sollten dies alle Personen sein, die nicht homozygot oder heterozygot für GSTZ1C sind? Sollten Kaukasier mit mindestens einer K-haltigen Variante (KRT, KGT, KGM) niedrigere DCA-Dosen erhalten als Menschen afrikanischer Abstammung, weil sie weniger GSTZ1-1 exprimieren? Welche Belege gibt es für eine geringe Expression von K-haltigen Varianten in anderen Populationen? Gibt es Populationsunterschiede in den Verteilungsmustern der wichtigsten Haplotypen? Haben pharmakogenetische Unterschiede bei Kindern dieselben Auswirkungen wie bei Erwachsenen? Kinder, die DCA einnehmen, bauen das Medikament in der Regel schneller ab als Erwachsene, aber mögliche pharmakogenetische Auswirkungen sind nicht umfassend untersucht worden. Zur Klärung dieser Fragen sind weitere Forschungsarbeiten erforderlich, um eine sichere und wirksame Dosierung von DCA zu gewährleisten.
Offenlegung finanzieller und konkurrierender Interessen
Die in diesem Manuskript besprochene Arbeit der Autoren wurde zum Teil vom US Public Health Service 1RO1 GM 099871, zum Teil von der Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore und University of Miami (NO1 HD90011) und zum Teil vom NIH/NCATS Clinical and Translational Science award to the University of Florida UL1 TR000064 finanziert. Die Autoren haben keine anderen relevanten Verbindungen oder finanziellen Beteiligungen zu Organisationen oder Einrichtungen, die ein finanzielles Interesse an oder einen finanziellen Konflikt mit den im Manuskript behandelten Themen oder Materialien haben, abgesehen von den offengelegten Verbindungen.
Bei der Erstellung dieses Manuskripts wurde keine Schreibhilfe in Anspruch genommen.
Zusammenfassung
- Die Auswahl einer wirksamen, aber nicht toxischen Dosis von Dichloracetat (DCA) ist wichtig für den langfristigen klinischen Einsatz von DCA zur Behandlung von Krebs und anderen Stoffwechselkrankheiten.
- Das Gen für das einzige Enzym, das dieses Medikament verstoffwechselt, GSTZ1-1, weist Polymorphismen auf, die nach einer Langzeitbehandlung mit DCA zu Phänotypen eines langsamen und eines schnellen Metabolisierers führen.
- SNPs in der kodierenden Region des GSTZ1-Gens führen zu fünf gemeinsamen Haplotypen mit unterschiedlicher Häufigkeit in den Populationen. Personen, die homozygot für den häufigsten, GSTZ1C, sind schnelle Metabolisierer. Es gibt Hinweise darauf, dass Haplotypen, die für GSTZ1-1-Proteine mit K an Position 32 kodieren, langsame Metabolisierer sind, aber andere Faktoren, die zu einem langsamen Metabolismus nach mehreren DCA-Dosen beitragen, sind nicht vollständig bekannt.
- Ein SNP in der Promotorregion bei Menschen mit kaukasischer, aber nicht afrikanischer Ethnizität ist mit einem SNP in der kodierenden Region verbunden, der zu einer Substitution von Lysin durch Glutaminsäure an Position 32 des GSTZ1-Proteins führt und mit einer verringerten Expression des Enzymproteins in der Leber einhergeht.
- In Gegenwart von DCA variiert die Stabilität des GSTZ1-1-Enzyms mit der Chloridkonzentration. Die GSTZ1A-Enzymvariante (KRT) wird unter physiologischen Chloridkonzentrationen in der Leber schneller inaktiviert als die übliche GSTZ1C-Variante (EGT).
REFERENZEN
1 Stacpoole PW. Überblick über die pharmakologischen und therapeutischen Wirkungen von Diisopropylammoniumdichloracetat (DIPA). J. Clin. Pharmacol. J. New Drugs 9(5), 282-291 (1969).2 Stacpoole PW. Die Pharmakologie von Dichloracetat. Stoffwechsel 38(11), 1124-1144 (1989).
3 Stacpoole PW, Felts JM. Diisopropylammoniumdichloracetat (DIPA) und Natriumdichloracetat (DCA): Wirkung auf den Glukose- und Fettstoffwechsel in normalem und diabetischem Gewebe. Stoffwechsel 19(1), 71-78 (1970).
4 Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N. Engl. J. Med. 298(10), 526-530 (1978).
5 Stacpoole PW, Harwood HJ Jr, Varnado CE. Regulation der Hydroxymethylglutaryl-Coenzym-A-Reduktase in der Rattenleber durch eine neue Klasse von nichtkompetitiven Inhibitoren. Auswirkungen von Dichloracetat und verwandten Carbonsäuren auf die Enzymaktivität. J. Clin. Invest. 72(5), 1575-1585 (1983).
6 Harwood HJ Jr, Bridge DM, Stacpoole PW. In vivo regulation of human mononuclear leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase. Studies in normal subjects. J. Clin. Invest. 79(4), 1125-1132 (1987).
7 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA, Stacpoole PW. Senkung des Serumcholesterins bei zwei Patienten mit homozygoter familiärer Hypercholesterinämie durch Dichloracetat. Atherosclerosis 33(3), 285-293 (1979).
8 Eichner HL, Stacpoole PW, Forsham PH. Treatment of streptozotocin diabetes with di-isopropylammonium dichloroacetate (DIPA). Diabetes 23(3), 179-182 (1974).
9 Backshear PJ, Holloway PA, Alberti KG. Metabolische Interaktionen von Dichloracetat und Insulin in experimenteller diabetischer Ketoazidose. Biochem. J. 146(2), 447-456 (1975).
10 Whitehouse S, Randle PJ. Aktivierung der Pyruvat-Dehydrogenase im perfundierten Rattenherz durch Dichloracetat (Kurzmitteilung). Biochem. J. 134(2), 651-653 (1973).
11 Whitehouse S, Cooper RH, Randle PJ. Mechanismus der Aktivierung von Pyruvatdehydrogenase durch Dichloracetat und andere halogenierte Carbonsäuren. Biochem. J. 141(3), 761-774 (1974).
12 Patel MS, Korotchkina LG. Regulierung des Pyruvatdehydrogenase-Komplexes. Biochem. Soc. Trans. 34(Pt 2), 217-222 (2006).
13 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Beweise für die Existenz einer gewebespezifischen Regulierung des Säugetier-Pyruvatdehydrogenase-Komplexes. Biochem. J. 329(Pt 1), 191-196 (1998).
14 Morten KJ, Caky M, Matthews PM. Stabilisierung der Pyruvatdehydrogenase E1alpha-Untereinheit durch Dichloracetat. Neurology 51(5), 1331-1335 (1998).
15 Han Z, Berendzen K, Zhong L et al. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol. Genet. Metab. 93(4), 381-387 (2008).
16 Evans OB, Stacpoole PW. Prolonged hypolactatemia and increased total pyruvate dehydrogenase activity by dichloroacetate. Biochem. Pharmacol. 31(7), 1295-1300 (1982).
17 Denton RM, Randle PJ, Bridges BJ et al. Regulation der Säugetier-Pyruvat-Dehydrogenase. Mol. Cell. Biochem. 9(1), 27-53 (1975).
18 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284(5), E855-E862 (2003).
19 Stacpoole PW, Nagaraja NV, Hutson AD. Die Wirksamkeit von Dichloracetat als laktatsenkendes Medikament. J. Clin. Pharmacol. 43(7), 683-691 (2003).
20 Kankotia S, Stacpoole PW. Dichloracetat und Krebs: neue Heimat für ein Arzneimittel für seltene Krankheiten? Biochim. Biophys. Acta 1846(2), 617-629 (2014).
21 Stacpoole PW. Das Dichloracetat-Dilemma: Umweltgefährdung oder therapeutische Goldmine – beides oder keines von beiden? Environ. Health Perspect. 119(2), 155-158 (2011).
22 Chu PI. Pharmakokinetik von Natriumdichloroacetat. Dissertation, Abteilung für Pharmazie, Universität von Florida, FL, USA (1987).
23 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmakokinetik, Metabolismus und Toxikologie von Dichloroacetat. Drug Metab. Rev. 30(3), 499-539 (1998).
24 Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. Plasmakonzentrationen und metabolische Wirkungen von intravenösem Natriumdichloracetat. Clin. Pharmacol. Ther. 37(1), 89-93 (1985).
25 Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G et al. Pharmacokinetics of oral dichloroacetate in dogs. J. Biochem. Mol. Toxicol. 27(12), 522-525 (2013).
26 Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolische Wirkungen und Pharmakokinetik von intravenös verabreichtem Dichloracetat beim Menschen. Diabetologia 19(2), 109-113 (1980).
27 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Behandlung der Laktatazidose mit Dichloroacetat. N. Engl. J. Med. 309(7), 390-396 (1983).
28 Henderson GN, Curry SH, Derendorf H, Wright EC, Stacpoole PW. Pharmakokinetik von Dichloroacetat bei erwachsenen Patienten mit Laktatazidose. J. Clin. Pharmacol. 37(5), 416-425 (1997).
29 Stacpoole PW, Kerr DS, Barnes C et al. Kontrollierte klinische Studie von Dichloracetat zur Behandlung der kongenitalen Laktatazidose bei Kindern. Pediatrics 117(5), 1519-1531 (2006).
30 Stacpoole PW, Gilbert LR, Neiberger RE et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121(5), e1223-e1228 (2008).
31 Kaufmann P, Engelstad K, Wei Y et al. Dichloracetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurologie 66(3), 324-330 (2006).
32 Stickler DE, Valenstein E, Neiberger RE et al. Periphere Neuropathie bei genetischen mitochondrialen Erkrankungen. Pediatr. Neurol. 34(2), 127-131 (2006).r
33 Kaufmann P, Pascual JM, Anziska Y et al. Nervenleitungsanomalien bei Patienten mit MELAS und der A3243G-Mutation. Arch. Neurol. 63(5), 746-748 (2006).
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Altersabhängige Kinetik und Metabolismus von Dichloracetat: mögliche Bedeutung für die Toxizität. J. Pharmacol. Exp. Ther. 324(3), 1163-1171 (2008).
35 James MO, Yan Z, Cornett R et al. Pharmakokinetik und Metabolismus von [14C]Dichloracetat in männlichen Sprague-Dawley-Ratten. Identifizierung von Glycinkonjugaten, einschließlich Hippurat, als Metaboliten von Dichloracetat im Urin. Drug Metab. Dispos. 26(11), 1134-1143 (1998).
36 Rowland A, Miners JO, Mackenzie PI. Die UDP-Glucuronosyltransferasen: ihre Rolle im Drogenmetabolismus und in der Entgiftung. Int. J. Biochem. Cell. Biol. 45(6), 1121-1132 (2013).
37 Vessey DA, Kelley M, Warren RS. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim. Biophys. Acta 1428(2-3), 455-462 (1999).
38 James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Glutathion-abhängige Umwandlung in Glyoxylat, ein Hauptweg der Biotransformation von Dichloracetat im hepatischen Zytosol von Menschen und Ratten, ist bei Dichloracetat-behandelten Ratten reduziert. Drug Metab. Dispos. 25(11), 1223-1227 (1997).
39 Tong Z, Brett PG, Anders MW. Glutathiontransferase zeta katalysiert die Oxygenierung des Karzinogens Dichloressigsäure zu Glyoxylsäure. Biochem. J. 331(Pt 2), 371-374 (1998).
49 Fernandez-Canon JM, Penalva MA. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J. Biol. Chem. 273(1), 329-337 (1998).
41 Blackburn AC, Woollatt E, Sutherland GR, Board PG. Charakterisierung und Chromosomenposition des Gens GSTZ1, das für die menschliche Glutathion-Transferase der Zeta-Klasse und die Maleylacetoacetat-Isomerase kodiert. Cytogenet. Cell. Genet. 83(1-2), 109-114 (1998).
42 Li W, James MO, Mckenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrium als neuer Ort der Dichloracetat-Biotransformation durch Glutathion-Transferase zeta 1. J. Pharmacol. Exp. Ther. 336(1), 87-94 (2011).
43 Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Immunhistochemische Lokalisierung und Aktivität der Glutathiontransferase zeta (GSTZ1-1) in Rattengeweben. Drug Metab. Dispos. 30(6), 616-625 (2002).
44 Shangraw RE, Fisher DM. Pharmakokinetik von Dichloracetat bei Patienten, die sich einer Lebertransplantation unterziehen. Anesthesiology 84(4), 851-858 (1996).
45 Cornett R, James MO, Henderson GN, Cheung J, Shroads AL, Stacpoole PW. Inhibition of glutathione S-transferase zeta and tyrosine metabolism by dichloroacetate: a potential unifying mechanism for its altered biotransformation and toxicity. Biochem. Biophys. Res. Commun. 262(3), 752-756 (1999).
46 Guo X, Dixit V, Liu H et al. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 34(1), 36-42 (2006).
47 Anderson WB, Board PG, Gargano B, Anders MW. Inaktivierung der Glutathiontransferase zeta durch Dichloressigsäure und andere fluorfreie Alpha-Halogenalkansäuren. Chem. Res. Toxicol. 12(12), 1144-1149 (1999).
48 Anderson WB, Liebler DC, Board PG, Anders MW. Massenspektrale Charakterisierung von Dichloressigsäure-modifizierter menschlicher Glutathiontransferase zeta. Chem. Res. Toxicol. 15(11), 1387-1397 (2002).
49 Dixit V. Inaktivierung von Glutathiontransferase zeta durch Dichloressigsäure. Doktorarbeit, Abteilung Medizinische Chemie, Universität Florida, FL, USA (2005).
50 Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. Die Dichloracetat-Laktatazidose-Studiengruppe. N. Engl. J. Med. 327(22), 1564-1569 (1992).
51 Abdelmalak M, Lew A, Ramezani R et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol. Genet. Metab. 109(2), 139-143 (2013).
52 Board PG, Baker RT, Chelvanayagam G, Jermiin LS. Zeta, eine neue Klasse von Glutathion-Transferasen in einer Reihe von Arten von Pflanzen bis zum Menschen. Biochem. J. 328(Pt 3), 929-935 (1997).
53 Blackburn AC, Tzeng HF, Anders MW, Board PG. Entdeckung eines funktionellen Polymorphismus in der menschlichen Glutathion-Transferase zeta durch Expressed Sequence Tag Database-Analyse. Pharmacogenetics 10(1), 49-57 (2000).
54 Blackburn AC, Coggan M, Tzeng HF et al. GSTZ1d: ein neues Allel der Glutathion-Transferase zeta und der Maleylacetoacetat-Isomerase. Pharmacogenetics 11(8), 671-678 (2001).
55 Shroads AL, Langaee T, Coats BS et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J. Clin. Pharmacol. 52(6), 837-849 (2012).
56 Li W, Gu Y, James MO et al. Prenatal and postnatal expression of glutathione transferase zeta 1 in human liver and the roles of haplotype and subject age in determining activity with dichloroacetate. Drug Metab. Dispos. 40(2), 232-239 (2012).Google Scholar
57 Board PG, Anders MW. Glutathiontransferase zeta: Entdeckung, polymorphe Varianten, Katalyse, Inaktivierung und Eigenschaften von Gstz1-/- Mäusen. Drug Metab. Rev 43(2), 215-225 (2011).
58 Polekhina G, Board PG, Blackburn AC, Parker MW. Crystal structure of maleylacetoacetate isomerase/glutathione transferase zeta reveals the molecular basis for its remarkable catalytic promiscuity. Biochemistry 40(6), 1567-1576 (2001).
59 Fang YY, Kashkarov U, Anders MW, Board PG. Polymorphismen im Promotor der menschlichen Glutathiontransferase zeta. Pharmacogenet. Genomics 16(5), 307-313 (2006).
60 Langaee TY, Zhong G, Li W et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics 25(5), 239-245 (2015).
61 Shroads AL, Coats BS, Mcdonough CW, Langaee T, Stacpoole PW. Haplotypvariationen in der Glutathiontransferase zeta 1 beeinflussen die Kinetik und Dynamik von chronischem Dichloracetat bei Kindern. J. Clin. Pharmacol. 55(1), 50-55 (2015).
62 Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Chlorid und andere Anionen hemmen die Dichloracetat-induzierte Inaktivierung von GSTZ1 in der menschlichen Leber in einer Haplotyp-abhängigen Weise. Chem. Biol. Interact. 215C, 33-39 (2014).
63 Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Chloridkonzentrationen in menschlichem Leberzytosol und Mitochondrien sind eine Funktion des Alters. Biochem. Biophys. Res. Commun. 459(3), 463-468 (2015).