Маргарет О Джеймс*,1 и Питер В Стейкпул2,3
1 Кафедра медицинской химии, Университет Флориды, Гейнсвилл, штат Флорида, 32610-0485, США
2 Кафедра медицины, Медицинский колледж, Университет Флориды, Гейнсвилл, штат Флорида, 32610-0485, США
Кафедра биохимии и молекулярной биологии, Университет Флориды, Гейнсвилл, штат Флорида, 32610-0485, США
Переписка: Тел: +1 352 273 7707 Email: [email protected]
Отправлено: 16 декабря 2015
Принято: 17 февраля 2016 г.
Опубликовано: 4 мая 2016 г
Аннотация
Исследуемый препарат дихлорацетат (ДХА) является регулятором обмена веществ, который успешно используется для лечения приобретенных и врожденных заболеваний обмена веществ, а в последнее время и солидных опухолей. Его клиническое применение выявило проблемы с подбором соответствующих доз. Хроническое применение DCA приводит к ингибированию метаболизма DCA и потенциальному накоплению до уровней, которые приводят к побочным эффектам. Это происходит потому, что превращение ДКА в глиоксилат катализируется одним ферментом, глутатионтрансферазой дзета 1 (GSTZ1-1), который инактивируется ДКА. SNPs в гене GSTZ1 приводят к экспрессии полиморфных вариантов фермента, которые отличаются по активности и скорости инактивации DCA в физиологических условиях: эти свойства приводят к значительным различиям между людьми в фармакокинетике DCA.
Ключевые слова: дихлорацетат; GSTZ1; фармакогенетика
Клиническое применение дихлорацетата
Для такой простой молекулы ДХА имеет удивительно богатый и разнообразный фармакологический портфель, насчитывающий почти столетие [1,2]. Однако его современное использование в качестве лекарственного средства началось в 1970 году, когда была обнаружена его избирательная способность снижать уровень глюкозы в крови у животных с диабетом, но не у недиабетиков [3], а затем подтверждена у людей [4]. В 1970-х и 1980-х годах были выявлены многие метаболические свойства ДКА, связанные с метаболизмом глюкозы и липидов. Однако большинство его фармакологических эффектов можно свести к нескольким основным участкам и механизмам действия. Во-первых, DCA является неконкурентным ингибитором фермента эндоплазматического ретикулума HMG CoA редуктазы, который катализирует лимитирующий шаг в биосинтезе холестерина. Ингибирующий эффект DCA наблюдается как в печени грызунов [5], так и в лейкоцитах человека [6]; вероятно, он объясняет связанное с приемом препарата снижение уровня общего холестерина и холестерина липопротеинов низкой плотности (ЛПНП) у пациентов с ЛПНП-рецептор-негативной гомозиготной семейной гиперхолестеринемией [7] и его назначение в качестве первого сиротского препарата для лечения этого редкого заболевания. Во-вторых, DCA подавляет печеночный синтез триглицеридов de novo у недиабетических грызунов [5] и снижает уровень циркулирующих триглицеридов и липопротеинов очень низкой плотности у пациентов с сахарным диабетом 2 типа [4]. Он также снижает уровень кетоновых тел в крови крыс с экспериментально вызванным диабетическим кетоацидозом [8,9]. Точные механизмы, лежащие в основе этих эффектов на синтез и окисление липидов, неизвестны. В-третьих, DCA стимулирует митохондриальный PDC, который необратимо окисляет пируват до ацетил-коэнзима А (ацетил-КоА) [10], свойство, общее для некоторых других галогенированных короткоцепочечных жирных кислот [11]. Именно способность DCA изменять активность PDC вызвала наибольшее количество экспериментальных и клинических исследований этой необычной молекулы.
PDC регулируется посттрансляционно в основном путем обратимого фосфорилирования одного или нескольких из трех остатков серина на субъединице E1α первого фермента PDH PDC, который декарбоксилирует пируват [12]. Четыре киназы PDH (PDK1-4) и две фосфатазы PDH (PDP 1 и 2) осуществляют этот регуляторный аспект PDC у человека, при котором фосфорилированный фермент каталитически неактивен. PDK по-разному экспрессируются в тканях, хотя PDK2 экспрессируется повсеместно [13]. Пируват, субстрат для реакции PDC, ингибирует PDKs, связываясь с небольшим карманом в N-конце киназы, что приводит к стимуляции активности PDC. Напротив, накопление продуктов реакции, ацетил-КоА и NADH, приводит к активации PDK и ингибированию PDC. DCA, структурный аналог пирувата, также присоединяется к пируват-связывающему сайту, что приводит к ингибированию PDK, причем порядок ингибирования следующий: PDK2>PDK1˜PDK4>>PDK3 [13]. Хроническое воздействие лекарств in vitro [14,15] и in vivo [16] может стабилизировать PDC и уменьшить его оборот, что обеспечивает второй механизм, посредством которого ДКА повышает активность комплекса, и объясняет его длительное фармакологическое действие после отмены препарата [4].
Хотя DCA является относительно слабым ингибитором PDK (Ki 0,2 мМ для PDK2), его влияние на аспекты промежуточного метаболизма очень велико и напрямую связано с ключевой ролью PDC в регуляции выбора топлива и биоэнергетики эукариотических клеток [17,18]. ДКА пока еще не одобрен Управлением по контролю за продуктами и лекарствами США. Однако его продолжают активно исследовать в качестве метаболического модулятора при ряде врожденных и приобретенных метаболических нарушений, основанных на его стимулирующем действии на PDC (табл. 1).
| Свойство | Механизм | Реф. |
|---|---|---|
| Увеличение OXPHOS и биоэнергетики | ↓ PDK ↑ PDC | [2,10] |
| Снижение уровня глюкозы в крови при голодании или диабете | ↓ PDK ↑ PDC | [4] |
| Снижение лактата в крови и ЦСЖ | ↓ ПДК ↑ ПДК | [19] |
| Снижение общего холестерина и холестерина ЛПНП в крови | ↓ HMG CoA редуктаза → ↓ синтез холестерина | [6,7] |
| Снижение уровня триглицеридов и холестерина ЛПНП в крови | ↓ печеночный синтез ТГ → ↓ синтез VLDL | [4] |
| Обращение вспять эффекта Варбурга при раке, ПАГ и других пролиферативных состояниях | ↓ PDK ↑ PDC | [20] |
ЦСЖ: цереброспинальная жидкость; ГМГ-КоА: Гидроксиметилглутарил коэнзим А; ЛПНП: липопротеин низкой плотности; OXPHOS: Окислительное фосфорилирование; PAH: Легочная артериальная гипертензия; PDC: Пируватдегидрогеназный комплекс; PDK: Киназа пируватдегидрогеназы; TG: Триглицерид; VLDL: Липопротеин очень низкой плотности.
Фармакокинетика и клиренс
Пероральный DCA быстро всасывается и имеет биодоступность, близкую к единице, как после перорального, так и парентерального приема [21,22]. Препарат можно обнаружить в плазме крови человека в течение 15 минут после перорального приема дозы 50 мг/кг. У ранее наивных людей период полураспада DCA при пероральном или внутривенном введении в плазме крови составляет приблизительно 1 ч [23]. Скорость метаболизма среди здоровых видов животных следующая: мышь>крыса>человек≥собака [22-25].
Ранние фармакокинетические исследования однократной дозы внутривенного ДКА описали нелинейную кинетику при дозах ≥35 мг/кг [26]. Позже было обнаружено, что скорость клиренса препарата из плазмы снижается при повторном внутривенном введении дозы 50 мг/кг массы тела у тяжелобольных взрослых с молочнокислым ацидозом [27,28]. Задержка плазменного клиренса препарата, введенного перорально в дозе 50 мг/кг, также наблюдалась у здоровых взрослых, которым потребовалось от нескольких недель до нескольких месяцев, чтобы достичь первоначальной скорости клиренса при повторном приеме [22].
Основной прогресс в понимании этой необычной особенности фармакокинетики ДКА был достигнут благодаря доказательным исследованиям его клинической токсикологии. Хронический прием DCA может вызывать обратимую периферическую нейропатию у разных видов животных [21,23], и этот эффект впервые был клинически отмечен 25 лет назад у 16-летнего мальчика, который получал 50 мг/кг/день в течение примерно 4 месяцев [7]. Напротив, в рандомизированном контролируемом исследовании (РКИ) 43 детей младшего возраста с различными типами первичных митохондриальных заболеваний (средний возраст при поступлении: 5,6 лет) пероральный прием DCA в дозе 12,5 мг/кг каждые 12 часов в течение 6 месяцев хорошо переносился и не вызывал негативных изменений в электрической проводимости периферических нервов по сравнению с плацебо [29], хотя при более длительном приеме препарата наблюдалось некоторое бессимптомное снижение нервной проводимости [30]. В отличие от этого, РКИ с участием 30 подростков и взрослых старшего возраста (средний возраст участников: 30 лет) с генетическим митохондриальным заболеванием, которые получали идентичную дозу ДКА и график дозирования, использовавшиеся в педиатрическом исследовании, было досрочно прекращено из-за ухудшения или нового появления периферической нейропатии [31], распространенного осложнения митохондриальных заболеваний [32,33]. При сравнении кинетики ДКА в этих двух группах пациентов было обнаружено, что клиренс хронического ДКА в плазме крови в старшей группе был заметно медленнее [34], что подтвердилось в исследованиях на крысах [34,35]. Эти данные послужили толчком к изучению энзимологии биотрансформации ДКА.
Важность глутатионтрансферазы дзета 1 в биотрансформации ДКА
Малые карбоновые кислоты часто подвергаются глюкуронидированию [36] или превращаются в производные коэнзима А, а затем в конъюгаты аминокислот [37], однако нет никаких доказательств того, что ДКК образует глюкуронид, производное коэнзима А или конъюгат аминокислот [2, James MO, неопубликованные данные]. Вместо этого DCA является субстратом для необычного фермента GST, GSTZ1-1, который превращает его в глиоксилат в реакции, требующей, но не потребляющей глутатион (GSH) [38,39]. Как и другие GST, GSTZ1-1 активен в димерной форме, а активный фермент представляет собой гомодимер. Дехлорирование ДКА с образованием глиоксилата, катализируемое GSTZ1-1, является единственным путем первичного метаболизма ДКА, наблюдаемым в печени [35]. Помимо катализации метаболизма ДКА, GSTZ1-1, также известный как MAAI, выполняет важную физиологическую функцию в катаболизме тирозина, где он изомеризует малеилацетоацетат (MAA) и малеилацетон (MA) в фумарилацетоацетат и фумарилацетон в реакциях, которые требуют, но не потребляют GSH (рис. 1) [40,41]. Биотрансформация ДКА в глиоксилат прекращает его фармакологически и терапевтически важные действия, которые обсуждались выше, поэтому активность GSTZ1-1 контролирует продолжительность действия ДКА.
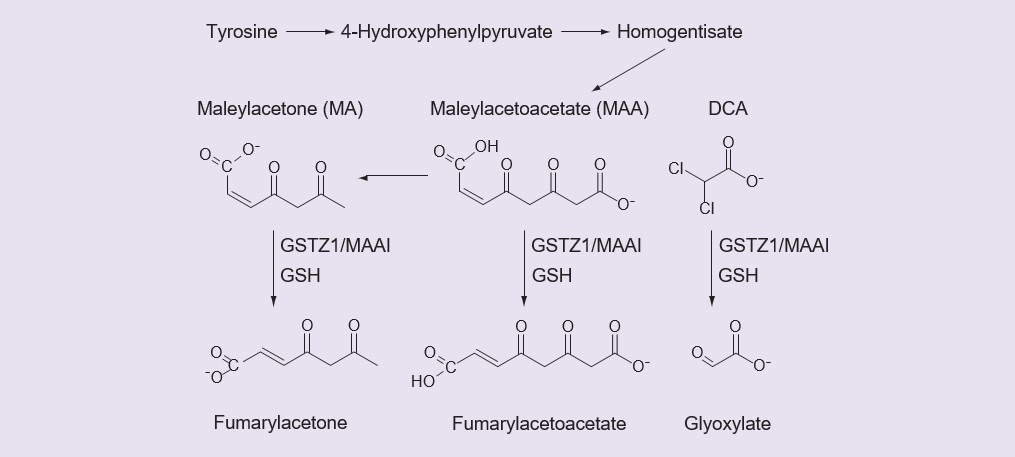
Неактивный метаболит DCA, глиоксилат, далее метаболизируется до углекислого газа, глицина и оксалата. Фумарилацетон и фумарилацетоацетат далее метаболизируются фумарилацетоацетат гидролазой. DCA: дихлорацетат: GSH: глутатион; MA: малеилацетон; MMA: Малеацетоацетат.
Печень является основным местом экспрессии GSTZ1-1, где он обнаруживается как в цитозоле, так и в митохондриальном матриксе [38,42]. Исследования на животных показали очень низкие уровни экспрессии GSTZ1-1 в почках, сердце, ЖКТ и мозге [43]. Роль внепеченочных тканей в метаболизме ДКА у людей подробно не изучалась, однако одно исследование показало, что ДКА не выводится из крови во время печеночной фазы у пациентов с пересадкой печени, которым ДКА вводили для борьбы с молочнокислым ацидозом, что указывает на отсутствие метаболизма [44]. Печеночная фаза была непродолжительной (в среднем 73 мин), поэтому данные были ограничены, однако это исследование предполагает незначительную роль других тканей, кроме печени, в выведении ДКА, по крайней мере, при однократном приеме.
Ингибирование GSTZ1-1 под действием ДКА
Исследования на животных и с использованием экспрессированных рекомбинантных белков GSTZ1-1 человека показали, что причина, по которой многократные дозы ДКА выводятся из организма гораздо медленнее, чем однократная доза, заключается в том, что ДКА является ингибитором GSTZ1-1 по механизму [25,34,45-47]. В процессе биотрансформации ДКА образуются аддукты к белку GSTZ1-1, которые инактивируют белок и, предположительно, запускают его протеолиз [48,49]. Введение ДКА крысам приводит к потере активности и экспрессии GSTZ1-1 в зависимости от дозы и времени: потеря функции GSTZ1-1 была более выраженной и сохранялась дольше у взрослых крыс по сравнению с молодыми [34,35]. Подобное влияние возраста на потерю функции GSTZ1-1 после введения ДКА наблюдалось и у людей, причем у детей наблюдалось меньшее увеличение периода полураспада в плазме и меньшее снижение клиренса, чем у взрослых [34]. Помимо влияния на фармакокинетику ДКА, потеря GSTZ1-1 (MAAI) приводит к накоплению физиологических субстратов MA и, предположительно, MAA, которые являются реактивными молекулами, способными образовывать аддукты с клеточными макромолекулами [34,45]. Ингибирование MAAI также приводит к отвлечению углерода тирозина на образование сукцинилацетона. Эта молекула ингибирует проксимальный этап синтеза гема, вызывая накопление реактивной молекулы δ-аминолевулината (δ-ALA). И МА, и δ-АЛА увеличиваются в крови и/или моче людей, хронически подвергающихся воздействию ДКА [34]: клиническое токсикологическое значение этого эффекта еще не доказано.
Другие метаболиты ДКА
Единственным другим известным первичным метаболитом ДКА является монохлоруксусная кислота (МХУ), которая иногда обнаруживается в следовых количествах в крови после приема ДКА [34]. МХУ, по-видимому, образуется из ДХУ в эритроцитах [34]. Нет никаких доказательств образования МКА из ДКА в печени. Для практических целей превращение ДКА в глиоксилат определяет фармакокинетику элиминации ДКА, поэтому знание факторов, влияющих на скорость и степень катализируемого GSTZ1-1 метаболизма ДКА у людей, важно для безопасного и эффективного дозирования пациентов.
Что касается последствий лечения ДКА, глиоксилат может быть далее метаболизирован до углекислого газа с помощью фермента карболигазы, до глицина с помощью одного или нескольких ферментов аминотрансфераз или может быть окислен до оксалата [35]. Доказательства в пользу всех трех путей были получены в ходе исследований, в которых крысы получали дозу DCA [35]. Из этих путей только образование оксалата может быть потенциально вредным для пациента, однако у крыс образование оксалата было второстепенным путем [35]. У тяжелобольных взрослых пациентов с молочнокислым ацидозом, получавших внутривенное введение DCA или плацебо в рандомизированном контролируемом клиническом исследовании, оценивали наличие оксалатных камней и токсичности, связанной с оксалатами; не было обнаружено никаких доказательств различий между группами DCA и плацебо [50]. Аналогичным образом, пероральный прием DCA в течение нескольких лет в исследовании детей с врожденными формами молочнокислого ацидоза не выявил признаков образования оксалатных камней или токсичности [51].
Полиморфные варианты GSTZ1
Человеческий GSTZ1 (NM_145870) подозревался в наличии аллельных вариантов с момента его открытия [52]. Последующие исследования выявили SNPs в кодирующей и некодирующей областях гена. Мы все еще изучаем влияние, если таковое имеется, большинства этих SNP на экспрессию, стабильность и активность GSTZ1 в отношении DCA. Это активная область исследований, поскольку эти факторы влияют на фармакокинетику и, в конечном счете, на безопасное применение ДКА. Внимание привлекают SNPs, приводящие к аминокислотным изменениям в экспрессируемом белке, а также SNPs, влияющие на трансляцию GSTZ1 и, в конечном итоге, на уровень экспрессии ферментного белка.
Варианты аминокислотной последовательности экспрессируемого ферментного белка
В кодирующей области GSTZ1 [53,54] были выявлены три распространенных SNP, которые, как было отмечено, дают пять гаплотипов (табл. 2). Эти пять гаплотипов встречаются у людей с разной частотой в зависимости от этнической принадлежности (рис. 2), однако наиболее часто встречающимся вариантом во всех исследованных на сегодняшний день популяциях, присутствующим примерно у 50% людей, является GSTZ1C [54-56]. Этот гаплотип также называют EGT, указывая на вариант аминокислот в позициях 32, 42 и 82 в белке фермента [54,55]. В настоящее время имеется относительно мало информации о частоте встречаемости пяти распространенных гаплотипов в популяциях, отличных от европеоидов. Экспрессированные рекомбинантные человеческие белки GSTZ1A-1A, 1B-1B, 1C-1C и 1D-1D были исследованы с DCA в качестве субстрата, и было установлено, что GSTZ1A-1A (KRT/KRT) имеет более высокую активность, чем другие варианты [54,57]. GSTZ1F — редкий вариант (табл. 2), и свойства рекомбинантного белка не изучались. Отдельный человек может быть гомо- или гетерозиготным по этим гаплотипам. Для гетерозигот неизвестно, экспрессируется ли один из вариантов преимущественно на белковом уровне, однако цитозольные фракции печени, полученные от людей, несущих вариант GSTZ1A по одному аллелю, обладают более высокой активностью in vitro в отношении ДКА, чем люди без этого аллеля [56], что указывает на возможность преимущественной экспрессии белка фермента варианта KRT.
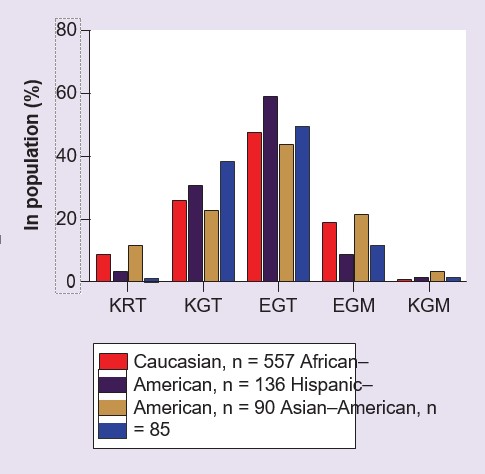
E: Глутаминовая кислота; G: Глицин; K: Лизин: М: метионин; Т: треонин.
| Вариант | Положение нуклеотида | Аминокислотная позиция | Процент в популяции (%)† | ||||
| 94 | 124 | 245 | 32 | 42 | 82 | ||
| GSTZ1A | A | A | C | Lys (K) | Arg (R) | Thr (T) | 1-10 |
| GSTZ1B | A | G | C | Lys (K) | Gly (G) | Thr (T) | 25-35 |
| GSTZ1C | G | G | C | Glu (E) | Gly (G) | Thr (T) | 45-55 |
| GSTZ1D | G | G | T | Glu (E) | Gly (G) | Мет (М) | 10-20 |
| GSTZ1F | A | G | T | Lys (K) | Gly (G) | Met (M) | <1 |
Полиморфные варианты GSTZ1-1 человека и их приблизительная частота в популяции.
†Частота встречаемости каждого гаплотипа зависит от этнической группы, см. рис. 2 и [54-56].
Glu (E): глутаминовая кислота; Gly (G): Глицин; Lys (K): Лизин: Met (M): Метионин; Thr (T): Треонин.
.
В исследованиях с экспрессированными рекомбинантными человеческими белками GSTZ1A-1A, 1B-1B, 1C-1C и 1D-1D также изучалась активность с физиологически важным субстратом, МА [54]. Скорость метаболизма МА отличалась от активности, измеренной с использованием DCA в качестве субстрата: вариант GSTZ1C-1C (EGT/EGT) имел самую высокую активность с МА, а GSTZ1A-1A — самую низкую [54].
Помимо часто встречающихся вариантов, описанных выше, имеются данные о двух других вариантах кодирующей последовательности. Один из них был найден в базе данных тегов экспрессированных последовательностей и назван GSTZ1E [54], однако этот вариант, в котором вместо лейцина в позиции 23 находится пролин, не был обнаружен у людей. Другой вариант был обнаружен у добровольца, участвовавшего в фармакокинетических исследованиях DCA. Этот человек, гетерозиготный по KGT/KGM (1B/1F), выводил даже одну дозу ДКА крайне медленно; секвенирование ДНК этого человека выявило новый SNP в пятом экзоне GSTZ1, в результате чего в положении 99 белка вместо валина был метионин [55]. Согласно кристаллической структуре GSTZ1B [58], положение 99 находится в α-спиральной связке. Несмотря на поиск этого варианта в образцах печени, которые демонстрировали низкую активность в отношении DCA [56], других людей с этим полиморфизмом пока не выявлено.
Полиморфизмы промоторной области
Десять SNP в образцах геномной ДНК африканских и австралийских европейцев были обнаружены в области, простирающейся на 1500 нуклеотидов вверх по течению от сайта начала транскрипции GSTZ1,
считается промоторной областью гена [59]. Два из этих SNP, -1002G>A и -289C>T, были связаны, соответственно, с уменьшением и увеличением промоторной активности, оцененной в клетках HepG2. Последующие исследования с использованием ДНК из образцов печени показали, что у американцев европейского происхождения, но не афроамериканцев, аллель А SNP rs 7975, который приводил к экспрессии лизина вместо глутаминовой кислоты в положении 32 белка, находился в неравновесии по связи с аллелем А SNP -1002 G>A промоторной области, rs7160195, и приводил к снижению экспрессии белка GSTZ1-1 с лизином (K) в положении 32 [60]. Сниженная экспрессия наблюдалась как у гетерозигот, так и у гомозигот по GSTZ1A (KRT), GSTZ1B (KGT) и GSTZ1F (KGM) у американцев кавказского происхождения [60]. В образцах печени, исследованных у афроамериканцев, экспрессия K-содержащих вариантов GSTZ1-1 не наблюдалась. По неясным пока причинам не было обнаружено различий в уровнях мРНК GSTZ1 между гаплотипами, что указывает на несоответствие между результатами по экспрессии белка и экспрессии мРНК [60]. Обнаружение более низкой экспрессии белка GSTZ1-1 для K-содержащих гаплотипов у кавказцев предполагает, что у этих людей (кроме тех, у кого есть KRT) будет снижена скорость начального метаболизма ДКА. Кроме того, это дает возможность предположить, что у кавказцев с К-содержащими гаплотипами, получающих лечение ДКА, ресинтез белка GSTZ1-1 будет происходить менее эффективно, что приведет к еще более медленной элиминации, чем у людей без этого гаплотипа.
Влияние вариантов GSTZ1 на фармакокинетику ДКА у людей
Если предположить, что побочные эффекты хронического лечения ДКА связаны с накоплением либо самого ДКА, либо реактивных эндогенных субстратов для GSTZ1, МАО и МА, то существует убедительный аргумент в пользу того, что безопасное клиническое применение ДКА должно основываться на понимании изменчивости элиминации препарата у людей во время хронического лечения. В этом случае дозу ДКА можно было бы снизить для тех, у кого наблюдается медленная элиминация, сохраняя при этом эффективный уровень препарата в крови. Если бы можно было предсказать, у кого ДКА будет выводиться медленно, основываясь на их генетических особенностях, можно было бы провести диагностический тест для определения генотипа GSTZ1, а дозировка могла бы быть основана на генотипе. Это было бы дешевле, чем многократное измерение уровня ДКА в крови. С этой целью фармакокинетика DCA была изучена на добровольцах и пациентах. В одном из исследований на добровольцах-людях участникам был присвоен генотип, и фармакокинетика ДКА изучалась после однократного приема 25 мг/кг или пяти последовательных ежедневных доз 25 мг/кг/день [55]. Как показано в таблице 3, у лиц, имеющих хотя бы один вариант EGT генотипа GSTZ1, клиренс ДКА после пяти доз был выше, чем у тех, кто не имел EGT в своем генотипе. Рисунок 3 иллюстрирует кинетику выведения ДКА из плазмы крови у одного быстрого и одного медленного метаболизатора ДКА из этого исследования [55]. Аналогичные результаты были обнаружены у пациентов с генетическим митохондриальным заболеванием, которые получали 12,5 мг/кг ДКА каждые 12 часов в течение 6, 12 или 30 месяцев [55,61]. В данной работе также показано, что уровни МА, выделяемого с мочой, — еще один показатель сниженной активности GSTZ1/MAAI — были выше у добровольцев и пациентов без EGT в генотипе (Таблица 3).
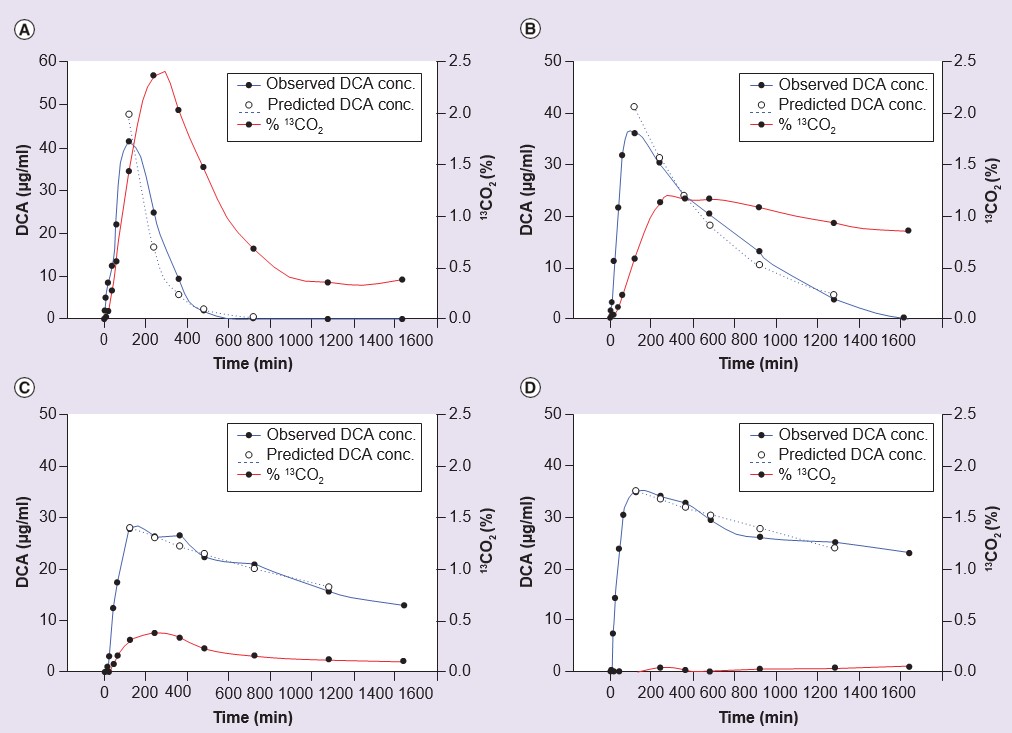
conc..: Концентрация; DCA: дихлорацетат.
| Sunject, продолжительность лечения ДКА | Плазменный клиренс, ммл/мин | Выведение малеилацетона, мкг/г креатинина | Ref. | ||
| Носитель EGT | Неноситель ЭГТ | Носитель ЭГТ | Носитель ЭГТ не носитель ЭГТ | ||
| Добровольцы, 5 дней | 2.22 ± 0.72 (7) | 0.73 ± 0.84* (5) | Не обнаружено | 7.2 ± 4.1* | [55] |
| Пациенты, 12 месяцев | 2.16 ± 0.99 (4) | 0.91, 0.17 | — | — | [55] |
| Пациенты, 6 месяцев | 1.90 ± 1.13 (11) | 0.53 ± 0.35* (6) | 1.2 ± 0.9 | 6.9 ± 2.6* | [61] |
| Пациенты, 30 месяцев | 2.08 ± 1.10 (11) | 0.67 ± 0.45* (6) | 1.9 ± 1.1 | 5.5 ± 1.2* | [61] |
Значения представлены как среднее ± стандартное отклонение (n) или индивидуальные значения, если n < 3.
* Значительно отличается от носителей ЭГТ, p < 0,05.
Этническая принадлежность добровольцев была следующей:
Носители ЭГТ: четыре европеоида-американца, два афроамериканца, один азиат-американец; не носители ЭГТ: пять европеоидов-американцев. Данные об этнической принадлежности пациентов отсутствуют.
DCA: дихлорацетат.
Анализ данных исследований фармакокинетики ДХА у добровольцев показал, что, как и in vitro (см. ниже), у лиц, несущих вариант GSTZ1A, кодирующий KRT, на одном аллеле, начальный клиренс ДХА был более быстрым, чем у других. У этих людей также наблюдалось большее снижение клиренса после приема пяти доз. Отношение клиренса от первой до пятой дозы составило 3,6 ± 0,8 (среднее ± стандартная ошибка среднего, n = 5) у лиц с вариантом GSTZ1C (кодирующим EGT), но не GSTZ1A, и 18,2 ± 9,3 (n = 5) у лиц с вариантом GSTZ1A хотя бы по одному аллелю. У человека, гомозиготного по GSTZ1A, наблюдалось наибольшее изменение от первой до пятой дозы, при этом клиренс снизился с 16,7 до 0,31 мл/мин [55]. Разница в среднем изменении клиренса не была статистически значимой из-за высокой вариабельности в группе, гомо- или гетерозиготной по GSTZ1A, однако она указывает на тенденцию, которая согласуется с исследованиями in vitro восприимчивости различных гаплотипов к инактивации DCA.
Исследования метаболизма ДКАin vitro
Дальнейшее понимание роли генотипа в метаболизме ДКА было получено в результате исследований экспрессии и активности GSTZ1 в отношении ДКА в цитозоле и митохондриях печени человека.
Онтогенез активности
Исследования цитозоля печени человека, полученного из 230 образцов печени доноров в возрасте от 42 дня беременности до 84 лет, показали очень низкую или не обнаруживаемую экспрессию и активность GSTZ1 пренатально, а также в первые 2 недели жизни: затем активность и экспрессия постепенно возрастают [56]. Экспрессия и активность достигают уровня взрослых к 7 годам, однако была обнаружена значительная межиндивидуальная вариабельность. Измерение активности с ДКА и экспрессии белка GSTZ1 в этих образцах показало, что образцы от людей с вариантом GSTZ1A на одном аллеле имели более высокое соотношение активность/экспрессия, чем все остальные (Рисунок 4). Это согласуется с результатами экспрессии рекомбинантного человеческого GSTZ1A-1A, которые показали более высокую активность в отношении ДКА для этого варианта [54], и с результатами фармакокинетических исследований на добровольцах, где у людей с одним геном GSTZ1A наблюдался быстрый начальный клиренс ДКА [55]. Хотя исследование развития показало, что активность у детей в возрасте до 7 лет была ниже, чем у взрослых, учет большего размера печени детей по отношению к массе тела означает, что на килограмм массы тела активность ДКА сходна у взрослых и детей в возрасте от 2 месяцев до 7 лет [56].
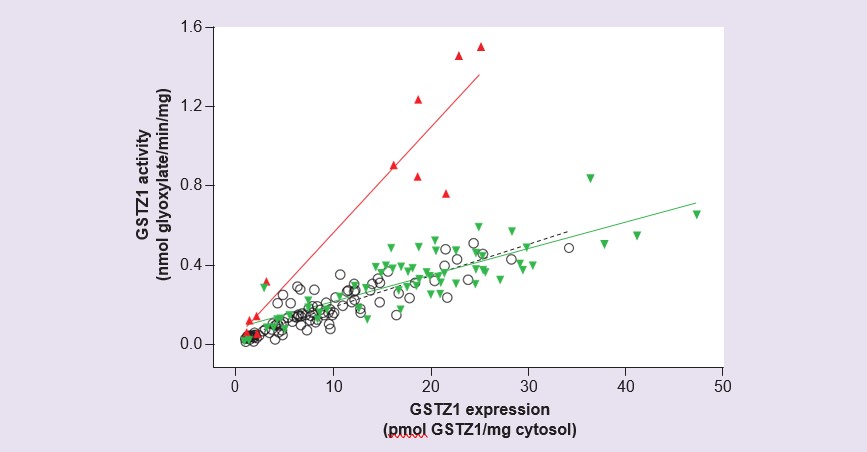
Влияние хлорида на инактивацию GSTZ1 под действием DCA
По-видимому, в противоречие с наблюдением, что белок KRT связан с более быстрым метаболизмом DCA in vitro [56] и более быстрым выведением одной дозы DCA [55], вступает тот факт, что у людей с вариантом GSTZ1A на одном аллеле наблюдается большее снижение клиренса после повторных доз DCA, чем у гомозиготных по GSTZ1C. Недавние данные о влиянии хлорида на инактивацию GSTZ1-1 могут объяснить эти расхождения. Было установлено, что хлорид защищает GSTZ1-1 от инактивации DCA в физиологически значимых концентрациях в печени [62,63], однако GSTZ1-1 в цитозоле печени у гетерозиготных по GSTZ1A был менее защищен от инактивации физиологическим (38 мМ) хлоридом, чем у пациентов с другими аллельными вариантами GSTZ1 (Таблица 4). Эффект хлорида был таков, что ферменты GSTZ1-1 в печени людей с вариантом GSTZ1A на одном аллеле инактивировались в два раза быстрее, чем у людей с другими гаплотипами. Точный механизм действия хлорида еще предстоит выяснить, но этот феномен является одним из объяснений более значительного снижения клиренса ДКА после хронического приема у лиц с одной или несколькими копиями варианта GSTZ1A. Как отмечалось выше, более низкая экспрессия фермента у лиц кавказской национальности с лизином (K) в положении 32 белка, вероятно, способствует очень низкому клиренсу.
.
| Гаплотип | EC50 мМ | Период полураспада инактивации, ч без Cl- | Период полураспада инактивации, ч с 38 мМ Cl- |
|---|---|---|---|
| EGT/EGT | 15 ± 3.1 (3) | 0.53, 0.49 | 5.73, 5.02 |
| КРТ/ЭГТ | 36 ± 2.2 (3) | 0.38, 0.38 | 2.66, 2.37 |
| ЭГМ/ЭГМ | 16.9 | — | 5.55 |
EC50 — концентрация хлорида, которая защищала половину цитозольного GSTZ1 от инактивации после инкубации в течение 2 ч с Na DCA, 0,5 мМ.
Заключение
Благодаря своей способности ингибировать киназу PDH, DCA может применяться для лечения рака и метаболических заболеваний. При хроническом введении ДКА людям его метаболизм и выведение из организма снижаются. Причина такого снижения клиренса после приема нескольких доз ДКА заключается в том, что единственный известный фермент, метаболизирующий ДКА, GSTZ1-1, инактивируется в процессе метаболизма ДКА. Такое ингибирование GSTZ1-1 под действием ДКА приводит к замедлению клиренса не только ДКА, но и эндогенных субстратов МАО и МА; вопрос о том, является ли накопление ДКА и/или реактивных катаболитов тирозина причиной обратимой периферической нейропатии, возникающей у некоторых пациентов, остается нерешенным. Другой нерешенной возможной причиной нейропатии является умеренное увеличение ∂-ALA, которое следует за хроническим лечением ДКА. Для понимания фармакокинетики ДКА важно то, что ген GSTZ1 имеет полиморфные варианты во всех изученных на сегодняшний день популяциях людей. Свойства ферментных белков вариантов GSTZ1-1 отличаются в отношении их способности метаболизировать ДКА и эндогенные субстраты МАА и МА, особенно при хроническом приеме. Хотя все варианты GSTZ1-1 инактивируются ДКА, один вариант, имеющий лизин вместо глутаминовой кислоты в положении 32 и аргинин вместо глицина в положении 42 белка, быстрее инактивируется, чем другие варианты, в присутствии физиологических концентраций хлорида. У кавказцев, но не у лиц африканской национальности, варианты GSTZ1, имеющие лизин в положении 32, связаны с SNP промоторной области в гене GSTZ1, что приводит к снижению экспрессии белка GSTZ1-1; это также может проявляться как фенотип медленного метаболизма. Понимание факторов, влияющих на фармакокинетику DCA при хроническом применении, необходимо для выбора правильной дозы и интервала дозирования, чтобы безопасно и эффективно назначать этот препарат пациентам.
Перспективы на будущее
Из проведенных на сегодняшний день исследований ясно, что гаплотип GSTZ1 является важным фактором, определяющим клиренс ДКА. Более глубокое понимание регуляции фермента в физиологических условиях и в присутствии ДКА приведет к более безопасному и эффективному применению препарата в будущем. Остается изучить несколько вопросов. У гетерозигот предпочтительно экспрессируется один из вариантов белка? Исследования активности, показавшие более высокую активность в отношении DCA для цитозольных фракций печени людей, гетерозиготных по GSTZ1A/GSTZ1C, позволяют предположить, что у гетерозигот предпочтительно экспрессируется вариант белка фермента 1A (KRT), но это не было доказано. Этот вопрос имеет практическое значение, поскольку вариант 1А инактивируется быстрее, чем вариант 1С. Следует ли назначать более низкую дозу ДКА лицам, у которых при хроническом приеме ДКА, скорее всего, будет выводиться медленнее, чем в среднем? Если да, то должны ли это быть все те, кто не гомозиготен или гетерозиготен по GSTZ1C? Следует ли кавказцам, имеющим хотя бы один К-содержащий вариант (KRT, KGT, KGM), назначать более низкие дозы ДКА, чем лицам африканской национальности, поскольку они экспрессируют меньше GSTZ1-1? Каковы доказательства низкой экспрессии K-содержащих вариантов в других популяциях? Существуют ли популяционные различия в распределении основных гаплотипов? Одинаково ли влияние фармакогенетических различий у детей и взрослых? Дети, принимающие ДКА, обычно быстрее выводят препарат из организма, чем взрослые, но возможные фармакогенетические эффекты не были детально изучены. Для решения этих вопросов необходимы дальнейшие исследования, чтобы обеспечить безопасное и эффективное дозирование ДКА.
Раскрытие финансовых и конкурирующих интересов
Работа авторов, обсуждаемая в данной рукописи, частично финансировалась Службой здравоохранения США 1RO1 GM 099871, частично — Банком мозга и тканей для лечения нарушений развития, Университетом Мэриленда, Балтимор и Университетом Майами (NO1 HD90011), и частично — грантом NIH/NCATS Clinical and Translational Science Award Университету Флориды UL1 TR000064. Авторы не имеют других соответствующих связей или финансового участия с какой-либо организацией или структурой, имеющей финансовый интерес или финансовый конфликт с предметом или материалами, обсуждаемыми в рукописи, кроме тех, которые были раскрыты.
При подготовке данной рукописи не использовалась помощь авторов.
Резюме
- Подбор эффективной, но нетоксичной дозы дихлорацетата (ДХА) важен для долгосрочного клинического применения ДХА в лечении рака и других метаболических заболеваний.
- Ген единственного фермента, метаболизирующего этот препарат, GSTZ1-1, характеризуется полиморфизмами, которые приводят к фенотипам медленного и быстрого метаболизма после длительного лечения ДХА.
- SNPs кодирующей области в гене GSTZ1 приводят к появлению пяти общих гаплотипов с различной частотой в популяциях. Люди, гомозиготные по самому распространенному из них, GSTZ1C, являются быстрыми метаболизаторами. Есть основания полагать, что гаплотипы, кодирующие белки GSTZ1-1 с K в положении 32, будут медленными метаболизаторами, однако другие факторы, способствующие медленному метаболизму после многократных доз ДКА, до конца не изучены.
- SNP промоторной области у людей кавказской, но не африканской национальности связан с SNP кодирующей области, который приводит к замене лизина на глутаминовую кислоту в положении 32 белка GSTZ1, и связан со снижением экспрессии белка фермента в печени.
- В присутствии DCA стабильность фермента GSTZ1-1 изменяется в зависимости от концентрации хлорида. Фермент варианта GSTZ1A (KRT) инактивируется быстрее, чем обычный вариант GSTZ1C (EGT) при физиологических концентрациях хлорида в печени.
ССЫЛКИ
1 Stacpoole PW. Обзор фармакологических и терапевтических эффектов диизопропиламмония дихлорацетата (DIPA). J. Clin. Pharmacol. J. New Drugs 9(5), 282-291 (1969).2 Stacpoole PW. Фармакология дихлорацетата. Метаболизм 38(11), 1124-1144 (1989).
3 Stacpoole PW, Felts JM. Диизопропиламмоний дихлорацетат (DIPA) и дихлорацетат натрия (DCA): влияние на метаболизм глюкозы и жира в нормальной и диабетической ткани. Метаболизм 19(1), 71-78 (1970).
4 Stacpoole PW, Moore GW, Kornhauser DM. Метаболические эффекты дихлорацетата у пациентов с сахарным диабетом и гиперлипопротеинемией. N. Engl. J. Med. 298(10), 526-530 (1978).
5 Stacpoole PW, Harwood HJ Jr, Varnado CE. Регулирование гидроксиметилглутарил коэнзим А редуктазы печени крысы новым классом неконкурентных ингибиторов. Влияние дихлорацетата и родственных карбоновых кислот на активность фермента. J. Clin. Invest. 72(5), 1575-1585 (1983).
6 Harwood HJ Jr, Bridge DM, Stacpoole PW. In vivo regulation of human mononuclear leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase. Исследования у нормальных людей. J. Clin. Invest. 79(4), 1125-1132 (1987).
7 Moore GW, Swift LL, Rabinowitz D, Crofford OB, Oates JA, Stacpoole PW. Снижение уровня холестерина в сыворотке крови у двух пациентов с гомозиготной семейной гиперхолестеринемией с помощью дихлорацетата. Atherosclerosis 33(3), 285-293 (1979).
8 Eichner HL, Stacpoole PW, Forsham PH. Лечение стрептозотоцинового диабета диизопропиламмоний дихлорацетатом (DIPA). Diabetes 23(3), 179-182 (1974).
9 Backshear PJ, Holloway PA, Alberti KG. Метаболические взаимодействия дихлорацетата и инсулина при экспериментальном диабетическом кетоацидозе. Биохим. J. 146(2), 447-456 (1975).
10 Whitehouse S, Randle PJ. Активация пируватдегидрогеназы в перфузированном сердце крысы дихлорацетатом (краткое сообщение). Biochem. J. 134(2), 651-653 (1973).
11 Whitehouse S, Cooper RH, Randle PJ. Механизм активации пируватдегидрогеназы дихлорацетатом и другими галогенированными карбоновыми кислотами. Биохим. J. 141(3), 761-774 (1974).
12 Патель МС, Корочкина Л.Г. Регуляция пируватдегидрогеназного комплекса. Biochem. Soc. Trans. 34(Pt 2), 217-222 (2006).
13 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Доказательства существования тканеспецифической регуляции пируватдегидрогеназного комплекса млекопитающих. Biochem. J. 329(Pt 1), 191-196 (1998).
14 Morten KJ, Caky M, Matthews PM. Стабилизация субъединицы пируватдегидрогеназы Е1альфа дихлорацетатом. Неврология 51(5), 1331-1335 (1998).
15 Han Z, Berendzen K, Zhong L et al. Комбинированный терапевтический подход к дефициту пируватдегидрогеназы с использованием самокомплементарных серотип-специфических векторов адено-ассоциированного вируса и дихлорацетата. Мол. Genet. Metab. 93(4), 381-387 (2008).
16 Evans OB, Stacpoole PW. Длительная гиполактатемия и повышение общей активности пируватдегидрогеназы под действием дихлорацетата. Биохим. Pharmacol. 31(7), 1295-1300 (1982).
17 Denton RM, Randle PJ, Bridges BJ et al. Regulation of mammalian pyruvate dehydrogenase. Мол. Cell. Biochem. 9(1), 27-53 (1975).
18 Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284(5), E855-E862 (2003).
19 Stacpoole PW, Nagaraja NV, Hutson AD. Эффективность дихлорацетата как препарата, снижающего лактат. J. Clin. Pharmacol. 43(7), 683-691 (2003).
20 Kankotia S, Stacpoole PW. Дихлорацетат и рак: новый дом для сиротского препарата? Biochim. Biophys. Acta 1846(2), 617-629 (2014).
21 Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine — both or neither? Environ. Health Perspect. 119(2), 155-158 (2011).
22 Chu PI. Фармакокинетика дихлорацетата натрия. Докторская диссертация, факультет фармацевтики, Университет Флориды, штат Флорида, США (1987).
23 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Фармакокинетика, метаболизм и токсикология дихлорацетата. Drug Metab. Rev. 30(3), 499-539 (1998).
24 Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin. Pharmacol. Ther. 37(1), 89-93 (1985).
25 Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G et al. Pharmacokinetics of oral dichloroacetate in dogs. J. Biochem. Mol. Toxicol. 27(12), 522-525 (2013).
26 Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 19(2), 109-113 (1980).
27 Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI. Лечение молочнокислого ацидоза дихлорацетатом. N. Engl. J. Med. 309(7), 390-396 (1983).
28 Henderson GN, Curry SH, Derendorf H, Wright EC, Stacpoole PW. Фармакокинетика дихлорацетата у взрослых пациентов с молочнокислым ацидозом. J. Clin. Pharmacol. 37(5), 416-425 (1997).
29 Stacpoole PW, Kerr DS, Barnes C и др. Контролируемое клиническое исследование дихлорацетата для лечения врожденного молочнокислого ацидоза у детей. Pediatrics 117(5), 1519-1531 (2006).
30 Stacpoole PW, Gilbert LR, Neiberger RE et al. Оценка долгосрочного лечения детей с врожденным молочнокислым ацидозом дихлорацетатом. Pediatrics 121(5), e1223-e1228 (2008).
31 Kaufmann P, Engelstad K, Wei Y et al. Дихлорацетат вызывает токсическую нейропатию при MELAS: рандомизированное контролируемое клиническое исследование. Неврология 66(3), 324-330 (2006).
32 Stickler DE, Valenstein E, Neiberger RE et al. Периферическая нейропатия при генетических митохондриальных заболеваниях. Pediatr. Neurol. 34(2), 127-131 (2006).r
33 Kaufmann P, Pascual JM, Anziska Y et al. Аномалии нервной проводимости у пациентов с MELAS и мутацией A3243G. Арх. Neurol. 63(5), 746-748 (2006).
34 Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Кинетика и метаболизм дихлорацетата в зависимости от возраста: возможное отношение к токсичности. J. Pharmacol. Exp. Ther. 324(3), 1163-1171 (2008).
35 James MO, Yan Z, Cornett R et al. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Идентификация глициновых конъюгатов, включая гиппурат, в качестве мочевых метаболитов дихлорацетата. Drug Metab. Dispos. 26(11), 1134-1143 (1998).
36 Rowland A, Miners JO, Mackenzie PI. UDP-глюкуронозилтрансферазы: их роль в метаболизме и детоксикации лекарств. Int. J. Biochem. Cell. Biol. 45(6), 1121-1132 (2013).
37 Vessey DA, Kelley M, Warren RS. Характеристика КоА-лигаз митохондрий печени человека, катализирующих активацию коротко- и среднецепочечных жирных кислот и ксенобиотических карбоновых кислот. Biochim. Biophys. Acta 1428(2-3), 455-462 (1999).
38 James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Глутатион-зависимое преобразование в глиоксилат, основной путь биотрансформации дихлорацетата в печеночном цитозоле человека и крыс, снижается у крыс, обработанных дихлорацетатом. Drug Metab. Dispos. 25(11), 1223-1227 (1997).
39 Tong Z, Board PG, Anders MW. Глутатионтрансфераза дзета катализирует окисление канцерогена дихлоруксусной кислоты до глиоксиловой кислоты. Biochem. J. 331(Pt 2), 371-374 (1998).
49 Fernandez-Canon JM, Penalva MA. Характеристика гена грибковой малеилацетоацетат изомеразы и идентификация ее человеческого гомолога. J. Biol. Chem. 273(1), 329-337 (1998).
41 Blackburn AC, Woollatt E, Sutherland GR, Board PG. Характеристика и хромосомное расположение гена GSTZ1, кодирующего человеческую глутатионтрансферазу класса Zeta и малеилацетоацетатную изомеразу. Цитогенет. Cell. Genet. 83(1-2), 109-114 (1998).
42 Li W, James MO, Mckenzie SC, Calcutt NA, Liu C, Stacpoole PW. Митохондрия как новое место биотрансформации дихлорацетата глутатионтрансферазой дзета 1. J. Pharmacol. Exp. Ther. 336(1), 87-94 (2011).
43 Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Иммуногистохимическая локализация и активность глутатионтрансферазы дзета (GSTZ1-1) в тканях крыс. Drug Metab. Dispos. 30(6), 616-625 (2002).
44 Shangraw RE, Fisher DM. Фармакокинетика дихлорацетата у пациентов, перенесших трансплантацию печени. Анестезиология 84(4), 851-858 (1996).
45 Cornett R, James MO, Henderson GN, Cheung J, Shroads AL, Stacpoole PW. Ингибирование глутатион S-трансферазы дзета и метаболизма тирозина дихлорацетатом: потенциальный объединяющий механизм его измененной биотрансформации и токсичности. Биохим. Biophys. Res. Commun. 262(3), 752-756 (1999).
46 Guo X, Dixit V, Liu H et al. Ингибирование и восстановление дзета-трансферазы глутатион S-трансферазы печени крыс и изменение метаболизма тирозина после воздействия дихлорацетата и его отмены. Drug Metab. Dispos. 34(1), 36-42 (2006).
47 Anderson WB, Board PG, Gargano B, Anders MW. Инактивация глутатионтрансферазы дзета дихлоруксусной кислотой и другими альфа-галоалкановыми кислотами, не содержащими фтор. Хим. Res. Toxicol. 12(12), 1144-1149 (1999).
48 Anderson WB, Liebler DC, Board PG, Anders MW. Масс-спектральная характеристика модифицированной дихлоруксусной кислотой человеческой глутатионтрансферазы дзета. Chem. Res. Toxicol. 15(11), 1387-1397 (2002).
49 Диксит В. Инактивация глутатионтрансферазы дзета дихлоруксусной кислотой. Докторская диссертация, кафедра медицинской химии, Университет Флориды, штат Флорида, США (2005).
50 Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. Группа по изучению дихлорацетата при молочнокислом ацидозе. N. Engl. J. Med. 327(22), 1564-1569 (1992).
51 Abdelmalak M, Lew A, Ramezani R et al. Долгосрочная безопасность дихлорацетата при врожденном молочнокислом ацидозе. Mol. Genet. Metab. 109(2), 139-143 (2013).
52 Board PG, Baker RT, Chelvanayagam G, Jermiin LS. Zeta, новый класс глутатионтрансфераз в ряде видов от растений до человека. Biochem. J. 328(Pt 3), 929-935 (1997).
53 Blackburn AC, Tzeng HF, Anders MW, Board PG. Обнаружение функционального полиморфизма в глутатионтрансферазе зета человека с помощью анализа базы данных тегов выраженной последовательности. Фармакогенетика 10(1), 49-57 (2000).
54 Blackburn AC, Coggan M, Tzeng HF et al. GSTZ1d: новый аллель глутатионтрансферазы дзета и малеилацетоацетатной изомеразы. Фармакогенетика 11(8), 671-678 (2001).
55 Shroads AL, Langaee T, Coats BS et al. Полиморфизмы человека в гене глутатионтрансферазы дзета 1/малеилацетоацетатной изомеразы влияют на токсикокинетику дихлорацетата. J. Clin. Pharmacol. 52(6), 837-849 (2012).
56 Li W, Gu Y, James MO et al. Пренатальная и постнатальная экспрессия глутатионтрансферазы дзета 1 в печени человека и роль гаплотипа и возраста субъекта в определении активности в отношении дихлорацетата. Drug Metab. Dispos. 40(2), 232-239 (2012).Google Scholar
57 Board PG, Anders MW. Глутатионтрансфераза дзета: открытие, полиморфные варианты, катализ, инактивация и свойства Gstz1-/- мышей. Drug Metab. Rev 43(2), 215-225 (2011).
58 Polekhina G, Board PG, Blackburn AC, Parker MW. Crystal structure of maleylacetoacetate isomerase/glutathione transferase zeta reveals the molecular basis for its remarkable catalytic promiscuity. Биохимия 40(6), 1567-1576 (2001).
59 Fang YY, Kashkarov U, Anders MW, Board PG. Полиморфизмы в промоторе дзета глутатионтрансферазы человека. Фармакогенет. Геномика 16(5), 307-313 (2006).
60 Langaee TY, Zhong G, Li W et al. Влияние вариаций гаплотипов гена GSTZ1 человека на экспрессию GSTZ1. Pharmacogenet Genomics 25(5), 239-245 (2015).
61 Shroads AL, Coats BS, Mcdonough CW, Langaee T, Stacpoole PW. Вариации гаплотипов глутатионтрансферазы дзета 1 влияют на кинетику и динамику хронического дихлорацетата у детей. J. Clin. Pharmacol. 55(1), 50-55 (2015).
62 Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Chloride and other anions inhibit dichloroacetate-induced inactivation of human liver GSTZ1 in a haplotype-dependent manner. Хим. Биол. Interact. 215C, 33-39 (2014).
63 Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Концентрация хлоридов в цитозоле и митохондриях печени человека зависит от возраста. Биохим. Biophys. Res. Commun. 459(3), 463-468 (2015).